
Rapid separation of bacteria from primary nasal samples using inertial microfluidics†
Jesus
Shrestha‡
 ab,
Sajad
Razavi Bazaz‡
a,
Lin
Ding
a,
Steven
Vasilescu
a,
Sobia
Idrees
c,
Bill
Söderström
d,
Philip M.
Hansbro
c,
Maliheh
Ghadiri
ab and
Majid
Ebrahimi Warkiani
*aef
ab,
Sajad
Razavi Bazaz‡
a,
Lin
Ding
a,
Steven
Vasilescu
a,
Sobia
Idrees
c,
Bill
Söderström
d,
Philip M.
Hansbro
c,
Maliheh
Ghadiri
ab and
Majid
Ebrahimi Warkiani
*aef
aSchool of Biomedical Engineering, University of Technology Sydney, Sydney, New South Wales 2007, Australia. E-mail: majid.warkiani@uts.edu.au
bWoolcock Institute of Medical Research, Respiratory Technology Group, University of Sydney, Sydney, New South Wales 2037, Australia
cCentre for Inflammation, Centenary Institute and University of Technology Sydney, Faculty of Science, School of Life Sciences, University of Technology Sydney, Sydney 2007, Australia
dAustralian Institute for Microbiology and Infection, Faculty of Science, University of Technology Sydney, New South Wales 2007, Australia
eInstitute for Biomedical Materials and Devices, Faculty of Science, University of Technology Sydney, New South Wales 2007, Australia
fInstitute of Molecular Medicine, Sechenov First Moscow State University, Moscow 119991, Russia
First published on 7th December 2022
Abstract
Microbial populations play a crucial role in human health and the development of many diseases. These diseases often arise from the explosive proliferation of opportunistic bacteria, such as those in the nasal cavity. Recently, there have been increases in the prevalence of these opportunistic pathogens displaying antibiotic resistance. Thus, the study of the nasal microbiota and its bacterial diversity is critical in understanding pathogenesis and developing microbial-based therapies for well-known and emerging diseases. However, the isolation and analysis of these populations for clinical study complicates the already challenging task of identifying and profiling potentially harmful bacteria. Existing methods are limited by low sample throughput, expensive labeling, and low recovery of bacteria with ineffective removal of cells and debris. In this study, we propose a novel microfluidic channel with a zigzag configuration for enhanced isolation and detection of bacteria from human clinical nasal swabs. This microfluidic zigzag channel separates the bacteria from epithelial cells and debris by size differential focusing. As such, pure bacterial cell fractions devoid of large contaminating debris or epithelial cells are obtained. DNA sequencing performed on the separated bacteria defines the diversity and species present. This novel method of bacterial separation is simple, robust, rapid, and cost-effective and has the potential to be used for the rapid identification of bacterial cell populations from clinical samples.
1. Introduction
Extensive misuse of antibacterial drugs has led to a massive global public health problem in the form of antimicrobial resistance (AMR).1,2 AMR impacts opportunistic and life-threatening infections, compounding their severity and leading to an increase in hospital stay, treatment costs, morbidity, and mortality rates, all of which place a tremendous burden on healthcare. Moreover, the prevalence of opportunistic pathogens in the nasal cavity with the ability to resist standard antibiotics is increasing.3 The development and exacerbation of diseases such as chronic rhinosinusitis (CRS), allergic rhinitis, asthma, otitis media, pneumonia, chronic obstructive pulmonary disease (COPD), and cystic fibrosis (CF) are related to these opportunistic pathogens.4–8 Standard clinical procedures to profile antibiotic resistance are phenotypic sample culture-dependent assays and drug resistance susceptibility tests, taking days to produce results. Standard practice for treating bacterial infections typically entails broad-spectrum antibiotics, which are usually inadequate, expensive, and sometimes fatal in cases of sepsis or patients with other underlying comorbidities.9,10 Moreover, these treatments contribute to the emergence of resistant organisms and increase AMR.11 Thus, it is critical to rapidly identify disease- or exacerbation-causing bacteria and the antibacterial resistance (ABR) profile without the need for culture. Microbial community studies can identify bacterial diversity and abundance, providing insights into the health status of the individual and initiating the most effective antibiotic regimens for improved clinical outcomes.5–7,12The application of numerous culture-based methods developed to identify bacteria in clinical samples (e.g., blood, sputum, faeces) is limited by time, cost, efficiency, sample dilution, and detection.8,10,13 Molecular diagnostic methods based on amplification, such as real-time polymerase chain reaction and fluorescence in situ hybridization, allow rapid bacterial detection and characterization.14,15 However, they still involve lengthy sample preparations and have a low sensitivity at typical bacterial levels.16,17 The biological samples used for these tests typically contain primary cells, proteins, and endogenous inhibitors that may interfere with polymerases used in these assays or interact with the extracted DNA, decreasing the efficiency of molecular detection techniques.9,18 This severely limits the reliability and usability of these methods, which can substantially benefit from sample purification prior to use. One of the most popular methods of bacterial enrichment is ultra-centrifugation. However, it is a time-consuming process that can generate loads of genetic materials from contaminated epithelial cells19 and even alter the interior structures of bacteria, including DNA.20 Alternative methods, such as chemical and magnetic sorting techniques, rely on targeting specific antigens, which can irreversibly damage bacterial cells or lose a subpopulation due to the low expression of certain markers.21 Moreover, prior knowledge of bacterial species is required to choose the proper markers, limiting the methods to identify unknown species.9,10 Primary samples often have small sample volumes with low bacterial counts. Thus, to enable effective and reliable omics analysis and profiling of bacterial populations, the bacterial separation process needs to be rapid, have high throughput, and be able to leave the mRNA and protein expression profiles of bacteria intact.22
Microfluidic-based particle separation technologies have proven to be well suited for isolating target cells from heterogeneous cell suspensions.21,23–26 However, most of these techniques are limited by low throughput and low separation efficiency. Also, they are often prone to clogging, hindering non-continuous operations.27,28 Recently, inertial microfluidics has been used for size-based particle separation by leveraging the effects of microfluidic channel structure and hydrodynamic forces. This has enabled the separation of various cell types such as circulating tumour cells, plasma, or platelets from whole blood, allowing continuous and high-throughput cell separation without clogging.29–35 Inertial microfluidics allows separated cell fractions to be quickly transferred for downstream analysis and post-processing. The flexibility of inertial microfluidics allows the separation of diagnostically and therapeutically significant target cells from body fluids such as blood, urine, vaginal secretions, semen, cerebrospinal fluid, and saliva.21 Often these devices are tailored towards separating relatively large cells and particulates above the range of bacterial cells. Thus, developing new microfluidic devices able to enrich bacterial cells is appealing. Microfluidic solutions for rapid separation of bacteria from human clinical samples such as swabs and aspirates are not well studied and investigated. This could have broad applicability and make bacterial profiling more accessible, time-efficient, and cost-effective.
Here, we present a novel microfluidic channel with a sharp zigzag pattern for enhanced enrichment of bacteria from human nasal swabs. This is the first report of DNA sequencing-based bacterial detection from clinical samples through the use of inertial microfluidics. Samples containing nasal epithelial cells, debris, and bacteria were collected from the inferior turbinate of both nares. A polydimethylsiloxane (PDMS)-made microfluidic zigzag channel was used to separate bacteria from epithelial cells and debris by size differential focusing on the device. This resulted in pure bacterial cell fractions devoid of contaminating debris or epithelial cells without requiring centrifugation, labeling with antibodies, or human intervention. DNA sequencing was then performed on separated bacteria to identify nasal microbiota. These results demonstrate that zigzag channels can be used to recover significant amounts of bacteria from clinical samples, producing pure bacteria within minutes. Rapid enrichment of bacteria from patient samples along with the identifying bacteria they contain and their ABR profile using microfluidic sample processing has the potential to enable prompt diagnosis and effective treatment in the future.
2. Experimental section
2.1. Mold and device fabrication
Devices were fabricated using a previously reported PDMS-based soft lithography technique.36 The mold was designed in SolidWorks 2016 and printed using a DLP 3D printer (MiiCraft Ultra 50, MiiCraft, Hsinchu, Taiwan).37 The mold was then washed with isopropyl alcohol (IPA) 3 times, dried with pressurised air, and cured under 385–405 nm UV light for 5 minutes. To ensure the removal of any resin residue on the mold's surface, the mold was dipped in IPA for 3 hours. Finally, the mold was plasma treated for 2 minutes (Basic Plasma cleaner PDC-002, Harrick Plasma) and left in a vacuum pot with 50 μL trichloro(1H,1H,2H,2H-perfluoro-octyl)silane (Sigma-Aldrich, Australia) for 6 hours. Following silanisation, PDMS (made in a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of polymer to cross-linker) was cast over the mold and the mold was incubated at 45 °C for 5 hours. The PDMS channel was then peeled from the mold, which was punched with a 1.0 mm biopsy punch and permanently bonded to a PDMS base with plasma treatment (Basic Plasma cleaner PDC-002, Harrick Plasma).
:1 ratio of polymer to cross-linker) was cast over the mold and the mold was incubated at 45 °C for 5 hours. The PDMS channel was then peeled from the mold, which was punched with a 1.0 mm biopsy punch and permanently bonded to a PDMS base with plasma treatment (Basic Plasma cleaner PDC-002, Harrick Plasma).
2.2. Device testing
The performance of the microfluidic zigzag channel was first characterised by observing the focusing behaviour of different-sized fluorescent microbeads. The cross-section dimensions of the zigzag channel were 120 μm × 40 μm (width × height). Fluorescent microparticles (1, 3, 10, 15, and 20 μm, MagSphere, USA) were diluted in 15 mL of MACS buffer (Miltenyi Biotec, Australia) with ∼500000 particles per mL and filled into 10 mL BD plastic syringes. 1 μm beads were used to represent bacteria, while beads 3 μm and above were used to represent epithelial cells and debris. The syringes were then loaded onto a syringe pump (Fusion 200, Chemyx Inc), and particles were pumped through the device at flow rates of 50, 70, 90, 150, and 200 μL min−1; the video for each was recorded using Camtasia (TechSmith, USA). Samples from the middle and side outlets were collected in 1.5 mL PCR tubes, and the distribution of particle sizes was determined using a CytoFLEX LX flow cytometer (Beckman Coulter, USA). The separation efficiency (SE) of the microbeads was calculated using the equation SE = Ntargetoutlet/(Ntargetoutlet + Notheroutlet), where N is the number of particles collected and counted from each outlet. The counting was repeated three times.
2.3. Ethics approval
The study was conducted according to approvals from the Royal Prince Alfred Human Research Ethics Committee (X19-0172) and in accordance with the Declaration of Helsinki, the Australian Code of Responsible Research (2007), and the ‘National Statement’ on Ethical Conduct in Human Research (2007). All subjects provided written informed consent.2.4. Cell culture and preparation
Epithelial cells (PD07i) were grown and maintained in 75 cm2 flasks in EPILIFE medium supplemented with human keratinocyte growth supplement (HKGS) and 100 μg ml−1 penicillin/streptomycin (Gibco, ThermoFisher, Australia). To introduce the cells in the channel, cultures were first trypsinized (incubated with trypsin + EDTA for 10 minutes, then stopped by 1× DTI) and subsequently washed twice and resuspended in 1× PBS. A sample with a concentration of ∼6 × 106 cells per mL was collected. The culture was stained with the membrane dye FM4-64 (3 μg mL−1) for 5 minutes and washed three times in 1× PBS before being fixed in 4% paraformaldehyde for 20 minutes on ice. Fixed cells were washed twice in 1× PBS and stored at 4 °C until needed.2.5. Bacterial growth and preparation
The uropathogenic E. coil (UPEC) strain UTI89 (serotype O18:K1:H7), originally isolated from a cystic patient,38 was transformed with plasmid pGI5 (ref. 39) to enable constitutive expression of sfGFP in the cytoplasm for visualization. Following overnight growth at 37 °C without shaking in rich LB medium, the cultures were back-diluted 1:50 in fresh medium (LB broth) and allowed to grow to a final concentration of ∼8 × 108 cells per mL. Cells were fixed as previously described;40 in short, cultures were mixed in 70% ice-cold methanol (f.c.) for one hour, washed three times in 1× PBS, and stored at 4 °C until needed.
2.6. Sample preparation
Nasal epithelial cells were collected from the inferior turbinate of both nares of 5 individuals using disposable cytology brushes (2 mm diameter; Olympus Medical Supplies, Japan). Donors were asked to blow their noses to clear excess fluid from the nasal passages before the collection. The cytology brushes were inserted until they reached the length half the distance between the donors' noses and ears. Once at the collecting site, the brushes were rotated back and forth for ∼5 seconds to collect epithelial samples. Caution was taken to avoid contact with the nares. Samples were immediately transferred into sterile Eppendorf tubes containing PBS without calcium or magnesium (PBS, Corning). The collected swabs were processed using the microfluidic zigzag channel.2.7. Bacterial cell separation
Bovine serum albumin (BSA, 0.1 wt%, Sigma-Aldrich) was added into the microfluidic device and incubated for 10 minutes before loading buffer solution with the clinical samples to avoid non-specific binding of nasal epithelial cells and bacteria to the channel walls and tubing surfaces. The chip and connecting tubes were also primed with BSA. The device was screened via a microscope to ensure that no air bubbles were trapped. Buffer solutions of clinical samples were loaded into 1 ml syringes and connected to a syringe pump (Fusion 200, Chemyx Inc.). A short ramp-up time was allowed until the focusing of epithelial cells in the central outlet was visualized with a high-speed camera (Phantom Camera) before collecting the solutions from the outlets in separate vials. Cells obtained from the central outlet were recorded using a high-speed camera and counted using a hemocytometer. Separated bacterial samples were further analyzed using microscopy. The primary nasal epithelial cells collected from the central outlet were immediately transferred into sterile tubes containing collection and transport medium (Medium199 supplemented with 1% penicillin/streptomycin (v/v); Gibco, USA). Cells were cultured in human collagen-coated containers (Sigma, USA) using bronchial epithelial growth medium (BEGM) supplemented with BEGM SingleQuots (Lonza, Switzerland). The medium was replenished every 48 hours.2.8. Microscopy
For visualization with optical microscopy, epithelial and bacterial cells were mixed just prior to microfluidic sorting and imaging. After flowing through the microfluidic zigzag channels, samples from the side and central outlets were placed on pre-made agarose pads (1–1.5% w/w) in M9 minimal medium in 65 μl gene frames (Thermo Scientific, AB0577), left to immobilize and imaged within 5 minutes. Epifluorescence imaging was performed on a Nikon (Ti2-E) N-STORM v5 with NIS v.5.30 using a 100× 1.49 NA oil objective. Cover glass slides were washed with 95% EtOH, air dried, cleaned for at least 3 minutes with a plasma cleaner (Harrick plasma, PDC-23G), and used within 15 minutes of cleaning. Excitation of fluorophores was performed using a Lumencor Spectra II module with a 20 ms acquisition time. GFP emission was collected through a FITC filter and FM4-64 emission through a Cy5 filter (Semrock), and images were captured using an sCMOS Flash 4.0 v3 (Hamamatsu) camera.For SEM analysis, one drop of samples from the side outlet channel was placed on coverslips and fixed for 1 hour with 4% paraformaldehyde. Samples were washed three times with PBS and then dehydrated in a series of graded ethanol solutions of 50%, 60%, 70%, 80%, and 90% and twice in 100% (vol/vol) for 10 min each and dried in a desiccator overnight. Samples were then mounted on SEM specimen stubs using conductive carbon tape and coated with a thin layer (10 nm) of Au using a sputter coater. The resulting samples were imaged using an SEM (Zeiss SUPRA 55 VP, USA).
2.9. DNA extraction
Qiagen DNeasy® PowerSoil® Pro-Kit along with Qiagen TissueLyser® II was used to extract DNA from the collected nasal samples using a modified procedure.41 Eluted DNA samples (60 μL) were stored at −80 °C before further processing. De-identified samples of extracted bacterial DNA were then sent for sequencing.2.10. 16S rRNA gene sequencing, profiling and analysis
3. Results and discussion
3.1. Evaluation of zigzag microfluidic device
Here, we have developed a zigzag microfluidic channel to isolate and enrich bacterial cells from clinical nasal swabs. The fundamentals and principles of particle focusing within a zigzag microchannel have been discussed by our group recently.44 This particular microfluidic device leverages the effects of inertial migration via properly adjusting the inertial lift and Dean drag forces within the zigzag microchannel to separate bacterial cells from larger background cell populations and debris. Inertial migration is defined as the positioning of randomly dispersed particles toward certain equilibrium positions after a sufficiently long particle migration process.45 Inertial focusing happens in intermediate Reynolds numbers (1 < Re < 100) (Re = ρUDh/μ, where ρ is the fluid density, μ is the dynamic viscosity, Dh is the hydraulic diameter of the channel cross-section, and U is the average flow), allowing the fluid flow to sit between Stokes and turbulent flow regimes. In straight channels, inertial migration results from two inertial forces: shear gradient and wall-induced lift forces. The shear gradient lift force pushes the particles toward the channel wall, while wall-induced lift forces repel particles from the channel walls. The balance of these two forces defines the equilibrium position of different particles.46 The overall inertial lift force (FL) applied on a particle can be calculated viaeqn (1),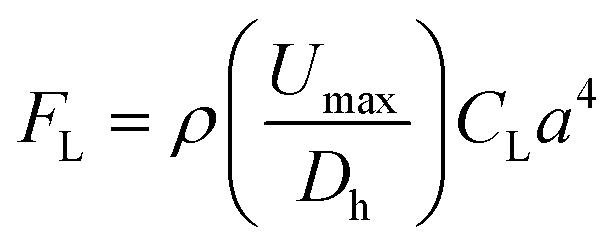 | (1) |
In zigzag channels, however, the mismatch between fluid momentums in the center and wall regions of the chip contributes to a pressure gradient along the transverse direction. Consequently, a secondary force called Dean force with alternating direction is introduced. Fluid near the centreline of the channel will tend to move outward due to the higher momentum than fluid near the outer wall of the channel. The introduction of the Dean force accelerates focusing and increases the separation efficiency of particles. Compared to other serpentine-type microchannels such as curvilinear and square-wave devices, zigzag microfluidics shows superior particle focusing accuracy and efficiency, as we have shown previously.44 This is because, in zigzag microchannels, cells and particles experience sudden cross-sectional changes (Lagrangian point of view) at the corners after passing each loop of the channel, resulting in alteration of Dean drag and inertial lift force applied to the particles, which is missing in square-wave and curvilinear microchannels, which have the same cross-section throughout the channel length. As the cross-section expands at the corners of zigzag microchannels, the channel aspect ratio increases, resulting in narrower focusing bands and more stable equilibrium positions. Also, zigzag channels have a smaller footprint, shorter particle positioning time, and more significant parallelization potential than straight channels.
Within the zigzag microchannel, there exist three focusing regimes. At a low flow rate, all particles occupy two focusing positions along the sidewalls of the channels. These two-sided focusing positions are in the Dean drag force dominant area since FD overcomes FL at a low flow rate. Increasing the flow rate enhances inertial lift forces; therefore, the particle focusing mode shifts towards one single line focusing position. The regime between these two focusing modes is called a transition regime. Also, particles with a larger diameter are more influenced by inertial lift force and require less length for successful focusing (FL ∝ a4). When a particle is in the inertial regime while another is in the drag-dominant regime, the separation of particles with different sizes occurs. We show that our channel made by PDMS with a cross-section of 120 μm (width) × 40 μm (height) is capable of collecting particles at the channel centerline with a diameter of more than 3 μm, while particles smaller than 3 μm are dispersed throughout the channel. In this study, we have adjusted the fluid resistance at the outlets to have the same volume as from the center and side outlets. This assists in pushing particles, cells, and debris ∼3 μm in diameter and above towards the center outlet, leaving a pure stream of bacteria to exit from the side outlets. Since the minimum length for successful particle migration is estimated by Lmin = 2πμDh3/ρUa3, we have selected a zigzag microchannel with 20 effective loops, as schematically illustrated in Fig. 1.
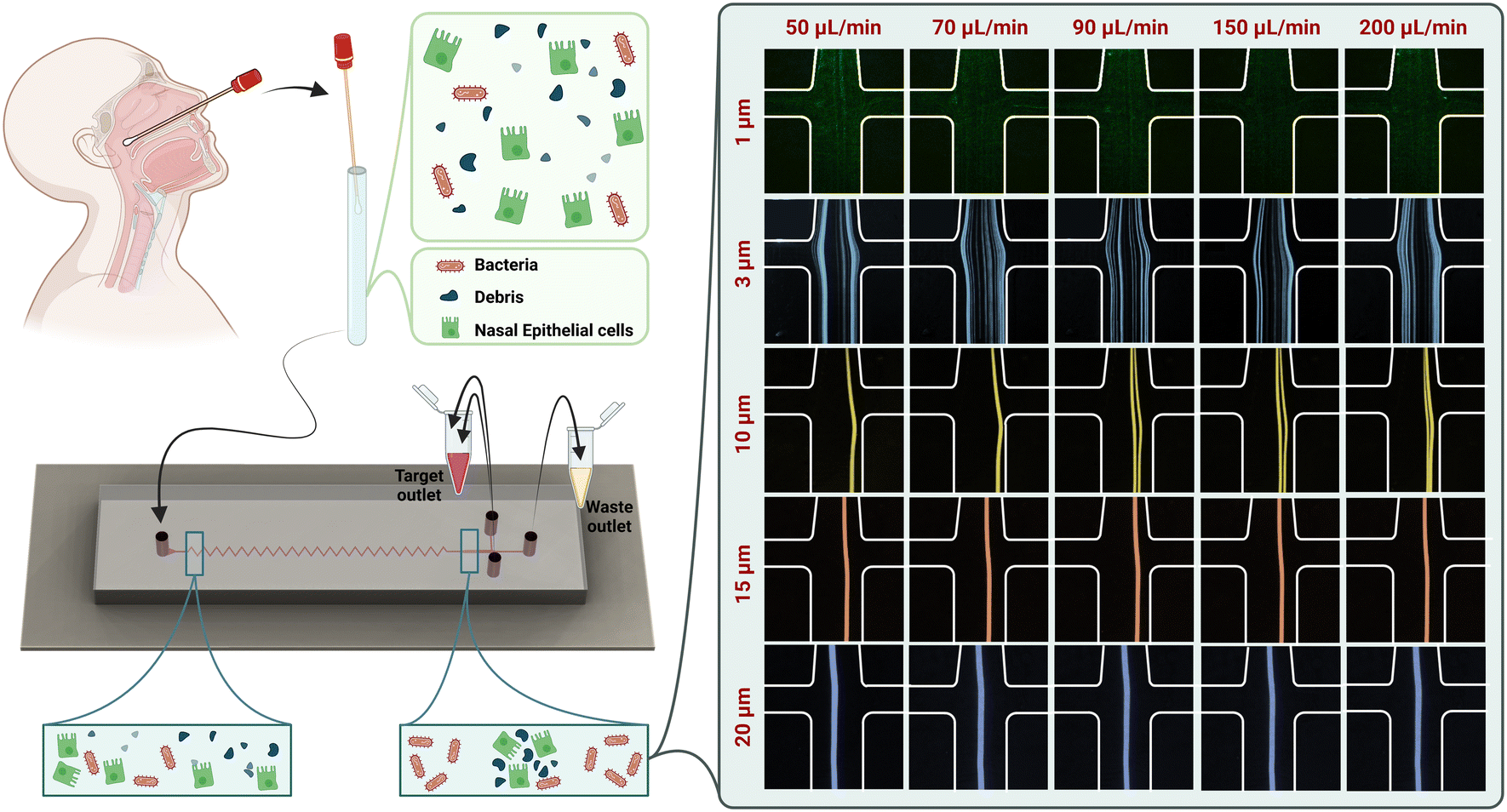 | ||
| Fig. 1 Experimental setup and use of the device: nasopharyngeal swabs are taken from the inferior turbinate of both nares and suspended in PBS for microfluidic separation of bacteria from epithelial cells and cellular debris. The sample is then introduced into the zigzag microchannel. Bacteria are collected from the channel side, while other debris and nasal epithelial cells are collected from the center channel. The device performance in the presence of different particles by increasing the flow rate from 50 to 200 μL min−1 is demonstrated. | ||
To assess the focusing behaviour of particles in our chip, we used fluorescent microbeads of various diameters at different flow rates to observe their focusing positions. The focusing positions of different microbeads in the outlet of our zigzag microfluidic device are different at different flow rates (Fig. 1). The diameters of beads chosen are representative of various bacteria, cells, and debris present in mixed sample cell suspensions. Beads of 1 μm, representing bacterial cells, were scattered in both central and side outlets at flow rates of 50, 70, 90, 150, and 200 μL min−1. The remaining 3, 10, 15, and 20 μm microbeads were all collected at the central outlet for each flow rate tested. Considering the microchannel performance in the presence of beads and the microchannel backpressure and stability, we have selected the flow rate of 150 μL min−1 to continue the sample processing.
A mixture of 1, 3, and 10 μm beads at the flow rate of 150 μL min−1 was tested to determine the separation efficiency of the microfluidic device. The number of particles from the input and central and side outlets was analyzed by flow cytometry, and the separation efficiency of each bead size was determined (Fig. 2A). More than 95% of 3 μm beads and almost 99% of 10 μm beads were collected from the central outlet (Fig. 2B). Of 1 μm size beads, 49.82 ± 4% were retrieved from the side outlet (Fig. 2C). Samples collected from the side outlet were recirculated through the microfluidic device; as a result, overall separation efficiency was further increased to 72.37 ± 5%.
 | ||
| Fig. 2 Evaluation of the separation efficiency of the zigzag channel. (A) Separation of 1, 3, and 10 μm particles with the separation efficiency at each outlet. The samples collected from the side outlet were passed through the device for the second time. The separation efficiency increased to 72.37 ± 5%. (B) Micrograph of the mixture of 1, 3, and 10 μm particles collected from the central outlet of the microfluidic zigzag channel (scale bar: 50 μm). (C) Micrograph of the 1 μm particles collected from the side outlets (scale bar: 50 μm). | ||
3.2. Bacterial separation from the actual sample
The performance of the zigzag microfluidic channel with actual samples was evaluated in the ability to separate bacteria from epithelial cells. For this experiment, pathogenic E. coli cells were mixed with epithelial cells. The size difference between the bacteria and the eukaryotic cells is about one order of magnitude. Since the epithelial cells are relatively large, they are influenced mainly by inertial lift forces, occupy the center of the channel cross-section, and are collected from the center outlet. However, bacteria are small enough not to be influenced by inertial lift or Dean drag forces and are dispersed in the channel. Almost 99% of the epithelial cells focused on the central outlet, whereas the E. coli cells showed about 50% extraction yield from the side outlets in the first run. The samples collected from the central outlet were rerun in the device, with an increased separation efficiency of E. coli up to 71.43% (Fig. 3A). Fig. 3C and D show brightfield and stained micrographs of epithelial cells with cellular debris collected from the center outlet and cell-free samples collected from the side outlets that contain pure bacteria. These results clearly demonstrated the efficacy of the zigzag channel. Based on these results, the device was tested with cell suspensions from primary nasopharyngeal swabs diluted in 500 μl of PBS and injected into the device at 150 μL min−1 flow rate. Almost 99% of nasal cells and cellular debris were collected from the central outlet (Fig. 3E and Video S1, ESI†). These results demonstrate the successful separation of nasal epithelial cells and debris in the central outlet and of bacterial samples devoid of cells in the side outlets. | ||
| Fig. 3 Evaluation of the bacterial separation using the zigzag channel. (A) Separation of the epithelial cells and bacteria with the separation efficiency in each outlet. The samples collected from the side outlet were passed through the device for the second time. The separation efficiency of the bacteria, i.e., E. coli, increased to 71.43%. (B) The focusing position of the epithelial cells and debris through the central outlet at 150 μL min−1 while bacteria are dispersed throughout the channel. (C) Brightfield images of epithelial cells focused in the central outlet with cellular debris and cell-free samples collected from the side outlets with pure bacteria (scale bar: 100 μm). (D) Micrographs of stained epithelial cells (magenta) and E. coli (green) collected from the central outlet and the side outlets. The E. coli express free sfGFP in the cytoplasm for visualization, while the epithelial cells are stained for membranes (with FM4-64). Scale bar: 20 μm. (E) Haemocytometer micrograph of primary nasal epithelial cells focused on the central outlet with no cells observed in the side outlets (scale bar: 100 μm). (F) Micrographs of brightfield and DAPI-stained cultured human nasal epithelial cells collected from the central outlet (scale bar: 50 μm). N = 3 for the number of cultured clinical samples. | ||
Traditionally, the microfluidic separation of submicron-sized particles like bacteria from such a small sample volume has been met with little success, especially when the bacterial count in the samples is relatively low. More importantly, to perform metagenomic or metaproteomic analyses, the separation process must be rapid with high throughput to prevent changes in mRNA or protein expression profiles.22 The microfluidic zigzag channel retrieved a visibly pure background-free fraction of concentrated bacteria from nasopharyngeal swabs in 4 minutes. After successfully separating the background cell population from the collected bacteria, the primary nasal epithelial cells from the central outlet were immediately transferred to 24-well cell culture plates. All samples were successfully cultured without contamination (Fig. 3F).
To identify the bacteria present in these complex clinical samples, it is essential to enrich them and remove the background larger particles. Since epithelial cells, cellular and environmental debris, hair, and dust particles constitute much of the sample volume, the removal efficiency of these particles needs to be high. Furthermore, the separation process needs to have high throughput to process samples rapidly without applying high shear forces that induce cellular damage. Such damage would cause the release of cell contents that induce bacterial damage and/or growth and host DNA contaminating bacterial DNA. In the microfluidic zigzag channels, the maximum force calculated was ∼2g, which is significantly lower than the force generated in ultracentrifuges: ∼5000–20000g.20 Size-dependent separation is an attractive solution for bacterial isolation, considering bacterial cells are typically 1 μm in diameter, ten times smaller than epithelial cells.47 The microfluidic zigzag channel successfully pushes any particles greater than 3 μm to the central outlet with >95% separation efficiency. Samples were diluted in 500 μl of PBS, and the flow rate was 150 μl min−1 and processed within 4 minutes. Thus, the microfluidic zigzag channel offers a rapid and low-cost separation method with high throughput and high recovery of bacteria from clinical samples.
The bacterial samples separated from the nasopharyngeal swab samples and collected from the side outlet of the device were observed with SEM imaging (Fig. 4A). The diversity of the bacteria in the collected samples was assessed in terms of alpha diversity by Chao1 and the Shannon index, which showed diverse populations in each donor sample (D) (Fig. 4B). We assessed the variation of the nasal microbiota at the family level and the samples collected were dominated by Staphylococcaceae, Streptococcaceae, Sphingomonadaceae, Enterobacteriaceae, Rhodobacteraceae, Propionibacteriaceae and in one sample, predominantly by Musa textilis family (Fig. S1, ESI†). The most common genera identified were Staphylococcus, Sphingomonas, Rubellimicrobium, Propionibacterium, Escherichia–Shigella, and Corynebacterium (Fig. S1, ESI†). At the species level, Musa textilis, Rubellimicrobium species, Propionibacterium acnes, Escherichia coli, Haemophilus parainfluenzae, and Pelomonas saccharophila were the most abundant species identified in the samples (Fig. 4C and D). These findings corroborated numerous studies showing similar taxonomic profiles at the genus and species levels and were consistent with our enrichment methods.12,48–50 The differential and relative abundance values are shown in Tables S1 and S2, ESI.†
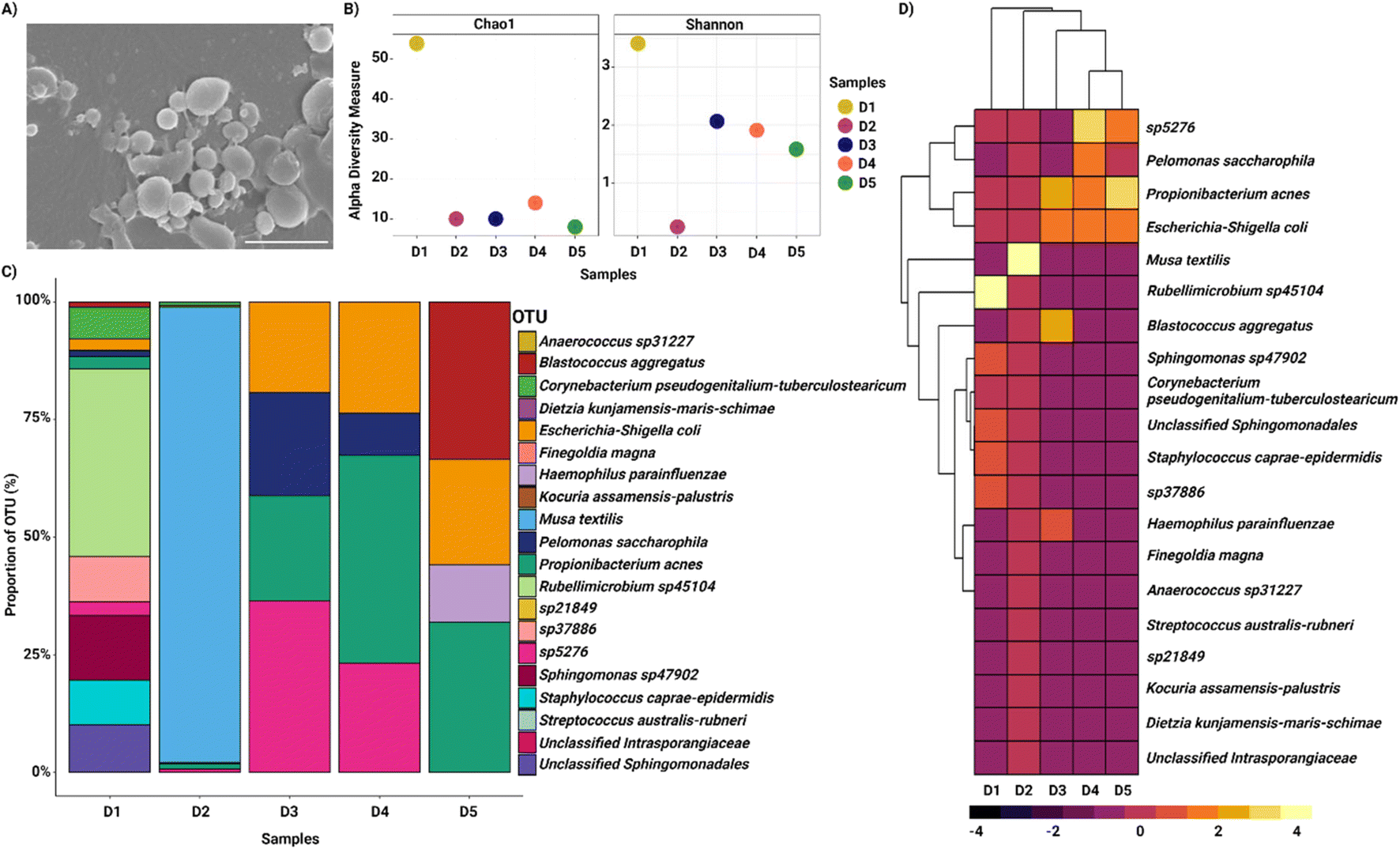 | ||
| Fig. 4 Microbial distribution in the nasopharyngeal swab samples collected from the side outlets of the microfluidic zigzag channels. (A) Representative scanning electron microscopy image of a sample collected from the side outlets of the zigzag channel showing various bacteria (scale bar: 2 μm). (B) Alpha diversity indicators, including Chao1 and Shanon indexes as per the donor samples, show diverse populations in each sample (D – donor). (C) Proportions of OTUs (%) belonging to the different bacterial species classes in the nasal swab samples. (D) Heatmap of abundance profile at the species level of microbiota collected using the microfluidic zigzag channel. The top 20 operational taxonomic units, where lighter shades represent highly abundant while darker shades represent low-abundant bacteria. | ||
Bacterial abundance and diversity are related to disease status.12 Microbial population alterations of dysbiosis are observed in diseases like CRS, CF, asthma, and COPD, which are debilitating and result in a poor quality of life.5,42,51 CF is a serious genetic disorder characterized by reduced mucociliary function and increased bacterial colonization, resulting in patients being more susceptible to chronic infections.52–54 Aggressive and prolonged antibiotic therapy is typically implemented to control infections in CF, which can contribute to developing ABR.55,56 However, with increases in S. aureus infections in CF patients, the prevalence of methicillin-resistant S. aureus has also risen significantly.57,58 Despite advances in the treatment of CF and a marked improvement in life expectancy over the last 20 years, respiratory infection remains the leading cause of morbidity and mortality in patients.57 Thus, improved knowledge of the microbial ecology of CF through culture-independent detection methods is expected to improve infection control initiatives and refined management strategies.57 The first step in refining infection control is the implementation of rapid and reliable sample processing methods to support diagnostics. The zigzag microfluidic channels we have developed can be used to rapidly separate and identify bacteria through molecular techniques and generate resistance profiles for individual patients. Based on the results obtained, selecting appropriate antibiotics and dose adjustments can provide the most effective treatment and ultimately prevent ABR. Furthermore, pre- and post-treatment analysis of microbial characteristics will improve the understanding of the disease status and treatment response.59 This will enable the development of novel therapeutics and the administration of patient-tailored treatments targeting the microbiota or their behaviour to improve health outcomes.
4. Conclusion
Bacterial detection in primary samples becomes an issue when host DNA contaminates bacterial DNA, limiting its recovery of reads during bioinformatics analysis. The ability to separate bacteria from primary clinical samples will enhance the extraction of enriched bacterial DNA, improving bacterial detection and sequencing. To enrich the limited bacteria in clinical samples for identification and further analysis, larger particles need to be removed efficiently and rapidly. The separation process also needs to be high throughput and gentle to prevent host cell lysis, alteration of DNA structures, or protein expression with time. Available methods suffer from the low separation efficiency of bacteria with ineffective removal of cells and debris, expensive labeling, and low throughput. To address these issues, we have developed a microfluidic device with an overall sharp zigzag configuration for separating bacteria from human clinical samples containing epithelial cells and cellular debris in an automated, fully contained process. The microfluidic zigzag channel successfully separated primary nasal epithelial cells and cellular debris in the central outlet with greater than 95% efficiency, achieving a negative selection of bacteria through side outlets. DNA sequencing on separated bacterial fractions revealed similar taxonomic profiles at the genus and species levels normally observed in the nasal cavity. This novel method of bacterial separation is simple, robust, rapid, and cost-effective with high throughput, providing enriched bacteria from human nasal samples. We are of the opinion that our microfluidic device will enable the rapid and accurate identification of bacterial cell populations from clinical samples without the use of centrifugation or filtration and will be applicable for numerous diagnostic and prognostic applications.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
M. E. W. would like to acknowledge the support of the Cancer Institute New South Wales through the Career Development Fellowship. Prof. Hansbro is a recipient of an NHMRC Investigator Grant (1175134) and support from the Rainbow Foundation. Dr. Ghadiri is a recipient of the Ann Woolcock Fellowship from the Woolcock Institute of Medical Research. Dr. Söderström acknowledges support from a data generation fund provided by the Australian Institute for Microbiology and Infection at UTS.References
- J. Y. Ang, E. Ezike and B. I. Asmar, Antibacterial resistance, Indian J. Pediatr., 2004, 71(3), 229–239 CrossRef PubMed.
- World Health Organization, Antimicrobial resistance: global report on surveillance, World Health Organization, 2014 Search PubMed.
- C. B. Creech, D. S. Kernodle, A. Alsentzer, C. Wilson and K. M. Edwards, Increasing rates of nasal carriage of methicillin-resistant Staphylococcus aureus in healthy children, Pediatr. Infect. Dis. J., 2005, 24(7), 617–621 CrossRef PubMed.
- S. Dimitri-Pinheiro, R. Soares and P. Barata, The Microbiome of the Nose—Friend or Foe?, Allergy Rhinol., 2020, 11, 2152656720911605 Search PubMed.
- K. F. Budden, S. D. Shukla, S. F. Rehman, K. L. Bowerman, S. Keely and P. Hugenholtz, et al., Functional effects of the microbiota in chronic respiratory disease, Lancet Respir. Med., 2019, 7(10), 907–920 Search PubMed.
- S. H. Chotirmall, S. L. Gellatly, K. F. Budden, M. Mac Aogáin, S. D. Shukla and D. L. Wood, et al., Microbiomes in respiratory health and disease: an Asia-Pacific perspective, Respirology, 2017, 22(2), 240–250 CrossRef PubMed.
- S. D. Shukla, K. F. Budden, R. Neal and P. M. Hansbro, Microbiome effects on immunity, health and disease in the lung, Clin. Transl. Immunol., 2017, 6(3), e133 Search PubMed.
- J. M. Leung, P. Y. Tiew, M. Mac Aogáin, K. F. Budden, V. F. L. Yong and S. S. Thomas, et al., The role of acute and chronic respiratory colonization and infections in the pathogenesis of COPD, Respirology, 2017, 22(4), 634–650 Search PubMed.
- W. G. Pitt, M. Alizadeh, G. A. Husseini, D. S. McClellan, C. M. Buchanan and C. G. Bledsoe, et al., Rapid separation of bacteria from blood—review and outlook, Biotechnol. Prog., 2016, 32(4), 823–839 Search PubMed.
- C. M. Buchanan, R. L. Wood, T. R. Hoj, M. Alizadeh, C. G. Bledsoe and M. E. Wood, et al., Rapid separation of very low concentrations of bacteria from blood, J. Microbiol. Methods, 2017, 139, 48–53 CrossRef CAS PubMed.
- CDC, Antibiotic Resistance Threats in the United States, 2019, CDC, U.S. Department of Health and Human Services, Atlanta, GA, 2019 Search PubMed.
- K. Biswas, M. Hoggard, R. Jain, M. W. Taylor and R. G. Douglas, The nasal microbiota in health and disease: variation within and between subjects, Front. Microbiol, 2015, 6, 134 Search PubMed.
- K. F. Budden, S. L. Gellatly, D. L. Wood, M. A. Cooper, M. Morrison and P. Hugenholtz, et al., Emerging pathogenic links between microbiota and the gut–lung axis, Nat. Rev. Microbiol., 2017, 15(1), 55–63 CrossRef CAS PubMed.
- R. Ruimy, M. Dos-Santos, L. Raskine, F. Bert, R. Masson and S. Elbaz, et al., Accuracy and potential usefulness of triplex real-time PCR for improving antibiotic treatment of patients with blood cultures showing clustered gram-positive cocci on direct smears, J. Clin. Microbiol., 2008, 46(6), 2045–2051 CrossRef CAS PubMed.
- H. Shen, J. Wang, H. Liu, Z. Li, F. Jiang and F.-B. Wang, et al., Rapid and selective detection of pathogenic bacteria in bloodstream infections with aptamer-based recognition, ACS Appl. Mater. Interfaces, 2016, 8(30), 19371–19378 Search PubMed.
- D.-K. Kang, M. M. Ali, K. Zhang, S. S. Huang, E. Peterson and M. A. Digman, et al., Rapid detection of single bacteria in unprocessed blood using Integrated Comprehensive Droplet Digital Detection, Nat. Commun., 2014, 5(1), 1–10 CrossRef PubMed.
- A. Niemz, T. M. Ferguson and D. S. Boyle, Point-of-care nucleic acid testing for infectious diseases, Trends Biotechnol., 2011, 29(5), 240–250 CrossRef CAS PubMed.
- J. Hedman and P. Rådström, Overcoming inhibition in real-time diagnostic PCR, PCR detection of microbial pathogens, Springer, 2013, pp. 17–48 Search PubMed.
- S. O. Majekodunmi, A review on centrifugation in the pharmaceutical industry, Am. J. Biomed. Eng., 2015, 5(2), 67–78 Search PubMed.
- B. W. Peterson, P. K. Sharma, H. C. van der Mei and H. J. Busscher, Bacterial cell surface damage due to centrifugal compaction, Appl. Environ. Microbiol., 2012, 78(1), 120–125 CrossRef CAS PubMed.
- A. J. Mach and D. Di Carlo, Continuous scalable blood filtration device using inertial microfluidics, Biotechnol. Bioeng., 2010, 107(2), 302–311 Search PubMed.
- Z. Wu, B. Willing, J. Bjerketorp, J. K. Jansson and K. Hjort, Soft inertial microfluidics for high throughput separation of bacteria from human blood cells, Lab Chip, 2009, 9(9), 1193–1199 RSC.
- J.-H. Lee, S.-K. Lee, J.-H. Kim and J.-H. Park, Separation of particles with bacterial size range using the control of sheath flow ratio in spiral microfluidic channel, Sens. Actuators, A, 2019, 286, 211–219 Search PubMed.
- M. A. Raoufi, H. A. N. Joushani, S. Razavi Bazaz, L. Ding, M. Asadnia and M. Ebrahimi Warkiani, Effects of sample rheology on the equilibrium position of particles and cells within a spiral microfluidic channel, Microfluid. Nanofluid., 2021, 25(9), 75 Search PubMed.
- F. Mirakhorli, S. S. Mohseni, S. R. Bazaz, A. A. Mehrizi, P. J. Ralph and M. E. Warkiani, Microfluidic Platforms for Cell Sorting, Sustainable Separation Engineering, 2022, pp. 653–695 Search PubMed.
- S. Hassanpour Tamrin, A. Sanati Nezhad and A. Sen, Label-Free Isolation of Exosomes Using Microfluidic Technologies, ACS Nano, 2021, 15(11), 17047–17079 Search PubMed.
- A. Dalili, E. Samiei and M. Hoorfar, A review of sorting, separation and isolation of cells and microbeads for biomedical applications: microfluidic approaches, Analyst, 2019, 144(1), 87–113 RSC.
- N. Rahmanian, M. Bozorgmehr, M. Torabi, A. Akbari and A.-H. Zarnani, Cell separation: Potentials and pitfalls, Prep. Biochem. Biotechnol., 2017, 47(1), 38–51 Search PubMed.
- K. Choi, H. Ryu, K. J. Siddle, A. Piantadosi, L. Freimark and D. J. Park, et al., Negative selection by spiral inertial microfluidics improves viral recovery and sequencing from blood, Anal. Chem., 2018, 90(7), 4657–4662 CrossRef CAS PubMed.
- D. Di Carlo, Inertial microfluidics, Lab Chip, 2009, 9(21), 3038–3046 Search PubMed.
- M. E. Warkiani, A. K. P. Tay, G. Guan and J. Han, Membrane-less microfiltration using inertial microfluidics, Sci. Rep., 2015, 5, 11018 CrossRef PubMed.
- N. Xiang, J. Wang, Q. Li, Y. Han, D. Huang and Z. Ni, Precise Size-Based Cell Separation via the Coupling of Inertial Microfluidics and Deterministic Lateral Displacement, Anal. Chem., 2019, 91(15), 10328–10334 CrossRef CAS PubMed.
- A. S. Rzhevskiy, A. Y. Kapitannikova, S. A. Vasilescu, T. A. Karashaeva, S. Razavi Bazaz and M. S. Taratkin, et al., Isolation of Circulating Tumor Cells from Seminal Fluid of Patients with Prostate Cancer Using Inertial Microfluidics, Cancers, 2022, 14(14), 3364 CrossRef CAS PubMed.
- H. M. Tay, S. Y. Leong, X. Xu, F. Kong, M. Upadya and R. Dalan, et al., Direct isolation of circulating extracellular vesicles from blood for vascular risk profiling in type 2 diabetes mellitus, Lab Chip, 2021, 21(13), 2511–2523 Search PubMed.
- H. M. Tay, S. Kharel, R. Dalan, Z. J. Chen, K. K. Tan and B. O. Boehm, et al., Rapid purification of sub-micrometer particles for enhanced drug release and microvesicles isolation, NPG Asia Mater., 2017, 9(9), e434 CrossRef.
- J. Shrestha, M. Ghadiri, M. Shanmugavel, S. R. Bazaz, S. Vasilescu and L. Ding, et al., A Rapidly Prototyped Lung-on-a-chip Model Using 3D-Printed Molds, Organs-on-a-Chip, 2020, 100001 Search PubMed.
- S. Razavi Bazaz, N. Kashaninejad, S. Azadi, K. Patel, M. Asadnia and D. Jin, et al., Rapid Softlithography Using 3D-Printed Molds, Adv. Mater. Technol., 2019, 4(10), 1900425 CrossRef CAS.
- M. A. Mulvey, J. D. Schilling and S. J. Hultgren, Establishment of a persistent Escherichia coli reservoir during the acute phase of a bladder infection, Infect. Immun., 2001, 69(7), 4572–4579 Search PubMed.
- G. Iosifidis and I. G. Duggin, Distinct Morphological Fates of Uropathogenic Escherichia coli Intracellular Bacterial Communities: Dependency on Urine Composition and pH, Infect. Immun., 2020, 88(9), e00884-19 CrossRef PubMed.
- B. Söderström, A. Ruda, G. Widmalm and D. O. Daley, An OregonGreen488-labelled d-amino acid for visualizing peptidoglycan by super-resolution STED nanoscopy, Microbiology, 2020, 166(12), 1129–1135 CrossRef PubMed.
- E. Copeland, K. Leonard, R. Carney, J. Kong, M. Forer and Y. Naidoo, et al., Chronic rhinosinusitis: potential role of microbial dysbiosis and recommendations for sampling sites, Front. Cell. Infect. Microbiol., 2018, 8, 57 CrossRef PubMed.
- K. L. Bowerman, S. F. Rehman, A. Vaughan, N. Lachner, R. Y. Kim and D. L. A. Wood, et al., Disease-associated gut microbiome and metabolome changes in patients with chronic obstructive pulmonary disease, Nat. Commun., 2020, 11(1), 5886 Search PubMed.
- B. J. Callahan, P. J. McMurdie, M. J. Rosen, A. W. Han, A. J. A. Johnson and S. P. Holmes, DADA2: high-resolution sample inference from Illumina amplicon data, Nat. Methods, 2016, 13(7), 581–583 CrossRef CAS PubMed.
- S. Razavi Bazaz, A. Mihandust, R. Salomon, H. A. N. Joushani, W. Li and H. A. Amiri, et al., Zigzag microchannel for rigid inertial separation and enrichment (Z-RISE) of cells and particles, Lab Chip, 2022, 22(21), 4093–4109 RSC.
- S. Razavi Bazaz, A. Mashhadian, A. Ehsani, S. C. Saha, T. Krüger and M. Ebrahimi Warkiani, Computational inertial microfluidics: a review, Lab Chip, 2020, 20(6), 1023–1048 Search PubMed.
- S. Razavi Bazaz, O. Rouhi, M. A. Raoufi, F. Ejeian, M. Asadnia and D. Jin, et al., 3D Printing of Inertial Microfluidic Devices, Sci. Rep., 2020, 10(1), 5929 CrossRef CAS PubMed.
- J. Devalia, R. Sapsford, C. Wells, P. Richman and R. Davies, Culture and comparison of human bronchial and nasal epithelial cells in vitro, Respir. Med., 1990, 84(4), 303–312 Search PubMed.
- C. M. Bassis, A. L. Tang, V. B. Young and M. A. Pynnonen, The nasal cavity microbiota of healthy adults, Microbiome, 2014, 2(1), 1–5 CrossRef PubMed.
- S. Savolainen, J. Ylikoski and H. Jousimies-Somer, The bacterial flora of the nasal cavity in healthy young men, Rhinology, 1986, 24(4), 249–255 Search PubMed.
- H. Jousimies-Somer, S. Savolainen and J. S. Ylikoski, Comparison of the nasal bacterial floras in two groups of healthy subjects and in patients with acute maxillary sinusitis, J. Clin. Microbiol., 1989, 27(12), 2736–2743 CrossRef CAS PubMed.
- D. L. Hamilos, Chronic rhinosinusitis: epidemiology and medical management, J. Allergy Clin. Immunol., 2011, 128(4), 693–707 Search PubMed.
- J. B. Lyczak, C. L. Cannon and G. B. Pier, Lung infections associated with cystic fibrosis, Clin. Microbiol. Rev., 2002, 15(2), 194–222 Search PubMed.
- R. L. Gibson, J. L. Burns and B. W. Ramsey, Pathophysiology and management of pulmonary infections in cystic fibrosis, Am. J. Respir. Crit. Care Med., 2003, 168(8), 918–951 CrossRef PubMed.
- M. Berkhout, E. Rijntjes, L. El Bouazzaoui, W. Fokkens, R. Brimicombe and H. Heijerman, Importance of bacteriology in upper airways of patients with cystic fibrosis, J. Cystic Fibrosis, 2013, 12(5), 525–529 CrossRef CAS PubMed.
- P. Lambert, Mechanisms of antibiotic resistance in Pseudomonas aeruginosa, J. R. Soc. Med., 2002, 95(Suppl 41), 22 CAS.
- L. J. Sherrard, M. M. Tunney and J. S. Elborn, Antimicrobial resistance in the respiratory microbiota of people with cystic fibrosis, Lancet, 2014, 384(9944), 703–713 CrossRef CAS PubMed.
- J. J. LiPuma, The changing microbial epidemiology in cystic fibrosis, Clin. Microbiol. Rev., 2010, 23(2), 299–323 CrossRef PubMed.
- E. L. Salsgiver, A. K. Fink, E. A. Knapp, J. J. LiPuma, K. N. Olivier and B. C. Marshall, et al., Changing epidemiology of the respiratory bacteriology of patients with cystic fibrosis, Chest, 2016, 149(2), 390–400 CrossRef PubMed.
- A. Bevivino, G. Bacci, P. Drevinek, M. T. Nelson, L. Hoffman and A. Mengoni, Deciphering the ecology of cystic fibrosis bacterial communities: towards systems-level integration, Trends Mol. Med., 2019, 25(12), 1110–1122 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2lc00794k |
| ‡ These authors contributed equally as the first author. |
| This journal is © The Royal Society of Chemistry 2023 |