
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidation of solid thin films of neonicotinoid pesticides by gas phase hydroxyl radicals†
B. J.
Finlayson-Pitts‡
 *,
A.
Anderson‡
,
P. S. J.
Lakey
,
W.
Wang
,
M. J.
Ezell
,
X.
Wang
,
L. M.
Wingen
,
V.
Perraud
and
M.
Shiraiwa
*
*,
A.
Anderson‡
,
P. S. J.
Lakey
,
W.
Wang
,
M. J.
Ezell
,
X.
Wang
,
L. M.
Wingen
,
V.
Perraud
and
M.
Shiraiwa
*
Department of Chemistry, University of California Irvine, Irvine, California 92697-2025, USA. E-mail: bjfinlay@uci.edu; m.shiraiwa@uci.edu; Fax: +1-949-824-2420; Tel: +1-949-824-7670
First published on 24th November 2022
Abstract
Neonicotinoids (NNs) are commonly found throughout the environment on surfaces such as seeds, soil, vegetation, and blowing dust particles. However, there is a paucity of data on the kinetics and oxidation products formed on contact with the atmosphere which limits understanding of their potentially far-reaching impacts. In this study, in situ attenuated total reflectance (ATR) FTIR spectroscopy was used to investigate the OH oxidation of thin films of three solid NNs, imidacloprid (IMD), dinotefuran (DNF) and clothianidin (CLD) at 295 ± 3 K. The experimentally measured reaction probabilities based on initial rates of NN loss are (1.6 ± 0.8) × 10−2 for IMD, (1.5 ± 0.6) × 10−2 for DNF and (0.9 ± 0.2) × 10−2 for CLD (±1σ), suggesting initial NN lifetimes with respect to OH of 10–17 days. The kinetics were interpreted using a multiphase kinetics model, KM-SUB, which showed that the OH uptake and reaction occurred primarily in the surface layer. Products identified by mass spectrometry included carbonyl-, alcohol- and olefin-containing species formed via hydrogen abstraction from aliphatic C–H groups. Additionally, carbonyl-containing desnitro and urea derivative products were observed from secondary reactions of the initially formed photodegradation products. Reaction with OH will contribute to NN loss both during the day as well as at night when there are non-photolytic sources of this radical. Thus, OH reactions with both the parent neonicotinoid and its photodegradation products should be considered in assessing their environmental impacts.
Environmental significanceNeonicotinoids (NNs) are the most widely used insecticides globally and their use is increasingly implicated in negative impacts on ecosystem services, particularly pollinator health. However, their environmental fates and effects remain undetermined. Here, the reaction rate constants and mechanisms of thin films of three solid NNs with gas-phase OH radicals, a primary oxidant in air, were investigated. Our study shows that OH oxidation contributes to the loss of the parent NN over days, and to subsequent loss of their desnitro and urea photolysis products. This oxidation will also play a significant role in NN loss at night when photolysis is not occurring but there are non-photolytic sources of OH. |
Introduction
Introduced in the 1990s, the use of neonicotinoid (NN) pesticides quickly grew due to their effectiveness across a broad range of insects, low toxicity to humans, and ability to be used as a seed coating.1–8 Their systemic nature allows for uptake and transport throughout a plant's vascular system, prophylactically protecting the plant from insects without the need for frequent treatment. Since 2003 NN use in the US has increased dramatically, particularly as a seed coating, an application type which is not always captured in traditional pesticide use surveys.9 As NN use has grown, however, so has increased concern about their negative impacts on ecosystem services, particularly pollinator health.7,10–18 Although the EU introduced a moratorium on three NNs (imidacloprid, clothianidin, and thiamethoxam) to protect honeybees,19 NNs remain a major part of the international and US agricultural system (although restrictions have been proposed recently by the California Department of Pesticide Regulation).20Due to their systemic nature and widespread use, both the parent NNs and their degradation products have accumulated in the environment; for example, studies conducted in Europe showed that NNs were still present in the soil even five years after the moratorium, with no clear declining trend.21 Globally, NNs have been found in 75% of honey samples,5 in non-target species further up the food chain such as birds,22,23 and perhaps not surprisingly then, are often detected in human biological samples.24–27 Therefore, NNs are a persistent threat to non-target species via continual exposure at sub-lethal concentrations.2,28
A major advantage of the NNs is that they have lower mammalian toxicity than previously used pesticides. While there is increasing concern regarding potential impacts on long-term exposure, there are at present relatively few data available on their health effects.25,28 However, there are some studies that suggest that at least some of the metabolites and oxidation products are significantly more toxic than the parent NN. For example, the binding of the desnitro-derivative of imidacloprid to the nicotinic acetylcholine receptor in mouse brain membranes, which is related to toxicity, is much stronger than that of imidacloprid itself.29
Of crucial importance in understanding the environmental impact of NNs is identifying the environmental degradation products so that a complete toxicity map on non-target species, including humans, can be assessed. An abundance of work is available on the reactions and fate of NNs in aqueous systems.2–4,26,30–41 In addition, previous studies from this lab have reported the heterogeneous photolysis of solid NNs42–45 as would be the case for example for NNs on leaf, soil, and dust surfaces, and the oxidation of the NN nitenpyram by gas phase ozone.46 However, little is known about the heterogeneous reactions of solid NNs with the hydroxyl (OH) radical, which is a major atmospheric oxidant that is ubiquitous in air.
Previous work on aqueous phase oxidation of NNs has largely focused on imidacloprid (IMD), the most widely used NN. These studies have applied a variety of oxidation techniques, in which OH radicals were generated from chemical reactions involving O3, H2O2, and photolysis,33–35,40 photo-Fenton reactions,38,39,41,47–50 and heterogeneous photocatalysis.36–38,41,51–54 While these studies have documented mineralization of IMD for removal of the NN from wastewater, the oxidation methods chosen were often not solely due to OH but rather to a combination of processes such as photolysis and catalysis. As a result, extrapolation of the kinetics, products and mechanisms to the reactions of NNs as a solid on environmental surfaces is fraught with uncertainty.
This work provides kinetics and products data on the heterogeneous reactions of three structurally unique solid-phase NNs, imidacloprid (IMD), dinotefuran (DNF), and clothianidin (CLD), with gas phase OH, a major atmospheric oxidant.55 While these three NNs are all nitroguanidines, they have a variety of C–H bonds, amino groups, and ring structures:

These three NNs are of interest as they are widely used, with relative amounts of each varying by location. For example, in Japan, dinotefuran has historically had the highest use, followed by imidacloprid and clothianidin, while in California, imidacloprid is the major NN used, with smaller amounts of clothianidin and dinotefuran.1 Similarly, they are found in a variety of environmental media worldwide in varying proportions, including rivers and streams,4 pollen and food such as honey.2,5 From the kinetics data, atmospheric lifetimes with respect to the OH reaction are estimated and these, along with the products and proposed mechanisms, provide a guide for future toxicity assessments of NNs in the environment.
Experimental
NN thin film preparation
The neonicotinoids used were IMD (Sigma-Aldrich, PESTANAL™ ≥ 98% purity), DNF (ChemService, 99.5% purity), and CLD (ChemService, 99.5% purity). 20 mM solutions of each were prepared in acetonitrile (Fisher Chemical, HPLC-grade). An aliquot of the solution was pipetted onto the surface of a ZnSe ATR (attenuated total reflectance) crystal and dried under a stream of O2, forming a thin film as the solvent evaporated. The initial amount of NN on the ATR crystal ranged from (0.6–3.5) × 1017 molecules. The ATR crystal (Pike Technologies) is trapezoidal shaped with dimensions of 80 mm (l) × 10 mm (w) × 4 mm (d), a 45° entrance angle, and 10 internal reflections. The calculated depth of penetration56 (dp) is 0.72–0.74 μm at 1615–1560 cm−1 where the NNs were monitored.The number of monolayers (ML) on the ATR crystal was calculated from the thickness and the surface area per molecule estimated using the volume per molecule derived from the densities (1.54 g cm−3 for IMD, 1.40 g cm−3 for DNF and 1.61 g cm−3 for CLD) and molecular masses of the NNs (255.7 g mol−1 for IMD, 202.2 g mol−1 for DNF and 249.7 g mol−1 for CLD). Assuming the average dimension was the same for all three directions, the average thickness of one molecule was the cube root of its volume and calculated to be ∼0.65–0.62 nm with ∼(2.4–2.6) × 1014 molecules cm−2 in one monolayer. This is similar to the surface concentrations that are calculated based on the crystal structures for IMD and DNF.57 From the total number of molecules on the crystal, the film thicknesses and number of monolayers were estimated to be 0.04–0.22 μm and 60–350 ML. All film thicknesses were less than the depth of penetration, assuring the entire film was probed by ATR-FTIR. The ATR crystal was mounted in a custom reaction cell58 (Fig. S1†) and held in a stream of O2 carrier gas for 15 minutes in the dark before reaction.
Kinetics studies of OH uptake on NN thin films
The custom reaction cell (Fig. S1†) contains gas inlets and outlets, an ATR crystal, a horizontally mounted quartz window on top, and zinc selenide end windows (ZnSe, Edmund Optics, 10 mm diameter × 1.5 mm thickness) for probing the gas phase above the crystal.45,58 Photolysis of isopropyl nitrite (IPN, Karl Industries, Ohio) was used to generate OH via a series of reactions ((R1)–(R3)) described in detail elsewhere.58,59| (CH3)2CHONO + hν → (CH3)2CHO˙ + NO | (R1) |
| (CH3)2CHO˙ + O2 → CH3C(O)CH3 + HO2 | (R2) |
| HO2 + NO → OH + NO2 | (R3) |
During the experiment, a high pressure Xe lamp (Ushio 300 W bulb, UXL-302-O) mounted in a housing with a power source (Newport model 69911) was used to photolyze the flow of IPN in the cell. The UV light beam first passed through a water filter to absorb infrared/heat and was directed at an angle onto the quartz window via a reflecting mirror. A 360 nm optical filter (Newport longpass colored glass alternative filter, 20CGA-360) was placed on top of the quartz window to remove light below 360 nm in order to reduce the photolysis of the NN in this region. All experiments were carried out at 295 ± 3 K.
The flow of O2 (AirGas, Ultrahigh purity, >99.993%) was controlled at 500 mL min−1 using a mass flow controller (Alicat, Model # MC500-SCCM-D/5M) and could either flow directly into the cell, or be routed over the headspace of a trap containing liquid IPN at 263 K. The gas phase IPN concentration was measured before and after each experiment using infrared spectroscopy (Mattson Galaxy 5020 FTIR) in a separate 10 cm gas cell with 16 coadded scans at a resolution of 0.5 cm−1. An absorption cross section of 1.61 × 10−18 cm2 molecule−1 (base e) at 1664 cm−1 was used,59 giving IPN concentrations ranging from (1.5–2.8) × 1016 molecules cm−3. The concentration of OH was measured by adding 13CO with an IPN/O2 mixture into the cell and monitoring formation of 13CO2 (Fig. S2†) from the reaction of 13CO with OH as a function of time under static conditions using the same reaction cell (Fig. S1†) in the transmission IR mode. Details of the OH measurement are described in the ESI.† The average steady state OH concentration was determined to be (8.2 ± 2.0) × 108 molecule cm−3.
The loss of NN and formation of surface-bound products were followed using ATR-FTIR with 16 coadded scans at a resolution of 0.5 cm−1. FTIR spectra were recorded before irradiation and at 20 or 30 minute intervals during the irradiation while IPN was continuously flowing over the NN films. Absorbance data were converted to a total number of NN molecules on the ATR crystal using calibrations generated by adding a known number of molecules (10 μL aliquots of 4–40 mM solutions) to the ATR crystal and recording the absorbance of the thin films after evaporation of the solvent.
After the reaction, the ATR crystal was removed from the holder, and the reacted film was analyzed via mass spectrometry, either directly on the ATR crystal by DART-MS or by UPLC-HESI-HRMS on extracted samples, as described below.
Product analysis by mass spectrometry
Products were identified using two approaches: (1) ultra-performance liquid chromatography coupled to an Orbitrap high resolution mass spectrometer system with heated electrospray ionization (UPLC-HESI-HRMS); and (2) a direct analysis in real time ambient ionization source interfaced to a triple quadrupole mass spectrometer (DART-MS).Kinetic modeling
The KM-SUB model has previously been described in detail and has been adapted for the current study to treat the mass transport and chemical reactions of OH, NNs and reaction products.60 The model consists of a number of layers: gas phase, near-surface gas phase, sorption layer, quasi-static surface layer, near-surface bulk, and a number of bulk layers. Mass transport processes included in the model are gas-phase diffusion, the reversible adsorption of OH to the surface, the reversible partitioning of OH into the bulk, and the Fickian diffusion of all species throughout the bulk. Three reactions ((R4)–(R6)) are treated at the interface and in the bulk as follows:| NN + OH → Products1-OH | (R4) |
| NN + hν → Productshν | (R5) |
| Productshν + OH → Products2-OH | (R6) |
The ensemble of products from the initial NN-OH reaction is designated Products1-OH, and that from the reactions of the photolysis products Products2-OH. Parameters used in the model are summarized in Table S1† and include the gas-diffusion coefficient, the boundary layer thickness adjacent to the surface, the surface accommodation coefficient of OH, the desorption lifetime of OH, the partitioning coefficient of OH, the bulk diffusion coefficient of OH, NNs and products as well as the surface and bulk reaction rate coefficients of reactions (R4)–(R6). Parameters were determined based on literature values and by fitting to the measurements. Explanations of the final parameter values are summarized in Table S1.† Rapid OH surface uptake and slow gas diffusion may cause depletion of OH concentrations in the near-surface gas phase. The true uptake coefficient is calculated as the ratio of net uptake and gas collision from near-surface NN bulk to the surface using modeled near-surface gas-phase concentrations, while the effective uptake coefficient is calculated as the ratio of net uptake and gas collision from the gas phase using the average gas-phase concentration determined in the experiments.
Results
Kinetics of oxidation
Two approaches were taken to define the oxidation kinetics. The first involved monitoring the loss with time of the NN from thin films. The second was application of a multiphase kinetics model to elucidate the contributions of diffusion and reactions as a function of time and depth in the film.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) S0/St, where S0 is the single beam of the initial NN and St is the single beam at the selected reaction time. Negative peaks indicate reactant loss and positive peaks indicate product formation. For all three NNs, absorption bands due to product formation are observed in the ∼1600–1800 region characteristic of C
S0/St, where S0 is the single beam of the initial NN and St is the single beam at the selected reaction time. Negative peaks indicate reactant loss and positive peaks indicate product formation. For all three NNs, absorption bands due to product formation are observed in the ∼1600–1800 region characteristic of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (1850–1550 cm−1), as well as CN (1690–1620 cm−1) and CC (1680–1620 cm−1) groups.61 Losses of C–H (3000–2800 cm−1) and N–H (3500–3300 cm−1) stretches61 are also observed. In the case of IMD, there is a broad absorption in the 3000–3300 cm−1 region characteristic of a hydrogen-bonded –C(O)OH.
O (1850–1550 cm−1), as well as CN (1690–1620 cm−1) and CC (1680–1620 cm−1) groups.61 Losses of C–H (3000–2800 cm−1) and N–H (3500–3300 cm−1) stretches61 are also observed. In the case of IMD, there is a broad absorption in the 3000–3300 cm−1 region characteristic of a hydrogen-bonded –C(O)OH.
 | ||
| Fig. 1 FTIR spectra of IMD, DNF and CLD (grey) and difference spectra after reaction, which show logS0/St where S0 is the single beam of the NN before reaction and St is the single beam at the selected reaction time. The blue trace shows difference spectra after reaction with OH for ∼16 hours, yellow is after exposure to UV and pink is after exposure to IPN in the dark. Initial absorbance spectra have been scaled by a factor of 0.5. | ||
To test for the loss of the NN in the absence of OH, difference spectra were also acquired in control studies of flowing IPN/O2 in the dark or photolysis of NN in the absence of IPN. No significant changes were detected under the NN + IPN/O2 flow in the dark but some small loss and product formation were observed under NN + UV with the 360 nm filter (yellow traces, Fig. 1).
The loss of the –NO2 asymmetric stretch (Fig. 1) during the OH reaction and the photolysis only was followed to generate plots of the number of NN molecules cm−2 (NNt) as a function of time (Fig. 2). To estimate the initial rate of loss as t → 0 where the surface is only NN, the data were fit using a third order polynomial fit of the form NNt = at3 + bt2 + ct + d, where a, b, c and d were obtained from the best fit to each data set. The initial slope (R0 = d(NNt)/dt) was obtained from the derivative of this polynomial with respect to time evaluated at t → 0 (i.e., the coefficient c):
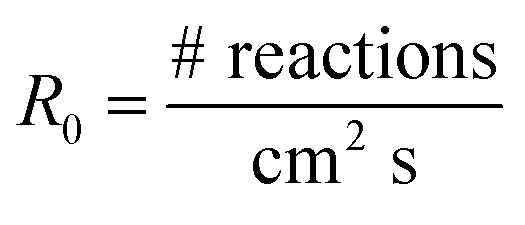 | (I) |
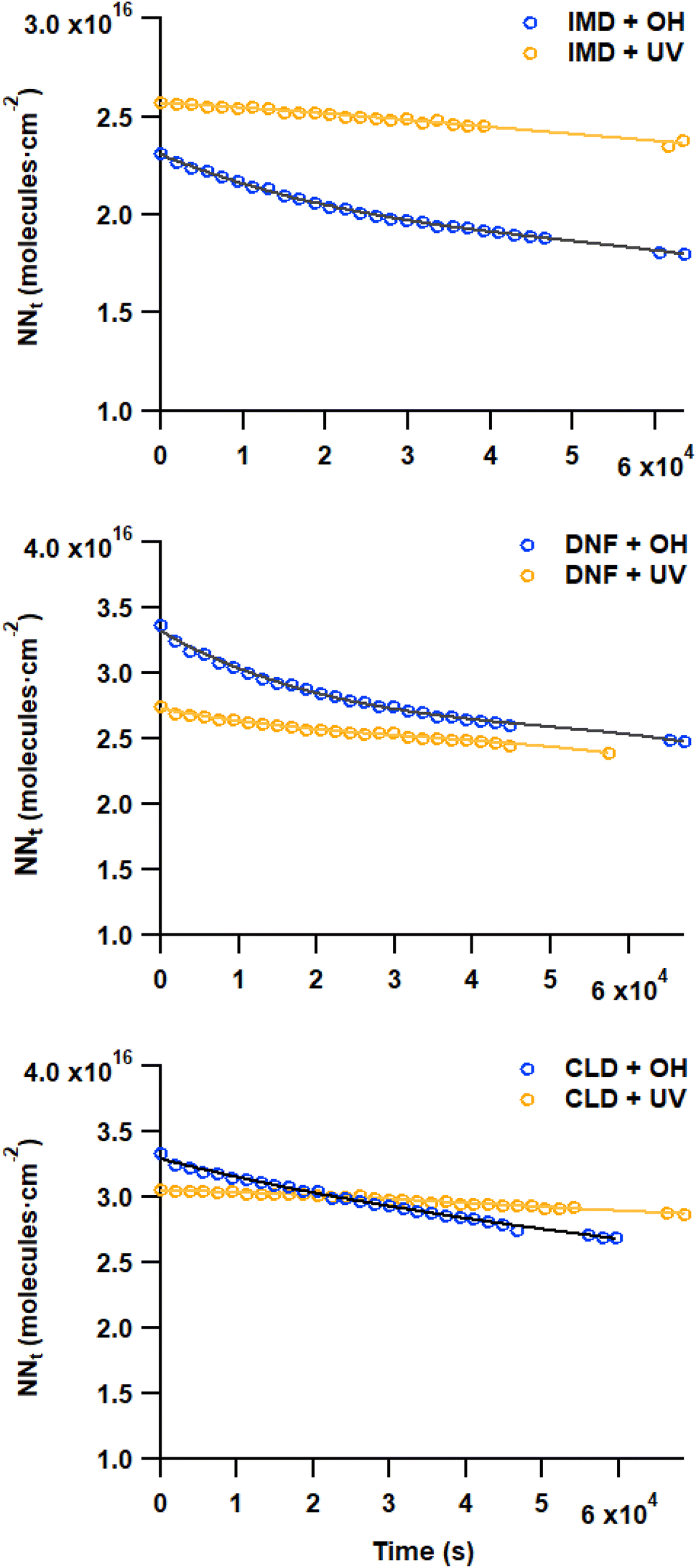 | ||
| Fig. 2 Typical loss of each NN using the peaks due to the –NO2 asymmetric stretch for IMD (1560 cm−1), DNF (1615 cm−1), and CLD (1615 cm−1) thin films during OH reaction (blue circles) and during photolysis in the absence of IPN/OH (orange circles). The slope as t → 0 provides the initial number of reactions cm−2 s−1 (R0). | ||
To determine the reaction probability (γ) for the NN + OH reaction, which is equal to the rate of loss of the NN normalized to the rate of gas-surface collisions of OH,55 the initial number of reactions cm−2 s−1 (R0), corrected for the loss due to photolysis in the absence of OH, is divided by the gas phase concentration of OH and the number of collisions per second from gas kinetic molecular theory:55,62
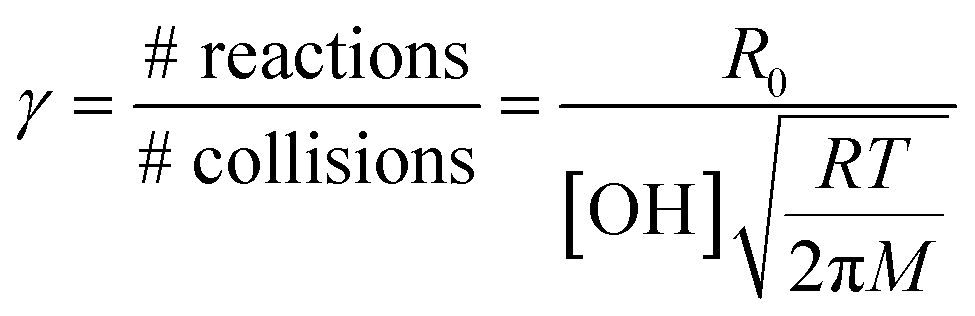 | (II) |
In eqn (II), [OH] is the steady state gas concentration of OH radicals (molecules cm−3), R is the gas constant, T is temperature (K) and M is molar mass of OH (g mol−1). As seen in Table 1, the average reaction probabilities (± one standard deviation) range from (0.9 ± 0.2) × 10−2 for CLD to (1.6 ± 0.8) × 10−2 for IMD. Note that these are likely lower limits to the initial reaction probabilities since the first data points are taken at 30 min. In addition, as discussed below, the multiphase kinetics modeling suggests that the reaction is sufficiently fast that the OH concentration in the near-surface gas phase is limited by gas diffusion so that the actual near-surface concentration is somewhat smaller than the measured bulk gas phase concentration. This also leads to these values being lower limits.
| NN (# experiments) | R 0 (initial # rxns cm−2 s−1 ± 1σ) | γ (±1σ) | γ ratio to IMD (±1σ) |
|---|---|---|---|
| a Corrected for the decay due to photolysis alone. | |||
| IMD (7) | (1.9 ± 1.0) × 1011 | (1.6 ± 0.8) × 10−2 | 1 |
| DNF (6) | (1.8 ± 0.7) × 1011 | (1.5 ± 0.6) × 10−2 | 0.94 ± 0.6 |
| CLD (4) | (1.1 ± 0.2) × 1011 | (0.9 ± 0.2) × 10−2 | 0.56 ± 0.15 |
Abstraction of hydrogens from C–H groups is expected to be the primary reaction step. Using known structure–activity relationships (SAR) from gas phase OH reactions with alkanes, relative reactivities for the hydrogen abstraction can be calculated for the three NNs,55,63,64 with the caveat that this assumes that the relative reactivities in the gas–solid reactions follow those in the gas phase. The SAR predict that 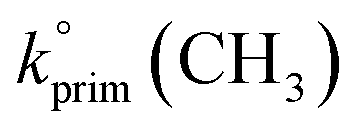 = 1.36 × 10−13 cm3 molecule−1 s−1,
= 1.36 × 10−13 cm3 molecule−1 s−1, 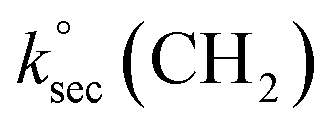 = 9.34 × 10−13 cm3 molecule−1 s−1 and
= 9.34 × 10−13 cm3 molecule−1 s−1 and 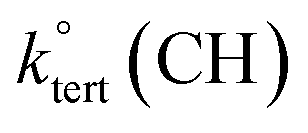 = 1.95 × 10−12 cm3 molecule−1 s−1 at 298 K, with neighboring group factors (F) for –CH2–, 〉CH– and 〉C〈 of 1.23, F(〉N–) of 10 and F(–O–) of 6.1. For a C–H in a 5-membered ring, F is 0.64.63,65 Using the sum of the reactivity of the aliphatic C–H groups predicts gas phase rate constants of 2.40 × 10−11, 2.50 × 10−11, and 1.07 × 10−11 cm3 molecule−1 s−1 for IMD, DNF, and CLD respectively. This gives ratios to IMD of 1.0 for DNF and 0.45 for CLD. These ratios for predicted gas phase reaction rate constants are within experimental uncertainties (1σ) of the experimentally measured ratios of the reaction probabilities of 0.94 ± 0.6 for DNF and 0.56 ± 0.15 for CLD (Table 1). This calculation ignores the potential reactivity of the N–H groups as there was no evidence from identified products (see below) for attack on the amino groups. The C–H groups in the aromatic ring structures of IMD and CLD were also not included as SAR predicts them to be less reactive, and no ring-opening products were observed.
= 1.95 × 10−12 cm3 molecule−1 s−1 at 298 K, with neighboring group factors (F) for –CH2–, 〉CH– and 〉C〈 of 1.23, F(〉N–) of 10 and F(–O–) of 6.1. For a C–H in a 5-membered ring, F is 0.64.63,65 Using the sum of the reactivity of the aliphatic C–H groups predicts gas phase rate constants of 2.40 × 10−11, 2.50 × 10−11, and 1.07 × 10−11 cm3 molecule−1 s−1 for IMD, DNF, and CLD respectively. This gives ratios to IMD of 1.0 for DNF and 0.45 for CLD. These ratios for predicted gas phase reaction rate constants are within experimental uncertainties (1σ) of the experimentally measured ratios of the reaction probabilities of 0.94 ± 0.6 for DNF and 0.56 ± 0.15 for CLD (Table 1). This calculation ignores the potential reactivity of the N–H groups as there was no evidence from identified products (see below) for attack on the amino groups. The C–H groups in the aromatic ring structures of IMD and CLD were also not included as SAR predicts them to be less reactive, and no ring-opening products were observed.
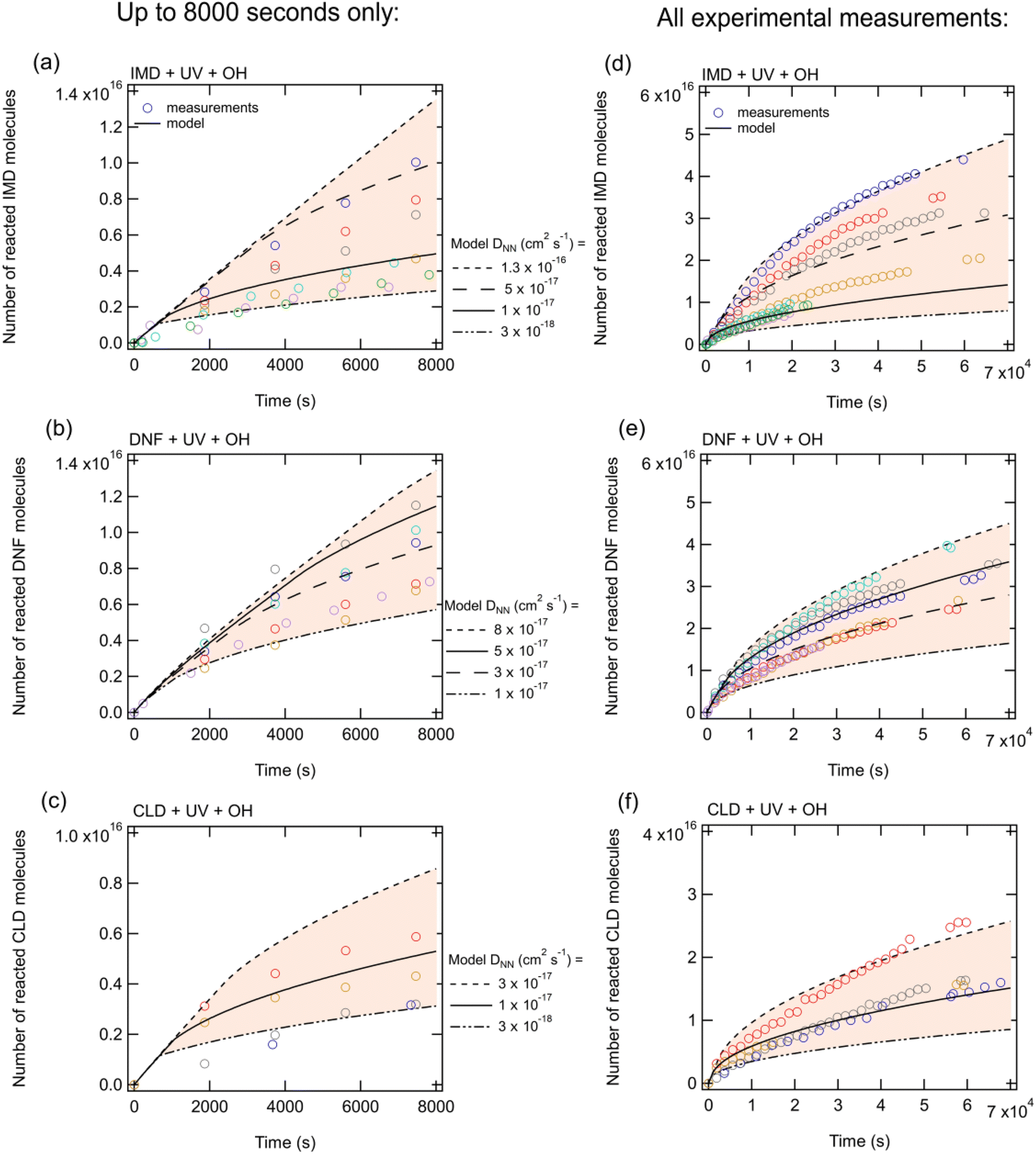 | ||
| Fig. 3 The number of neonicotinoid molecules reacted away in (a and d) IMD, (b and e) DNF and (c and f) CLD films which have been spread over a surface area of 4 cm2 in the presence of UV and an OH gas phase concentration of 8 × 108 molecules cm−3. Panels (a–c) show the NNs reacted away up to 8000 seconds while panels (d–f) show the entire measurement range. Experimental measurements are shown by markers and KM-SUB model output is shown by the lines. The different lines represent different model NN bulk diffusion coefficients while the shading shows the range that these lines encapsulate. | ||
Sensitivity simulations by varying each parameter (Table S1†) indicated that the most sensitive parameters include the NN surface rate coefficient due to photolysis in the absence of OH, the NN bulk diffusion coefficient and the boundary layer thickness. Gas-phase diffusion of OH radicals across a boundary layer limits the decay of NN at early times (approximately <1000 seconds) as it causes OH radical concentrations close to the surface to be significantly reduced compared to the bulk average gas-phase concentrations. Hydroxyl radical concentrations in the bulk of the solid NN are small and therefore the majority of the NN loss in the presence of OH occurs via reaction at the surface rather than in the bulk. This is seen by the common curves in Fig. 3 for the first ∼1000 s regardless of the diffusion coefficients of NN, DNN, in the bulk. NN loss at later times is limited by the rate at which NN can diffuse to the surface, and hence the curves in Fig. 3 diverge for different modelled NN diffusion coefficients. Our results are consistent with the study by Arangio et al.66 where the decay of levoglucosan was limited by bulk-to-surface diffusion of the organic under high OH concentrations.
There are a number of uncertainties that impact the comparison between the experimental data and model predictions. Inputs to the KM-SUB model assumed that the film was evenly distributed on the ATR crystal surface, i.e., had a uniform thickness, which is likely not to be the case. Measuring the OH concentration under the same conditions as the NN photolysis is also challenging, and we estimate the uncertainty in the OH concentration to be ±25%. Experimental measurements show that direct photolysis is not occurring throughout the entire bulk as this would lead to more molecules being lost for thicker films (Fig. S3†). (This is in contrast to earlier studies of the photolysis of thin films of the NN43–45 where the light source was different and irradiation was directly from above the film; in the present studies, the light beam from a different lamp was reflected from a mirror at an angle onto the film which was likely quite irregular at the surface.) Thus, it was assumed in the model that all of the photolytic loss occurred in the surface layer. However, it should be noted that even if the photolysis occurs throughout the entire bulk, the impact on the best fit diffusion coefficients of NN is relatively small. For example, the best fit to the data for this scenario was obtained by decreasing the NN bulk diffusion coefficient by approximately a factor of 1.5–3, and at the same time decreasing the photolysis rate coefficients in both the bulk and the surface by a factor of 200–400 compared to the values in Table S1.†
The scatter in the experimental data combined with different combinations in the model inputs that can provide a reasonable match to the data preclude assigning unique, accurate values to some model input parameters such as bulk diffusion coefficients. However, these uncertainties do not affect the overall conclusions and insights from the model that the reaction occurs mainly at the surface and is initially limited by gas-phase diffusion of OH to the surface and then limited by diffusion of NN from the bulk into the surface region.
Products and mechanisms of oxidation
The nature of the products and likely mechanisms responsible are similar for the three NNs. We thus describe the data in some detail for imidacloprid oxidation, with more abbreviated summaries for DNF and CLD that have similar products and reaction paths.N–NO2 chromophore, resulting in a CNH (desnitro) or CO (urea) group respectively.42,45
 | ||
| Fig. 4 Typical UPLC-HESI(+)-HRMS total ion chromatograms (TIC) for (a) IMD, (b) DNF, and (c) CLD after reaction with OH (blue traces) for 16 hours, as well as control studies of before reaction (grey traces) and after photolysis (green traces). Chromatogram peaks are labeled with the major ion observed within that peak except for 212/272 in (a) and 236/266 in (c) with 2 major ions. | ||
Table 2 summarizes the measured accurate masses of the [M + H]+ ions from the IMD-OH reaction, the corresponding molecular formulae, the errors between the measured accurate masses and the exact masses, and the molecular structures of the products proposed based on the MS/MS fragmentation patterns summarized in the table and reasonable reaction mechanisms. In all chromatograms, the [M + H]+ ion of the parent IMD at m/z 256 is observed. Products of the OH reaction include the IMD structure with a carbonyl (m/z 270) or alcohol (m/z 272) group, desnitro (m/z 211) and urea (m/z 212) derivatives that are also seen to a smaller extent than under UV irradiation only, and their carbonyl oxidation products (m/z 225 and 226). Additionally, products observed at m/z 131 and 158 are identified in HRMS as nitroguanidine and 6-chloronicotinic acid which have been observed as oxidation products in a number of aqueous oxidation studies.34,37–39,47,51 Products with alkene groups are also observed (m/z 209 and 254).
| Detected ion [M+ H]+ | Ion formula | Error (ppm) | Proposed product structurea | MS/MS fragments |
|---|---|---|---|---|
| a Only one isomer shown for simplicity. The MS/MS for the other isomers were consistent with the alternate structures. b Also reported in Schippers and Schwack73 on tomato plant surfaces. c IMD photolysis product. d MS/MS matched authentic standard. e MS/MS is consistent with the fragmentation reported by Fusetto et al.85 | ||||
| 256.0594 | IMD, [C9H10O2N5Cl + H]+ | −0.75 |

|
209 (–HNO2), 175 (–NO2, –Cl), 128, 126, 84 |
| 131.0562 | [C3H6O2N4 + H]+ | −1.17 |

|
85 (–NO2) |
| 158.0003 | 6-Chloronicotinic acid, [C6H4O2NCl + H]+ | −0.27 |
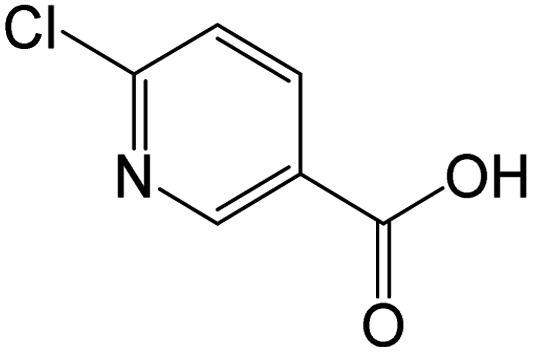
|
140 (–H2O), 112 (–H2O, –CO), 78 |
| 185.0592b | [C7H9N4Cl + H]+ | 1.66 |
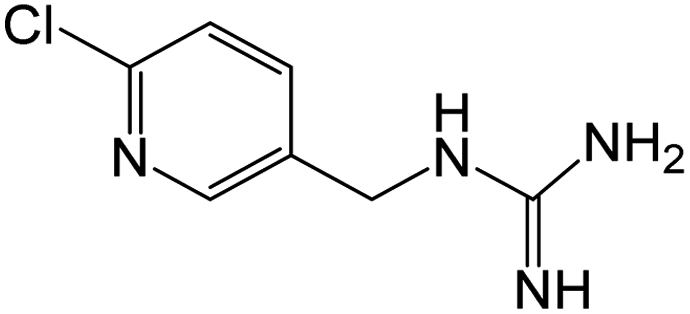
|
168 (–NH3), 143 (–C(NH)2), 126, 107 |
| 209.0585 | [C9H9N4Cl + H]+ | 1.47 |
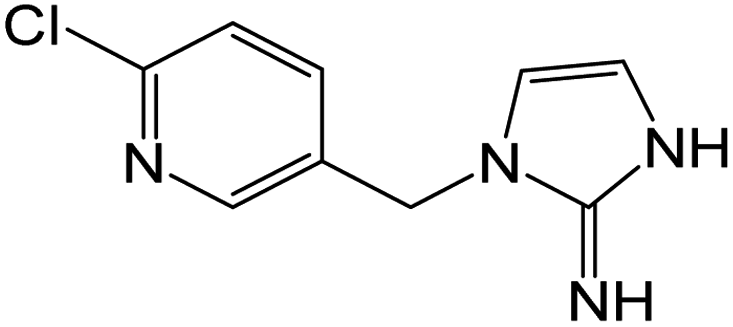
|
173 (–HCl), 126 |
| 211.0745b,c,d | Desnitro IMD, [C9H11N4Cl + H]+ | 0.11 |

|
175 (–HCl), 133, 126, 84 |
| 212.0585b,c,d | IMD urea, [C9H10ON3Cl + H]+ | 0.01 |

|
175, 128, 126, 99 |
| 225.0535 | [C9H9ON4Cl + H]+ | −0.11 |

|
126 |
| 226.0378b | [C9H8O2N3Cl + H]+ | −0.20 |
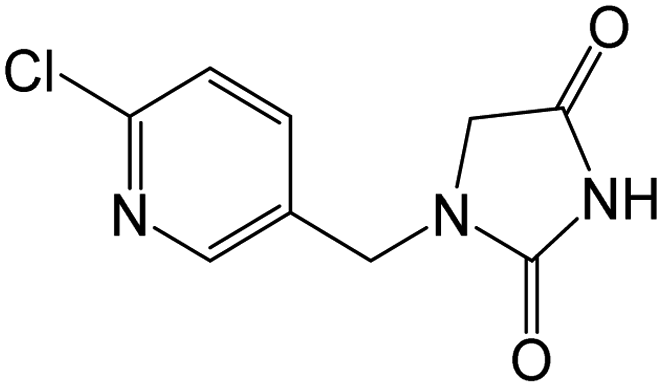
|
169, 126 |
| 254.0438e | [C9H8O2N5Cl + H]+ | −0.72 |
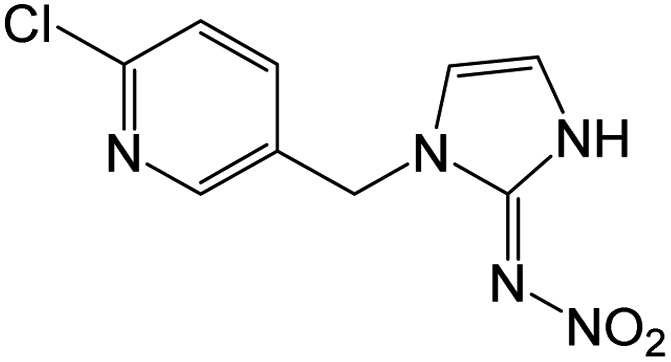
|
236 (–H2O), 207 (–HNO2), 206 (–H2O, –NO), 205 (–H2O, –HNO), 171 (–H2O, –NO, –Cl), 167, 126 |
| 270.0387 | [C9H8O3N5Cl + H]+ | −0.54 |
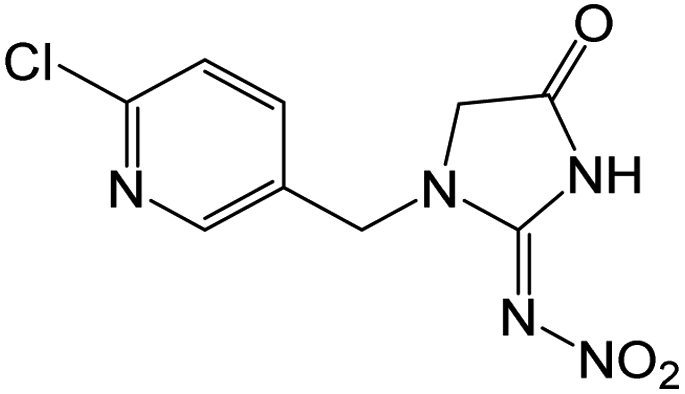
|
226 (–N2O), 223 (–HNO2), 189 (–NO2, –Cl), 161 (–NO2, –Cl, –CO), 126 |
| 272.0543b | [C9H10O3N5Cl + H]+ | −0.52 |

|
228 (–N2O), 209 (–NO2, –OH), 173 (–NO2, –Cl, –H2O), 144 |
Multiple peaks often appear at the same mass due to different isomers. These can be more clearly seen in extracted ion chromatograms (EIC) for selected values of m/z. For example, Fig. 5 shows the EIC for m/z at 256 for the parent IMD as well as 270, 272, 254 and 225 due to products of the OH reaction. One significant peak is seen for m/z 254, two for m/z 272 and 225, and three for m/z 270. Attack of OH at the C–H groups as discussed above could occur at three different sites, giving three different isomeric products. However, the isomers representing attack on the two sites on the five-membered ring are sufficiently similar that the products may co-elute, leading to two peaks rather than three. One of the peaks for m/z 225 has a larger area, indicating either one isomer is formed preferentially and the third is not formed at all, or more likely, the larger peak intensity is due to the contribution of both 5-membered ring isomers.
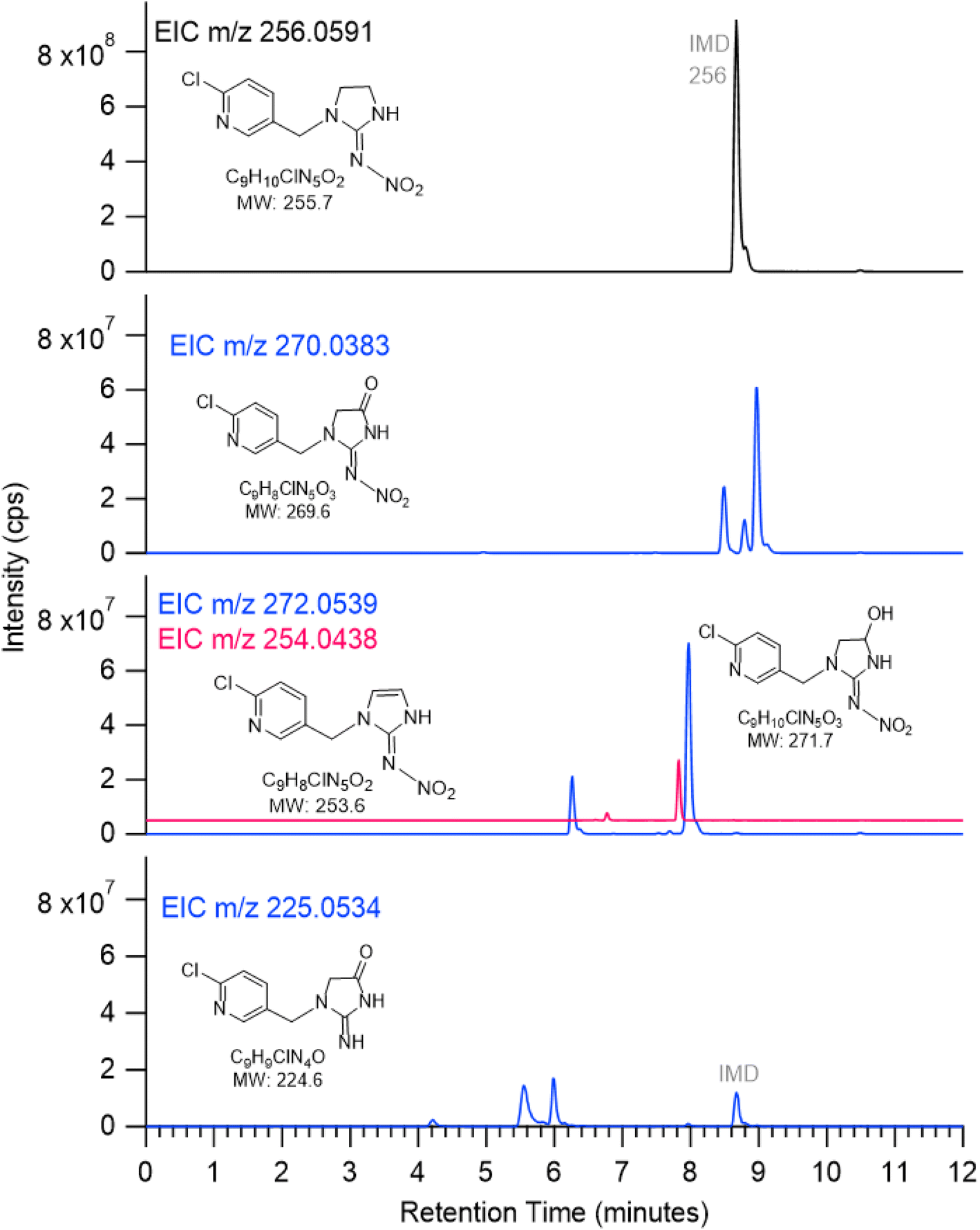 | ||
| Fig. 5 ESI(+) extracted ion chromatograms (EIC) of species with m/z 256 (IMD), 270, 272, 254 (offset for clarity) and 225 for IMD + OH reaction. | ||
Most of the product peaks are also seen in DART-MS spectra (Fig. 6). There is not an exact correspondence in terms of their mass-to-charge ratios and their relative intensities, as some compounds may not survive the heating used in DART-MS to volatilize the sample. Thus, DART-MS detects gas phase species while ESI detects soluble species. For example, the peak at m/z 272 assigned to the alcohol derivative of IMD is the largest product peak in the UPLC chromatogram (Fig. 4a) but is much smaller in the DART-MS spectrum (Fig. 6). This is likely due to fragmentation of the alcohol at the higher temperatures in DART to yield an ion at m/z 254 due to [M + H − H2O]+. However, the EIC data showed that the alcohol (m/z 272) and alkene (m/z 254) chromatographically separated (Fig. 5), indicating that the olefin is formed as its own unique product of the OH reaction. To the best of our knowledge, this is the first observation of this alkene product from OH reactions, although the desnitro-olefin product (Table 2, m/z 209) has been reported in oxidations in aqueous solution.33,37,47 From an environmental impacts perspective, it is interesting that the IMD–olefin has been widely observed as a plant metabolite and is more toxic to insects than IMD itself.67–69 This product is expected to undergo additional oxidation processes in air. Many of the oxidation products identified here have been observed in soils at pesticide manufacturing sites in China.70
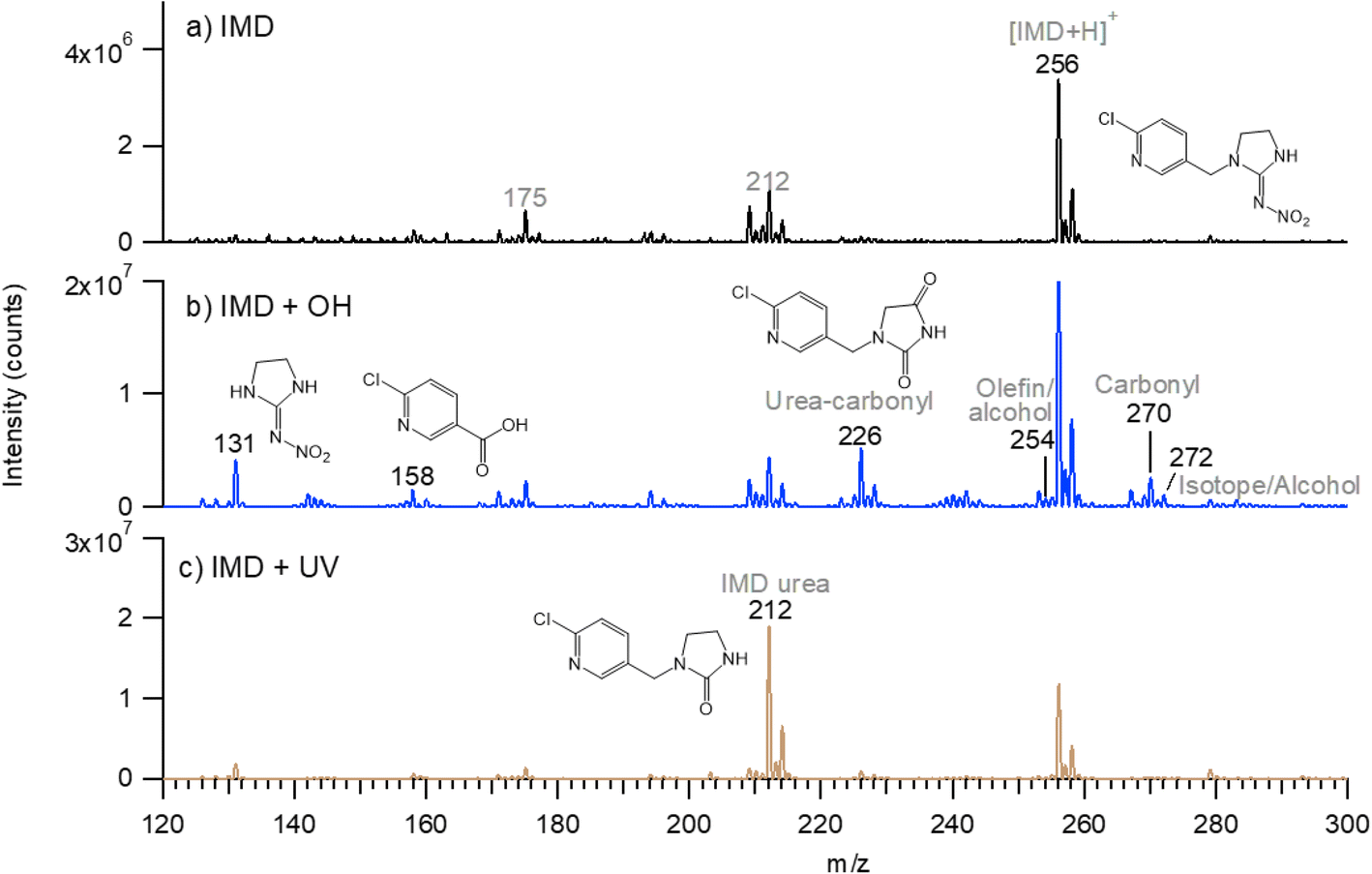 | ||
| Fig. 6 Typical DART-MS spectra of IMD samples. (a) Pure IMD on ZnSe crystal before reaction; (b) after 15 hours reaction with OH; (c) after 20 hours of photolysis in the absence of IPN/OH. Spectra of room air background have been subtracted. | ||
The ATR-FTIR spectra are consistent with the mass spectrometry data. The difference spectrum on reaction with OH (Fig. 1a) shows loss of the parent compound and the formation of oxidized products with absorption bands in the 1600–1800 cm−1 region. These infrared bands are assigned to CN and CO groups, the latter group being present in many of the products identified by HRMS and DART-MS. The broad feature in the 3000–3300 cm−1 region reflects a hydrogen-bonded OH, for example from 6-chloronicotinic acid (m/z 158) which was also identified from HRMS and DART-MS.
There are a great deal of data for oxidation of NN in aqueous media. For example, the most commonly observed aqueous product for IMD is 6-chloronicotinic acid,34,37–39,47,51 which is also observed in this work. The carbonyl products at m/z 270 were observed in several other studies.33,34,51 Alcohol products at m/z 272 have also been observed in the aqueous studies.34,36,37,47,51 IMD urea is frequently observed in aqueous reactions,33,37,38,41,47,51 and desnitro IMD has also been reported.47 However in the aqueous reactions, there are other potential processes occurring in addition to OH oxidation, such as photolysis, ozonolysis, and catalytic reactions that may be contributing to product formation. Many smaller products (MW 100–157) are commonly observed in the aqueous reactions,37,39–41 as the goal is often complete mineralization of the NN.
Most relevant to the current studies are photodegradation reactions on tomato plant surfaces that were conducted outdoors in ambient air.71 Under these conditions, it is likely that OH was present and contributed to the oxidation. The most important products identified were the IMD-alcohol and a nitroso-derivative, along with the desnitro and urea products which were likely formed at least in part by direct photolysis. Other studies of the photodegradation on solid surfaces72,73 identified very similar products to those observed here (Table 2).
Fig. 7 summarizes proposed mechanisms for the formation of the major products. Both the kinetics and products point to abstraction of a hydrogen from a C–H bond as being the initial step. This produces an alkyl radical which in the presence of O2 forms a peroxy radical.55 Both RO2 radicals and NO are generated in the gas phase during the IPN/O2 photolysis, and the NN peroxy radicals may react with these gas phase species to form alkoxy radicals, alcohols, and carbonyls. The oxidized desnitro and urea carbonyl products identified in these studies are likely due to secondary reactions of the OH with the desnitro and urea products.
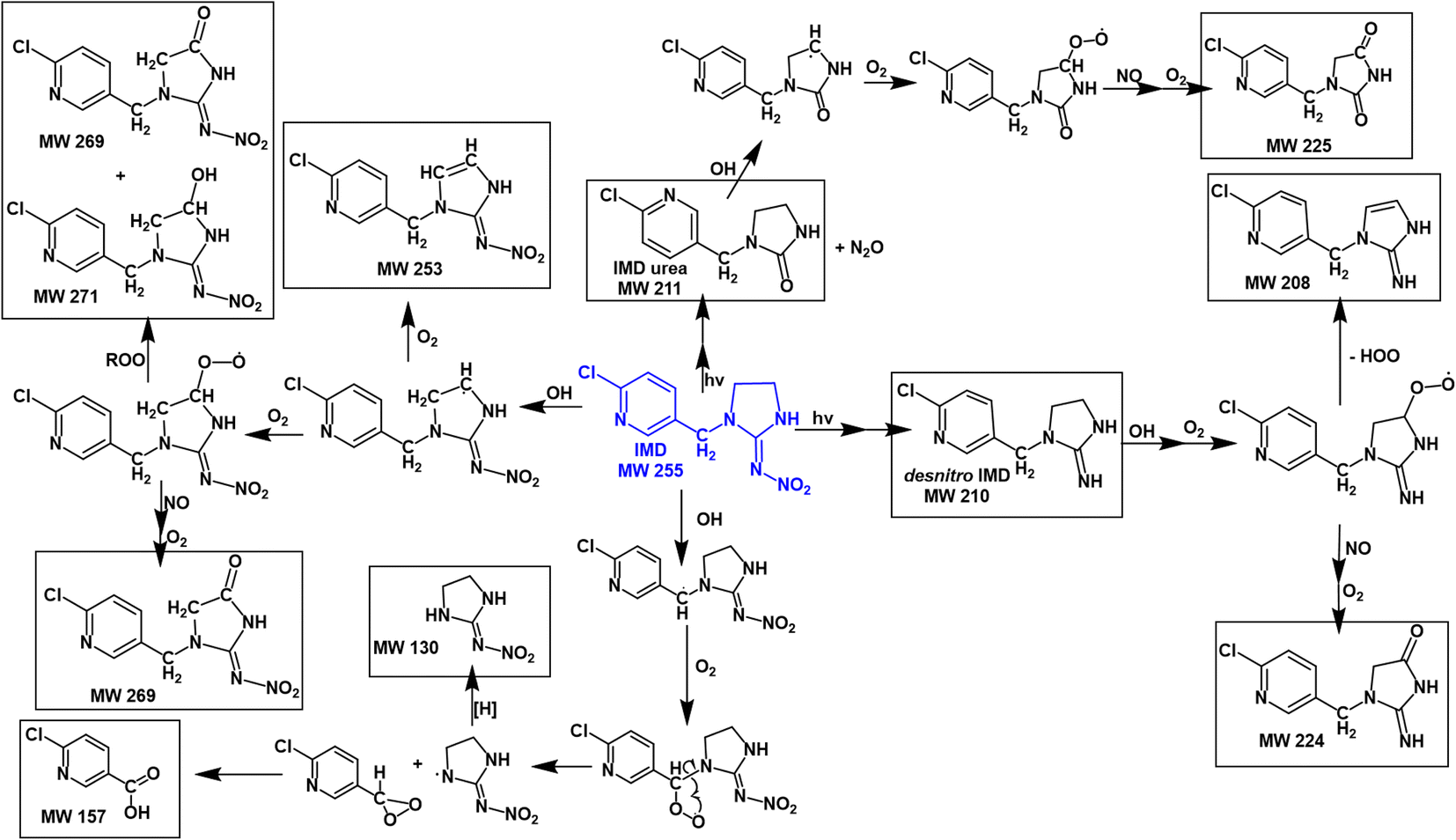 | ||
| Fig. 7 Proposed mechanisms for formation of OH + IMD reaction products (where more than one isomer is possible, only one is shown for simplicity). | ||
| Detected ion [M + H]+ | Ion formula | Error (ppm) | Proposed structurea | MS/MS fragments |
|---|---|---|---|---|
| a Only one isomer shown for simplicity. The MS/MS for the other isomers were consistent with the alternate structures. b DNF photolysis product. | ||||
| 203.1142 | DNF, [C7H14O3N4 + H]+ | 1.4 |
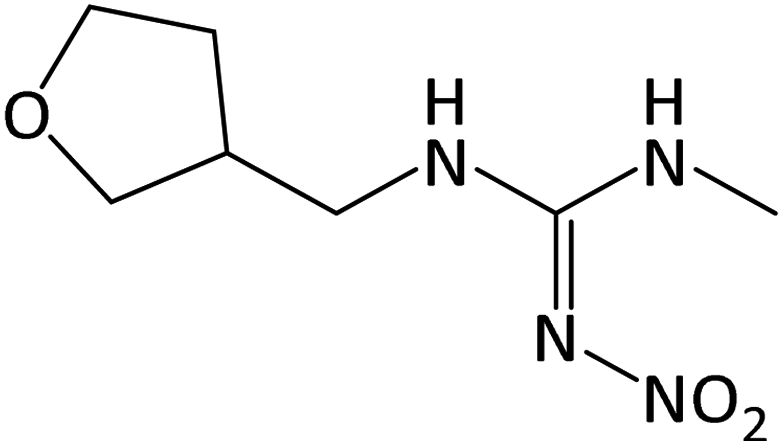
|
129, 114, 100, 87, 73 |
| 154.0976 | [C7H11ON3 + H]+ | 0.2 |

|
69 |
| 156.1128b | [C7H13ON3 + H]+ | −1.14 |

|
83 |
| 158.1284b | Desnitro DNF, [C7H15ON3 + H]+ | −1.09 |
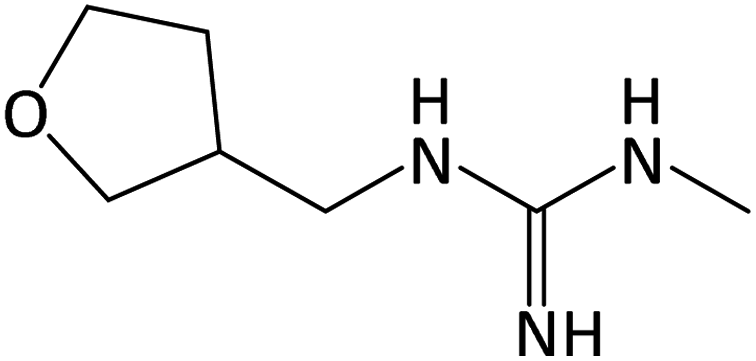
|
102 |
| 159.1124b | DNF urea, [C7H14O2N2 + H]+ | −1.13 |

|
102, 85, 67 |
| 172.108 | [C7H13O2N3 + H]+ | −0.83 |

|
155, 116, 57 |
| 189.0982 | [C6H12O3N4 + H]+ | 0.42 |

|
131, 115, 99, 73 |
| 201.0988 | [C7H12O3N4 + H]+ | 2.30 |

|
187 (–CH2), 155 (–NO2), 127, 112 |
| 217.0928 | [C7H12O4N4 + H]+ | −1.09 |

|
171 (–NO2), 127, 112, 87, 73 |
| 219.1083 | [C7H14O4N4 + H]+ | −1.33 |
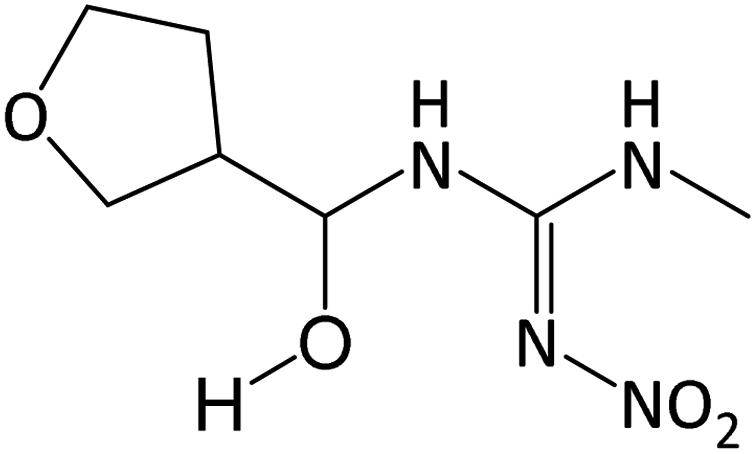
|
144, 129, 114, 87 |
| Detected ion [M + H]+ | Ion formula | Error (ppm) | Proposed structurea | MS/MS fragments |
|---|---|---|---|---|
| a Only one isomer shown for simplicity. The MS/MS for the other isomers were consistent with the alternate structures. b CLD photolysis product. | ||||
| 250.0158 | CLD, [C6H8O2N5ClS + H]+ | −0.89 |

|
169 (–NO2, –Cl), 132, 113 |
| 205.0311b | Desnitro CLD, [C6H9N4ClS + H]+ | 0.93 |

|
132, 113 |
| 206.0152b | CLD urea, [C6H8ON3ClS + H]+ | 1.27 |

|
175, 132, 120, 113, 86 |
| 219.0101 | [C6H7ON4ClS + H]+ | −0.66 |
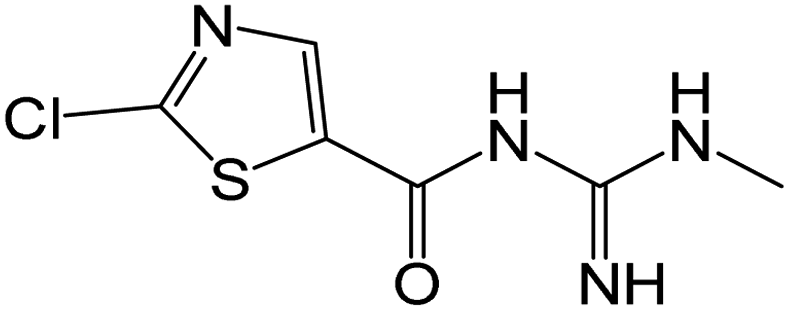
|
202, 163, 146, 75, 57 |
| 219.9940 | [C6H6O2N3ClS + H]+ | −1.04 |

|
189, 163, 146, 120 |
| 236.0002 | [C5H6O2N5ClS + H]+ | −0.64 |
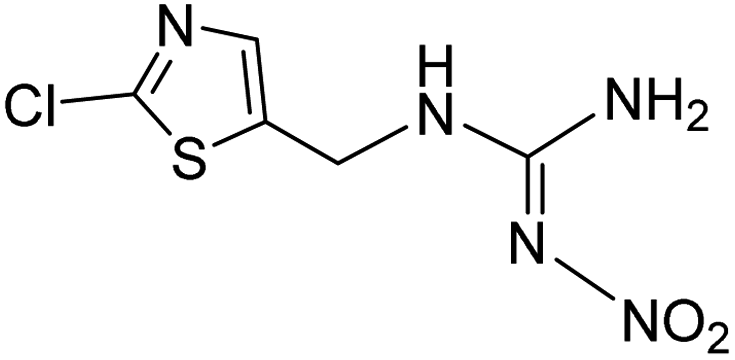
|
155 (–NO2, –Cl), 132, 113, 96 |
| 263.9950 | [C6H6O3N5ClS + H]+ | −1.13 |
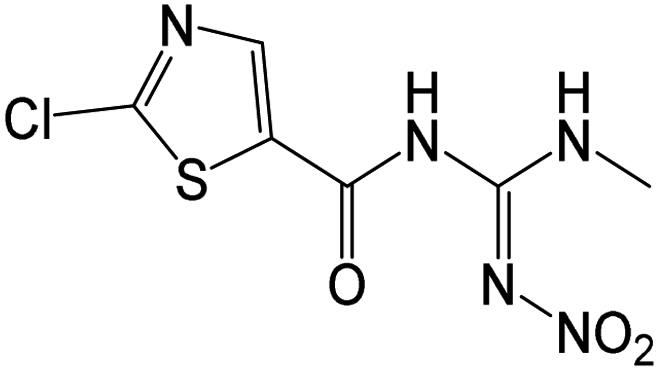
|
183 (–NO2, –Cl), 145 |
| 266.0108 | [C6H8O3N5ClS + H]+ | −0.42 |
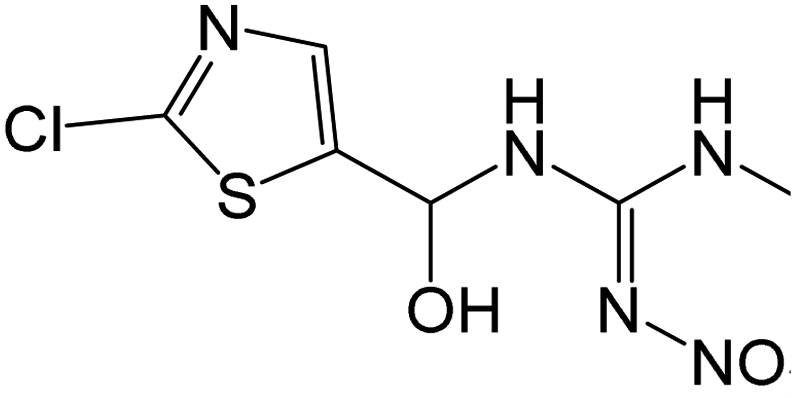
|
222, 203, 191, 167, 147, 129, 86 |
 | ||
| Fig. 8 Proposed mechanisms for formation of OH + DNF reaction products (where more than one isomer is possible, only one is shown for simplicity). | ||
 | ||
| Fig. 9 Proposed mechanisms for formation of OH + CLD reaction products (where more than one isomer is possible, only one is shown for simplicity). | ||
While IMD has three potential locations for aliphatic C–H hydrogen abstraction, DNF has six (four CH2 groups, one CH3 group, and one CH group) and CLD has two, a secondary CH2 group and a primary CH3 group. The fact that the extracted ion chromatogram for a carbonyl product of CLD oxidation at m/z 264 shows two isomers of similar intensities (Fig. S6†) suggests that hydrogen abstraction occurs efficiently from both the CH2 and CH3 groups. Evidence of hydrogen abstraction on the CH3 group was also observed in the DNF–OH reaction with a product at m/z 189 (Fig. 4b). This was surprising because the SAR for gas phase reactions predict that the terminal CH3 group will be less reactive. However, results from a combination of experiments and theory show that the bond dissociation energy (BDE) of a primary C–H group adjacent to an amine nitrogen is about 4 kJ mol−1 larger than that for a secondary C–H adjacent to a nitrogen,77,78 much less than a difference of ∼13 kJ mol−1 in BDE for primary vs. secondary C–H groups in alkanes.77 This suggests that the difference between abstraction from a terminal –CH3 group and a secondary CH2 group may be less in the case of amines, resulting in relatively more oxidation of the terminal methyl groups in DNF and CLD.
The ATR-FTIR spectra of the reaction products (Fig. 1) are consistent with the mass spectrometry identification of products summarized in Tables 3 and 4. A variety of carbonyl-containing products lead to absorption bands in the 1700–1800 cm−1 region, but unlike IMD, there are no significant broad peaks in the 3000–3300 cm−1 region. This is consistent with the identification of 6-chloronicotinic acid as the source of a hydrogen-bonded carboxylic acid only in the IMD reaction since corresponding acids cannot be formed in the DNF and CLD reactions.
Environmental implications
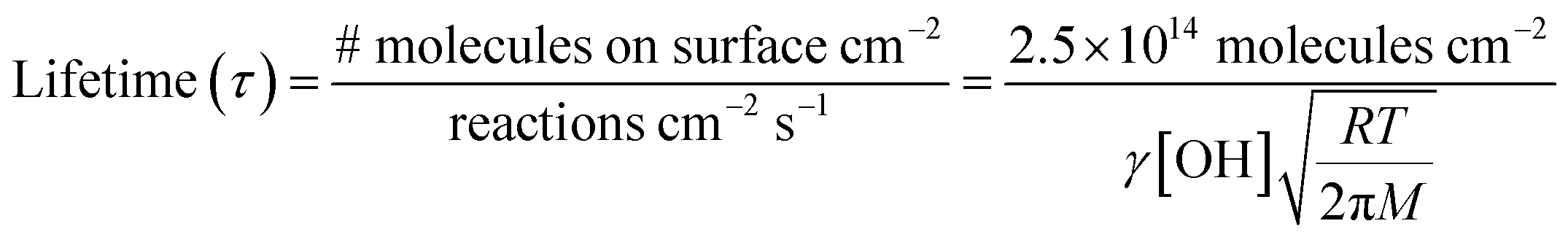 | (III) |
In air, the NN peroxy radical intermediate on the surface, RO2(surf), is likely to react with gas phase R′O2 radicals or NO via reactions (R7) or (R8):
 | (R7) |
 | (R8) |
In California's agriculturally-dominant San Joaquin Valley, for example, the NO concentration varies seasonally with an average of ∼8.2 × 108 molecules cm−3 in the summer and ∼1.5 × 1011 molecules cm−3 in the winter.80 Typical R′O2(g) concentrations measured at various locations around the world peak at ∼5 × 108 molecules cm−3 at midday.81–84 Using typical rate constants for reactions (R7) and (R8),55 first order rates are k7[R′O2(g)] = 5 × 10−4 s−1 and k8[NO] = 0.007–1.35 s−1. Thus, the NO reaction with RO2 to form carbonyl products, of both the parent NN and the desnitro and urea photolysis products, is likely favored under environmentally realistic conditions, even at low NO concentrations. The reaction with NO forms an alkoxy radical that depending on its structure, can decompose, react with O2 or isomerize. In the solid phase, reaction of alkoxy with O2 to form the carbonyl compounds shown in Fig. 7–9 seems most likely. Although not shown in the mechanisms, some formation of organic nitrates from the RO2 reactions with NO may also occur.55
In short, studies of the environmental fates and impacts of NN should take into account not only the parent compound but also its oxidation and photolysis products.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was funded by NSF (Grant #1404233, #2002909 and #1654104), the Ridge to Reef NSF Research Traineeship (award DGE-1735040), Army Research Office DURIP equipment grant (#W911NF2010064), and NSF Major Research Instrumentation Program (Grant #1920242 and #1337080). The authors thank Adam Thomas for assistance with experiments.References
- N. Simon-Delso, V. Amaral-Rogers, L. P. Belzunces, J. M. Bonmatin, M. Chagnon, C. Downs, L. Furlan, D. W. Gibbons, C. Giorio, V. Girolami, D. Goulson, D. P. Kreutzweiser, C. H. Krupke, M. Liess, E. Long, M. McField, P. Mineau, E. A. D. Mitchell, C. A. Morrissey, D. A. Noome, L. Pisa, J. Settele, J. D. Stark, A. Tapparo, H. Van Dyck, J. Van Praagh, J. P. Van der Sluijs, P. R. Whitehorn and M. Wiemers, Systemic insecticides (neonicotinoids and fipronil): trends, uses, mode of action and metabolites, Environ. Sci. Pollut. Res., 2015, 22, 5–34 CrossRef CAS.
- J. M. Bonmatin, C. Giorio, V. Girolami, D. Goulson, D. P. Kreutzweiser, C. Krupke, M. Liess, E. Long, M. Marzaro, E. A. D. Mitchell, D. A. Noome, N. Simon-Delso and A. Tapparo, Environmental fate and exposure; neonicotinoids and fipronil, Environ. Sci. Pollut. Res., 2015, 22, 35–67 CrossRef CAS PubMed.
- C. A. Morrissey, P. Mineau, J. H. Devries, F. Sanchez-Bayo, M. Liess, M. C. Cavallaro and K. Liber, Neonicotinoid contamination of global surface waters and associated risk to aquatic invertebrates: A review, Environ. Int., 2015, 74, 291–303 CrossRef CAS.
- J. F. Borsuah, T. L. Messer, D. D. Snow, S. D. Comfort and A. R. Mittelstet, Literature review: Global neonicotinoid insecticide occurrence in aquatic environments, Water, 2020, 12, 3388 CrossRef CAS.
- E. A. D. Mitchell, B. Mulhauser, M. Mulot, A. Mutabazi, G. Glauser and A. Aebi, A worldwide survey of neonicotinoids in honey, Science, 2017, 358, 109–111 CrossRef CAS.
- P. Jeschke, R. Nauen, M. Schindler and A. Elbert, Overview of the status and global strategy for neonicotinoids, J. Agric. Food Chem., 2011, 59, 2897–2908 CrossRef CAS PubMed.
- D. Goulson, Review: An overview of the environmental risks posed by neonicotinoid insecticides, J. Appl. Ecol., 2013, 50, 977–987 CrossRef.
- A. Elbert, M. Haas, B. Springer, W. Thielert and R. Nauen, Applied aspects of neonicotinoid uses in crop protection, Pest Manage. Sci., 2008, 64, 1099–1105 CrossRef CAS PubMed.
- M. R. Douglas and J. F. Tooker, Large-scale deployment of seed treatments has driven rapid increase in use of neonicotinoid insecticides and preemptive pest management in US field crops, Environ. Sci. Technol., 2015, 49, 5088–5097 CrossRef CAS PubMed.
- C. A. Mullin, M. Frazier, J. L. Frazier, S. Ashcraft, R. Simonds, D. vanEngelsdorp and J. S. Pettis, High levels of miticides and agrochemicals in North American apiaries: implications for honey bee health, PLoS One, 2010, 5, 1–19 CrossRef PubMed.
- L. W. Pisa, V. Amaral-Rogers, L. P. Belzunces, J. M. Bonmatin, C. A. Downs, D. Goulson, D. P. Kreutzweiser, C. Krupke, M. Liess, M. McField, C. A. Morrissey, D. A. Noome, J. Settele, N. Simon-Delso, J. D. Stark, J. P. Van der Sluijs, H. Van Dyck and M. Wiemers, Effects of neonicotinoids and fipronil on non-target invertebrates, Environ. Sci. Pollut. Res., 2015, 22, 68–102 CrossRef CAS PubMed.
- D. Goulson, E. Nicholls, C. Botias and E. L. Rotheray, Bee declines driven by combined stress from parasites, pesticides, and lack of flowers, Science, 2015, 347, 1255957 CrossRef PubMed.
- M. L. Hladik, A. R. Main and D. Goulson, Environmental risks and challenges associated with neonicotinoid insecticides, Environ. Sci. Technol., 2018, 52, 3329–3335 CrossRef CAS PubMed.
- O. Lundin, M. Rundlof, H. G. Smith, I. Fries and R. Bommarco, Neonicotinoid insecticides and their impacts on bees: A systematic review of research approaches and identification of knowledge gaps, PLoS One, 2015, 10, 0136928 Search PubMed.
- N. Tsvetkov, O. Samson-Robert, K. Sood, H. S. Patel, D. A. Malena, P. H. Gajiwala, P. Maciukiewicz, V. Fournier and A. Zayed, Chronic exposure to neonicotinoids reduces honey bee health near corn crops, Science, 2017, 356, 1395–1397 CrossRef CAS.
- T. J. Wood and D. Goulson, The environmental risks of neonicotinoid pesticides: a review of the evidence post 2013, Environ. Sci. Pollut. Res., 2017, 24, 17285–17325 CrossRef CAS PubMed.
- B. A. Woodcock, J. M. Bullock, R. F. Shore, M. S. Heard, M. G. Pereira, J. Redhead, L. Ridding, H. Dean, D. Sleep, P. Henrys, J. Peyton, S. Hulmes, L. Hulmes, M. Sarospataki, C. Saure, M. Edwards, E. Genersch, S. Knabe and R. F. Pywell, Country-specific effects of neonicotinoid pesticides on honey bees and wild bees, Science, 2017, 356, 1393–1395 CrossRef CAS PubMed.
- R. Schmuck and G. Lewis, Review of field and monitoring studies investigating the role of nitro-substituted neonicotinoid insecticides in the reported losses of honey bee colonies (Apis mellifera), Ecotoxicology, 2016, 25, 1617–1629 CrossRef CAS PubMed.
- European Commission, Journal, Commission Implementing Regulation (EU) 2018/783 of 29 May 2018, Implementing Regulation (EU) No 540/2011 as regards the conditions of approval of the active substance imidacloprid, clothianidin, and thiamethoxam, 2018, L, 31–44.
- California Department of Pesticide Regulation, Neonicotinoids, https://www.cdpr.ca.gov/docs/registration/reevaluation/chemicals/neonicotinoids.htm, accessed 27 April 2022 Search PubMed.
- D. Wintermantel, J. F. Odoux, A. Decourtye, M. Henry, F. Allier and V. Bretagnolle, Neonicotinoid-induced mortality risk for bees foraging on oilseed rape nectar persists despite EU moratorium, Sci. Total Environ., 2020, 704, 135400 CrossRef CAS.
- S. Humann-Guilleminot, S. Clement, J. Desprat, L. J. Binkowski, G. Glauser and F. Helfenstein, A large-scale survey of house sparrows feathers reveals ubiquitous presence of neonicotinoids in farmlands, Sci. Total Environ., 2019, 660, 1091–1097 CrossRef CAS PubMed.
- G. G. Distefano, R. Zangrando, M. Basso, L. Panzarin, A. Gambaro, A. Volpi Ghirardini and M. Picone, The ubiquity of neonicotinoid contamination: Residues in seabirds with different trophic habits, Environ. Res., 2022, 206, 112637 CrossRef CAS PubMed.
- Q. Zhang, Z. Li, C. H. Chang, J. L. Lou, M. R. Zhao and C. Lu, Potential human exposures to neonicotinoid insecticides: A review, Environ. Pollut., 2018, 236, 71–81 CrossRef CAS PubMed.
- W. Han, Y. Tian and X. Shen, Human exposure to neonicotinoid insecticides and the evaluation of their potential toxicity: An overview, Chemosphere, 2018, 192, 59–65 CrossRef CAS PubMed.
- J. M. Bonmatin, E. A. D. Mitchell, G. Glauser, E. Lumawig-Heitzman, F. Claveria, M. B. van Lexmond, K. Taira and F. Sanchez-Bayo, Residues of neonicotinoids in soil, water and people’s hair: A case study from three agricultural regions of the Philippines, Sci. Total Environ., 2021, 757, 143822 CrossRef CAS PubMed.
- D. Zhang and S. Lu, Human exposure to neonicotinoids and the associated health risks: A review, Environ. Int., 2022, 163, 107201 CrossRef CAS.
- D. A. Thompson, H. J. Lehmler, D. W. Kolpin, M. L. Hladik, J. D. Vargo, K. E. Schilling, G. H. LeFevre, T. L. Peeples, M. C. Poch, L. E. LaDuca, D. M. Cwiertny and R. W. Field, A critical review on the potential impacts of neonicotinoid insecticide use: current knowledge of environmental fate, toxicity, and implications for human health, Environ. Sci.: Processes Impacts, 2020, 22, 1315–1346 RSC.
- S. L. Chao and J. E. Casida, Interaction of imidacloprid metabolites and analogs with the nicotinic acetylcholine receptor of mouse brain in relation to toxicity, Pestic. Biochem. Physiol., 1997, 58, 77–88 CrossRef CAS.
- D. Pietrzak, J. Kania, E. Kmiecik, G. Malina and K. Wator, Fate of selected neonicotinoid insecticides in soil-water systems: Current state of the art and knowledge gaps, Chemosphere, 2020, 255, 126981 CrossRef CAS.
- K. L. Klarich, N. C. Pflug, E. M. DeWald, M. L. Hladik, D. W. Kolpin, D. M. Cwiertny and G. H. LeFevre, Occurrence of neonicotinoid insecticides in finished drinking water and fate during drinking water treatment, Environ. Sci. Technol. Lett., 2017, 4, 168–173 CrossRef CAS.
- G. Mahai, Y. J. Wan, W. Xia, A. Z. Wang, L. S. Shi, X. Qian, Z. Y. He and S. Q. Xu, A nationwide study of occurrence and exposure assessment of neonicotinoid insecticides and their metabolites in drinking water of China, Water Res., 2021, 189, 116630 CrossRef CAS PubMed.
- B. S. Baghirzade, U. Yetis and F. B. Dilek, Imidacloprid elimination by O3 and O3/UV: kinetics study, matrix effect, and mechanism insight, Environ. Sci. Pollut. Res., 2021, 28, 24535–24551 CrossRef CAS PubMed.
- M. Bourgin, F. Violleau, L. Debrauwer and J. Albet, Ozonation of imidacloprid in aqueous solutions: Reaction monitoring and identification of degradation products, J. Hazard. Mater., 2011, 190, 60–68 CrossRef CAS PubMed.
- S. Chen, J. Deng, Y. Deng and N. Gao, Influencing factors and kinetic studies of imidacloprid degradation by ozonation, Environ. Technol., 2019, 40, 2127–2134 CrossRef CAS.
- X. Liu, X. L. Wu, Z. Long, C. Zhang, Y. Q. Ma, X. H. Hao, H. Y. Zhang and C. P. Pan, Photodegradation of imidacloprid in aqueous solution by the metal-free catalyst graphitic carbon nitride using an energy-saving lamp, J. Agric. Food Chem., 2015, 63, 4754–4760 CrossRef CAS.
- Q. H. Kan, K. Lu, S. P. Dong, D. L. Shen, Q. G. Huang, Y. Tong, W. Wu, S. X. Gao and L. Mao, Transformation and removal of imidacloprid mediated by silver ferrite nanoparticle facilitated peroxymonosulfate activation in water: Reaction rates, products, and pathways, Environ. Pollut., 2020, 267, 115438 CrossRef CAS PubMed.
- S. Malato, J. Caceres, A. Aguera, M. Mezcua, D. Hernando, J. Vial and A. R. Fernandez-Alba, Degradation of imidacloprid in water by photo-Fenton and TiO2 photocatalysis at a solar pilot plant: A comparative study, Environ. Sci. Technol., 2001, 35, 4359–4366 CrossRef CAS PubMed.
- C. F. Z. Lacson, M. D. G. de Luna, C. D. Dong, S. Garcia-Segura and M. C. Lu, Fluidized-bed Fenton treatment of imidacloprid: Optimization and degradation pathway, Sustainable Environ. Res., 2018, 28, 309–314 CrossRef CAS.
- M. Turabik, N. Oturan, B. Gözmen and M. A. Oturan, Efficient removal of insecticide “imidacloprid” from water by electrochemical advanced oxidation processes, Environ. Sci. Pollut. Res., 2014, 21, 8387–8397 CrossRef CAS.
- V. Kitsiou, N. Filippidis, D. Mantzavinos and I. Poulios, Heterogeneous and homogeneous photocatalytic degradation of the insecticide imidacloprid in aqueous solutions, Appl. Catal., B, 2009, 86, 27–35 CrossRef CAS.
- A. Rohrbacher, M. J. Ezell, V. Perraud and B. J. Finlayson-Pitts, Probing matrix effects on the heterogeneous photochemistry of neonicotinoid pesticides, dinotefuran and nitenpyram, ACS Earth Space Chem., 2021, 5, 1196–1209 CrossRef CAS.
- W. Wang, K. Z. Aregahegn, S. T. Andersen, A. Z. Ni, A. F. Rohrbacher, O. J. Nielsen and B. J. Finlayson-Pitts, Quantum yields and N2O formation from photolysis of solid films of neonicotinoids, J. Agric. Food Chem., 2019, 67, 1638–1646 CrossRef CAS.
- K. Z. Aregahegn, M. J. Ezell and B. J. Finlayson-Pitts, Photochemistry of solid films of the neonicotinoid nitenpyram, Environ. Sci. Technol., 2018, 52, 2760–2767 CrossRef CAS PubMed.
- K. Z. Aregahegn, D. Shemesh, R. B. Gerber and B. J. Finlayson-Pitts, Photochemistry of thin solid films of the neonicotinoid imidacloprid on surfaces, Environ. Sci. Technol., 2017, 51, 2660–2668 CrossRef CAS PubMed.
- W. H. Wang, M. J. Ezell, P. S. J. Lakey, K. Z. Aregahegn, M. Shiraiwa and B. J. Finlayson-Pitts, Unexpected formation of oxygen-free products and nitrous acid from the ozonolysis of the neonicotinoid nitenpyram, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 11321–11327 CrossRef CAS PubMed.
- W. C. Wang, D. Y. Huang, D. X. Wang, M. X. Tan, M. Y. Geng, C. Y. Zhu, N. Chen and D. M. Zhou, Extensive production of hydroxyl radicals during oxygenation of anoxic paddy soils: Implications to imidacloprid degradation, Chemosphere, 2022, 286, 131565 CrossRef CAS PubMed.
- C. Segura, C. Zaror, H. D. Mansilla and M. A. Mondaca, Imidacloprid oxidation by photo-Fenton reaction, J. Hazard. Mater., 2008, 150, 679–686 CrossRef CAS PubMed.
- D. D. D. Nguyen, K. A. Huynh, X. H. Nguyen and T. P. Nguyen, Imidacloprid degradation by electro-Fenton process using composite Fe3OT Mn3O4 nanoparticle catalyst, Res. Chem. Intermed., 2020, 46, 4823–4840 CrossRef CAS.
- M. Sedaghat, B. Vahid, S. Aber, M. H. Rasoulifard, A. Khataee and N. Daneshvar, Electrochemical and photo-assisted electrochemical treatment of the pesticide imidacloprid in aqueous solution by the Fenton process: effect of operational parameters, Res. Chem. Intermed., 2016, 42, 855–868 CrossRef CAS.
- G. Rozsa, M. Nafradi, T. Alapi, K. Schrantz, L. Szabo, L. Wojnarovits, E. Takacs and A. Tungler, Photocatalytic, photolytic and radiolytic elimination of imidacloprid from aqueous solution: Reaction mechanism, efficiency and economic considerations, Appl. Catal., B, 2019, 250, 429–439 CrossRef CAS.
- Y. H. Sun and X. Liu, Efficient visible-light photocatalytic degradation of imidacloprid and acetamiprid using a modified carbon nitride/tungstophosphoric acid composite induced by a nucleophilic addition reaction, Appl. Surf. Sci., 2019, 485, 423–431 CrossRef CAS.
- W. Hayat, Y. Q. Zhang, I. Hussain, X. D. Du, M. M. Du, C. H. Yao, S. B. Huang and F. Si, Efficient degradation of imidacloprid in water through iron activated sodium persulfate, Chem. Eng. J., 2019, 370, 1169–1180 CrossRef CAS.
- F. Soltani-nezhad, A. Saljooqi, T. Shamspur and A. Mostafavi, Photocatalytic degradation of imidacloprid using GO/Fe3O4/TiO2-NiO under visible radiation: Optimization by response level method, Polyhedron, 2019, 165, 188–196 CrossRef CAS.
- B. J. Finlayson-Pitts and J. N. Pitts, Chemistry of the Upper and Lower Atmosphere: Theory, Experiments, and Application, Academic Press, San Diego, 2000 Search PubMed.
- N. J. Harrick, Internal Reflection Spectroscopy, Harrick Scientific Corporation, Ossining, New York, 3rd edn, 1979 Search PubMed.
- J. Y. Le Questel, J. Graton, J. P. Cerón-Carrasco, D. Denis Jacquemin, A. Planchat and S. H. Thany, New insights on the molecular features and electrophysiological properties of dinotefuran, imidacloprid and acetamiprid neonicotinoid insecticides, Bioorg. Med. Chem., 2011, 19, 7623–7634 CrossRef CAS PubMed.
- S. G. Moussa and B. J. Finlayson-Pitts, Reaction of gas phase OH with unsaturated self-assembled monolayers and relevance to atmospheric organic oxidations, Phys. Chem. Chem. Phys., 2010, 12, 9419–9428 RSC.
- J. D. Raff and B. J. Finlayson-Pitts, Hydroxyl radical quantum yields from isopropyl nitrite photolysis in air, Environ. Sci. Technol., 2010, 44, 8150–8155 CrossRef CAS PubMed.
- M. Shiraiwa, C. Pfrang and U. Pöschl, Kinetic multi-layer model of aerosol surface and bulk chemistry (KM-SUB): the influence of interfacial transport and bulk diffusion on the oxidation of oleic acid by ozone, Atmos. Chem. Phys., 2010, 10, 3673–3691 CrossRef CAS.
- G. Socrates, Infrared and Raman Characteristic Group Frequencies: Tables and Charts, John Wiley & Sons Ltd, West Sussex, England, 3rd edn, 2001 Search PubMed.
- A. C. Vander Wall, P. S. J. Lakey, E. R. Molina, V. Perraud, L. M. Wingen, J. Xu, D. Soulsby, R. B. Gerber, M. Shiraiwa and B. J. Finlayson-Pitts, Understanding interactions of organic nitrates with the surface and bulk of organic films: implications for particle growth in the atmosphere, Environ. Sci.: Processes Impacts, 2018, 20, 1593–1610 RSC.
- R. Atkinson, A Structure-Activity Relationship for the estimation of rate constants for the gas-phase reactions of OH radicals with organic-compounds, Int. J. Chem. Kinet., 1987, 19, 799–828 CrossRef CAS.
- R. Atkinson, Kinetics and mechanisms of the gas-phase reactions of the hydroxyl radical with organic-compounds under atmospheric conditions, Chem. Rev., 1985, 86, 69–201 CrossRef.
- R. Atkinson, D. L. Baulch, R. A. Cox, R. F. J. Hampson, J. A. Kerr, M. J. Rossi and J. Troe, Evaluated kinetic and photochemical data for atmospheric chemistry: Supplement VI. IUPAC subcommittee on gas kinetic data evaluation for atmospheric chemistry, J. Phys. Chem. Ref. Data, 1997, 26, 1329–1499 CrossRef CAS.
- A. M. Arangio, J. H. Slade, T. Berkemeier, U. Pöschl, D. A. Knopf and M. Shiraiwa, Multiphase chemical kinetics of OH radical uptake by molecular organic markers of biomass burning aerosols: humidity and temperature dependence, surface reaction, and bulk diffusion, J. Phys. Chem. A, 2015, 119, 4533–4544 CrossRef CAS PubMed.
- R. Nauen, K. Tietjen, K. Wagner and A. Elbert, Efficacy of plant metabolites of imidacloprid against Myzus persicae and Aphis gossypii (Homoptera: Aphididae), Pestic. Sci., 1998, 52, 53–57 CrossRef CAS.
- E. P. Benton, J. F. Grant, R. J. Webster, R. J. Nichols, R. S. Cowles, A. F. Lagalante and C. I. Coots, Assessment of imidacloprid and its metabolites in foliage of Eastern hemlock multiple years following treatment for hemlock Woolly Adelgid, Adelges tsugae (Hemiptera: Adelgidae), in forested conditions, J. Econ. Entomol., 2015, 108, 2672–2682 CrossRef CAS PubMed.
- M. Seifrtova, T. Halesova, K. Sulcova, K. Riddellova and T. Erban, Distributions of imidacloprid, imidacloprid-olefin and imidacloprid-urea in green plant tissues and roots of rapeseed (Brassica napus) from artificially contaminated potting soil, Pest Manage. Sci., 2017, 73, 1010–1016 CrossRef CAS PubMed.
- Y. Tao, J. Liu, Y. Xu, H. Liu, G. Yang, Y. He, J. Xu and Z. Lu, Suspected screening “known unknown” pesticides and transformation products in soil at pesticide manufacturing sites, Sci. Total Environ., 2022, 808, 152074 CrossRef CAS PubMed.
- K. Scholz and F. Reinhard, Photolysis of imidacloprid (NTN 33893) on the leaf surface of tomato plants, Pestic. Sci., 1999, 55, 652–654 CrossRef CAS.
- N. Schippers and W. Schwack, Photochemistry of imidacloprid in model systems, J. Agric. Food Chem., 2008, 56, 8023–8029 CrossRef CAS PubMed.
- N. Schippers and W. Schwack, Phototransformation of imidacloprid on isolated tomato fruit cuticles and on tomato fruits, J. Photochem. Photobiol., B, 2010, 98, 57–60 CrossRef CAS PubMed.
- S. Kurwadkar, A. Evans, D. DeWinne, P. White and F. Mitchell, Modeling photodegradation kinetics of three systemic neonicotinoidsdinotefuran, imidacloprid, and thiamethoxamin aqueous and soil environment, Environ. Toxicol. Chem., 2016, 35, 1718–1726 CrossRef CAS PubMed.
- Y. Li, Y. D. Li, Y. M. Liu and T. J. Ward, Photodegradation of clothianidin and thiamethoxam in agricultural soils, Environ. Sci. Pollut. Res., 2018, 25, 31318–31325 CrossRef CAS PubMed.
- R. Zabar, T. Komel, J. Fabjan, M. B. Kralj and P. Trebse, Photocatalytic degradation with immobilised TiO2 of three selected neonicotinoid insecticides: Imidacloprid, thiamethoxam and clothianidin, Chemosphere, 2012, 89, 293–301 CrossRef CAS PubMed.
- T. J. Burkey, A. A. Castelhano, D. Griller and F. P. Lossing, Heats of formation and ionization potentials of some α-aminoalkyl radicals, J. Am. Chem. Soc., 1983, 105, 4701–4703 CrossRef CAS.
- D. D. M. Wayner, K. B. Clark, A. Rauk, D. Yu and D. A. Armstrong, C-H Bond dissociation energies of alkyl amines: Radical structures and stabilization energies, J. Am. Chem. Soc., 1997, 119, 8925–8932 CrossRef CAS.
- M. A. H. Khan, M. J. Ashfold, G. Nickless, D. Martin, L. A. Watson, P. D. Hamer, R. P. Wayne, C. E. Canosa-Mas and D. E. Shallcross, Night-time NO3 and OH radical concentrations in the United Kingdom inferred from hydrocarbon measurements, Atmos. Sci. Lett., 2008, 9, 140–146 CrossRef.
- California Air Resources Board (CARB), Air Quality Data Query Tool, accessed April 19, 2021 Search PubMed.
- D. Mihelcic, F. Holland, A. Hofzumahaus, L. Hoppe, S. Konrad, P. Müsgen, H.-W. Pätz, H.-J. Schäfer, T. Schmitz, A. Volz-Thomas, K. Bächmann, S. Schlomski, U. Platt, A. Geyer, B. Alicke and G. K. Moortgat, Peroxy radicals during BERLIOZ at Pabstthum: Measurements, radical budgets and ozone production, J. Geophys. Res.: Atmos., 2003, 108, 8254 CrossRef.
- P. S. Stevens, J. H. Mather, W. H. Brune, F. Eisele, D. Tanner, A. Jefferson, C. Cantrell, R. Shetter, S. Sewall, A. Fried, B. Henry, E. Williams, K. Baumann, P. Goldan and W. Kuster, HO2/OH and RO2/HO2 ratios during the Tropospheric OH Photochemistry Experiment: Measurement and theory, J. Geophys. Res.: Atmos., 1997, 102, 6379–6391 CrossRef CAS.
- N. Carslaw, D. J. Creasey, D. E. Heard, A. C. Lewis, J. B. McQuaid, M. J. Pilling, P. S. Monks, B. J. Bandy and S. A. Penkett, Modeling OH, HO2, and RO2 radicals in the marine boundary layer: 1. Model construction and comparison with field measurements, J. Geophys. Res.: Atmos., 1999, 104, 30241–30255 CrossRef CAS.
- D. Mihelcic, P. Müsgen and D. H. Ehhalt, An improved method of measuring tropospheric NO2 and RO2 by matrix isolation and electron spin resonance, J. Atmos. Chem., 1985, 3, 341–361 CrossRef CAS.
- R. Fusetto, J. M. White, C. A. Hutton and R. A. J. O'Hair, Structure of olefin-imidacloprid and gas-phase fragmentation chemistry of its protonated form, Org. Biomol. Chem., 2016, 14, 1715–1726 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Reaction cell (Fig. S1); details of OH measurements, including observed 13CO2 formation during photolysis of IPN in the presence of 13CO (Fig. S2); experimental details for UPLC-HESI-HRMS and DART-MS measurement; KM-SUB model results (Fig. S3) and summary of parameters used as model inputs (Table S1); extracted ion chromatograms (EIC) for multiply oxidized products measured using UPLC-HESI-HRMS and DART-MS for DNF + OH (Fig. S4 and S5), and CLD + OH (Fig. S6 and S7). See DOI: https://doi.org/10.1039/d2ea00134a |
| ‡ Co-first authors. |
| This journal is © The Royal Society of Chemistry 2023 |