
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new microfluidic platform for the highly reproducible preparation of non-viral gene delivery complexes†
Giovanni
Protopapa
a,
Nina
Bono
 a,
Roberta
Visone
b,
Fabio
D'Alessandro
a,
Marco
Rasponi
b and
Gabriele
Candiani
*a
a,
Roberta
Visone
b,
Fabio
D'Alessandro
a,
Marco
Rasponi
b and
Gabriele
Candiani
*a
aDepartment of Chemistry, Materials and Chemical Engineering “G. Natta”, Politecnico di Milano, Milan, Italy. E-mail: gabriele.candiani@polimi.it
bDepartment of Electronics, Information and Bioengineering, Politecnico di Milano, Milan, Italy
First published on 30th November 2022
Abstract
Transfection describes the delivery of exogenous nucleic acids (NAs) to cells utilizing non-viral means. In the last few decades, scientists have been doing their utmost to design ever more effective transfection reagents. These are eventually mixed with NAs to give rise to gene delivery complexes, which must undergo characterization, testing, and further refinement through the sequential reiteration of these steps. Unfortunately, although microfluidics offers distinct advantages over the canonical approaches to preparing particles, the systems available do not address the most frequent and practical quest for the simultaneous generation of multiple polymer-to-NA ratios (N/Ps). Herein, we developed a user-friendly microfluidic cartridge to repeatably prepare non-viral gene delivery particles and screen across a range of seven N/Ps at once or significant volumes of polyplexes at a given N/P. The microchip is equipped with a chaotic serial dilution generator for the automatic linear dilution of the polymer to the downstream area, which encompasses the NA divider to dispense equal amounts of DNA to the mixing area, enabling the formation of particles at seven N/Ps eventually collected in individual built-in tanks. This is the first example of a stand-alone microfluidic cartridge for the fast and repeatable preparation of non-viral gene delivery complexes at different N/Ps and their storage.
1. Introduction
Non-viral gene delivery (also called transfection), the process of introducing exogenous nucleic acids (NAs) into target cells, is now at the cornerstone of (nano)medicine, for the prevention1 or treatment at a genetic level of several diseases,2–5 and (nano)biotechnology, for the production of recombinant proteins.6 In the last couple of decades, we have been witnessing a good deal of effort devoted to designing and developing cutting-edge tools and materials to transfer NAs into cells.7Cationic lipids and polymers, collectively called transfection reagents, are very popular because they are reasonably effective in transfecting cells and the most user-friendly. Indeed, they spontaneously assemble with anionic NAs in a vial to form lipoplexes and polyplexes, respectively, which are next dripped onto cell cultures to deliver their payload intracellularly.4,8
Although the preparation of non-viral gene delivery complexes is basically an easy task, slight yet often unintended changes in its execution may result in disparate transfection behavior.9 Altogether, this contributes to the considerable heterogeneity of results observed across studies.
Polyplexes are typically prepared in a vial, following one of these procedures: (i) dripping the solution containing one component into the other (i.e., bulk mixing of transfection reagent and NA solutions), eventually followed by (ii) vortexing the test tube, or (iii) manual mixing of the two solutions through pipetting.9,10 Besides, intra- and inter-individual imprecision in pipetting is likely to be another source of poor data reproducibility to be seriously reckoned with in daily laboratory practice.11 Considering these drawbacks and because of the clinical quest for an ever-larger quantity of nanocomplexes, the development of tools for scaling up and standardizing the production of gene delivery particles cannot be delayed any longer.1,12–16
Microfluidic systems are notable for predictable fluid behavior and are ideal tools for controlling the physical interaction of reagents at the microscale.17 As microfluidics allows the most accurate manipulation of small amounts of fluids by simply tuning the experimental parameters such as species concentrations and flow rates, this technology has thus been extensively used to produce optimally tailored nanoparticles.18–22 Another advantage is the miniaturization of the mixing process, which breaks down experimental costs because it minimizes the consumption of reagents and shortens the time for preparing nanoassemblies.23–27 By way of example only, the physicochemical properties of lipoplexes and polyplexes have been improved through the control of the flow rates of single reagents in devices fitted with T- or Y-shaped junctions.28–34 Besides, hydrodynamic focusing-based microfluidic systems have been used to produce specifically sized gene delivery assemblies through the fine control of the mixing of cation–anion pairs.26,35,36 Furthermore, a range of microfluidic schemes, such as Tesla structured channels37 and chaotic mixers,38,39 has been devised to ease the mixing of these two components. Other approaches rely on flash-nanocomplexation and nanoprecipitation through rapid mixing in confined impingement jet mixers (CIJMs) or multiple inlet vortex mixers (MIVMs) to control the physical interaction of the components.40 These and other remarkable examples disclose microfluidics as a powerful tool to spur the development of gene delivery particles through the strict control of specific complexation parameters.41 In this light, the number of publications on gene delivery devices is steadily growing and the elegant microfluidic schemes devised are increasingly complex.42–46 Nevertheless, to the best of our knowledge, none has addressed the most impellent quest for the simultaneous generation of multiple polymer-to-NA ratios (N/Ps). Specifically, N/P is the mole ratio of the amino groups (N) borne by the cationic polymer used to complex the phosphate groups (P) of a given amount of NA.6
Driven by the need to fill this gap and motivated to address the lack of standardization in test procedures underpinning the low reproducibility of results,47,48 we herein describe the development and validation of a polydimethylsiloxane (PDMS)-made microfluidic cartridge. This platform enables the fast and repeatable preparation of non-viral gene delivery complexes at different N/Ps, their recovery, and storage until use.
2. Experimental
2.1. Materials
L929 (murine fibroblasts from subcutaneous connective tissue; ATCC®-CCL-1), HeLa (human epithelial ovarian carcinoma cells; ATCC®-CCL-2.2), and Jurkat (T lymphoblasts, TIB-152) cell lines were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). 25 kDa linear PEI (lPEI) was from PolySciences (Eppelheim, Germany) and 25 kDa branched PEI (bPEI) was from Merck Life Science (Milan, Italy), while jetPEI-fluoF® was purchased from Polyplus Transfection (Euroclone, Milan, Italy).pGL3 (pDNA encoding the modified firefly luciferase, pGL3-Control Vector; 5256 bp) and the Luciferase Assay System were obtained from Promega (Milan, Italy), while pGLuc (pDNA encoding the secreted Gaussia luciferase, pGLuc-Basic 2 Vector; 4988 bp) and the BioLux® Gaussia Luciferase Assay Kit were purchased from New England BioLabs (Ipswich, MA, USA). pGL3 and pGLuc were amplified, isolated, purified, and diluted in 0.1× TE buffer (1 mM Tris, pH 8; 0.1 mM EDTA).
The AlamarBlue® cell viability assay was purchased from Life Technologies Italia (Monza, Italy). PDMS (Sylgard® 184, Dow Corning) was purchased from Ellsworth Adhesives GmbH (Burlafingen, Germany). Microslides (Microscope Slides Premiere, 75 mm × 50 mm × 1 mm, Dow Corning) were purchased from VWR (Milano, Italy). All the fluidic connections (Øinner = 0.5 mm) were made of Tygon from Saint-Gobain Performance Plastics (Courbevoie, France). Any other reagents were from Merck Life Science (Rome, Italy) unless otherwise stated.
2.2. Architecture of the microfluidic device
The chip layout was designed using CAD software (AutoCAD, Autodesk Inc.). Details about the design are in the ESI.† Briefly, the device encompasses four different units (Fig. 1): i) a serial dilution generator (SDG) for the increasing dilution of the polymer solution; ii) a NA divider to dispense the DNA solution to a downstream region; iii) a mixing area made up of seven mixing units in which the polymer and DNA, coming respectively from the SDG and the NA divider, are mixed to give polyplexes. These are collected within iv) a storage area consisting of seven different tanks, which are eventually punched to make the outlet ports (O1–O7) and recover the polyplex suspensions. | ||
| Fig. 1 The microfluidic cartridge consists of i) an upstream 5-level serial dilution generator (SDG) integrated with herringbone grooves (HBGs), ii) a NA divider, iii) a mixing area, and iv) a storage area. The polymer solution and the water-based medium are injected into the two main inlets (inlets 1 and 2, I1 and I2) through the SDG and moved to the downstream area. An additional inlet (I3) allows for the addition and distribution of the DNA solution to the seven mixing units, where the polymer and DNA are mixed to give rise to polyplexes. These are collected into the storage tanks. Every inlet has a debubbling port. The middle right panel is a magnified view of the staggered HBG units integrated into the cartridge channels. | ||
2.3. Fabrication of the microfluidic device
The master mold was patterned through standard photolithography and consisted of integrated two layers of SU8 negative photoresist (MicroChem, Westborough, MA, USA) composed of i) a 100 μm-thick layer for fluidic channels and tanks and ii) a 36 μm-thick layer for herringbone grooves (HBGs), placed on the top of i). Microfluidic devices were obtained by replica molding of PDMS on the master mold. Briefly, PDMS was cast on the mold (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (w/w) of pre-polymer to curing agent). After degassing at room temperature (r.t) and curing at 65 °C for 3 h, the input (I1–I3) and output (O1–O7) ports were obtained using a biopsy puncher with a 1.5 mm-diameter tip (Harris Uni-Core™). PDMS was bonded to a glass slide (75 mm × 50 mm × 1 mm) through an air plasma treatment (Harrick Plasma), and next heated on a hot plate at 80 °C for 15 min. Production yields were about 90%.
:1 (w/w) of pre-polymer to curing agent). After degassing at room temperature (r.t) and curing at 65 °C for 3 h, the input (I1–I3) and output (O1–O7) ports were obtained using a biopsy puncher with a 1.5 mm-diameter tip (Harris Uni-Core™). PDMS was bonded to a glass slide (75 mm × 50 mm × 1 mm) through an air plasma treatment (Harrick Plasma), and next heated on a hot plate at 80 °C for 15 min. Production yields were about 90%.
2.4. Validation of the microfluidic device
The devices were soaked in sterile PBS in a vacuum chamber connected to a pump and kept under vacuum (at −25 bar) for 15 min and perfused with sterile PBS to remove air bubbles just before use. Any operation was performed under sterile conditions.
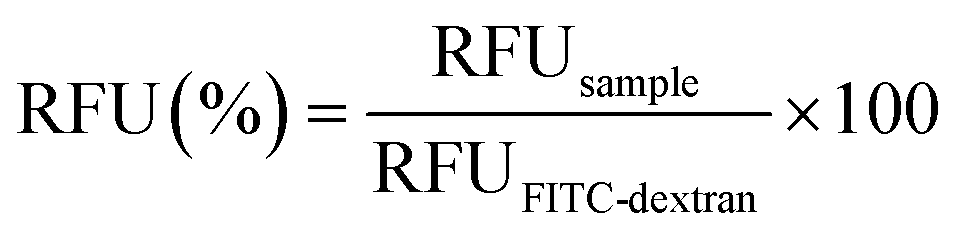 | (1) |
The MI was calculated according to the following equation (eqn (2)):49
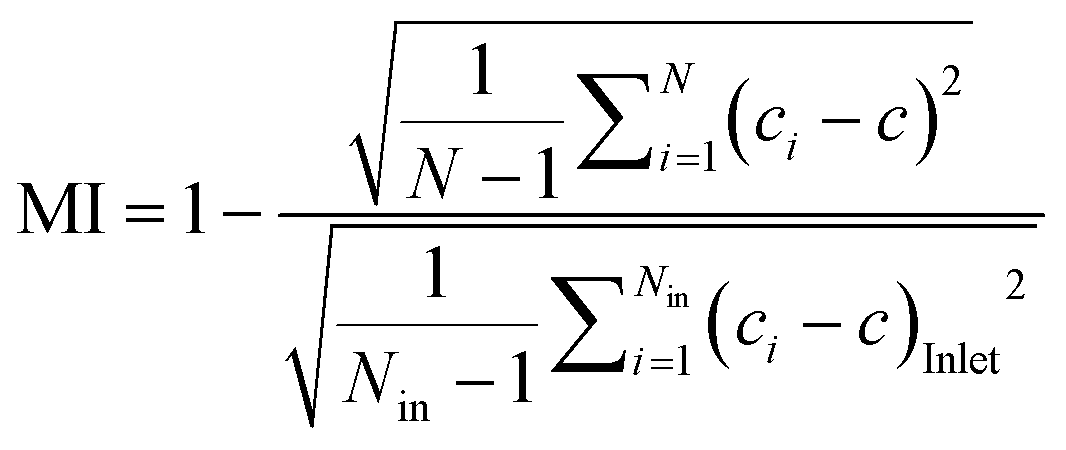 | (2) |
Further, the proper mixing of a cationic polymer solution within the SDG was assessed using the commercial transfection reagent jetPEI-FluoF® (λex = 490 nm, λem = 520 nm). Briefly, equal volumes of the polymer solution (prepared by adding 5 μL of jetPEI-FluoF® to 995 μL of 150 mM NaCl) and 150 mM NaCl were injected through I1 and I2 (flow rates of 25 μL min−1), respectively, and then the fluorescence intensities of the polymer solutions recovered from O1–O7 were measured. Data were expressed in terms of RFU and normalized to the fluorescence value of the jetPEI-FluoF® solution in the syringe, which was taken as 100%, as follows (eqn (3)):
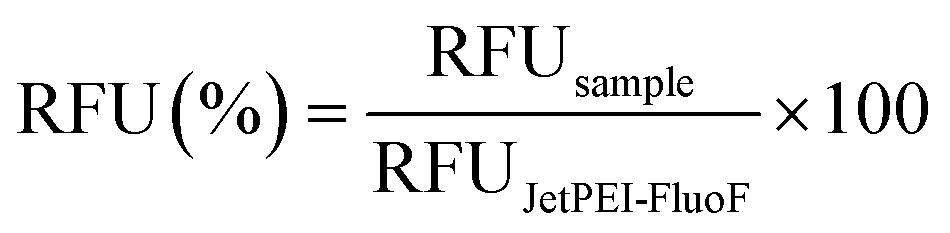 | (3) |
:1 (v/v) salmon sperm DNA (ssDNA; 1 μg μL−1 in 0.1× TE buffer) with 200× SYBR Green I dye (λex = 497 nm, λem = 520 nm), and pumped through I3 into the NA divider utilizing a single-channel syringe pump set at a flow rate of 50 μL min−1, while 10 mM HEPES buffer was injected through I1 and I2 using a two-channel syringe pump (flow rates = 25 μL min−1). Next, 20 μL of the DNA solution were recovered from O1–O7 and analyzed as reported here above. Data were expressed in terms of relative fluorescence units (RFU) and normalized to the fluorescence value of the DNA solution loaded into the syringe, which was taken as 100%, as follows (eqn (4)): | (4) |
To obtain more insight into the proper polymer/DNA mixing, every mixing unit length was measured and compared with each other. Briefly, digital images of the mixing units were acquired using a stereomicroscope (Leica S9, Leica Microsystems, Milan, Italy), and the lengths were measured with ImageJ software.
2.5. Polyplex preparation with the microfluidic device
To ascertain the polyplex formation within the microfluidic device, the polymer/DNA complexation process was monitored by the fluorophore-exclusion titration assay. Briefly, equal volumes of a 25 kDa lPEI solution (prepared at a concentration of 0.017 μg μL−1 in 10 mM HEPES, corresponding to an amine concentration ([N]) of 0.4 mM) and 10 mM HEPES were first injected into the chip through I1 and I2 (flow rates of 25 μL min−1), respectively, and then flowed through the SDG to obtain solutions at seven different polymer concentrations. Such polymer solutions and a fluorescent DNA solution, injected through I3 (flow rate = 50 μL min−1), were simultaneously mixed, as described here above, to give rise to polyplex suspensions at N/Ps from 0 to 6 (hereafter referred to as operation mode 1). Right after complexation, micrographs of the mixing units containing the polyplexes were taken by means of a fluorescence microscope and processed using ImageJ software. Next, 20 μL of polyplexes were recovered from O1–O7 and analyzed as reported here above. Data were expressed in terms of relative fluorescence units (RFU) and normalized to the fluorescence value of the fluorescent DNA solution loaded into the syringe, which was taken as 100%, as follows (eqn (5)):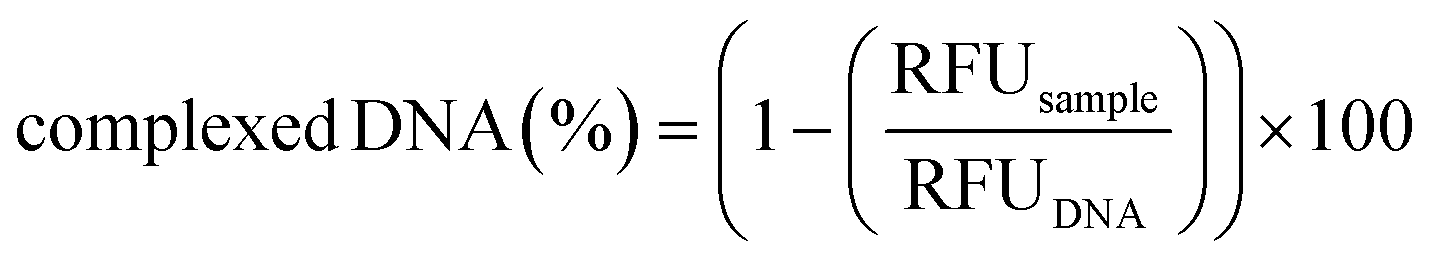 | (5) |
2.6. Biophysical characterization of polyplexes
The size (expressed in terms of hydrodynamic diameter, DH), the polydispersity index (PDI), and the surface charge (expressed in terms of zeta potential, ζP) of the complexes were measured at 25 °C by dynamic light scattering (DLS) and laser doppler micro-electrophoresis, respectively, using Malvern Zetasizer Nano ZS instruments (Malvern PANalytical, Malvern, UK), fitted with a 5 mV HeNe laser (λ = 633 nm) and a scattering angle of 173°. Briefly, 50 μL of polyplex suspensions at N/P 40 (1 μg of DNA) (hereafter referred to as operation mode 2) were prepared with the cartridge as described above, and then diluted to 1:10 (v/v) in 10 mM HEPES. The samples were allowed to equilibrate at 25 °C for 5 min prior to measurements.
The physicochemical properties of polyplexes prepared with the microfluidic device were compared with those of complexes prepared manually (hands-on preparation). Details about the manual preparation of polyplexes are in the ESI.†
2.7. In vitro transfection experiments
Twenty-four hours after polyplex delivery, the cytotoxicity was evaluated by means of the AlamarBlue® assay, according to the manufacturer's instructions. Briefly, the culture medium was replaced with 100 μL per well of 1× resazurin dye solution in complete culture medium. Next, plated cells were kept in the dark for 2 h under standard culture conditions, and then the fluorescence was read by means of a Synergy H1 reader (λex = 540 nm; λem = 595 nm). The viability of non-transfected cells (negative control) was assigned to 100% and the cytotoxicity was determined according to the following equation (eqn (6)):
| cytotoxicity [%] = 100% − viability [%] | (6) |
The transfection efficiencies of complexes prepared with the cartridge were compared to those of manually-prepared polyplexes.
2.8. Statistical analysis
Statistical analysis was carried out with Minitab® 21.1.1 statistical software. All data were initially analyzed using the Kolmogorov–Smirnov normality test. As an indicator of reproducibility and repeatability of the results, comparisons among groups (operation mode 1) were performed with the test for two variances using Bonett's method to test whether variances of transfection efficiencies and cytotoxicities were homogeneous or not. Comparisons among the seven outlets (operation mode 2) were performed by the one-way analysis of variance (ANOVA). Statistical significance was retained when the p-value < 0.05. Every experiment was independently repeated at least in triplicate.3. Results and discussion
3.1. Design of the microfluidic device
In recent years, microfluidics has emerged as an attractive technology for the semi-automated generation of different kinds of nanoparticles.18,19,40 Given the great potential of microfluidics for the preparation of non-viral gene delivery (nano)carriers, the number of publications related to this topic has been steadily increasing.42–45,51–54 Nevertheless, to the best of our knowledge, no one has tackled the daunting quest for the simultaneous generation of gene delivery complexes over a range of N/Ps. As this is the very first practical problem that scientists have to face to gauge the performance of transfection reagents,6 we decided to address this issue. Another equally important yet often neglected issue in science is the reliability and reproducibility of results55 that we sought to tackle utilizing the microfluidic chip we herein devise.The cartridge encompasses four functionally distinct modules: i) a chaotic serial dilution generator (SDG); ii) a NA divider; iii) a mixing area; iv) a storage region (Fig. 1). The i) SDG features two input ports (inlets 1 and 2, I1 and I2) and is based on a resistive flow model consisting of a symmetric microfluidic network25 arranged on 5 levels of 240 μm-wide and 100 μm-high channels. Their length was properly dimensioned (refer to ESI† S1.1) to mix and split the stock solutions (i.e., an aqueous medium and a polymer solution) and give rise to seven linearly diluted solutions (from 100% stock to 0% in steps of 16.7%), independently addressed to the iii) downstream mixing area.
Optimized staggered HBGs56 are integral to the ceiling of each channel (refer to the ESI† S1.2), and each channel encompasses 12 serial HBG units (Table S1†).
Besides, the ii) NA divider area was based on a one-input (I3) resistive flow model of seven microfluidic channels, designed and properly sized to enable equal partition of the stock DNA solution into the mixing area. As for the SDG, the seven polymer–NA mixing units that make up the mixing area were dimensioned in length (refer to ESI† S1.3) to mix the polymer solutions at different concentrations from the SDG with those from the NA divider. Each mixing unit is a 1.64 mm-long channel encompassing 12 serial HBG units (Fig. S3†). Overall, the design of the SDG and the mixing area ensured uniform mixing, that is, a mixing efficiency (ME) > 95%, as previously estimated in a previous paper from our group.56
Furthermore, a iv) storage area comprising seven parallelepiped-shaped reservoirs was designed downstream to collect the polyplex suspensions. The microfluidic cartridge accounted for the simultaneous and repeated generation of non-viral gene delivery assemblies at seven different N/Ps and their automated collection into storage tanks.
3.2. Validation of the SDG and the NA divider
To thoroughly validate the entire chip, we decided to break the whole task down into manageable chunks and assess the operation of functional areas individually, and eventually spot single points of failure.The functionality of the SDG was evaluated qualitatively in terms of the ability to mix highly diffusive dyes and generate a linear concentration gradient. The blue and yellow dyes were injected into the SDG through the inlets and properly mixed within and split between the microchannels to give a palette of seven distinct colors (Fig. 2A).
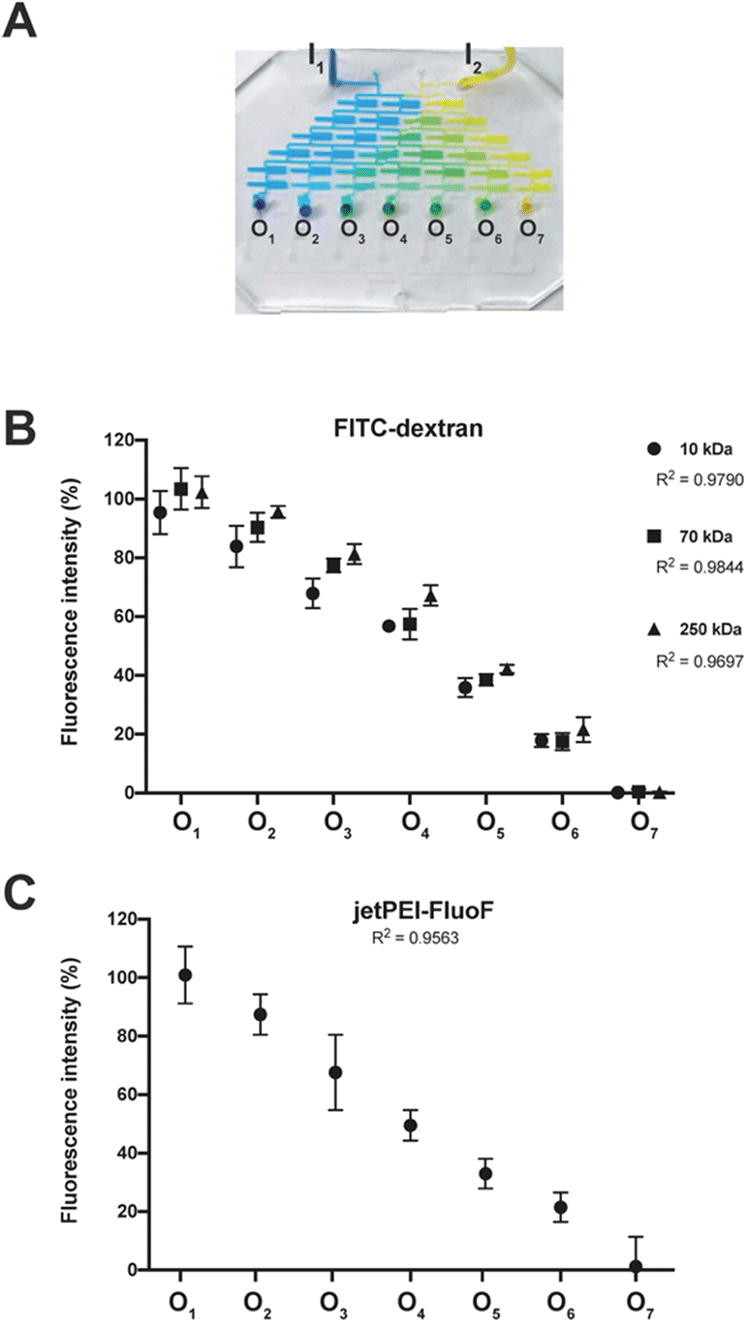 | ||
| Fig. 2 A) The device was perfused with two edible dyes (blue and yellow) from the inlets I1 and I2, and the generation of gradient was visually inspected. Fluorescence was recorded at each of the seven outlets (O1–O7) of the SDG after injecting (B) the solutions of FITC-dextran at different molecular weights (MW: 10 kDa, 70 kDa, and 250 kDa) and (C) the jetPEI-FluoF® solution. Fluorescence intensities were expressed as percentage fluorescence loss in comparison to the fluorescence of FITC-dextran and jetPEI stock solutions. Results are expressed as mean ± SD (n = 3). | ||
As no leaks were observed and the results were highly reproducible on repeated testing and different devices, this was the first anecdotal evidence that the SDG was fit for purpose from a practical standpoint.
Such results spurred us to further validate the function of the SDG with (macro)molecules with sizes of practical relevance. To accomplish this, we availed of an array of three fluorescently-labeled dextrans and evaluated the effectiveness of the SDG to generate a linear gradient of polymers which differ by more than one order of magnitude in size. Of note, as shown in Fig. 2B, the linear decrease in fluorescence observed from O1 to O7 and the high coefficients of determination (R2) of 0.98, 0.98, and 0.97 for 10 kDa, 70 kDa, and 250 kDa FITC-dextran, respectively, are the stark evidence of the SDG suitability for mixing and serially diluting any kinds of FITC-dextran. Moreover, to further assess the effective mixing capability of the device, we did estimate the characteristic MI of the SDG. Specifically, the characteristic MI of a single serpentine resulted to be equal to 0.915, thus confirming the reliability of the mixing.
Besides, as the SDG was designed to mix and split (poly)cations, but dextrans are essentially neutral molecules, the proper function of this area was assessed using a solution of the polymeric transfection reagent jetPEI-FluoF®. As the fluorescence signals of the polymer stock solution injected through I1 and the polymer solution at the exit from O1 were identical, we concluded that the transfection reagent did not significantly adsorb to the PDMS channels. This finding implies that we could gain strict control of the polymer concentrations at the cartridge outlets by injecting a specific polymer concentration upstream. Additionally, the linear decrease in fluorescence observed from O1 to O7 (R2 = 0.96) (Fig. 2C) was evidence that the polymer was linearly diluted while passing through the SDG. Besides, as the function of the mixing area depends on uniform mixing across the different mixing units, these have to be equal to each other to work properly. Of note, the seven mixing units showed a comparable length of ∼ 25 mm (p > 0.05) (Table S2†).
These results highlight that the device was well suited to serially dilute the transfection reagents to be next used to prepare polyplexes at different N/Ps.
To assess the effectiveness of the NA divider to dispense a DNA stock solution to each of the seven polymer–DNA mixing units, the DNA was made detectable using the SYBR Green I fluorescent dye. Of note, we noticed no significant difference in fluorescence intensity between the DNA solutions harvested from O1–O7 (Fig. S4†), meaning that the NA divider operated properly.
In summary, we validated the function of the individual chip areas, which are operated by microsyringe pumps controlling the flow rate of the solutions injected into the cartridge.
3.3. Assessment of the polyplex preparation with the microfluidic device
We took advantage of the DNA complexation assay to assess the effectiveness of the microfluidic cartridge for the simultaneous production and recovery of polyplexes at different N/Ps (Fig. 3). We hypothesized that the cartridge could profitably be used to investigate the NA complexation behavior of transfection reagents in place of classical assays carried out in multiwell plates.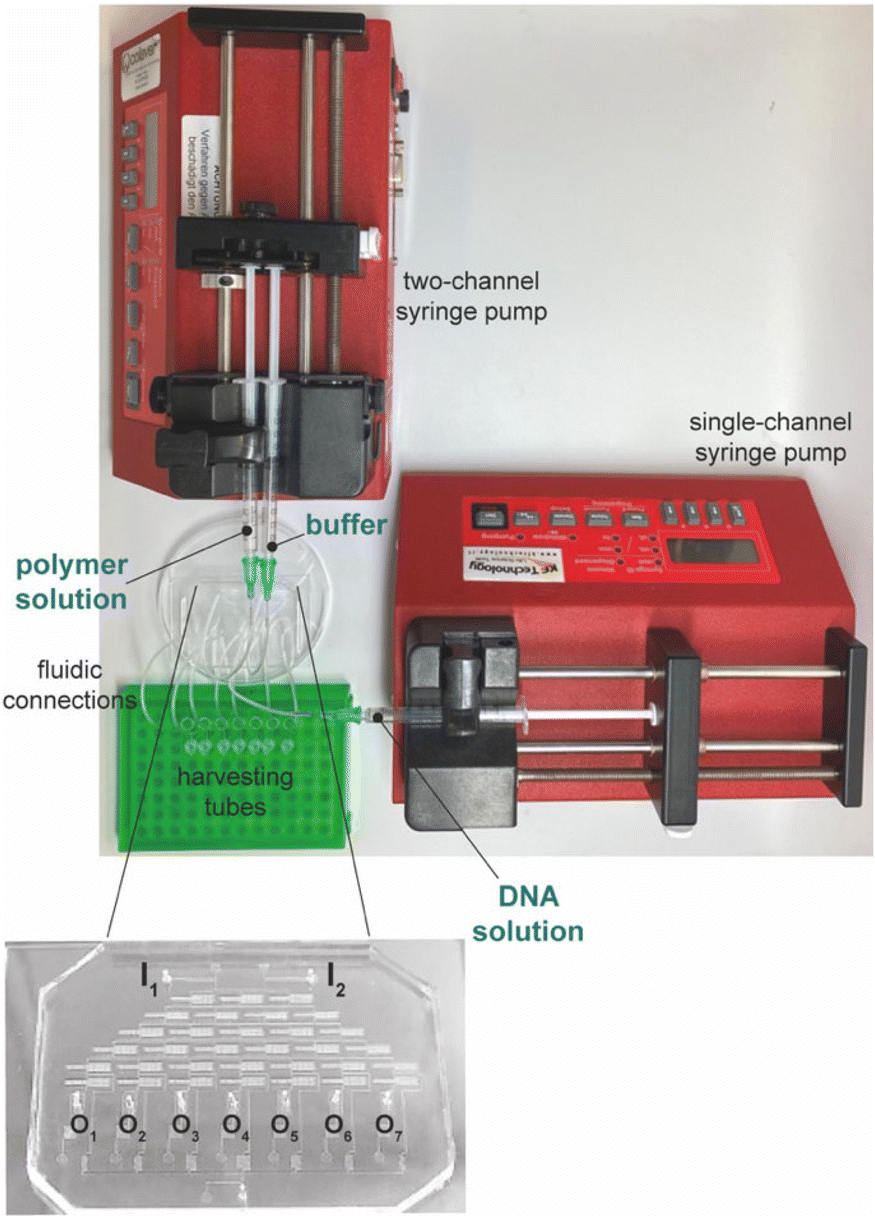 | ||
| Fig. 3 The two-channel syringe pump (in the top left of the picture) holding the two syringes filled with i) the polymer solution and ii) the buffer was connected to the microfluidic cartridge through the I1 and I2 inlet ports. The single-channel syringe driver in the bottom right corner holding a syringe containing DNA solution was connected to I3 of the mixing area through fluidic connections. The polyplex suspensions were recovered into seven harvesting microvials held in a test tube rack because storage tanks were punched with a hole puncher to allow polyplexes to spill from the different outlet ports (O1–O7) into the vials. The inset in the lower-left-hand corner of the picture displays the magnified photo of the microfluidic chip. | ||
The polymer used in the present study is a gold standard transfection reagent known to fully complex the NAs at N/P ≥ 5. Under these conditions, the polymer completely displaces the fluorochrome while binding to the DNA, such that no fluorescence is detected once the samples are excited.6,57,58
To spot any difference in DNA complexation between the mixing units, we imaged them using a fluorescence microscope (Fig. 4A), and the fluorescence signal of every polyplex suspension recovered from the cartridge was quantified with a spectrofluorometer (Fig. 4B). Of note, as lPEI and DNA were blended and the complexation took place while moving downwards through the mixing units, the fluorescence steadily decreased (by way of example, refer to N/P 2 in Fig. 4A). In good agreement with the literature,10,58,59 N/Ps 6 and 5, which correspond to the mixing units 1 and 2, showed maximal DNA complexation due to the relatively large excess of the cationic polymer (Fig. 4).
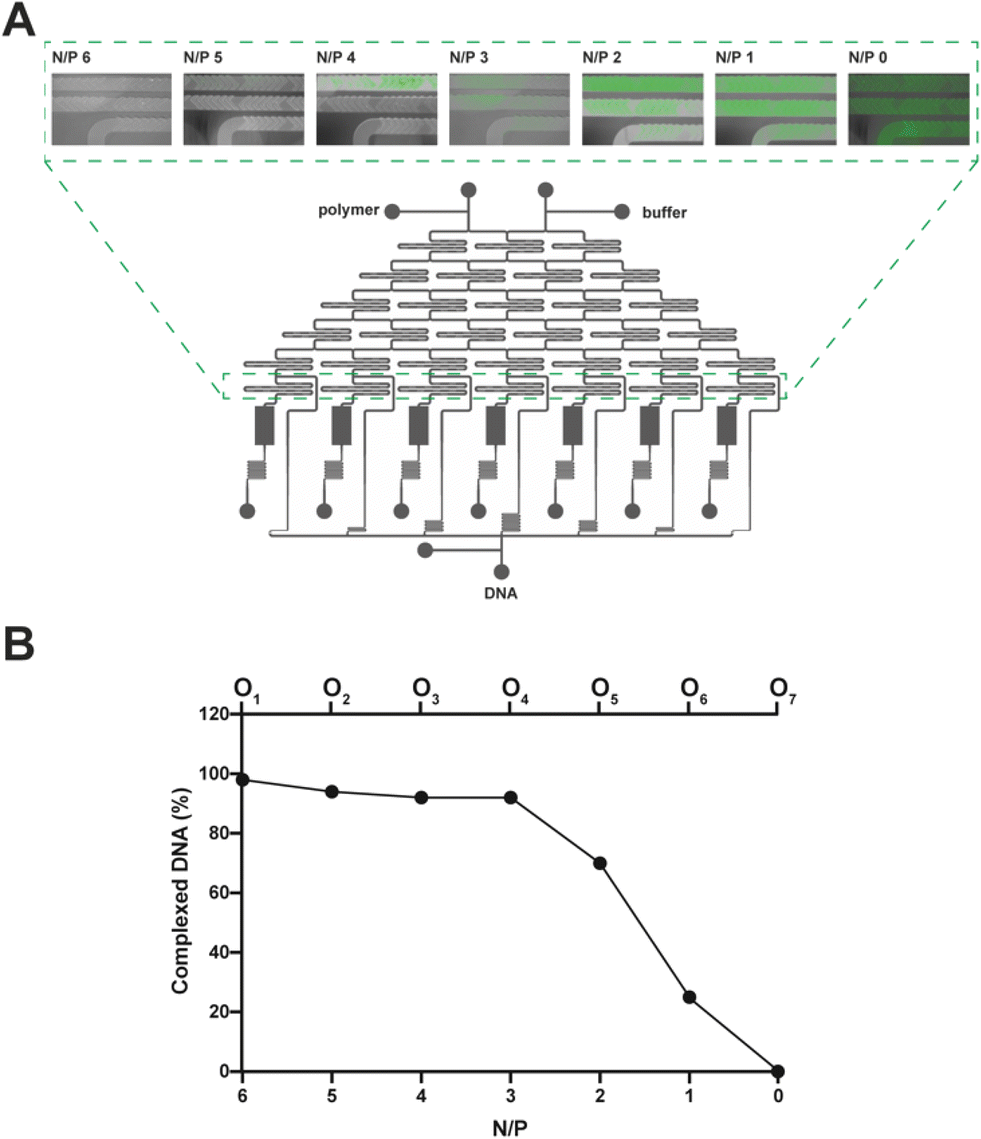 | ||
| Fig. 4 A) Acquisition of fluorescence images of the seven different mixing units. The fluorescence increases as the N/P decreases. Polyplexes at different N/Ps are next collected in the downstream storage tanks that may feature outlet ports (O1–O7). B) Complexed DNA (%) in each outlet port was evaluated by monitoring the fluorescence as a function of the N/P (n = 3). | ||
It is worth noting that the extent of DNA complexation was also rather high at N/Ps 4 and 3, as the residual fluorescence was less than 10% of the pristine DNA shown in O7 (N/P 0) (Fig. 4B).6,57 Afterward, the percentage of complexed DNA decreased as the N/P decreased, meaning that there was more and more uncomplexed DNA in O5 and O6.
These results point to the potential of using the microfluidic cartridge described herein to obtain gene delivery complexes over a given range of N/Ps.
3.4. Validation of the chip function through the biophysical characterization of polyplexes
At need, the microfluidic cartridge can be used in two distinct operation modes. One type of operation (operation mode 1) ensures the concurrent preparation of polyplexes at seven N/Ps. Besides, it also allows one to identify the most effective polymer-to-NA ratio in complexation, and transfection eventually. Accordingly, the DH, ζP, and PDI of polyplexes prepared at different N/Ps through the microfluidic cartridge were compared with each other (Table S3†). However, as polyplexes prepared at varying N/Ps are intrinsically different from one another and have different behaviors, we could not rule out any difference in the function among the chip units. Nevertheless, the data reported in Table S3† highlighted the suitability of the microfluidic cartridge in obtaining narrower polyplex populations if compared to manual counterparts.On the other hand, operation mode 2 would enable the production of significant volumes of polyplex suspensions at a pre-defined N/P. We took advantage of the latter set-up to verify the proper function of the single units and validate the platform for such use. This experimental setting allows for the injection of the cationic polymer solution through both I1 and I2, and the NAs through I3, to produce polyplexes at a single polymer-to-NA ratio, which here was the very effective N/P 40.57 Of note, polyplexes were invariably small (DH ≈ 145 nm) and cationic (ζP ≈ +29 mV). Most importantly, the polyplex populations were monodisperse (PDI = 0.2) and very similar across the outlets (Table S4†). Conversely, manually-prepared complexes obtained by pipetting and mixing PEI and NAs were slightly bigger (DH ≈ 176 nm) and less charged (ζP ≈ +7 mV), and definitely polydisperse (PDI = 0.5), in good agreement with previous findings.9
These results disclose this microfluidic cartridge as an effective tool to obtain narrower populations of non-viral gene delivery particles in a semi-automated and more repeatable manner than the standard way of mixing NAs and transfection reagents.
3.5. Evaluation of polyplex efficacy in vitro
To further validate and provide evidence of the practical usefulness of the device, we carried out cell transfection assays with polyplexes prepared using the microfluidic cartridge. Polyplexes were prepared with lPEI and bPEI transfection reagents because their behavior is extremely well-known.60–6325 kDa lPEI and the pGLuc plasmid encoding the secreted luciferase enzyme were mixed through the chip following operation mode 2 to give rise to seven polyplex suspensions invariably at N/P 40. L929 fibroblasts were the recipient cells used to assess the transfection behavior of single polyplex preparations and compare the effectiveness of the chip units with each other. Besides, we benchmarked each of them against polyplexes prepared by manual pipetting. To best visualize any difference among groups, the results are displayed as box-and-whisker plots (Fig. S5†) because the center, spread, and overall range of each distribution are immediately apparent.
Of note, there were no significant differences in cytotoxicity and transfection efficiency among polyplexes from the different device units (p > 0.05 with ANOVA) (Fig. S5†). This result is another piece of evidence of the high repeatability of the device in preparing non-viral gene delivery assemblies.
On the other hand, the polyplex populations produced in every cartridge unit were very similar and monodisperse, resulting in narrower distributions of the transfection efficiencies and cytotoxicities than the polydisperse, manually prepared complexes (p < 0.05 with test for two variances, on-chip vs. manual preparations).
Since these results were highly reproducible across independent testing, we decided to substantiate the benefits of using the microfluidic cartridge to repeatably generate polyplexes over a range of N/Ps (operation mode 1). To do so comprehensively, 25 kDa lPEI and 25 kDa bPEI were used to complex plasmids encoding different luciferase enzymes (pGLuc and pGL3) and transfect three cell lines. The different pDNAs were complexed through the device and manually using different polymer concentrations to yield a linear N/P scale spanning from 0 to 60 with N/P steps of 10 (Fig. 5 and S6†) and dripped onto L929 cells (Fig. 5A and B), HeLa cells (Fig. 5C and D), and Jurkat cells (Fig. S6†) in culture. Again, regardless of the N/P, the cytotoxicities (refer to empty boxes in Fig. 5A and C, and S6A†) and transfection efficiencies given by polyplexes produced with the microfluidic cartridge were remarkably homogeneous (refer to empty boxes in Fig. 5B and D, and S6B†). Indeed, it is apparent at a glance that each dataset shows very short whiskers and small box sizes, indicating data points consistently hovering around the center value. Besides, the results of microfluidic preparations were more repeatable than the manually-prepared counterparts (Fig. 5), even using two different plasmids and cells (p < 0.05 in test for two variances).
 | ||
| Fig. 5 A and C) Cytotoxicity and (B and D) transfection efficiency of polyplexes prepared with the microfluidic mixing (operation mode 1) (empty boxes) and by manual pipetting (red striped boxes), mixing variable amounts of linear PEI (lPEI) and a constant concentration of pGLuc (A and B) or pGL3 (C and D) in 10 mM HEPES buffer to give different N/Ps (0, 10, 20, 30, 40, 50, and 60). Results on L929 (A and B) and HeLa cells (C and D) are displayed as box and whisker plots (n ≥ 3). *p < 0.05, **p < 0.01, and ***p < 0.001 between microfluidic and manual preparations (in test for two variances). | ||
Overall, these results disclose this microfluidic platform as a reliable and effective tool for the concurrent generation of non-viral gene delivery assemblies over a wide range of N/Ps.
4. Conclusions
Recent years have witnessed the use of microfluidics to shape different types of nanoparticles, such as gene delivery complexes. This work addresses the on-chip production of non-viral gene delivery assemblies across a range of N/Ps. Here above, we described the development and validation of a user-friendly, dual-mode microfluidic cartridge that enables the preparation of non-viral gene delivery assemblies in a reproducible manner. In particular, the chaotic mixing of the polymer and DNA solutions through the cartridge results in the rapid self-assembly of reagents. This enables the effortless and repeatable preparation of polyplex suspensions at distinct (operation mode 1) or single (operation mode 2) N/P with an exceptionally narrow size distribution. Gene delivery complexes are next automatically harvested in built-in, dedicated storage units. These features make this cartridge the first example of a stand-alone, disposable device to produce, recover, and store non-viral gene delivery particles at desired N/Ps. Also, easy change of parameters (e.g., temperature, pH and concentration of reagents and flow velocity) allows for better control of the particle production process. In perspective, the daily use of this device would assist scholars in fast-tracking of the screening of transfection reagents through error-free, simultaneous production of gene delivery assemblies at different N/Ps.Future work will address the effects of in-cartridge storage conditions (i.e., temperature, time) of non-viral gene delivery complexes on their stability and effectiveness. Besides, the microfluidic cartridge will be validated with other kinds of NAs and transfection reagents.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Politecnico di Milano for financial support. The device manufacture was partially performed at Polifab, the micro- and nanofabrication facility of Politecnico di Milano.Notes and references
- M. Lim, A. Z. M. Badruddoza, J. Firdous, M. Azad, A. Mannan, T. A. Al-Hilal, C. S. Cho and M. A. Islam, Pharmaceutics, 2020, 12, 1–30 Search PubMed.
- I. Lostalé-Seijo and J. Montenegro, Nat. Rev. Chem., 2018, 2, 258–277 CrossRef.
- R. Ni, R. Feng and Y. Chau, Life, 2019, 9, 1–28 CrossRef PubMed.
- H. Zu and D. Gao, AAPS J., 2021, 23, 78 CrossRef PubMed.
- C. K. Chen, P. K. Huang, W. C. Law, C. H. Chu, N. T. Chen and L. W. Lo, Int. J. Nanomed., 2020 DOI:10.2147/IJN.S222419.
- N. Bono, et al. , Pharmaceutics, 2020, 12(2), 183 CrossRef CAS PubMed.
- S. Mirón-Barroso, E. B. Domènech and S. Trigueros, Int. J. Mol. Sci., 2021, 22, 8537 CrossRef PubMed.
- D. Pezzoli and G. Candiani, J. Nanopart. Res., 2013, 15, 1523 CrossRef.
- D. Pezzoli, E. Giupponi, D. Mantovani and G. Candiani, Sci. Rep., 2017, 7, 1–11 CrossRef PubMed.
- Y. Yue, F. Jin, R. Deng, J. Cai, Y. Chen, M. C. M. Lin, H. F. Kung and C. Wu, J. Controlled Release, 2011, 155, 67–76 CrossRef CAS PubMed.
- G. Lippi, G. Lima-Oliveira, G. Brocco, A. Bassi and G. L. Salvagno, Clin. Chem. Lab. Med., 2017, 55, 962–966 CAS.
- F. Sousa, L. Passarinha and J. A. Queiroz, Biotechnol. Genet. Eng. Rev., 2009, 26, 83–116 CrossRef.
- E. Farris, K. Heck, A. T. Lampe, D. M. Brown, A. E. Ramer-Tait and A. K. Pannier, Curr. Opin. Biomed. Eng., 2018, 7, 51–57 CrossRef PubMed.
- A. Tejeda-Mansir, A. García-Rendón and P. Guerrero-Germán, Biotechnol. Genet. Eng. Rev., 2019, 35, 46–68 CrossRef CAS PubMed.
- A. Tejeda-Mansir and R. Montesinos, Recent Pat. Biotechnol., 2008, 2, 156–172 CrossRef CAS PubMed.
- R. Yang, Y. Deng, B. Huang, L. Huang, A. Lin, Y. Li, W. Wang, J. Liu, S. Lu, Z. Zhan, Y. Wang, R. A. W. Wang, P. Niu, L. Zhao, S. Li, X. Ma, L. Zhang, Y. Zhang, W. Yao, X. Liang, J. Zhao, Z. Liu, X. Peng, H. Li and W. Tan, Signal Transduction Targeted Ther., 2021, 6, 213 CrossRef CAS PubMed.
- G. M. Whitesides, Nature, 2006, 442, 368–373 CrossRef CAS PubMed.
- Q. Feng, J. Sun and X. Jiang, Nanoscale, 2016, 8, 12430–12443 RSC.
- N. Hao, Y. Nie and J. X. J. Zhang, Int. Mater. Rev., 2018, 63, 461–487 CrossRef CAS.
- W. J. Duncanson, T. Lin, A. R. Abate, S. Seiffert, R. K. Shah and D. A. Weitz, Lab Chip, 2012, 12, 2135–2145 RSC.
- L. Capretto, D. Carugo, S. Mazzitelli, C. Nastruzzi and X. Zhang, Adv. Drug Delivery Rev., 2013, 65, 1496–1532 CrossRef CAS PubMed.
- F. Tian, L. Cai, C. Liu and J. Sun, Lab Chip, 2022, 22, 512–529 RSC.
- J. El-Ali, P. K. Sorger and K. F. Jensen, Nature, 2006, 442, 403–411 CrossRef CAS PubMed.
- J. Melin and S. R. Quake, Annu. Rev. Biophys. Biomol. Struct., 2007, 36, 213–231 CrossRef CAS PubMed.
- N. L. Jeon, S. K. W. Dertinger, D. T. Chiu, I. S. Choi, A. D. Stroock and G. M. Whitesides, Langmuir, 2000, 16, 8311–8316 CrossRef CAS.
- J. Kim, I. Hwang, D. Britain, T. D. Chung, Y. Sun and D. H. Kim, Lab Chip, 2011, 11, 3941–3948 RSC.
- M. A. Tomeh and X. Zhao, Mol. Pharmaceutics, 2020, 17, 4421–4434 CrossRef CAS PubMed.
- E. Quagliarini, S. Renzi, L. Digiacomo, F. Giulimondi, B. Sartori, H. Amenitsch, V. Tassinari, L. Masuelli, R. Bei, L. Cui, J. Wang, A. Amici, C. Marchini, D. Pozzi and G. Caracciolo, Pharmaceutics, 2021, 13, 1–12 CrossRef PubMed.
- J. C. Kasper, D. Schaffert, M. Ogris, E. Wagner and W. Friess, Eur. J. Pharm. Biopharm., 2011, 77, 182–185 CrossRef CAS PubMed.
- J. A. Kulkarni, J. L. Myhre, S. Chen, Y. Y. C. Tam, A. Danescu, J. M. Richman and P. R. Cullis, Nanomedicine, 2017, 13, 1377–1387 CrossRef CAS PubMed.
- D. Zukancic, E. J. A. Suys, E. H. Pilkington, A. Algarni, H. Al-Wassiti and N. P. Truong, Pharmaceutics, 2020, 12, 1068 CrossRef CAS PubMed.
- X. Li, M. Aghaamoo, S. Liu, D.-H. Lee, A. P. Lee, X. Li, M. Aghaamoo, D. Lee, A. P. Lee and S. Liu, Small, 2018, 14, 1802055 CrossRef PubMed.
- D. M. Loy, R. Krzysztoń, U. Lächelt, J. O. Rädler and E. Wagner, Process., 2021, 9, 129 CrossRef CAS.
- S. J. Shepherd, C. C. Warzecha, S. Yadavali, R. El-Mayta, M. G. Alameh, L. Wang, D. Weissman, J. M. Wilson, D. Issadore and M. J. Mitchell, Nano Lett., 2021, 21, 5671–5680 CrossRef CAS PubMed.
- G. K. Chee, X. Kang, Y. Xie, Z. Fei, J. Guan, B. Yu, X. Zhang and L. J. Lee, Mol. Pharmaceutics, 2009, 6, 1333–1342 CrossRef PubMed.
- L. Digiacomo, S. Palchetti, D. Pozzi, A. Amici, G. Caracciolo and C. Marchini, Biochem. Biophys. Res. Commun., 2018, 503, 508–512 CrossRef CAS PubMed.
- P. M. Valencia, P. A. Basto, L. Zhang, M. Rhee, R. Langer, O. C. Farokhzad and R. Karnik, ACS Nano, 2010, 4, 1671–1679 CrossRef CAS PubMed.
- N. M. Belliveau, J. Huft, P. J. Lin, S. Chen, A. K. Leung, T. J. Leaver, A. W. Wild, J. B. Lee, R. J. Taylor, Y. K. Tam, C. L. Hansen and P. R. Cullis, Mol. Ther.–Nucleic Acids, 2012, 1, e37 CrossRef PubMed.
- A. D. Stroock, S. K. W. Dertinger, A. Ajdari, I. Mezić, H. A. Stone and G. M. Whitesides, Science, 2002, 295, 647–651 CrossRef CAS PubMed.
- H. Hu, C. Yang, M. Li, D. Shao, H. Q. Mao and K. W. Leong, Mater. Today, 2021, 42, 99–116 CrossRef CAS PubMed.
- J. A. Jarrell, A. A. Twite, K. H. W. J. Lau, M. N. Kashani, A. A. Lievano, J. Acevedo, C. Priest, J. Nieva, D. Gottlieb and R. S. Pawell, Sci. Rep., 2019, 9, 1–11 CrossRef CAS PubMed.
- H. Aghaei and A. R. S. Nazar, Ind. Eng. Chem. Res., 2019, 58, 23032–23045 CrossRef CAS.
- S. J. Shepherd, D. Issadore and M. J. Mitchell, Biomaterials, 2021, 274, 120826 CrossRef CAS PubMed.
- P. C. O. S. Firmino, S. S. V. Vianna, O. M. M. M. da Costa, A. A. Malfatti-Gasperini, A. L. Gobbi, R. S. Lima and L. G. de la Torre, Lab Chip, 2021, 21, 2971–2985 RSC.
- A. Bokare, A. Takami, J. H. Kim, A. Dong, A. Chen, R. Valerio, S. Gunn and F. Erogbogbo, ACS Omega, 2019, 4, 4650–4657 CrossRef CAS PubMed.
- E. Giupponi, R. Visone, P. Occhetta, F. Colombo, M. Rasponi and G. Candiani, Biotechnol. Bioeng., 2018, 115, 775–784 CrossRef CAS PubMed.
- A. Casadevall and F. C. Fang, Infect. Immun., 2010, 78, 4972–4975 CrossRef CAS PubMed.
- A. L. Plant and R. J. Hanisch, Harvard Data Science Review, 2018, pp. 1–21 Search PubMed.
- I. H. Oevreeide, A. Zoellner and B. T. Stokke, Micromachines, 2021, 12(5) DOI:10.3390/mi12050556.
- I. H. Oevreeide, A. Zoellner, M. M. Mielnik and B. T. Stokke, J. Micromech. Microeng., 2021, 31(1) DOI:10.1088/1361-6439/abc820.
- D. P. Feldmann, J. Heyza, C. M. Zimmermann, S. M. Patrick and O. M. Merkel, Molecules, 2020, 25, 1–12 Search PubMed.
- D. M. Loy, P. M. Klein, R. Krzysztoń, U. Lächelt, J. O. Rädler and E. Wagner, PeerJ Mater. Sci., 2019, 1, e1 CrossRef.
- B. Carvalho, B. Ceccato, M. Michelon, S. Han and L. de la Torre, Pharmaceutics, 2022, 14, 141 CrossRef CAS PubMed.
- D. P. Feldmann, Y. Xie, S. K. Jones, D. Yu, A. Moszczynska and O. M. Merkel, Nanotechnology, 2017, 28, 224001 CrossRef PubMed.
- S. Laraway, S. Snycerski, S. Pradhan and B. E. Huitema, Perspect. Behav. Sci., 2019, 42, 33–57 CrossRef PubMed.
- P. Occhetta, N. Glass, E. Otte, M. Rasponi and J. J. Cooper-White, Integr. Biol., 2016, 8, 194–204 CrossRef CAS PubMed.
- C. Malloggi, D. Pezzoli, L. Magagnin, L. De Nardo, D. Mantovani, E. Tallarita and G. Candiani, Polym. Chem., 2015, 6, 6325–6339 RSC.
- N. Bono, C. Pennetta, A. Sganappa, E. Giupponi, F. Sansone, A. Volonterio and G. Candiani, Int. J. Pharm., 2018, 549, 436–445 CrossRef CAS PubMed.
- D. K. Bonner, X. Zhao, H. Buss, R. Langer and P. T. Hammond, J. Controlled Release, 2013, 167, 101–107 CrossRef CAS PubMed.
- F. Ponti, N. Bono, L. Russo, P. Bigini, D. Mantovani and G. Candiani, J. Nanobiotechnol., 2022, 20, 1–19 CrossRef PubMed.
- P. Erbacher, J. S. Remy and J. P. Behr, Gene Ther., 1999, 61, 138–145 CrossRef PubMed.
- A. P. Pandey and K. K. Sawant, Mater. Sci. Eng., C, 2016, 68, 904–918 CrossRef CAS PubMed.
- E. V. B. Van Gaal, R. Van Eijk, R. S. Oosting, R. J. Kok, W. E. Hennink, D. J. A. Crommelin and E. Mastrobattista, J. Controlled Release, 2011, 154, 218–232 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2lc00744d |
| This journal is © The Royal Society of Chemistry 2023 |