
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn electrochemical immunosensor based on a carboxylated multiwalled carbon nanotube-silver nanoparticle-chitosan functional layer for the detection of fipronil
Wen-Chien
Huang
 *,
You-Ning
Hsiung
and
Chia-Ling
Li
*,
You-Ning
Hsiung
and
Chia-Ling
Li
Department of Chemical and Materials Engineering, Chung Cheng Institute of Technology, National Defense University, Taoyuan, 33551, Taiwan. E-mail: wenchien2@gmail.com
First published on 3rd November 2023
Abstract
Fipronil (FP) is a very effective phenylpyrazole insecticide and is now widely used in agriculture. At the same time, the water and soil in the environment are polluted by FP. For the rapid detection of FP toxicants in food and the environment, we have designed an entirely novel electrochemical immunosensor that employs the combined functionalities of a cMWCNTs-AgNPs-CS-FAb-BSA layer to modify an SPCE by the freeze-drying technique. The high porosity of chitosan (CS) coupled with an excellent electron transfer enabled by the cMWCNTs and AgNPs increased the surface area for anti-fipronil (FAb) antibody immobilization and enhanced the current signal of the immunosensor. Cyclic voltammetry (CV) was applied for the quantitative determination of FP under optimized conditions (0.1 M PBS, pH 7.5, 35 °C incubation temperature, and 40 min incubation duration). The modified electrochemical immunosensor displayed excellent analytical performance, including a wide linear concentration range from 0.1 to 1000 ng mL−1 with a very low limit of detection of 0.021 ng mL−1 and good reproducibility (RSD = 2.58%, n = 6), stability (80.4% sensitivity after 5 days), and selectivity. Not only could the modified electrochemical immunosensor be applied in the FP residue analysis of agricultural products, but the present immobilization strategy can also potentially be applied to different biomolecules.
1. Introduction
The role of pesticides in agricultural production toward saving crops from plant diseases and pests is significant. Fipronil (FP) is widely used in agriculture and is a very effective phenylpyrazole insecticide. However, excessive use of pesticides can cause their residues to readily accumulate in the environment and the body. The water and soil in the environment are being polluted due to the widespread use of FP. Due to this, people and animals can be exposed to a range of severe health risks, including those affecting the nervous system and blood biochemistry.1 As a result, it is critical to detect the presence of FP traces in the environment.Currently, thin-layer chromatography (TLC),2,3 gas chromatography-mass spectrometry (GC-MS),4,5 liquid chromatography-tandem mass spectrometry (LC-MS/MS),6,7 and high-performance liquid chromatography (HPLC)8,9 are the most common techniques used for detecting FP. However, complicated sample pretreatment, high cost, and reliance on well-trained personnel are some unfavourable characteristics of these detection techniques. Thus, it is urgently needed to design a simple, quick, and sensitive technique for detecting FP residues. For comparison, electrochemical methods with many merits, including fast response, accuracy, simplicity, portability, and low cost, have been researched in recent years. Electrochemical immunosensors are a new class of sensors with the advantages of both electrochemical and immunosensor techniques.
Electrochemical immunoassays based on antigen–antibody interactions have recently attracted considerable interest due to their high sensitivity, specificity, low cost, and fast response.10,11 Electrochemical immunoassays combine electrochemical analyses with immunoassays by first immobilizing the antibodies on the electrode surface and analyzing the signal changes before and after the reaction via electrochemical methods for qualitative or quantitative detection. In addition, screen-printed carbon electrodes (SPCEs) have been demonstrated as suitable alternatives to expensive conventional electrodes due to their advantages, such as simple fabrication, low cost, easy modification with nanomaterials, and high sensitivity.12,13 To increase the sensitivity of SPCEs, different kinds of carbon nanomaterials and technologies have been employed for amplifying the signal of antigen–antibody interactions, such as graphene,14–16 reduced graphene oxides17–19 and single-wall carbon nanotubes (SWCNTs).20–22 Large surface area and excellent electron transfer capability are the key electrochemical features of carbon nanomaterials. Simultaneously, metal nanoparticles exhibit a variety of physical, chemical, and biological properties, and bind to antibodies through coordinated covalent bonds.
In this work, for the determination of FP, we designed a modified electrochemical immunosensor based on an SPCE modified with carboxylated multi-walled carbon nanotubes (cMWCNTs), silver nanoparticles (AgNPs) and chitosan (CS) and incorporated with anti-FP polyclonal antibodies (FAb), which were directly absorbed on the modified electrode surface. The cMWCNTs-AgNPs-CS-FAb-BSA/SPCE was prepared by using a freeze-drying technique to modify the electrode. The high porosity of CS coupled with the excellent electron transfer properties of cMWCNTs and AgNPs increased the surface area for FAb immobilization and enhanced the current signal of the sensor. The new strategy has three excellent features: (i) the loose structure of the cMWCNTs-AgNPs-CS nanocomposite leads to a high specific area, which is useful for biomolecule adsorption; (ii) the probability of the coupling reaction between silver nanoparticles and anti-FP antibodies (FAb) is improved; and (iii) the detection sensitivity of the developed electrochemical biosensor toward the analyte is enhanced because of the higher electrochemical performance of the electrode. Various experimental parameters, including pH, temperature and incubation duration, were optimized, and the electrochemical immunosensor proved to have good sensitivity, reproducibility, stability and selectivity in various experiments. Therefore, the modified electrochemical immunosensor could be applied to FP residue analysis of agriculture products, such as leafy vegetables, corn, rice, mango, cucumber, eggplant, red beans, egg and tea. Our sensor meets the international testing standards as its FP residue detection range was between 0.1–1000 ng mL−1.
2. Experimental details
2.1. Materials and apparatus
Silver nitrate (AgNO3), potassium ferricyanide (K3[Fe(CN)6]) and trisodium citrate dehydrate (Na3CA) were purchased from J. T. Baker Chemical Inc. (New Jersey, USA). MWCNTs (>95% carbon basis, 20–40 nm in diameter and 5–15 m in length), chitosan (medium molecular weight), glacial acetic acid (>99.7%), sodium borohydride (NaBH4) and potassium ferrocyanide (K4[Fe(CN)6]) were purchased from Sigma-Aldrich (Missouri, USA). Fipronil (FP), anti-FP polyclonal antibodies (FAb), ractopamine (RAC), pyraclofos and ethiprole were obtained from Taiwan Advance Bio-Pharmaceutical Inc. (Taipei, Taiwan). The tea leaf samples were purchased from a local supermarket. Polyoxyethylene (20) sorbitan monolaurate (tween 20) was obtained from Bio Basic Canada Inc. (Markham Ontario, Canada). Bovine serum albumin (BSA) was obtained from Jackson ImmunoResearch Laboratories Inc. (Pennsylvania, USA). Trehalose was obtained from Alfa Aesar (Massachusetts, USA). The chemicals were of analytical grade and used without further purification. All solutions were prepared using ultrapure water with resistivity not less than 18.2 MΩ cm (Milli-Q purification system). 0.1 M phosphate-buffered saline (PBS) (pH 7.4, 8.0 g NaCl, 0.2 g KCl, 1.42 g Na2HPO4 and 0.27 g KH2PO4) and the PBST solution were prepared (0.1% Tween-20 in 10 mM PBS). The UV-visible absorption spectra were measured using a CT-8600 UV-vis spectrophotometer (ChromTech, Germany). The crystalline phase of the cMWCNTs-AgNPs-CS composites was identified using an X-ray diffractometer (D2 PHASER, Bruker, Karlsruhe, Germany). The information about the chemical bonds and molecular structure of the material was obtained using Fourier transform infrared (FT-IR) spectrometry (PerkinElmer Spectrum 100). Morphological imaging studies, energy-dispersive spectroscopy (EDS), and elemental mapping of the MWCNTs-AgNPs-CS composites and the modified electrode were performed using a field-emission scanning electron microscope (JEOL, JSM-7600F, Japan). Transmission scanning electron microscopy (TEM) images were obtained at 300 kV using a Philips Tecnai F30 from The Netherlands. The composition and SERS spectra of the modified electrode were recorded and analyzed using X-ray photoelectron spectroscopy (XPS, VG Scientific ESCALAB 250, East Grinstead, UK) and Raman spectroscopy (TRIAX 550, Horiba Ltd, Japan). The cyclic voltammetry (CV) curves were measured in the scanning range of −1.6 V to 1.6 V and at a scanning rate of 100 mV s−1 in a 0.1 M PBS solution (pH 7.5) containing 10 mM [Fe(CN)6]3−/4−. The electrochemical impedance spectra (EIS) were obtained at the scanning frequency range of 0.1–1000 Hz and a pulse amplitude of 100 mV s−1 in a 0.1 M PBS solution (pH 7.5) containing 10 mM [Fe(CN)6]3−/4−. All electrochemical measurements were performed on a CHI 6045D electrochemical workstation (6045D, CH Instruments, Inc., USA).2.2. Fabrication of the cMWCNTs-AgNPs-CS nanocomposite
First, pristine MWCNTs (0.50 g) were dispersed in 150 mL of concentrated nitric acid under stirring and refluxed for 30 h at 100 °C. The cMWCNTs were separated by centrifugation and washed thoroughly with ultrapure water until a pH of about 7.0 was obtained. Finally, the cMWCNTs were dried at 120 °C for 24 h. Secondly, the dried cMWCNTs (0.01 g) were dispersed in 10 mL of an AgNO3 solution (0.01 M) by ultrasonication for 5 min, followed by magnetic stirring for 24 h (200 rpm). 30 mL of a 0.019 M NaBH4 solution was added to this solution at the rate of about 1 drop per second under stirring. Simultaneously, 2 mL of a 0.02 M TSC solution was added under vigorous stirring for 24 h. The cMWCNTs-AgNPs composite was obtained. Finally, the products were re-dispersed in 2 mL of a 1 wt% chitosan solution, and a homogeneous black suspension was obtained after magnetic stirring for 20 h (400 rpm). The cMWCNTs-AgNPs-CS composite was preserved in a refrigerator at 4 °C before use.2.3. Preparation of the modified immunosensor electrode
Fig. 1 shows the fabrication procedure of the electrochemical immunosensor. First, 8 μL of the cMWCNTs-AgNPs-CS composite solution were dropped on the SPCE surface and dried at room temperature for 2.5 h. Further, the cMWCNTs-AgNPs-CS/SPCE was processed by the freeze-drying method for 24 h. 8 μL of the FAb solution (40 μg mL−1) was dropped on the cMWCNTs-AgNPs-CS/SPCE at 37 °C and dried for 2 h. The cMWCNTs-AgNPs-CS-FAb/SPCE was then washed with 200 μL each of PBST and PBS solutions to remove unbound FAb. 8 μL of a BSA solution (1.0 wt%) was then dropped on the cMWCNTs-AgNPs-CS-FAb/SPCE at 37 °C for 1 h to eliminate the nonspecific binding effect and block the remaining active groups. Finally, the immunosensor was washed with 200 μL each of PBST and PBS and stored at 4 °C when not in use.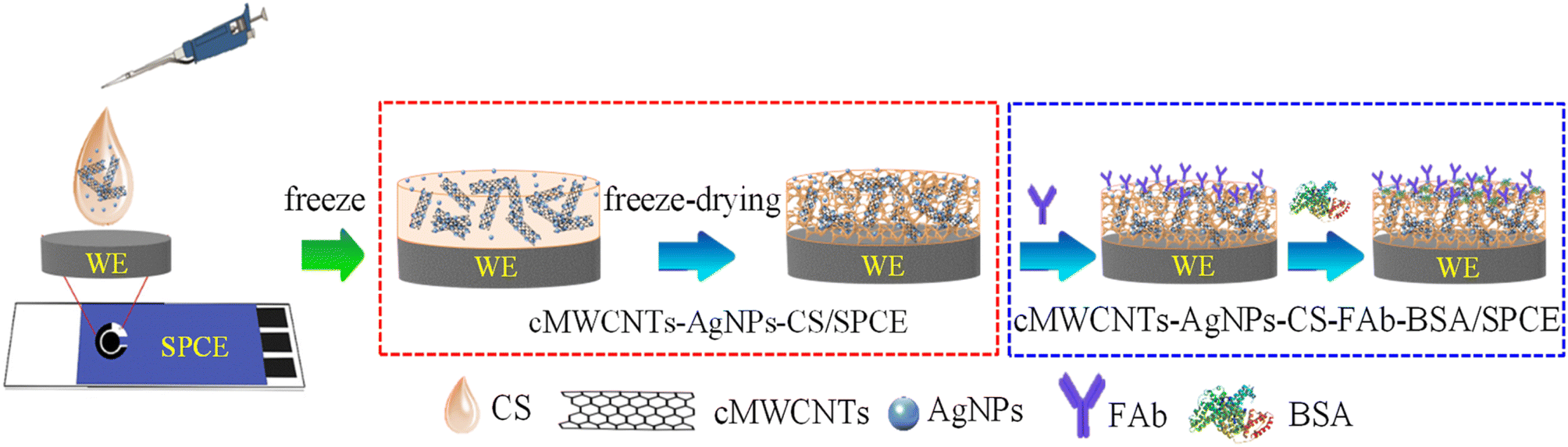 | ||
| Fig. 1 Schematic illustration of the preparation of cMWCNTs-AgNPs-CS-Fab-BSA/SPCE. | ||
2.4. Optimization of the detection conditions
The target FP was prepared in PBS solutions at six different pH values (FP: 100 ng mL−1; pH: 6.0, 6.5, 7.0, 7.5, 8.0, 8.5). The FP solutions of various pH (10 μL) were dropped on the cMWCNTs-AgNPs-CS-FAb-BSA/SPCE surface at 35 °C for 30 min. After incubation, each electrode was analysed by CV. To identify the optimal temperature, the target FP (8 μL, 100 ng mL−1, pH 7.5) was dropped on the cMWCNTs-AgNPs-CS-FAb-BSA/SPCE surface and incubated at five different temperatures (25, 30, 35, 40, and 45 °C). After incubation, the electrodes were analysed by CV. The ideal incubation time was identified by dropping the target FP (8 μL, 100 ng mL−1, pH 7.5) on the cMWCNTs-AgNPs-CS-FAb-BSA/SPCE surface and incubating it at 35 °C for different durations (10, 20, 30, 40, and 50 min). After incubation, the electrodes were analysed by CV.2.5. Electrochemical measurements
The modified electrode was incubated with standard FP solutions at different concentrations (10 μL, pH 7.5) in the range from 0.1 to 1000 ng mL−1 at 35 °C for 40 min. After washing with PBS (pH 7.5) to remove unattached FP, CV was used for further analysis. The CV curves were recorded three times for each concentration of FP, and the average values of peak currents are shown with SD for n = 3. The error bars represent the standard deviation, and the center of each error bar expresses the mean value of the corresponding three independent experiments.23 To verify the reproducibility of the FP response on cMWCNTs-AgNPs-CS-FAb-BSA/SPCE, six different cMWCNTs-AgNPs-CS-FAb-BSA-modified electrodes fabricated independently by the same procedure were tested for the detection of 1 ng per mL FP in 0.1 M PBS (pH 7.5) by CV. To verify the stability of the FP response of cMWCNTs-AgNPs-CS-FAb-BSA/SPCE, six different cMWCNTs-AgNPs-CS-FAb-BSA-modified electrodes fabricated independently by the same procedure were stored at 4 °C. After 5 days, the cMWCNTs-AgNPs-CS-FAb-BSA/SPCEs were tested for the detection of 1 ng per mL FP in 0.1 M PBS (pH 7.5) by CV.2.6. Preparation of real samples
Analyte detection in real samples is a key step in evaluating the practical applicability of immunosensors. The tea leaf sample was weighed (0.5 g) and added to a 10 mL centrifuge tube. The detection of FP in tea leaf samples was performed using the standard addition method, and 5 ng mL−1, 50 ng mL−1 and 150 ng per mL FP solutions were added to the tea leaf samples. Then, 3 mL of an acetonitrile–acetone extraction solution (volume ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was added to the spiked samples. The mixture was sonicated and centrifuged for 20 and 30 min, respectively. The clear supernatant was separated and transferred into test tubes and dried using nitrogen. Finally, the extracts were dissolved in 1 mL of PBS (pH 7.5) and then stored at 4 °C until detection.
:1) was added to the spiked samples. The mixture was sonicated and centrifuged for 20 and 30 min, respectively. The clear supernatant was separated and transferred into test tubes and dried using nitrogen. Finally, the extracts were dissolved in 1 mL of PBS (pH 7.5) and then stored at 4 °C until detection.
3. Results and discussion
3.1. Characterization of the cMWCNTs-AgNPs-CS nanocomposite
To further investigate the cMWCNTs-AgNPs-CS solution, the prepared solution was characterized by UV absorption spectroscopy (Fig. 2A). An obvious absorption peak originating from the structure of the cMWCNTs appeared at approximately 244 nm (curve a). The absorption peak of cMWCNTs-AgNPs-CS was influenced by the absorption of the AgNPs, which showed a redshift relative to that of the cMWCNTs. At the same time, the AgNPs formed in the presence of cMWCNTs were black-green in color, and their absorption maximum (λmax) was at ∼398 nm (curve c), whereas, in the absence of cMWCNTs, they were yellowish in color with a λmax of ∼392 nm (curve b). The above discussions support that the AgNPs were attached to the cMWCNTs.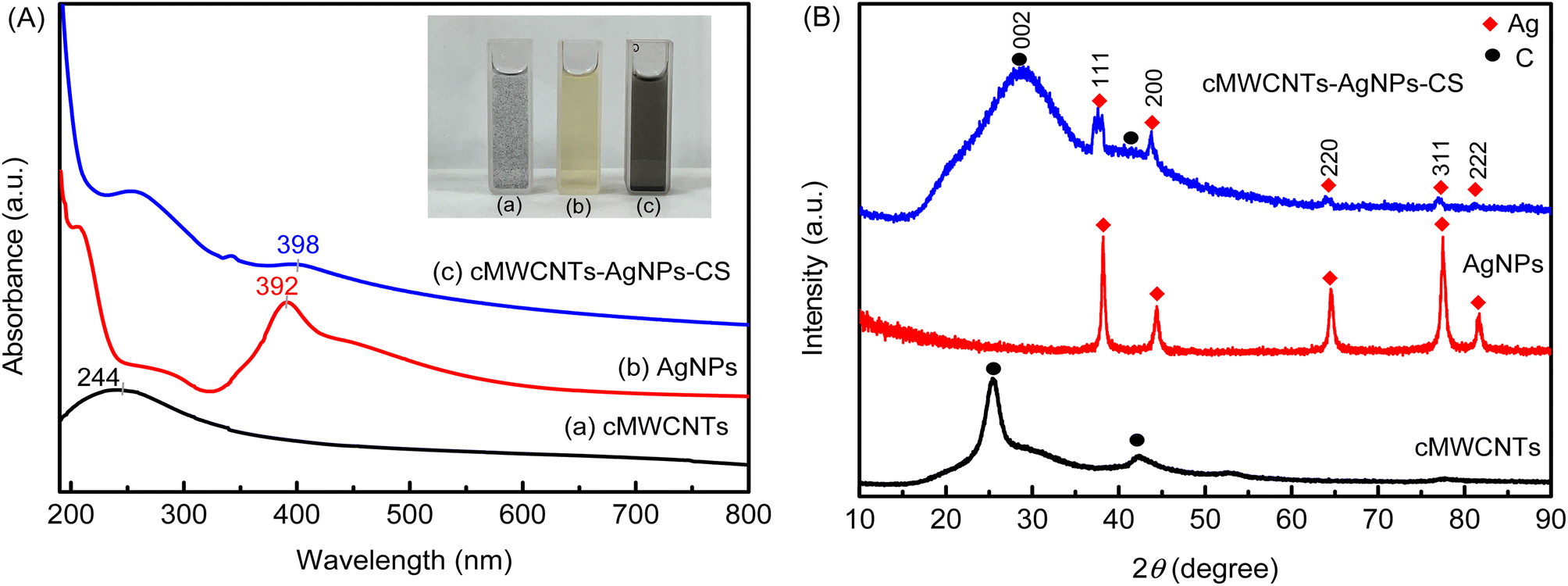 | ||
| Fig. 2 (A) The UV-visible spectra and photograph of cMWCNTs, AgNPs, and the cMWCNTs-AgNPs-CS composite. (B) The XRD spectra of cMWCNTs, AgNPs, and the cMWCNTs-AgNPs-CS composite. | ||
The crystalline nature of AgNPs in the cMWCNTs-AgNPs-CS composite was confirmed by the XRD analysis. The powder XRD patterns of the cMWCNTs, AgNPs and cMWCNTs-AgNPs-CS composite are shown in Fig. 2B. The XRD pattern of cMWCNTs-AgNPs-CS shows Bragg's reflections related to crystalline silver at 38.2°, 44.4°, 64.6°, 77.5° and 81.6°, representing the (111), (200), (220), (311) and (222) planes of face-centered cubic (FCC) crystalline silver nanoparticles (JCPDS No. 04-0783).24
Fig. 3A depicts the FT-IR spectra of the cMWCNTs, cMWCNTs-AgNPs, CS and cMWCNTs-AgNPs-CS composite. The FT-IR characteristic spectrum of cMWCNTs is shown as curve a. After acid functionalization, the peaks belonging to the –OH stretching vibrations and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations of the carboxyl groups appeared respectively at 3362 cm−1 and 1717 cm−1.25 The stretching bands of the conjugated CC bonds in the carbon skeleton appeared at approximately 1624 and 1569 cm−1. The peaks between 1000 and 1420 cm−1 belong to the C–O stretching and –OH bending modes of the alcoholic, phenolic and carboxylic groups. These confirm that the carboxyl groups on the surface of the cMWCNTs were formed by acid treatment.
O stretching vibrations of the carboxyl groups appeared respectively at 3362 cm−1 and 1717 cm−1.25 The stretching bands of the conjugated CC bonds in the carbon skeleton appeared at approximately 1624 and 1569 cm−1. The peaks between 1000 and 1420 cm−1 belong to the C–O stretching and –OH bending modes of the alcoholic, phenolic and carboxylic groups. These confirm that the carboxyl groups on the surface of the cMWCNTs were formed by acid treatment.
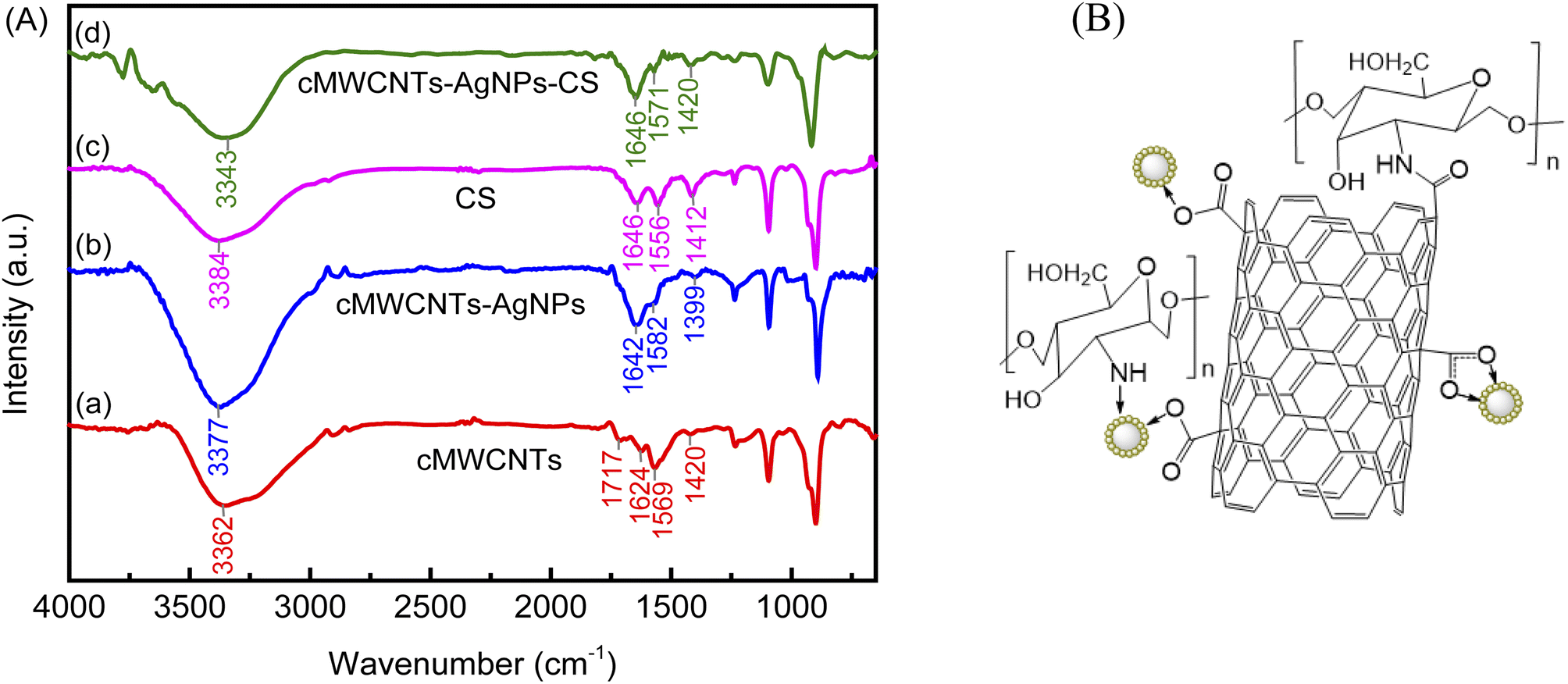 | ||
| Fig. 3 (A) The FTIR spectra and (B) structure of the cMWCNTs-AgNPs-CS composite. | ||
Curve b is the FT-IR characteristic spectrum of cMWCNTs-AgNPs, which shows a decrease in band intensity at 1717 cm−1 (OC–OH) and 1420 cm−1 (O–H). Because coordinate covalent bonds were generated between the carboxyl groups on the surface of the cMWCNTs and the silver ions in AgNPs, the absorption bands at 1642/1582 (νas(COO−)) and 1399 cm−1 (νs(COO−)) could be assigned to the asymmetric and symmetric stretching of COO− in the cMWCNTs. The coordinate covalent bond type can be distinguished based on the extent of Δν = νas(COO−) − νs(COO−).26 The Δν (νas − νs) values of the monodentate and bridging bidentate type bonding structures of cMWCNTs-AgNPs were 243 (νas(1642) − νs(1399)) and 183 (νas(1582) − νs(1399)) cm−1, respectively.
Curve c is the characteristic FT-IR spectrum of CS. The broad and strong peak at 3384 cm−1 belongs to the stretching of O–H. The peaks at 1646 cm−1 and 1556 cm−1 belong to the amide I (CO) and amide II (N–H) groups of CS, respectively. The peak at 1412 cm−1 belongs to stretching vibrations of the secondary (CH–OH) alcoholic structures in CS.
Curve d shows the FT-IR peaks of the cMWCNTs-AgNPs-CS composite at 1646 cm−1, 1571 cm−1 and 1420 cm−1. In addition, the blueshift of the amide II peak from 1556 cm−1 to 1571 cm−1 may be caused by the interaction between Ag+ in cMWCNTs-AgNPs and –NH2 in CS.27 These findings confirm the presence of CS on the cMWCNTs-AgNPs composite.
The surface morphological studies of the cMWCNTs, cMWCNTs-AgNPs and cMWCNTs-AgNPs-CS nanocomposites were performed using an emission scanning electron microscope (FESEM) and an energy-dispersive X-ray spectrometer (EDS). The MWCNTs were successfully functionalized with nitric acid and a tube-like morphology was observed for the functionalized cMWCNTs (Fig. 4A). Fig. 4B presents the elemental maps of cMWCNTs obtained by EDS, from which it can be observed that the weight percentages of C and O in the cMWCNTs were 94.41% and 5.59%, respectively. The EDS elemental mapping of the C and O elements clearly shows that the O element was evenly distributed on the surface of the cMWCNTs (Fig. 4C). The spherical spots found in the SEM image manifested successful AgNP formation on the surface of the cMWCNTs-AgNPs composite, as shown in Fig. 5A. Fig. 5B shows the EDS elemental maps of cMWCNTs-AgNPs, from which it can be observed that the weight percentages of C, O and Ag in the cMWCNTs-AgNPs sample were 43.26%, 5.41% and 51.33%, respectively. The EDS elemental mapping of the C, O and Ag elements clearly shows that the Ag element was evenly distributed on the surface of the cMWCNTs-AgNPs (Fig. 5C). As shown in Fig. 6A, the cMWCNTs-AgNPs-CS composite displayed a nano-porous structure. This nano-porous structure would increase the effective surface area of the microelectrodes and reduce the impedance, thus proving beneficial to increasing the current response. In Fig. 6B, the elemental maps of cMWCNTs-AgNPs-CS obtained by EDS are presented, from which it can be observed that the weight percentages of C, O, Ag and N in the sample were 65.67%, 10.57%, 12.59% and 11.17%, respectively. The EDS elemental mapping of the C, O, Ag and N elements clearly shows that the N element was evenly distributed on the surface of cMWCNTs-AgNPs-CS (Fig. 6C).
 | ||
| Fig. 4 (A) The SEM images of the cMWCNTs (inset: 150000× magnification). (B) The EDS spectrum of the cMWCNTs. (C) The EDS mapping of C and O elements in cMWCNTs. | ||
 | ||
| Fig. 5 (A) The SEM images of the cMWCNTs-AgNPs (inset: 200000× magnification). (B) The EDS spectrum of cMWCNTs-AgNPs. (C) The EDS mapping of C, O and Ag elements in cMWCNTs-AgNPs. | ||
 | ||
| Fig. 6 (A) The SEM images of the cMWCNTs-AgNPs-CS composite (inset: 200000× magnification). (B) The EDS spectrum of cMWCNTs-AgNPs-CS. (C) The EDS mapping of C, O, Ag, and N elements in cMWCNTs-AgNPs-CS. | ||
Characterization of the morphology of the cMWCNTs-AgNPs-CS composite was performed by TEM imaging. The oxygen-containing functional groups on the cMWCNTs provide nucleation sites for both the homing of Ag+ and the growth of evenly distributed AgNPs.28 As confirmed by the TEM photographs, the AgNPs were highly dispersed and uniformly distributed on the surface of the cMWCNTs (Fig. 7A). Fig. 7B shows a magnified section of the micrograph in Fig. 7A, and it can be clearly seen that AgNPs were anchored on the surface of the cMWCNTs. The average diameter of the AgNPs was approximately 10 nm. The Fig. 7B inset shows that the plane spacing value of 0.23 is consistent with the (111) facets of silver (JCPDS Card No. 04-0783).29,30 Overall, the AgNPs were well-distributed over the surface of the cMWCNTs, as assessed by TEM.
 | ||
| Fig. 7 The TEM images of the cMWCNTs-AgNPs-CS composite at (A) 50 nm and (B) 5 nm scales. | ||
3.2. Characterization of the modified immunosensor electrode
The thickness and the high-magnification micrographs of the surface and cross-section of the SPCE before and after modification were obtained by FESEM. The bare unmodified SPCE surface at 50000× magnification is shown in Fig. 8A. Evident flaws and surface roughness were observed on the surface of the bare unmodified SPCE. Fig. 8B shows the cross-section of the bare unmodified SPCE, and the thickness of the carbon film was approximately 63.58 μm. In Fig. 8B inset, it can be clearly seen that the irregular carbon particles appear the same as seen on the surface. The cMWCNTs-AgNPs-CS/SPCE surface at 30000× magnification is shown in Fig. 8C. It can be seen that the surface of the modified SPCE is smooth, with a significant reduction in defects and surface roughness. Fig. 8D shows the cross-section of the modified SPCE, and the thickness of the carbon films and cMWCNTs-AgNPs-CS films were approximately 63.58 and 18.57 μm, respectively. Fig. 8D inset clearly shows that the porous structure composed of randomly entangled cMWCNTs-AgNPs was penetrated by CS agglomerates. Due to the viscous characteristic of CS, the cMWCNTs-AgNPs in cMWCNTs-AgNPs-CS/SPCE increased not only the dispersion but also the specific surface area.
 | ||
| Fig. 8 SEM images of the top view and the side view (100000× magnification) of (A and B) unmodified SPCE and (C and D) cMWCNTs-AgNPs-CS/SPCE. | ||
The structural changes in the SPCE after the addition of the cMWCNTs-AgNPs-CS composite were examined by the changes in the Raman spectrum (Fig. 9A). The bare SPCE showed two Raman peaks (D and G bands) at 1356 and 1580 cm−1 when a 532 nm laser was used for excitation. The D band is associated with the double resonant Raman vibrations of the sp2 atoms and is usually used to quantify the structural disorder arising from amorphous carbon, while the G band corresponds to the tangential stretching vibrations of the carbon atoms. The ratio between the intensities of the D and G bands, ID/IG, can be used to compare the level of disorder in the carbon material structure.31 The ID/IG of bare SPCE was calculated as 0.3463. The ID/IG of the cMWCNTs-AgNPs-CS composite was calculated as 0.9748. The prominent D peak with intensity comparable to that of the G peak, as well as their large bandwidth, are indicative of significant structural disorder in the cMWCNTs-AgNPs-CS composite. At the same time, the ID/IG of cMWCNTs-AgNPs-CS/SPCE was calculated as 0.6489. The high ID/IG value of the cMWCNTs-AgNPs-CS/SPCE confirms the presence of significant defects or disorder in these nanostructures compared with bare SPCE. The above discussions further verify that SPCE was coated with the cMWCNTs-AgNPs-CS composite.
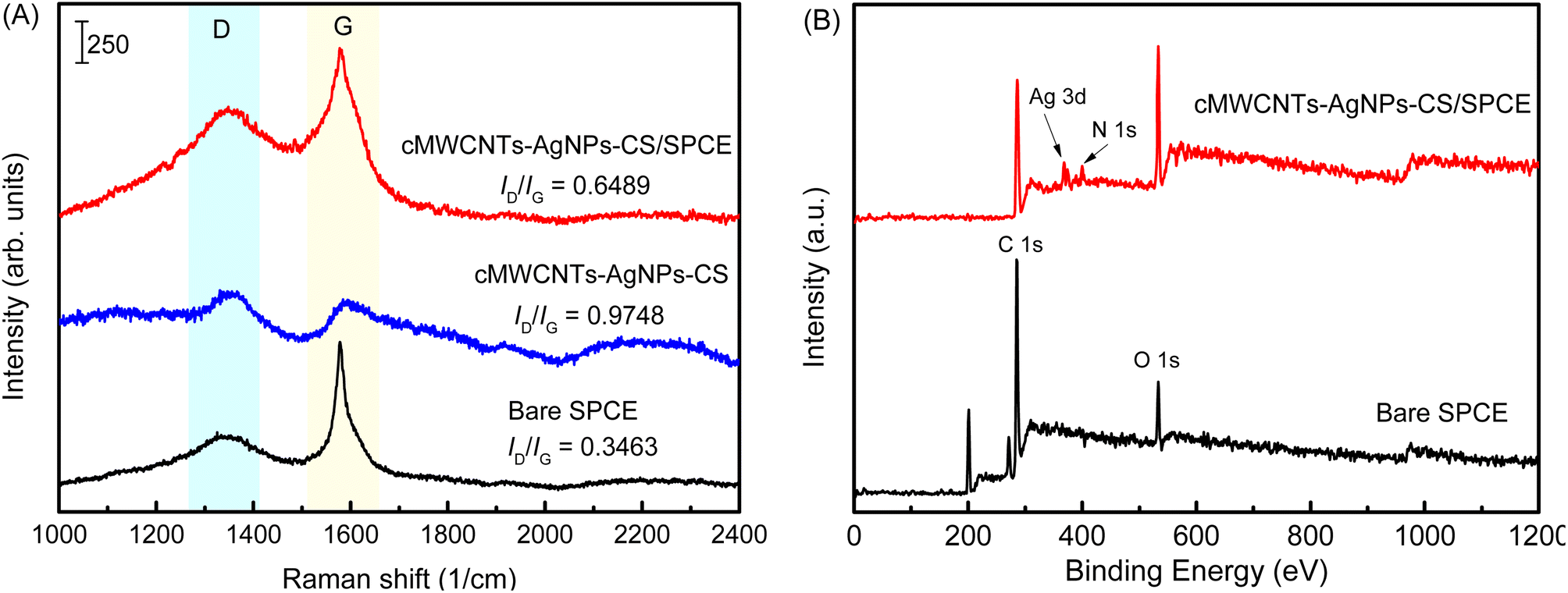 | ||
| Fig. 9 (A) The Raman spectra of bare SPCE, cMWCNTs-AgNPs-CS, and cMWCNTs-AgNPs-CS/SPCE. (B) The full-range XPS spectra of bare SPCE and cMWCNTs-AgNPs-CS/SPCE. | ||
The full-range XPS spectra of bare SPCE and cMWCNTs-AgNPs-CS/SPCE are depicted in Fig. 9B, which illustrate the binding energies of all elements, including Ag, C, O and N. In the full-range XPS spectrum of SPCE, we observed characteristic C 1s and O 1s peaks. The full-range XPS spectrum of cMWCNTs-AgNPs-CS/SPCE showed strong signals for C and O, along with weak Ag and N peaks corresponding to the AgNPs and CS bound to the surface of the SPCE, indicating the presence of the cMWCNTs-AgNPs-CS composite.
As shown in Fig. 10A, the assembly process of the electrochemical immunosensor was monitored by CV experiments in the potential range from −1.6 V to 1.6 V at a scan rate of 100 mV s−1 in a 0.1 M PBS buffer solution (pH 7.5) containing 10 mM [Fe(CN)6]3−/4−. A pair of redox peaks could be seen on the curve of bare SPCE (curve a). A pair of increasingly well-defined redox peaks were observed with the addition of the cMWCNTs-AgNPs-CS composite (curve b) due to its contribution to electron transmission. As expected, there was an obvious decrease in peak current with the addition of FAb and BSA (curve c and curve d) due to their insulating effect, which hindered the redox probe from reaching the electrode surface. Finally, the peak current decreased significantly with the addition of the FP antigen solution (curve e and curve f). This phenomenon indicates that an FP/FAb immunocomplex is formed between FP and FAb immobilized on cMWCNTs-AgNPs-CS-FAb-BSA/SPCE. The FP/FAb immunocomplex serves as a mass transfer blocking layer. Thus, the transport of electrons and the spread of the redox probe to the electrode interface are significantly blocked.32
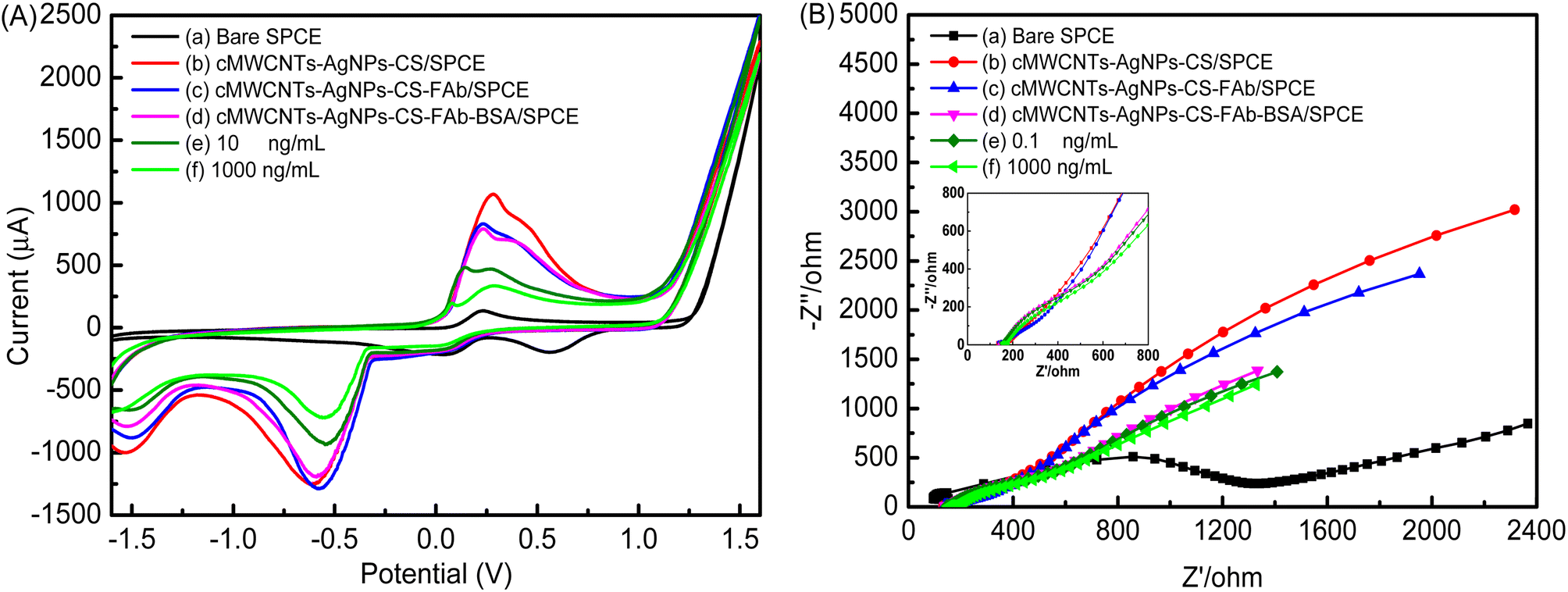 | ||
| Fig. 10 (A) The CV curves and (B) EIS diagrams produced by the immunosensor at different stages of its fabrication. | ||
Furthermore, the assembly process of the electrochemical immunosensor was also evaluated by EIS. Fig. 10B shows the EIS curve at each step of the electrode modification process. The semicircle portion observed at higher frequencies represents the electron transfer-limited process. The linear part is characteristic of the lower-frequency range and corresponds to the diffusion-limited electron-transfer process.33 The semicircle diameter equals the polarization resistance. There is a proportional relationship between the diameter of the semicircle and the impedance. With the addition of the cMWCNTs-AgNPs-CS composite, a sharp decrease in resistance was observed (curve b). This phenomenon indicates the enhanced specific surface area and excellent electron transfer of cMWCNTs-AgNPs-CS. Thus, the transport of electrons and the spread of the redox probe to the electrode interface would be promoted. With FAb and BSA hindering the electron transfer of the electrochemical probe, the value of resistance increased in their presence (curve c and curve d), indicating the successful immobilization of FAb onto the cMWCNTs-AgNPs-CS/SPCE. Additionally, the resistance increased further after the capture of the FP antigen (curve e and curve f), which shows that the modified electrode responds well to different concentrations of FP.
3.3. Optimization of detection conditions
The influence of pH on the oxidation peak of [Fe(CN)6]3−/4− was studied in a 0.1 M PBS buffer solution in the pH range from 6.0 to 8.5 (Fig. 11A). It was observed that upon increasing the pH from 6.0 to 7.5, the current value declined. The reason might be that the specific immunoreaction between FP and FAb on the cMWCNTs-AgNPs-CS-FAb-BSA/SPCE reaches equilibrium. The current value increased with an increment in pH value from 7.5 to 8.5. These results indicate that the stability of the proteins and the lifetime of the electrochemical immunosensor can be affected by highly acidic or alkaline environments.32 The optimum response was obtained at about pH 7.5. Therefore, pH 7.5 was selected as the optimal incubation pH in this work.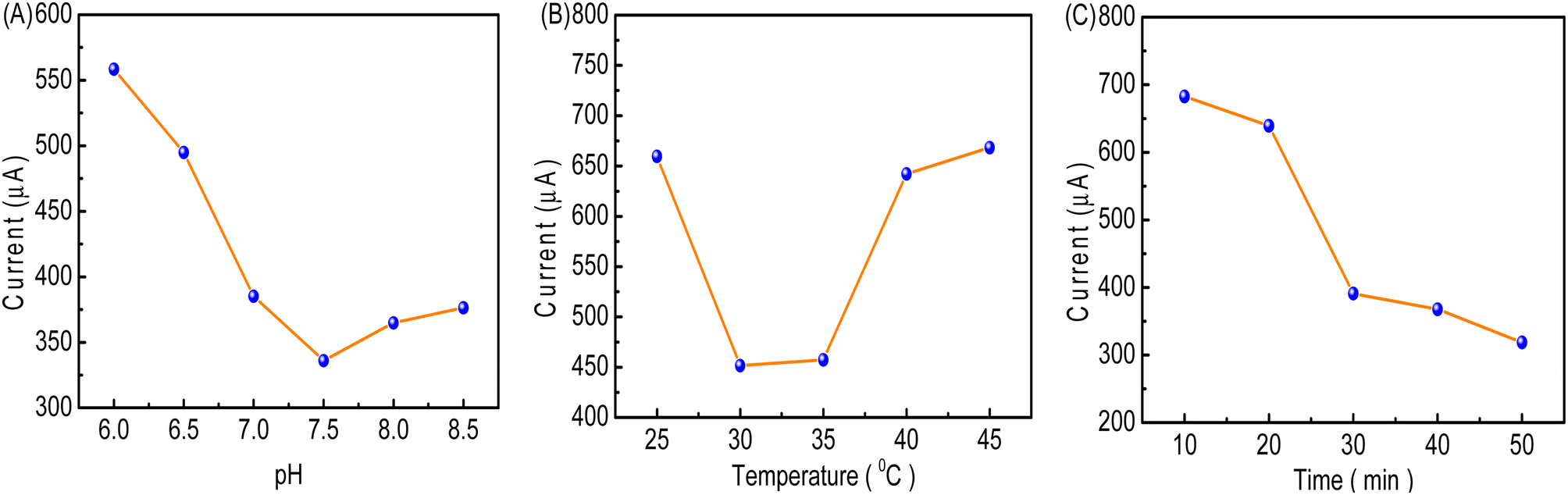 | ||
| Fig. 11 (A) The effect of pH on the CV anodic peak current of 100 ng per mL FP in the pH range from 6.0 to 8.5. (B) The effect of temperature on the CV anodic peak current of 100 ng per mL FP in the temperature range from 25 to 45 °C. (C) Effect of time on the CV anodic peak current of 100 ng per mL FP in the time range from 10 to 50 min. | ||
As shown in Fig. 11B, with increasing incubation temperature, the oxidation peak current of [Fe(CN)6]3−/4− initially showed a rapid decrease (from 25 to 30 °C) owing to the specific immunoreaction between FP and FAb. Then, the change in oxidation peak current of [Fe(CN)6]3−/4− tended to be minimal from 30 to 35 °C due to the specific immunoreaction achieving an equilibrium between FP and FAb. The current value increased with an increment in temperature from 35 to 45 °C. At temperatures above 35 °C, the FAb antibody gradually loses its antigen-binding activity. Thus, the optimal incubation temperature of 35 °C was chosen for use in subsequent experiments.34
The peak current of cMWCNTs-AgNPs-CS-FAb-BSA/SPCE initially showed a rapid decrease (from 10 to 30 min) with increasing incubation duration (Fig. 11C). After 30 min, the current value did not change as much. The decrease in current could be owing to the increase in the FP/FAb immunocomplex formation between FP and FAb on cMWCNTs-AgNPs-CS-FAb-BSA/SPCE. The immunoreaction was basically completed within 40 min. Thus, 40 min was chosen as the optimal incubation time in this work.35
3.4. Sensitivity of the immunosensor
Fig. 12A shows the relationship between the concentration of FP and the peak current. Immunocomplexes were formed between FP and FAb on the cMWCNTs-AgNPs-CS-FAb-BSA/SPCE. Thus, the transport of electrons and the spread of the redox probe to the electrode interface were blocked. As illustrated, the peak current decreased as the concentration of FP increased. As shown in Fig. 12B, the logarithmic values of CFP and ΔIpa of the FP immunosensor positively correlated across the FP concentration range of 0.1–1000 ng mL−1, and the correlation coefficient R2 was 0.998. The equation of the fitting line was ΔIpa = 132.603 + 77.703logCFP. The minimum FP detection limit of the immunosensor was 0.021 ng mL−1 (3σ). In addition, Table 1 compares the analytical performances of various electrochemical sensors in terms of the detection limit of FP, indicating that our electrochemical immunosensor is comparable with other electrochemical sensors reported in the literature.
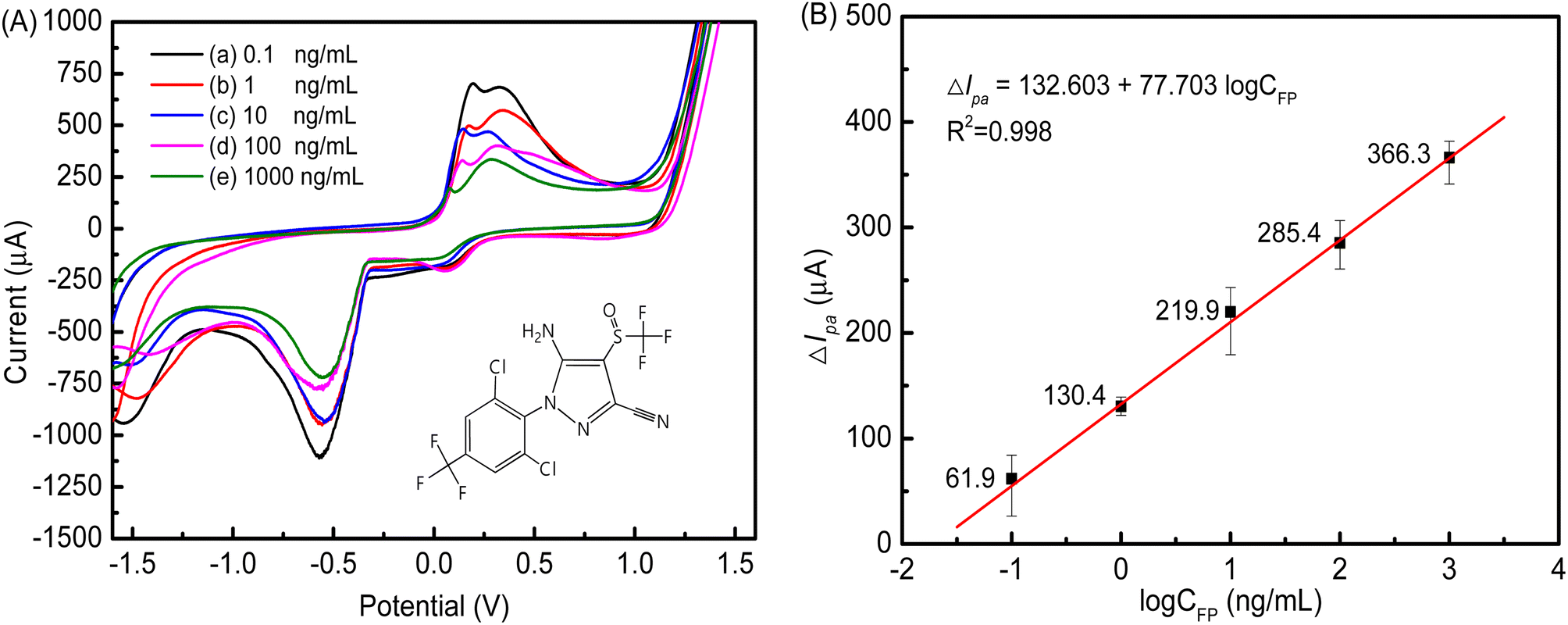 | ||
| Fig. 12 (A) The CV responses of the cMWCNTs-AgNPs-CS-FAb-BSA/SPCE immunosensor in 0.1 M PBS (pH 7.5) containing various concentrations of FP (a–e: 0.1, 1, 10, 100 and 1000 ng mL−1) (inset: the chemical structure of FP). (B) The calibration curve of current change (ΔIpa) against log FP concentration. | ||
| Materials | Detection technique | Linear range | LOD (ng mL−1) | RSD (%) | Ref. |
|---|---|---|---|---|---|
| Graphite-polyurethane (GPU) | SWVS | 0–2.5 × 10−6 mol L−1 | 0.8 | — | 36 |
| TiO2-CPE | CV | 10−6–10−1 μM | 0.148631 | — | 37 |
| FeO·TiO2/CPE | CV | 0.001–0.01 μM | 0.52458 | 0.173 | 38 |
| Al(III)–TiO2/graphene | CV | 0.01–0.09 μg L−1 | 0.0164 | 5 | 39 |
| Apt/primer/AuNPs/PLL-BP/GCE | DPV | 0.1–10000 ng mL−1 |
0.074 | — | 1 |
| TiO2-PTI/SPE | SWV | 0.01–10 μM | 3.680803 | — | 40 |
| Gr/TiO2@MIP-PANi | CV | 1.0–8.0 mM | 3.23491 × 105 | — | 41 |
| cMWCNTs-AgNPs-CS-FAb-BSA/SPCE | CV | 0.1–1000 ng mL−1 | 0.021 | 2.58 | This work |
3.5. Reproducibility, stability and selectivity of the immunosensor
Fig. 13A shows the reproducibility of the cMWCNTs-AgNPs-CS-FAb-BSA/SPCE immunosensor. The results indicate that the current responses stayed at the same level after 6 tests, which shows that FAb was immobilized stably on the cMWCNTs-AgNPs-CS-FAb-BSA/SPCE. The RSD for the oxidation peak current responses to 1 ng per mL FP in 0.1 M PBS buffer solution (pH 7.5) was 2.58%. This demonstrates that the novel electrochemical immunosensor has good reproducibility (Fig. 13B).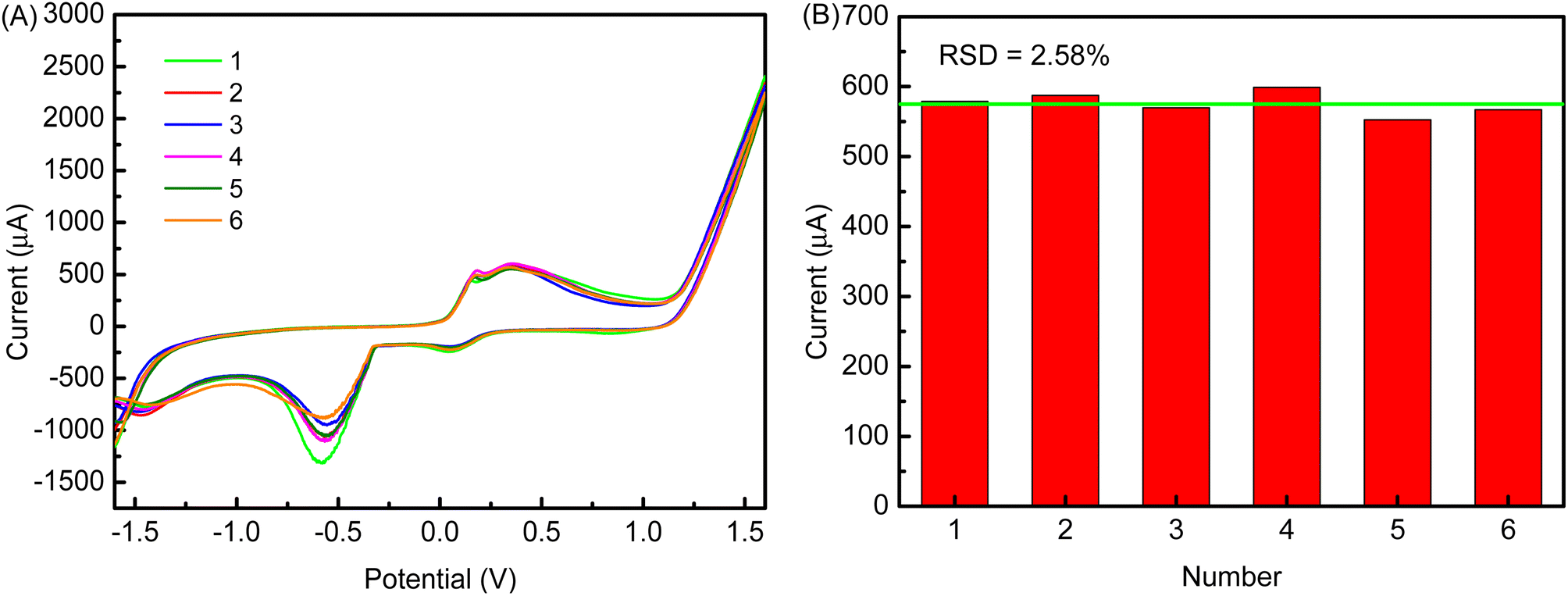 | ||
| Fig. 13 Reproducibility of the immunosensor. (A) The CV responses of six cMWCNTs-AgNPs-CS-FAb-BSA/SPCE devices fabricated under the same conditions toward 1 ng per mL FP in 0.1 M PBS solution (pH 7.5). (B) The histogram of their CV anodic peak currents. | ||
After 5 days of storage at 4 °C, the current response of the cMWCNTs-AgNPs-CS-FAb-BSA/SPCE reached 80.4% of the initial response in the 1 ng per mL FP solution. Such good stability may be ascribed to the effective protection of the bioactivity of FAb due to the coordination of the carbonyl groups of FAb with the outer-sphere silver atoms. At the same time, a biocompatible microenvironment is provided by the cMWCNTs and chitosan.42
To evaluate the selectivity of the immunosensor, some potential interferents were added to 1 ng mL−1 of FP, including RAC (10 ng mL−1), pyraclofos (10 ng mL−1) and ethiprole (10 ng mL−1). Compared with the current response to pure FP (Fig. 14), the variations in the amperometric response caused by the interfering substances RAC, pyraclofos and ethiprole were 3.6%, 6.5% and 3.7%, respectively. The change in the current signal was less than 7%, which suggests that the immunosensor has good anti-interference capability.
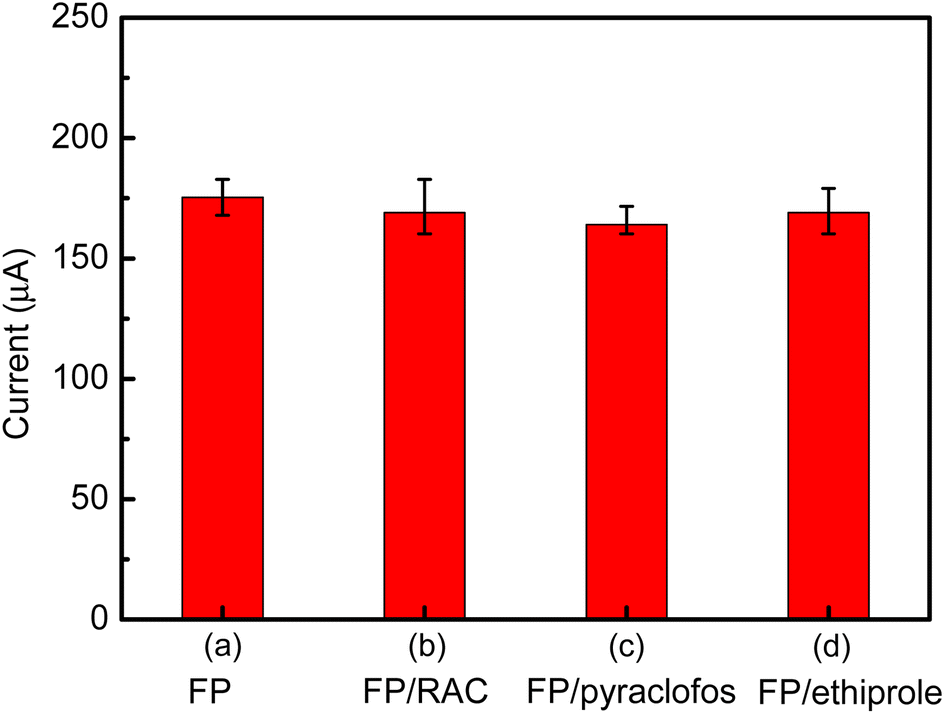 | ||
| Fig. 14 Selectivity of the immunosensor. The anodic peak currents of the immunosensor in the presence of 1 ng per mL FP and various interfering substances (10 ng mL−1) in a 0.1 M PBS solution (pH 7.5) containing 10 mM [Fe(CN)6]3−/4−: (a) 1 ng per mL FP; (b) 1 ng per mL FP + 10 ng per mL RAC; (c) 1 ng per mL FP + 10 ng per mL pyraclofos, and (d) 1 ng per mL RAC + 10 ng per mL ethiprole. | ||
3.6. Analysis of real samples
To demonstrate the practical application of the immunosensor, the detection of FP in tea leaf samples was performed using the standard addition method. The recovery and RSD (n = 5) values of FP are summarized in Table 2. According to the calibration curve, the FP recovery rates were between 95.64–104.32%. The RSD ranged from 2.91% to 8.79%. The results confirm that the procedure can be used for effective quantitative detection of FP in actual samples.| Amount added (ng mL−1) | Found (ng mL−1) | Recovery (%) | RSD (%) | ||||
|---|---|---|---|---|---|---|---|
| 5 | 4.44 | 4.95 | 4.95 | 4.44 | 5.58 | 95.64 | 8.79 |
| 50 | 46.40 | 51.78 | 58.29 | 51.78 | 46.40 | 104.32 | 8.43 |
| 150 | 158.73 | 159.19 | 168.41 | 162.05 | 154.09 | 101.37 | 2.91 |
4. Conclusions
In this study, we combined the advantages of cMWCNTs, AgNPs and CS to fabricate an electrochemical immunosensor sensitive to FP by the freeze-drying technique. The highly porous CS cryogel was decorated by the cMWCNTs-AgNPs composite. The modified SPCE with a large surface area was highly conductive, which enabled a high load of Fab, enhanced electron transfer and improved the current signal amplification. The cMWCNTs-AgNPs-CS-FAb-BSA-modified electrode had a higher electrocatalytic activity towards [Fe(CN)6]3−/4− oxidation than the bare SPCE, as shown by CV and EIS. In a concentration range of 0.1–1000 ng per mL FP, the correlation coefficient R2 was 0.998, and the detection limit of the sensor was 0.021 ng mL−1. At the same time, the immunosensor displayed great reproducibility, stability and selectivity. Therefore, the modified electrochemical immunosensor of cMWCNTs-AgNPs-CS-FAb-BSA/SPCE could be applied for FP residue analysis of agricultural products. In addition, different biomolecules can also be immobilized using the current immobilization strategy.Author contributions
Wen-Chien Huang: conceptualization, methodology, investigation, data curation, writing – original article, writing – review and edition. You-Ning Hsiung: acquisition of data, analysis and interpretation of data. Chia-Ling Li: acquisition of data, analysis of data.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Science and Technology Council for supporting this work (MOST 110-2222-E-606-001-).References
- H. Huang, C. Zhang, J. Zhou, D. Wei, T. Ma, W. Guo, X. Liu, S. Li and Y. Deng, Biosensors, 2022, 12, 775 CrossRef CAS PubMed.
- Y. Fukunaga and T. Okada, Anal. Chem., 2022, 94, 13507–13515 CrossRef CAS PubMed.
- I. Cabezudo, M. O. Salazar, I. A. Ramallo and R. L. E. Furlan, Food Chem., 2022, 390, 132937 CrossRef CAS PubMed.
- T. Chen, J. Hu, Y. Chen, Y. Liu, Y. Li and H. Xu, Anal. Chim. Acta, 2022, 1190, 339263 CrossRef CAS PubMed.
- R. Özcan, Ç. Büyükpınar and S. Bakırdere, J. Environ. Sci. Health, Part B, 2022, 55, 1041–1047 CrossRef PubMed.
- S. Wu, J. Zhao, J. Fang and H. Li, LCGC North Am., 2021, 39, 554–560 Search PubMed.
- Z. Wang, J. Chen, T. Zhan, X. He and B. Wang, Ecotoxicol. Environ. Saf., 2020, 189, 110002 CrossRef CAS PubMed.
- L. Paolini, N. Hausser and T. Zhang, Chirality, 2022, 34, 473–483 CrossRef CAS PubMed.
- G. L. Scheel and C. R. T. Tarley, J. Mol. Liq., 2020, 297, 111897 CrossRef CAS.
- I. H. Cho, J. Lee, J. Kim, M. S. Kang, J. K. Paik, S. Ku, H. M. Cho, J. Irudayaraj and D. H. Kim, Sensors, 2018, 18, 207 CrossRef PubMed.
- D. Lee, J. Bhardwaj and J. Jang, Sci. Rep., 2022, 12, 2311 CrossRef CAS PubMed.
- G. C. Chen, C. H. Liu and W. C. Wu, Anal. Chim. Acta, 2021, 1143, 84–92 CrossRef CAS PubMed.
- G. Ibáñez-Redín, D. Wilson, D. Gonçalves and O. N. Oliveira Jr, Adv. Colloid Interface Sci., 2018, 515, 101–108 CrossRef PubMed.
- N. A. Badruzaman, M. A. M. Azmi and N. A. M. Said, Key Eng. Mater., 2020, 833, 171–175 Search PubMed.
- C. Thunkhamrak, P. Chuntib, K. Ounnunkad, P. Banet, P. H. Aubert, G. Saianand, A. I. Gopalan and J. Jakmunee, Talanta, 2020, 208, 120389 CrossRef CAS PubMed.
- V. Primpray, W. Kamsong, S. Pakapongpan, K. Phochakum, A. Kaewchaem, A. Sappat, A. Wisitsoraat, T. Lomas, A. Tuantranont and C. Karuwan, Talanta Open, 2022, 6, 100155 CrossRef PubMed.
- N. Z. M. Azman, P. N. S. Zainal and S. A. Alang, PLoS One, 2020, 15, e0234148 CrossRef CAS PubMed.
- S. Chanarsa, J. Jakmunee and K. Ounnunkad, Front. Chem., 2021, 9, 631571 CrossRef CAS PubMed.
- S. Mahari and S. Gandhi, Bioelectrochemistry, 2022, 144, 108036 CrossRef CAS PubMed.
- N. X. Viet, N. X. Hoana and Y. Takamura, Mater. Chem. Phys., 2019, 227, 123–129 CrossRef.
- W. Yu, Y. Sang, T. Wang, W. Liu and X. Wang, Food Sci. Nutr., 2020, 8, 1001–1011 CrossRef CAS PubMed.
- P. Carneiro, J. A. Loureiro, C. Delerue-Matos, S. Morais and M. D. CarmoPereir, Talanta, 2023, 252, 123838 CrossRef CAS PubMed.
- S. Zhu, Y. Cao, Y. Xu, Y. Yin and G. Li, Int. J. Mol. Sci., 2013, 14, 10298–10306 CrossRef PubMed.
- V. Singh, A. Shrivastava and N. Wahi, Afr. J. Adv. Biotechnol., 2015, 14, 2554–2567 CrossRef.
- S. Mishra, S. N. Tripathi, V. Choudhary and B. D. Gupta, Plasmonics, 2015, 10, 1147–1157 CrossRef CAS.
- K. H. Wu, W. C. Huang, S. C. Chang and R. H. Shyu, RSC Adv., 2022, 12, 13035 RSC.
- S. Velmurugan, S. Palanisamy, T. C.-K. Yang, M. Gochoo and S. W. Chen, Ultrason. Sonochem., 2020, 62, 104863 CrossRef CAS PubMed.
- Y. Seo, J. Hwang, J. Kim, Y. Jeong, M. P. Hwang and J. Choi, Int. J. Nanomedicine, 2014, 9, 4621–4629 Search PubMed.
- F. Ahmed, M. B. Kanoun, C. Awada, C. Jonin and P. F. Brevet, Crystals, 2021, 11, 1488 CrossRef CAS.
- A. S. Lanje, S. J. Sharma and R. B. Pode, J. Chem. Pharm. Res., 2010, 2, 478–483 CAS.
- R. S. Costa, A. Guedes, A. M. Pereira and C. Pereira, J. Mater. Sci., 2020, 55, 10121–10141 CrossRef CAS.
- Y. Zhang, H. Ma, D. Wu, Y. Li, B. Du and Q. Wei, J. Electroanal. Chem., 2015, 741, 14–19 CrossRef CAS.
- A. A. Ansari, G. Sumana, M. K. Pandey and B. D. Malhotra, J. Mater. Res., 2009, 24, 1667–1673 CrossRef CAS.
- Y. Akazawa-Ogawa, H. Nagai and Y. Hagihara, Biophys. Rev., 2018, 10, 255–258 CrossRef CAS PubMed.
- C. Gu, P. Ren, F. Zhang, G. Zhao, J. Shen and B. Zhao, ACS Omega, 2020, 5, 5548–5555 CrossRef CAS PubMed.
- F. Okumura, R. Amaral, E. Orestes, A. D. Silva and L. H. Mazo, J. Braz. Chem. Soc., 2016, 27, 925–932 CAS.
- M. Maulidiyah, T. Azis, L. Lindayani, D. Wibowo, L. O. A. Salim, A. Aladin, M. Nurdin and M. Nurdin, J. Electrochem. Sci. Technol., 2019, 10, 394–401 CrossRef CAS.
- M. Nurdin, O. A. Prabowo, Z. Arham, D. Wibowo, M. Maulidiyah, S. K. M. Saad and A. A. Umar, Surf. Interfaces, 2019, 16, 108–113 CrossRef CAS.
- M. Nurdin, Z. Arham, S. Rahayu, L. O. A. Salim and M. Maulidiyah, J. Rekayasa Kimia Lingkungan, 2020, 15, 71–78 CrossRef.
- S. El-Akaad, R. Morozov, M. Golovin, O. Bol'shakov, S. Saeger and N. Beloglazova, Talanta, 2022, 238, 123025 CrossRef CAS PubMed.
- M. Nurdin, Z. Arham, W. O. Irna, M. Maulidiyah, K. Kurniawan, L. O. A. Salim, I. Irwan and A. A. Umar, Mater. Sci. Semicond. Process., 2022, 151, 106994 CrossRef CAS.
- K. H. Wu, W. C. Huang, R. H. Shyu and S. C. Chang, J. Inorg. Biochem., 2020, 210, 111163 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |