DOI:
10.1039/D2OB01644C
(Review Article)
Org. Biomol. Chem., 2023,
21, 24-38
Iodonium ylides: an emerging and alternative carbene precursor for C–H functionalizations
Received
9th September 2022
, Accepted 10th November 2022
First published on 23rd November 2022
Abstract
The metal-catalyzed successive activation and functionalization of arene/heteroarene is one of the most fundamental transformations in organic synthesis and leads to privileged scaffolds in natural products, pharmaceuticals, agrochemicals, and fine chemicals. Particularly, transition-metal-catalyzed C–H functionalization of arenes with carbene precursors via metal carbene migratory insertion has been well studied. As a result, diverse carbene precursors have been evaluated, such as diazo compounds, sulfoxonium ylides, triazoles, etc. In addition, there have been significant developments with the use of iodonium ylides as carbene precursors in recent years, and these reactions proceed with high efficiencies and selectivities. This review provides a comprehensive overview of iodonium ylides in C–H functionalizations, including the scope, limitations, and their potential synthetic applications.
1. Introduction
Transition-metal-catalyzed C–H functionalizations have proven to be a successful strategy for the formation of various C–X bonds (X = C, N, O, etc.) in the field of organic synthesis.1 The classical cross-couplings have been extensively studied by researchers for the formation of C–C-bond-forming reactions; however, these reactions demand pre-functionalized (hetero)arenes as substrates.2 In this context, an alternative strategy would appear to be the direct functionalization of (hetero)arene substrates by activation of an inert C–H bond, which would exclude the unwanted pre-functionalization of coupling reagents. Therefore, these transformations are economical, efficient, and environmentally friendly to synthesize complex scaffolds. In this protocol, the heteroatom of the organic molecule coordinates with the metal catalyst, which directs the cleavage of a proximal C–H bond and leads to formation of a kinetically or thermodynamically favoured cyclometallated complex, which further reacts with another coupling partner, and subsequent insertion and reductive elimination affords the corresponding product along with regeneration of the metal catalyst.3 Based on this strategy, diverse metal-catalyzed C–H functionalizations have been explored, and the topic has been covered by some excellent reviews.4
Carbene precursors have attracted great attention recently from synthetic organic chemists due to their unique reactivities.5 Interestingly, they have been extensively explored in transition-metal-catalyzed C–H functionalizations, such as alkylations, alkenylations, annulations, etc.6 Over the course of the past decade, many carbene precursors have been a part of these transformations, such as diazo compounds, hydrazones, triazoles, enynones, sulfoxonium ylides, etc. Based on that, several groups have developed elegant approaches toward achieving complex molecule synthesis, and this topic has been extensively reviewed.7 Despite the many impressive advances in this field, there are still many unmet challenges that remain unexplored. Hence, developing novel precursors to assemble interesting molecular frameworks remains highly desirable.
Hypervalent iodonium compounds have witnessed significant advances in organic chemistry due to their characteristic features, such as thermal stability, solubility in common organic solvents, and eco-friendly nature.8 Among them, iodonium ylides are known to show good stability and significant synthetic strategies via metalcarbenoid intermediates.9 Recently, iodonium ylides have been introduced as coupling partners in C–H functionalizations and have grown rapidly, and a large number of synthetically challenging transformations have been disclosed in the recent literature. Herein, iodonium ylide acts as a carbene precursor, which is a safe and alternative carbene coupling partner to the metalcarbenoids in previous literature. This review article will describe the synthesis of iodonium ylides and their implementation in various synthetic transformations towards the rapid assembly of diverse alkenylations and annulations and their biological and natural product applications. The following sections give an overview of all these different transformations.
2. Synthesis of iodonium ylides
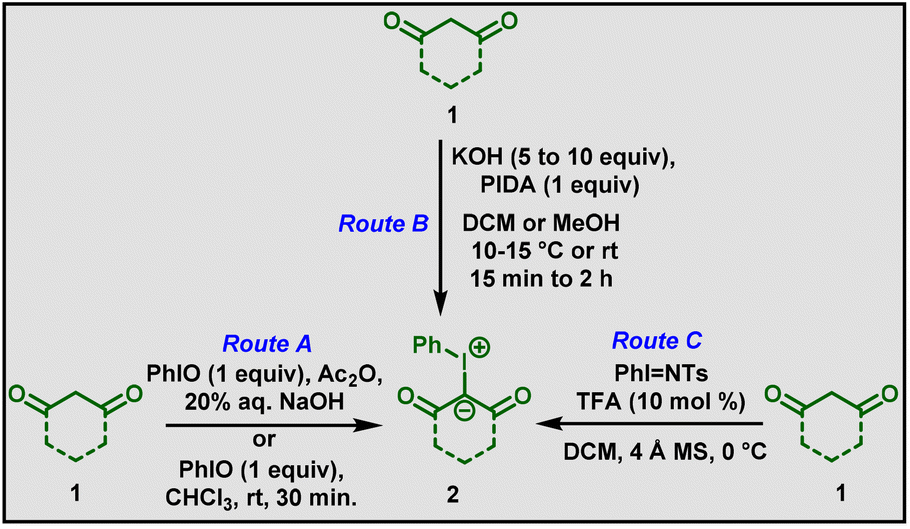
Iodonium ylides (2) can be synthesized by three routes (Scheme 1). The first report was published by Neiland and co-workers in 1957.10 The ylides were prepared from (difluoroiodo)benzene and dimedone. Later, Hayasi et al., reported the synthesis of iodonium ylides from the condensation of 1,3-dicarbonyl compounds with iodosobenzene in the presence of acetic anhydride (Route A, Scheme 1).11 Modification of the Hayasi conditions was then carried out. In 1975, Koser and co-workers found that a stoichiometric amount of (diacetoxyiodo)benzene (PIDA) in dichloromethane solvent could accelerate the formation of iodonium ylides.12 Based on that, many methods have been developed by changing the reaction conditions, such as solvent, temperature, base, and equivalents of (diacetoxyiodo)benzene (Route B, Scheme 1).13 Additionally, in 2014, Chan and co-workers reported that N-tosyliminobenzyliodinane could undergo iodonium ylide transfer in presence of trifluoroacetic acid (Route C, Scheme 1).14 Although all these synthetic routes generated iodonium ylides, Route B has gained much more attention and has been utilized in recent years.
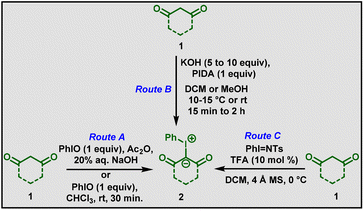 |
| | Scheme 1 Synthesis of iodonium ylides. | |
3. Synthesis of oxygen-related heterocycles
Oxygen-containing heterocyclic compounds are arguably the most important structural motifs in organic chemistry and are widely present in biologically active compounds, agrochemicals, ligands, natural products, etc.15 Therefore, novel, practical, and sustainable methods for synthesizing oxygen-bearing heterocycles remain a significant interest in synthetic chemistry.16 In this scenario, catalytic activation of the arene C–H bond followed by annulation with carbene precursors has become one of the most attractive methods to access the valuable heteroarene moieties. Recently, iodonium ylides are also involved in synthesis of oxygen heterocycles via transition metal catalyzed C–H functionalizations.
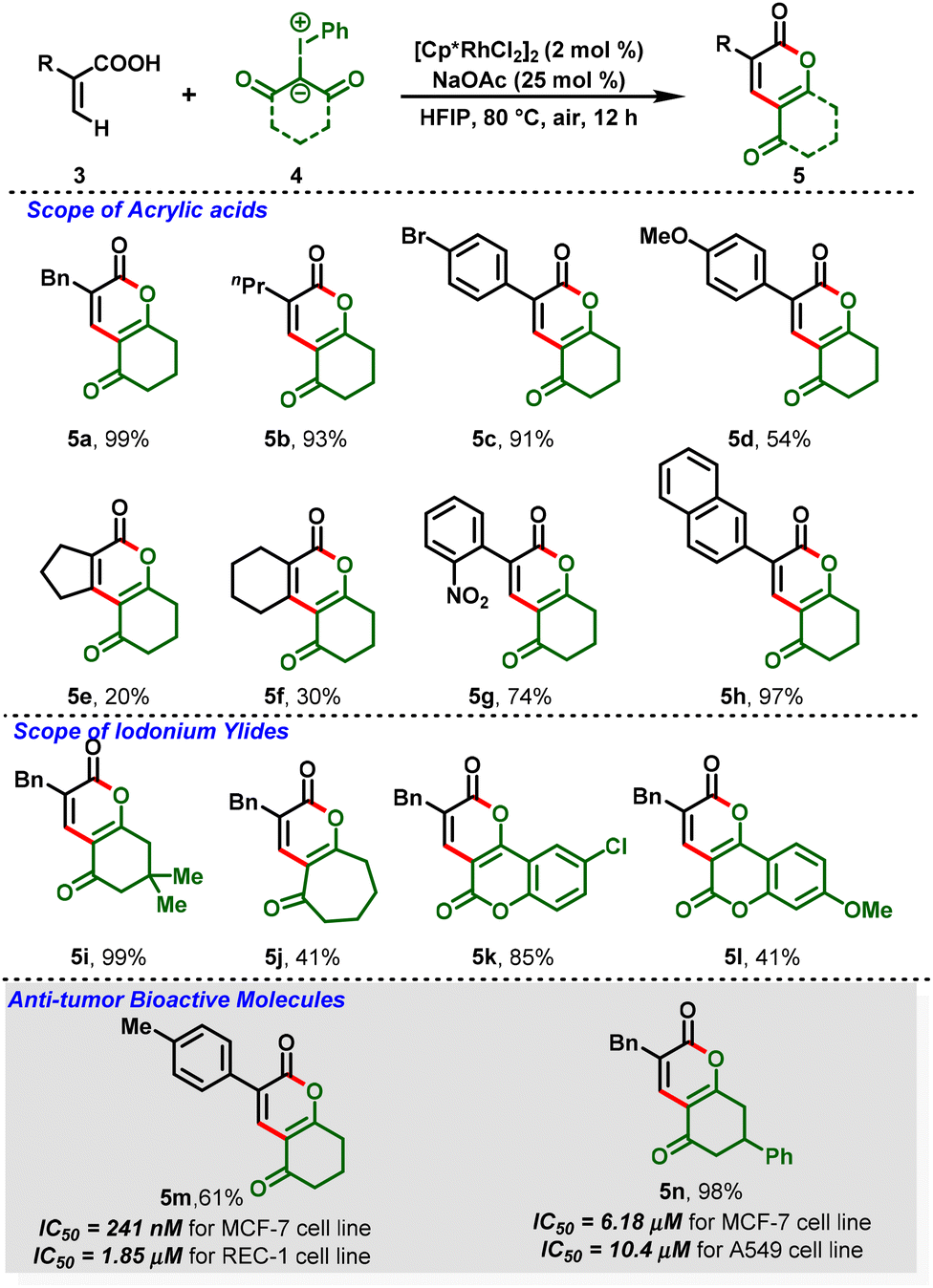
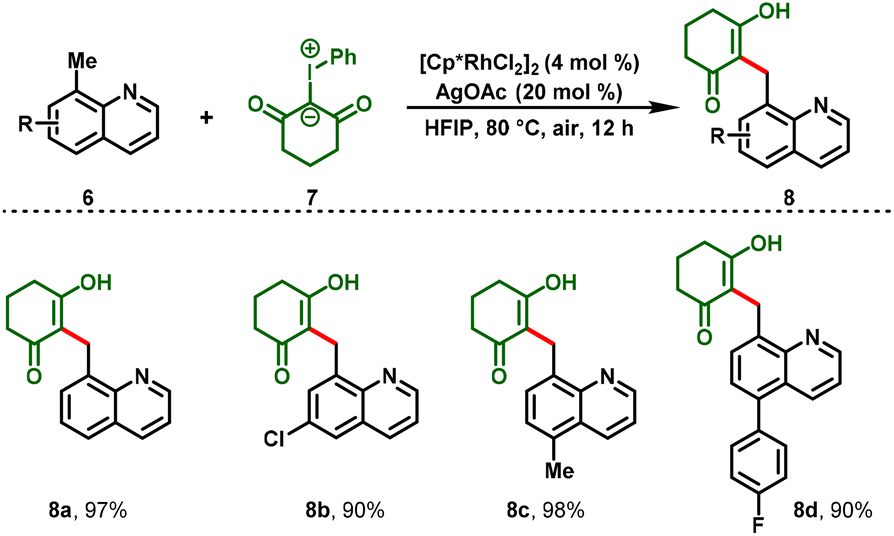
Notably, for the first time, Li and co-workers reported a Rh(III)-catalyzed C–H functionalization between iodonium ylides (4) and acrylic acids (3) leading to the formation of dihydro-2H-chromene derivatives (5), in which iodonium ylides are utilized as a carbene precursor.17 This protocol proceeded under mild and redox-neutral conditions, and the reaction system exhibits relatively high efficiency with broad substrate compatibility. Further, the [3 + 3]-annulated products showed anti-tumor activity in different human cancer cells at the micro-molar or nano-molar level (5m & 5n) (Scheme 2). Additionally, benzylic C–H functionalization was also investigated after a simple optimization. They showed that 8-methylquinolines (6) can react with iodonium ylides (7) under the optimized conditions to afford the C(sp3)–H functionalization products (8) in decent-to-excellent yields (Scheme 3). Furthermore, the authors tested the generality of iodonium ylides in diverse arene C–H activations. Interestingly, arenes containing either an electrophilic directing group (imine) or a nucleophilic directing group (NH, oxime, or OH) proceeded smoothly and provided the corresponding heterocyclic scaffolds in moderate-to-high yields.
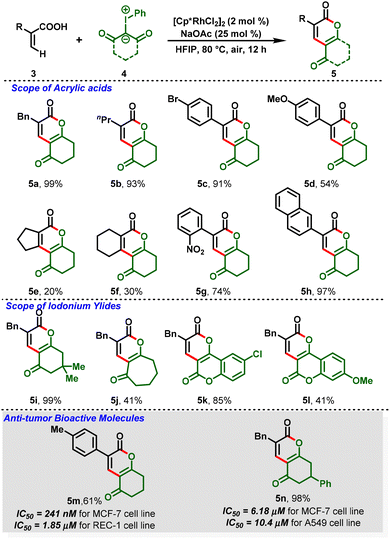 |
| | Scheme 2 Synthesis of dihydro-2H-chromenes. | |
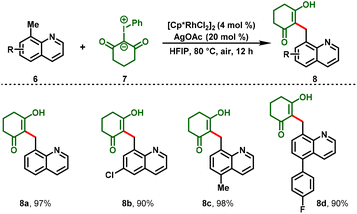 |
| | Scheme 3 Synthesis of benzyl C–H functionalizations. | |
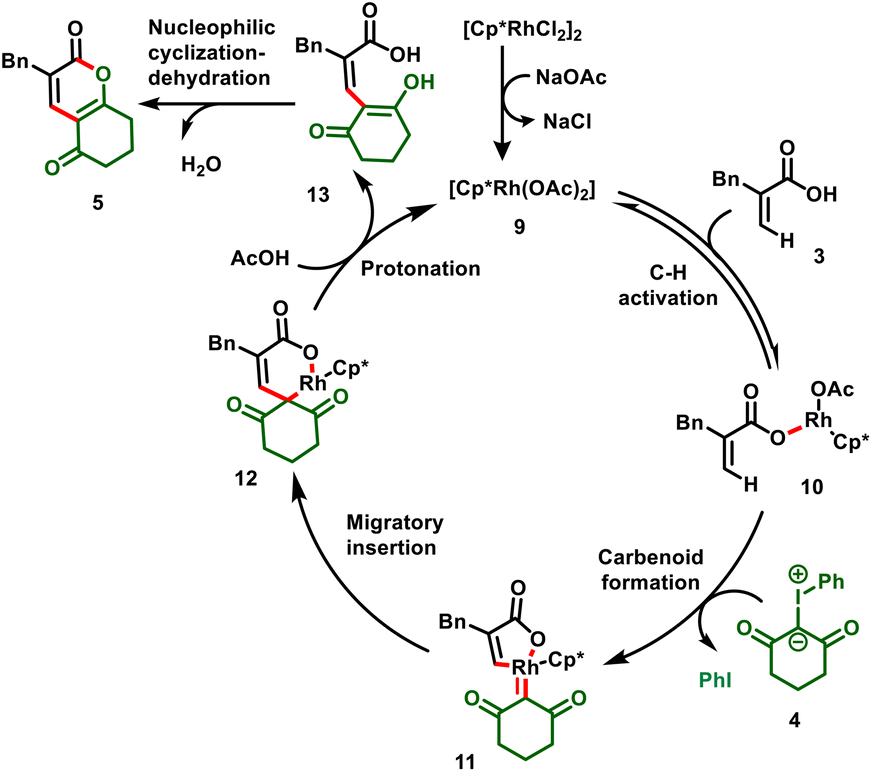
The authors elaborated a plausible mechanism, which is depicted in Scheme 4. The active catalyst 9 is achieved by ligand exchange between base and acrylate, which activates the vinyl C–H bond (10), and subsequent coordination with iodonium ylide, followed by elimination of iodobenzene, forms a 5-membered Rh-carbenoid intermediate (11). Intermediate 11 further undergoes migratory insertion followed by protonation to give the C–C coupled product (13) with the regeneration of active Rh catalyst 9. Compound 13 eventually undergoes nucleophilic cyclization–dehydration to furnish the corresponding dihydro-2H-chromene-diones (5).
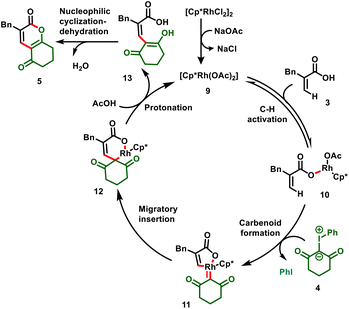 |
| | Scheme 4 Proposed mechanism. | |
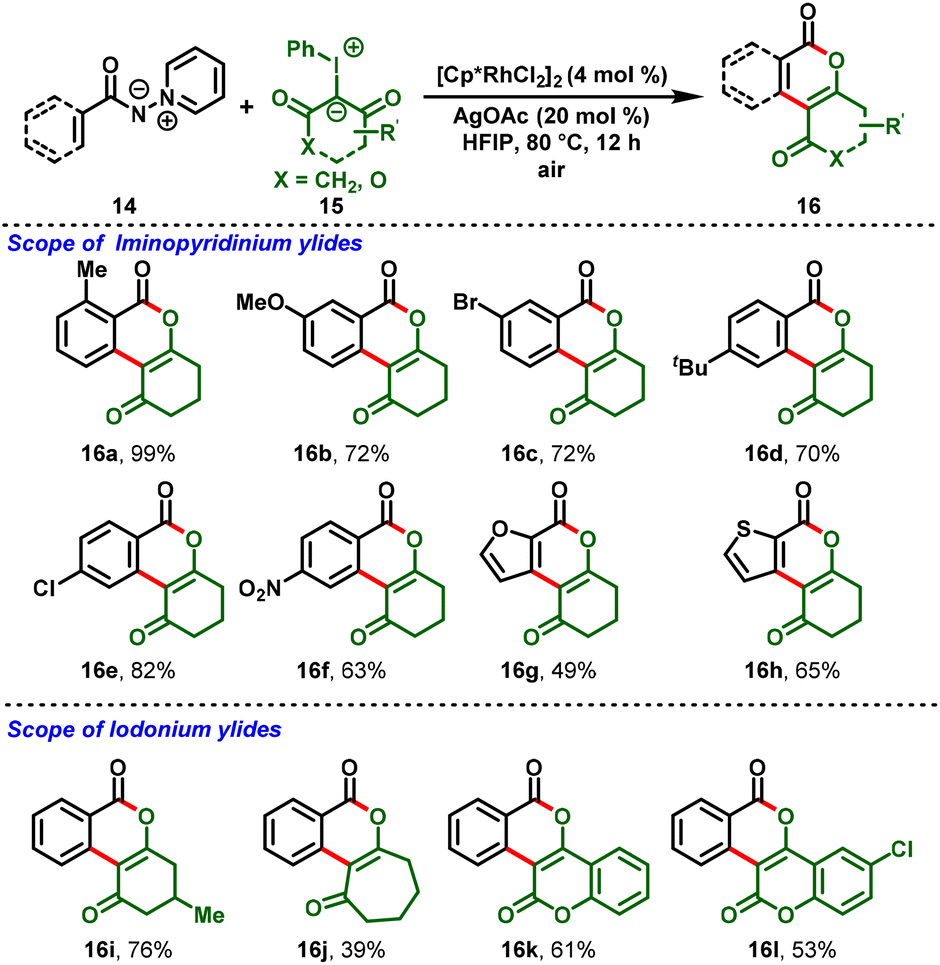
In 2021, Liu and his group realized the Rh(III)-catalyzed redox-neutral annulation of iminopyridinium ylides (14) with iodonium ylides (15). Herein, iminopyridinium ylides act as a directing group, and iodonium ylides as carbene coupling partners. The reaction proceeded smoothly to afford the biologically active isocoumarin skeletons (16) via cleavage of the C–N bond in the ylide directing group. With the aid of the mild reaction conditions, diverse iminopyridinium ylides were well-tolerated and delivered the corresponding products with moderate-to-excellent yields (Scheme 5).18
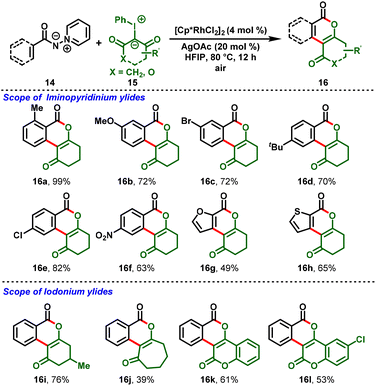 |
| | Scheme 5 Rh(III)-catalyzed C–H functionalization of iminopyridinium ylides. | |
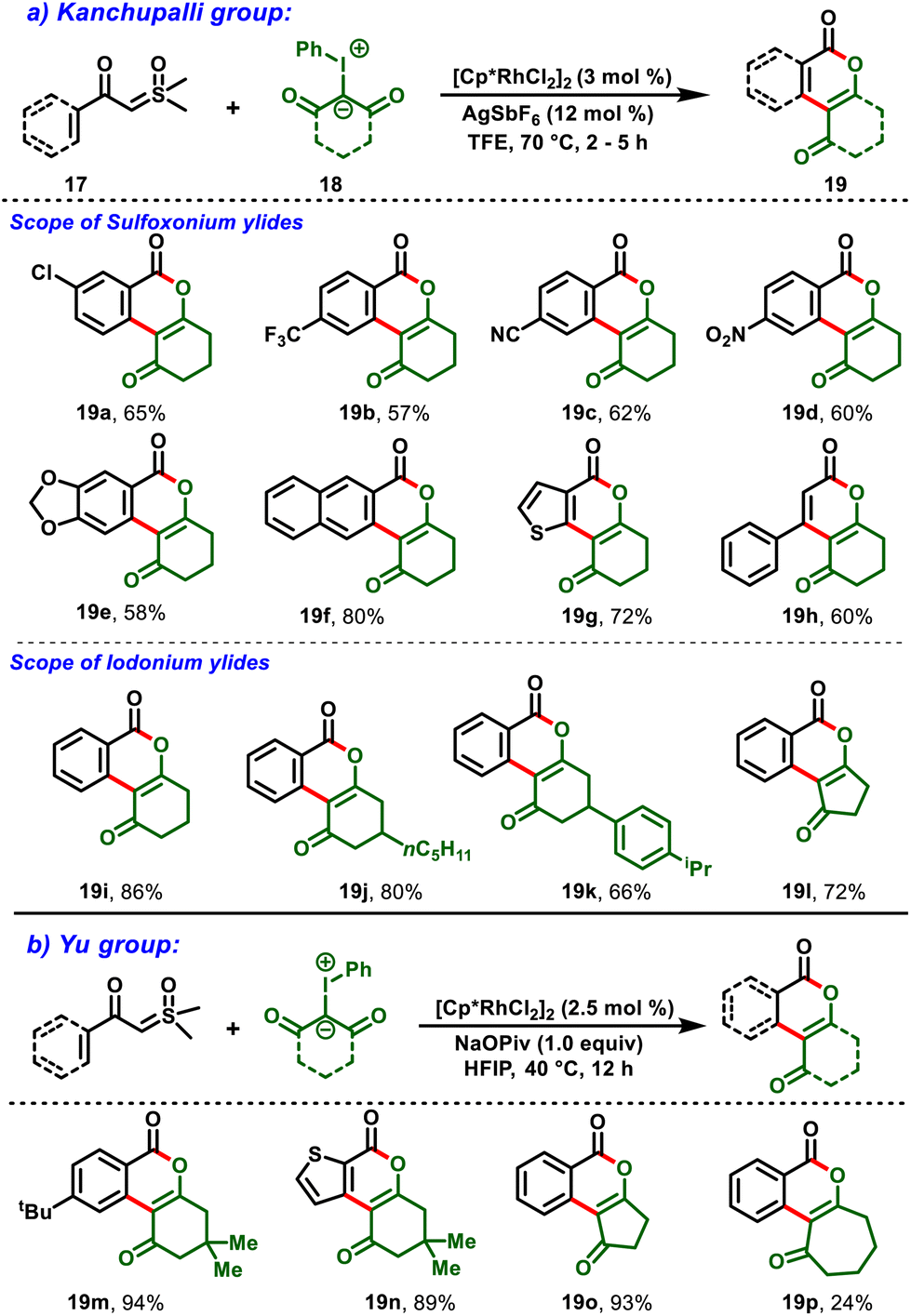
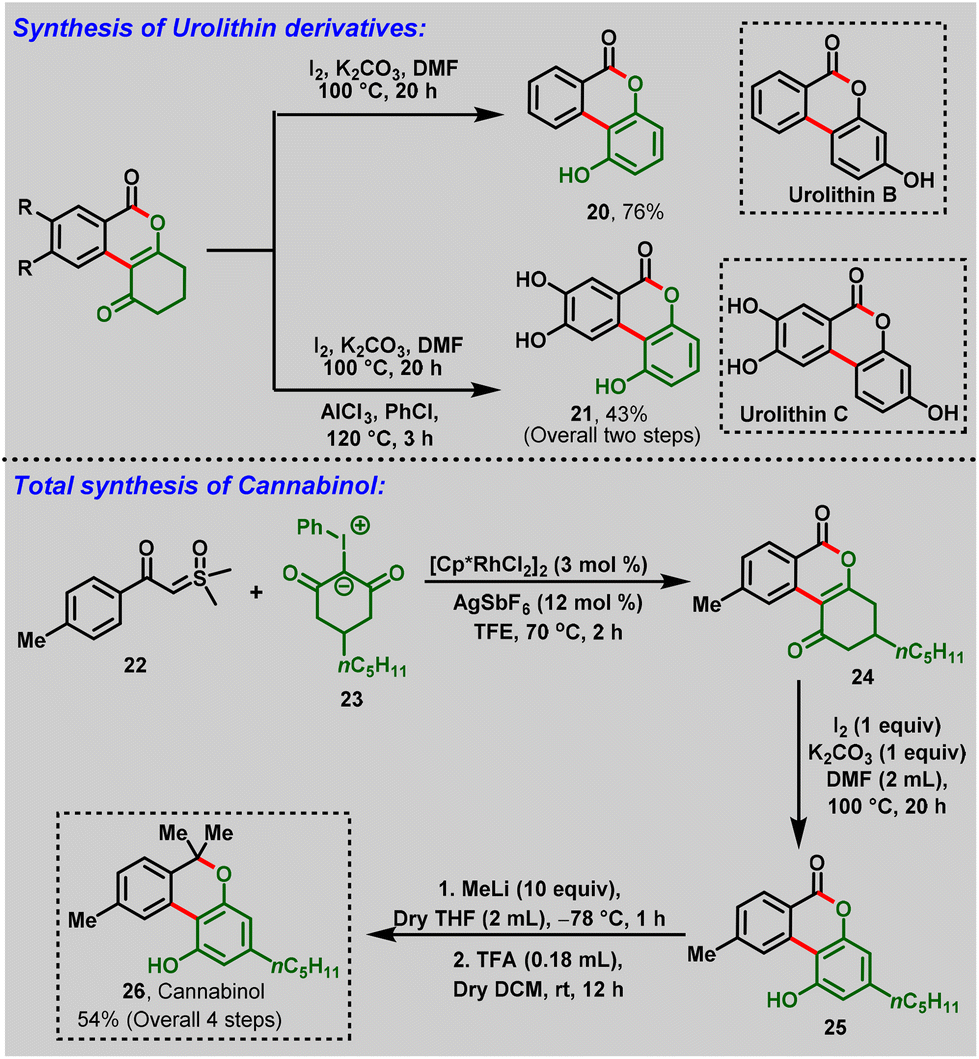
Later, Kanchupalli et al., realized an Rh(III)-catalyzed cross-coupling reaction between sulfoxonium ylides (17) and iodonium ylides (18) to access the biologically important dihydrobenzo[c]chromen-6-one frameworks (19), in which iodonium ylides act as carbene surrogates, and sulfoxonium ylides serve as a traceless directing group functionality in the catalytic system. Under mild conditions, the compatibility of this strategy was validated with various functional groups with moderate-to-excellent yields (Scheme 6a). Notably, the following approach was further extended to biologically potent urolithin derivatives (20 & 21) and a step-economic total synthesis of the natural product cannabinol (CBN) (26) (Scheme 7).19,20
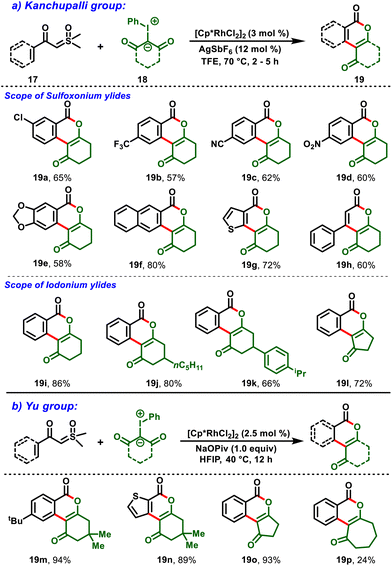 |
| | Scheme 6 Synthesis of dihydrobenzo[c]chromen-6-one frameworks. | |
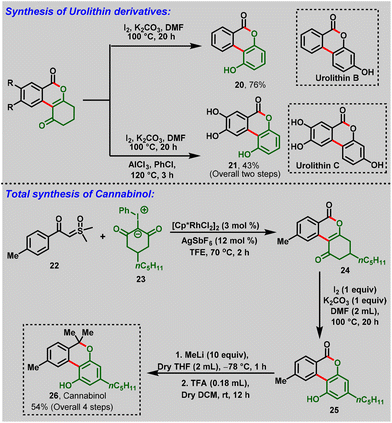 |
| | Scheme 7 Synthetic transformations of dihydrobenzo[c]chromen-6-ones. | |
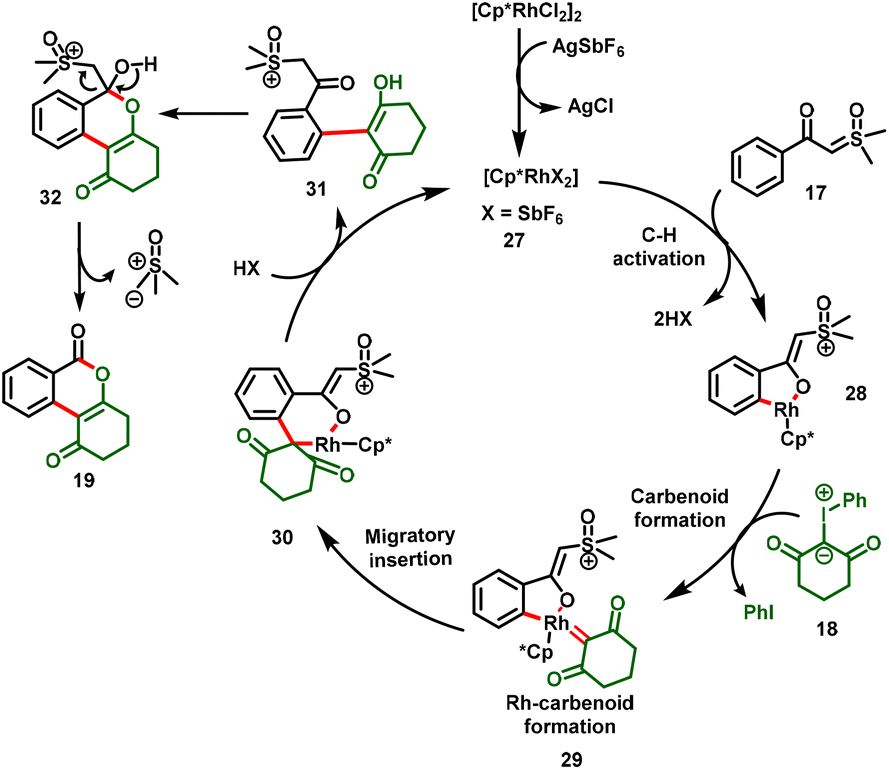
As illustrated in Scheme 8, sequential C–H activation and coordination of iodonium ylides followed by extrusion of aryl iodide generated the Rh carbenoid intermediate (29). Subsequent migratory insertion followed by proto-demetallation yielded the alkenylation product (31). Eventually, intermediate 31 underwent intramolecular nucleophilic addition/cyclization by the subsequent loss of Corey–Chaykovsky reagent (sulfoxonium methylide) to deliver the desired product (19). Within a short period, a similar protocol was demonstrated by Yu et al., (Scheme 6b). Notably, the [3 + 3]-cyclization proceeded under oxidant-free conditions, furnished the isocoumarin derivatives in moderate-to-good yields and exhibited excellent functional group tolerance.21
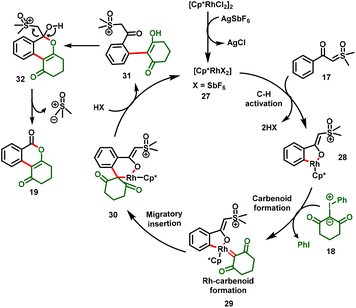 |
| | Scheme 8 Plausible mechanism for Rh(III)-catalyzed cross-coupling reaction. | |
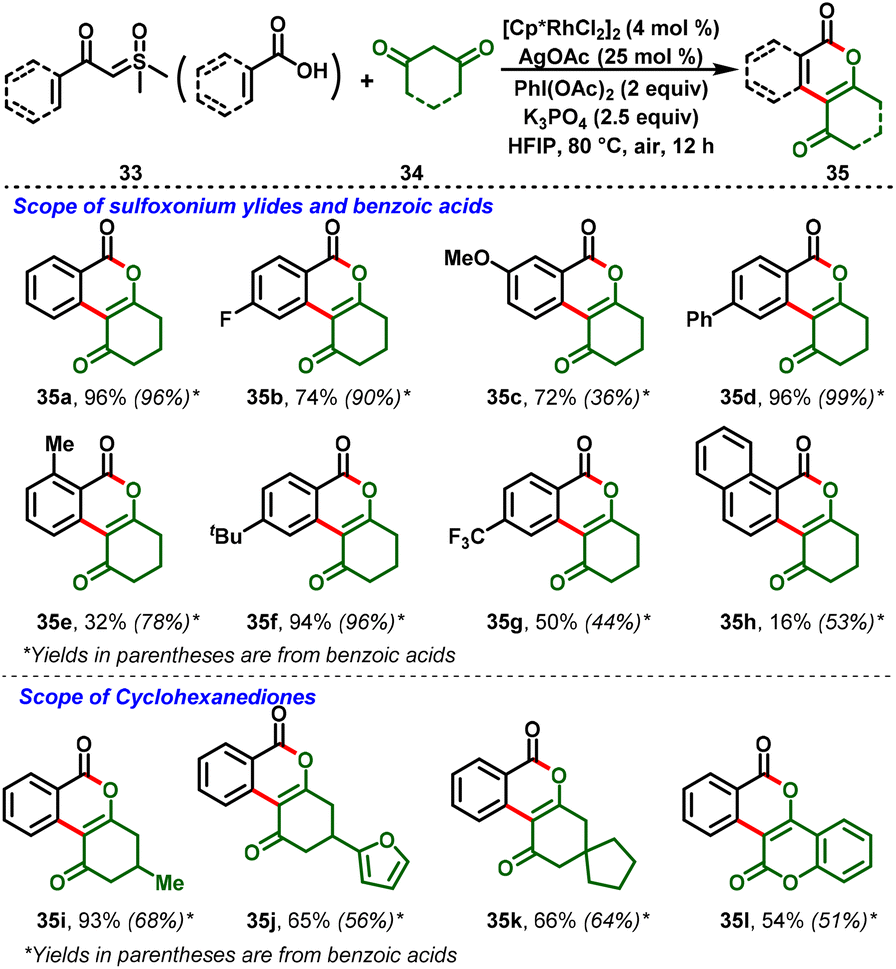
Carboxylic acids have also participated in the Rh(III)-catalyzed cross-coupling strategy. An interesting hetero-coupling reaction between aromatic carboxylic acids (33) or sulfoxonium derivatives with cyclohexadienone (34) under Rh-catalyzed conditions was reported by Liu et al., (Scheme 9). In this process, the key reaction step for the reaction to proceed was the in situ formation of iodonium ylides from 1,3-cyclohexadienone with (diacetoxy)iodobenzene. It further undergoes the C–H functionalization mechanism to achieve a broad range of isocoumarin skeletons (35). Moreover, control experiments and isolation of intermediates revealed that the in situ generation of iodonium ylides is mandatory for the reaction system.22
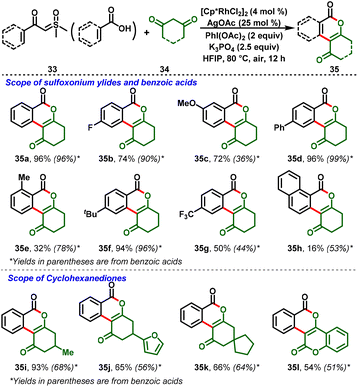 |
| | Scheme 9 Rh(III)-catalyzed efficient synthesis of isocoumarins from cyclohexanediones. | |
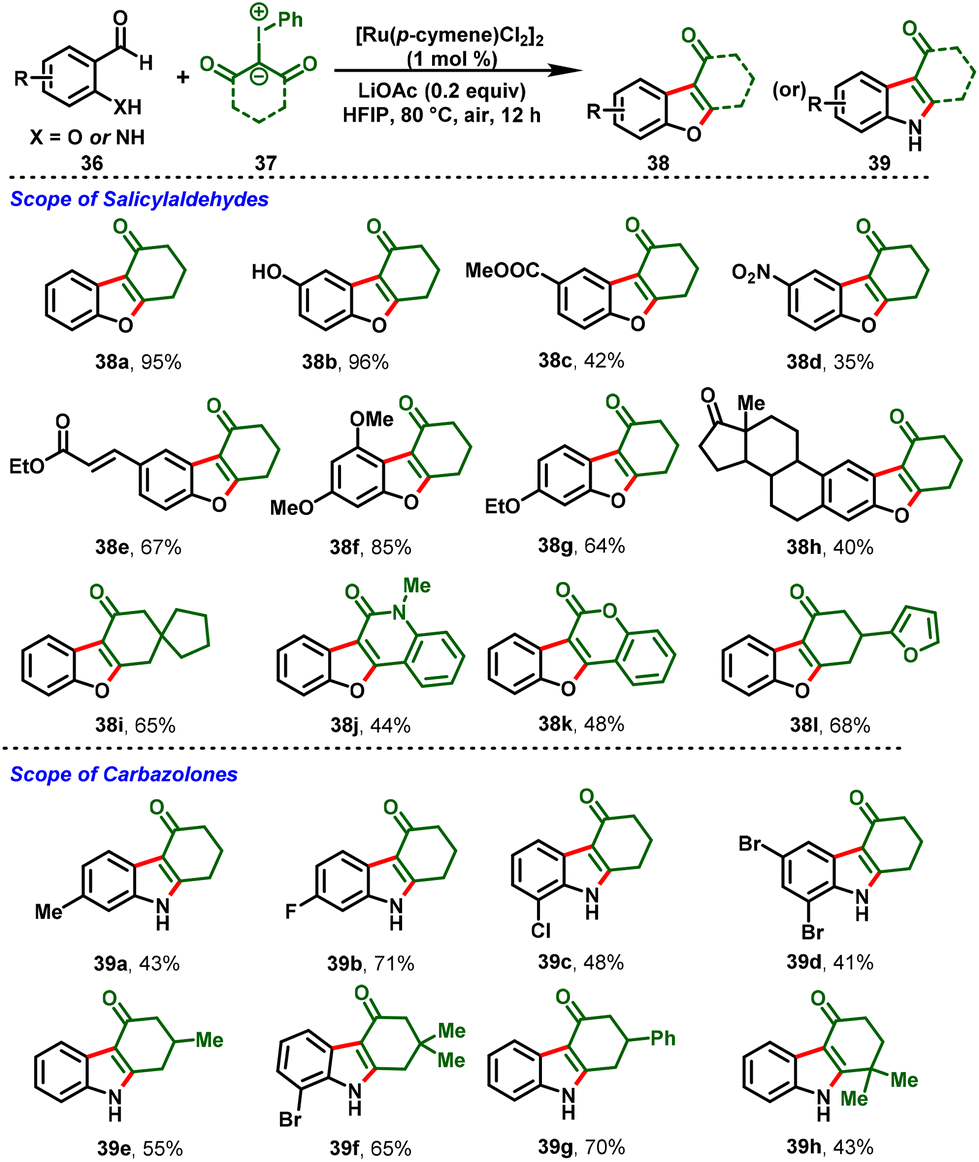
Recently, aldehydes have also proved to be practicable coupling partners in terms of C–C-bond-forming reactions using the decarbonylative strategy. Based on that, the group of Li disclosed Ru(II)-catalyzed chelation-assisted decarbonylation, and cyclization reactions furnishing dibenzo[b,d]furan (38) or carbazolone skeletons (39) using 2-hydroxy benzaldehydes or 2-amino benzaldehydes (36) with iodonium ylides (37) under a mild reaction system and low catalyst loading (Scheme 10). In these reactions, the hydroxy and free amino groups of the benzaldehyde derivatives serve as directing groups and achieve good functional group tolerance with excellent-to-moderate yields. Notably, this strategy extended to late-stage functionalization, scale-up reactions, and diverse synthetic transformations towards C–H bond alkenylation and reduction.23
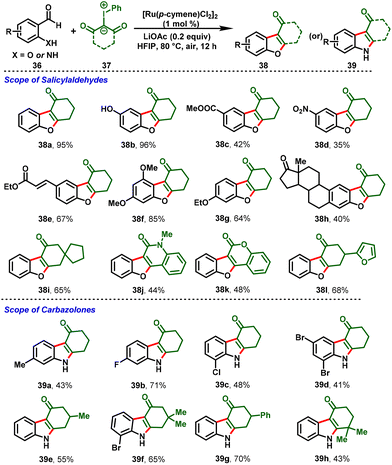 |
| | Scheme 10 Ru(II)-catalyzed decarbonylative annulations of benzaldehydes with iodonium ylides. | |
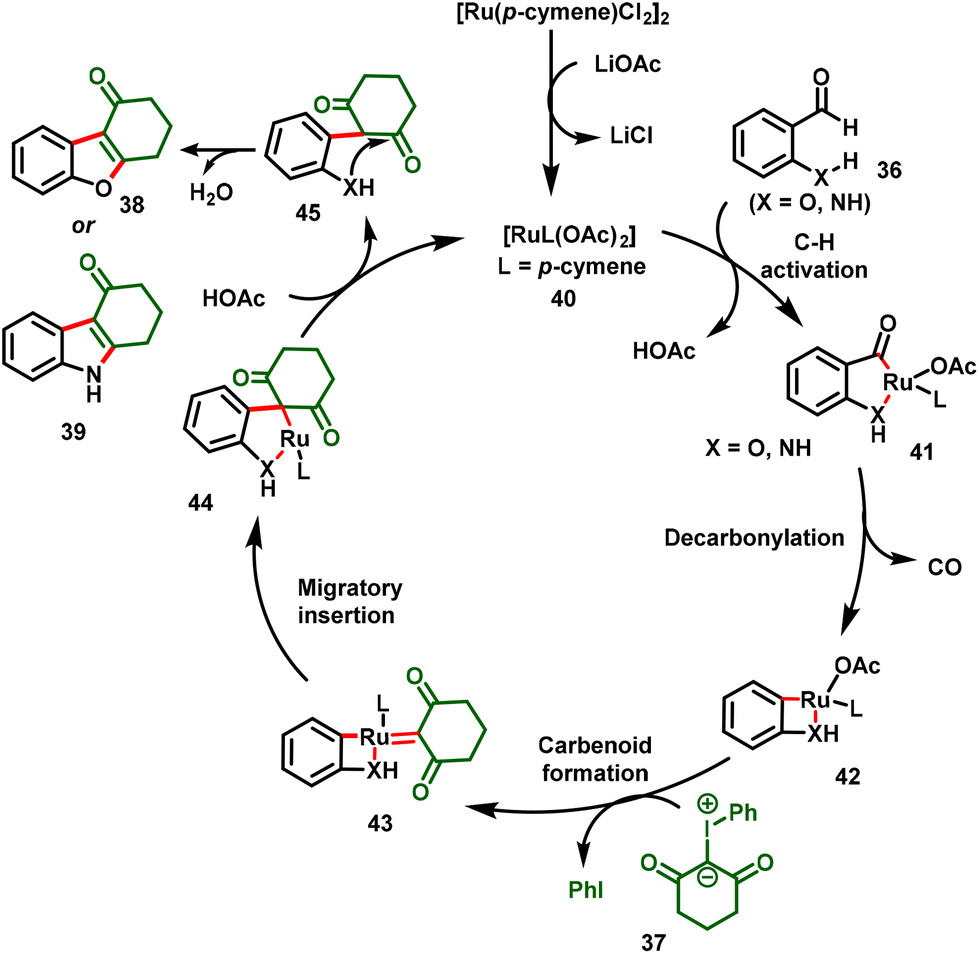
The proposed mechanism is shown in Scheme 11. The mechanism involved the initial C–H activation of 2-hydroxybenzaldehyde or 2-aminoaldehyde with the active Ru catalyst (40) to form five-membered cyclometallated complex 41, which underwent decarbonylation to afford the four-membered ruthenacycle species 42. Subsequently, metalcarbenoid 43 was formed via iodonium ylide coordination along with the release of iodobenzene. Migratory insertion followed by protonolysis then gave intermediate 45. Finally, intramolecular nucleophilic addition followed by dehydration provided the corresponding benzofuran or indole derivatives (38 or 39).
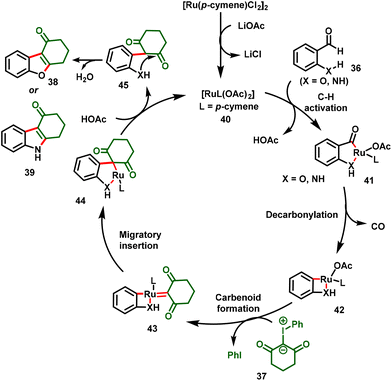 |
| | Scheme 11 Proposed mechanism for 2,3-difunctionalized benzofurans or 2,3-difunctionalized indoles. | |
4. Alkylations and alkenylations
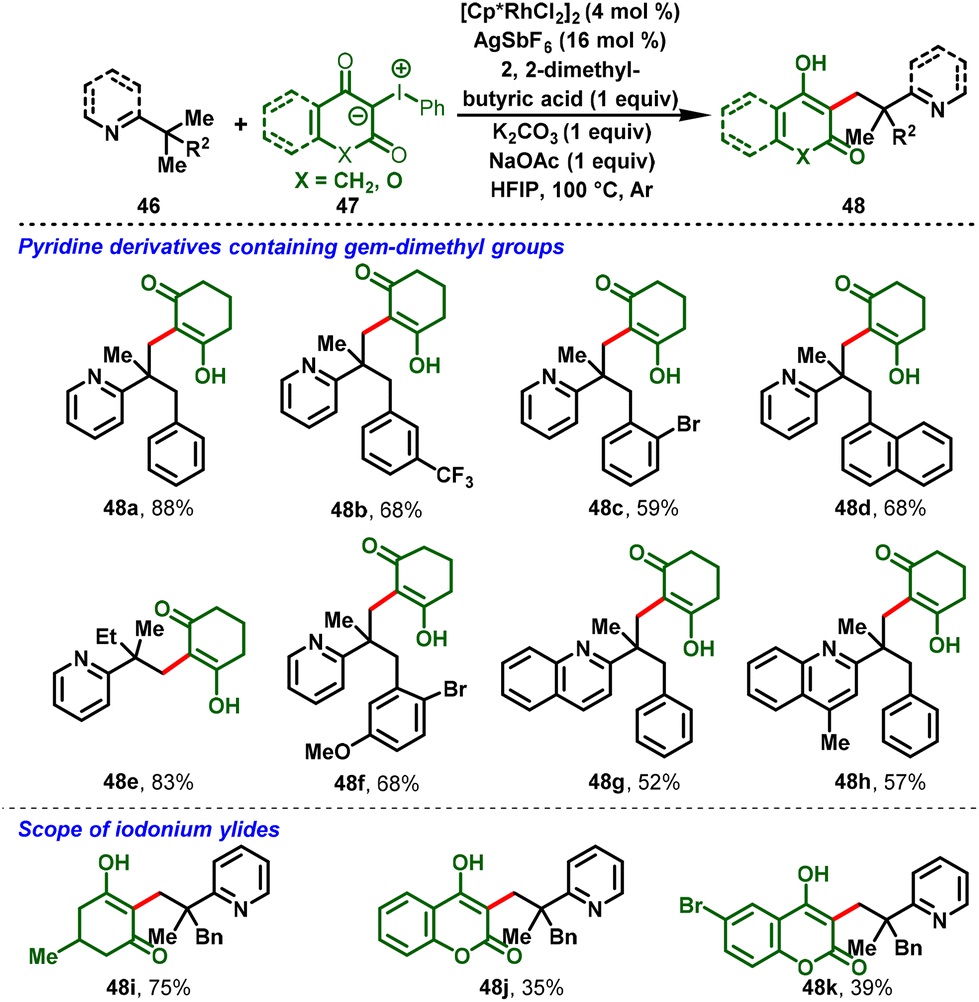
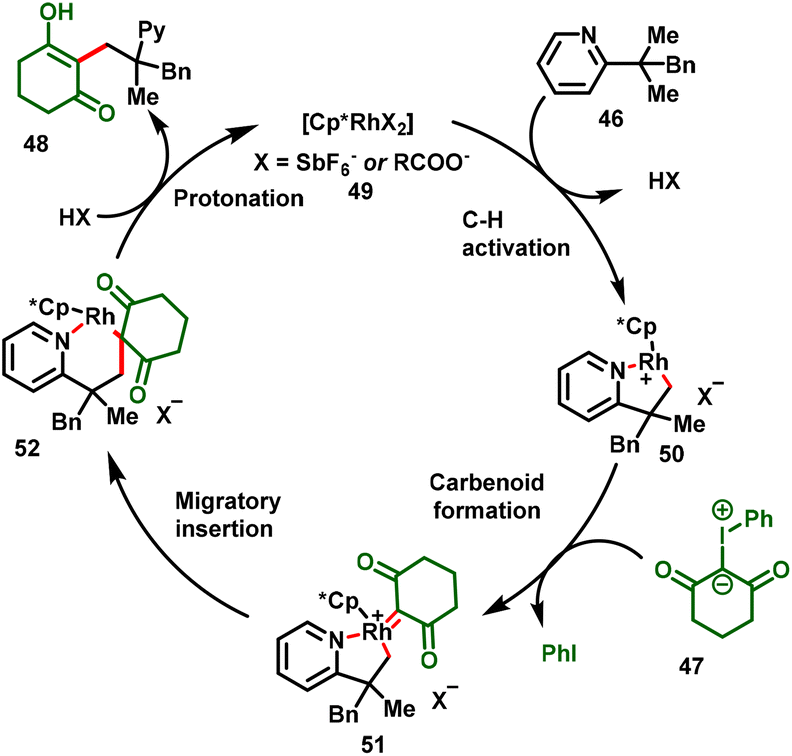
Liu and co-workers succeeded in developing directed C–H alkylations of pyridine containing a gem-dimethyl group (46) with iodonium ylides (47) under catalytic amounts of [Cp*RhCl2]2. In this strategy, pyridine and quinoline were employed as the directing group. The reaction proceeds via the inner-sphere C(sp3)–H carbene insertion reaction leading to C–C coupling products, which was the key to the success of this transformation. The reaction proceeded at 100 °C and gave moderate-to-good yields of the monoalkylation product (48) exclusively (Scheme 12).24
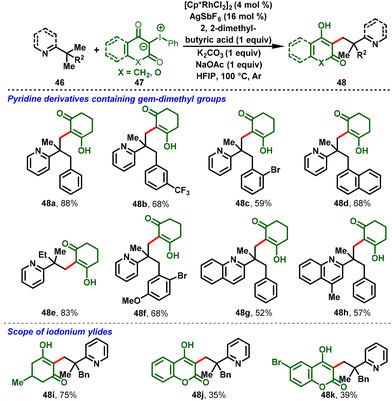 |
| | Scheme 12 Rh(III)-catalyzed C–C coupling of unactivated C(sp3)–H bonds with iodonium ylides. | |
The reaction presumably proceeds via a pyridine-assisted activation of the inert methyl C(sp3)–H (50)/carbene coordination/carbenoid formation (51)/migratory insertion (52)/protonation cascade process achieving the all-carbon-quaternary-centre product (48) (Scheme 13). In this strategy, iodonium ylides proved to be highly reactive C1 synthons for efficient C–C bond formation. Furthermore, the proposed mechanism was successfully demonstrated by the treatment of the carbene precursor with the isolated Rh-intermediate, which afford the corresponding product in good yield.
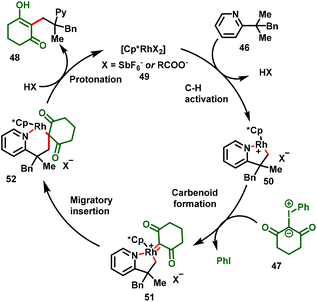 |
| | Scheme 13 Proposed mechanism. | |
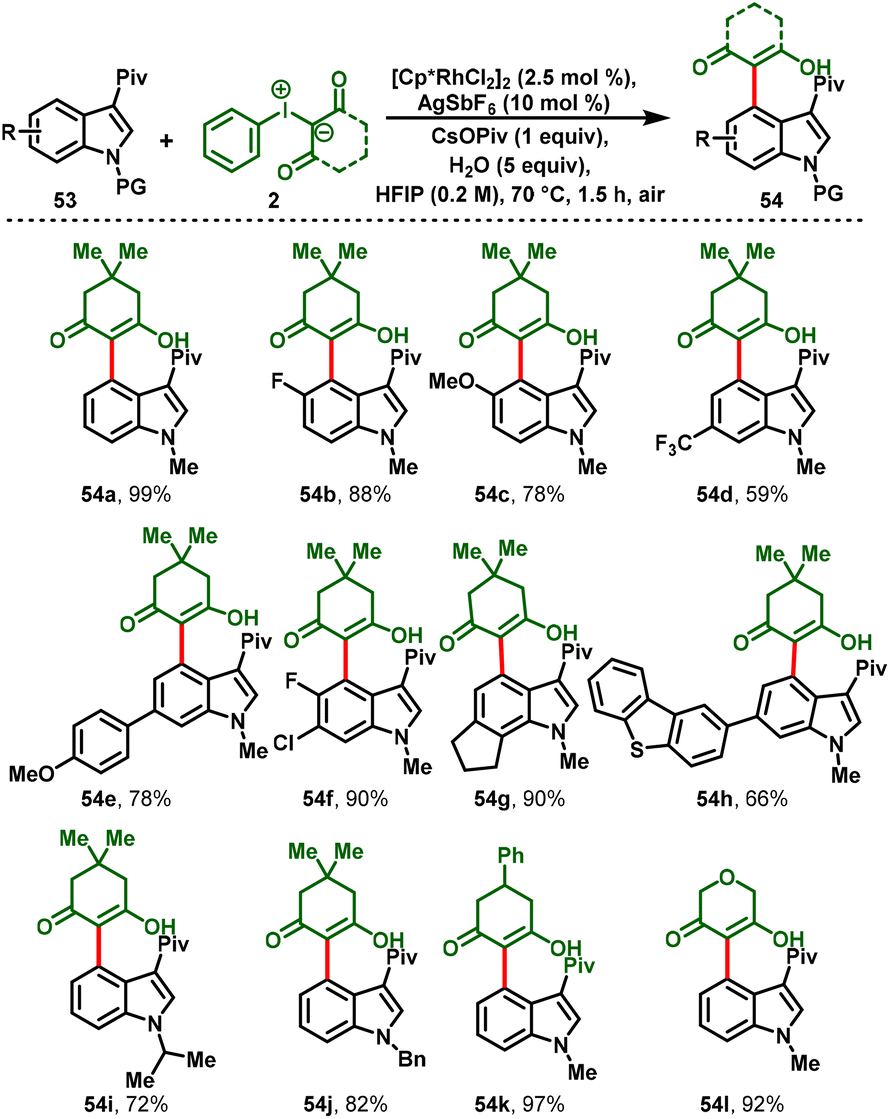
Later, Zhang and co-workers realized the Rh(III)-catalyzed C4-selective functionalization of N1-protected and 3-pivaloyl-attached indoles (53) by employing iodonium ylides (2) as carbene precursors. This method was carried out under redox-neutral conditions to achieve C4 functionalization of indoles (54). The strategy has various salient features, such as good functional group tolerance with high yields, one-pot synthesis, scale-up synthesis, and mechanistic studies (Scheme 14).25
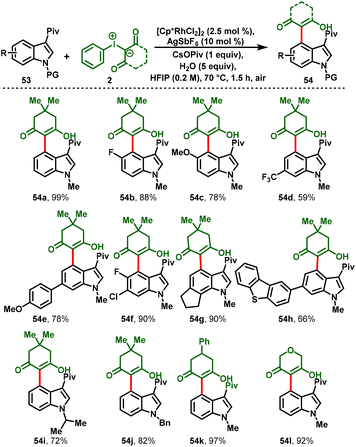 |
| | Scheme 14 Rhodium(III)-catalyzed regioselective C(sp2)–H activation of indoles with iodonium ylides. | |
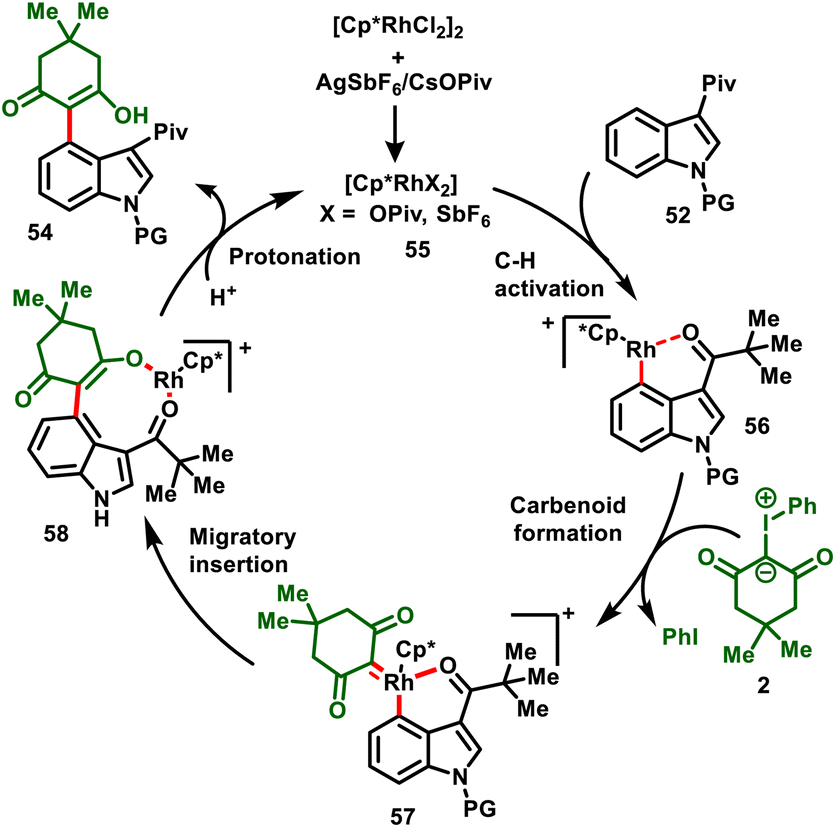
The proposed mechanism is depicted in Scheme 15. The ligand exchange with AgSbF6/CsOPiv forms the active rhodium catalyst 55, which undergoes a pivaloyl (Piv) group directed C–H activation process to activate the C-4 position of the indole via a concerted metalation–deprotonation (CMD) system to obtain the Rh complex intermediate 56. Later, coordination of iodonium ylide with intermediate 56 and elimination of iodobenzene delivers metal–carbene complex 57. Subsequently, 1,1-migratory insertion affords cyclic intermediate 58, followed by protonation in the presence of the solvent HFIP or H2O to furnish the desired product (54) and regenerate the catalyst (55).
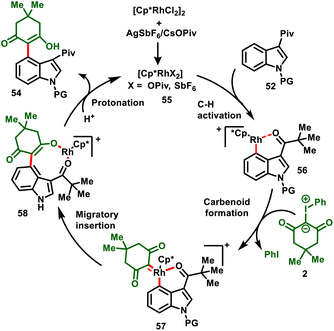 |
| | Scheme 15 Proposed mechanism for C4 functionalization of indole with iodonium ylide. | |
5. Synthesis of nitrogen-related heterocycles
Nitrogen-containing heterocycles have received a great deal of attention from chemists in the last few decades due to their significant importance in drug discovery, agrochemicals, and materials, and are also widely found in a large number of natural products.26 A variety of classical methods have been explored. In addition to classic reactions, the past decade has witnessed the rapid development of transition-metal-catalyzed C–H functionalizations followed by annulations for nitrogen heterocycles.27
5.1 Indoles and their derivatives
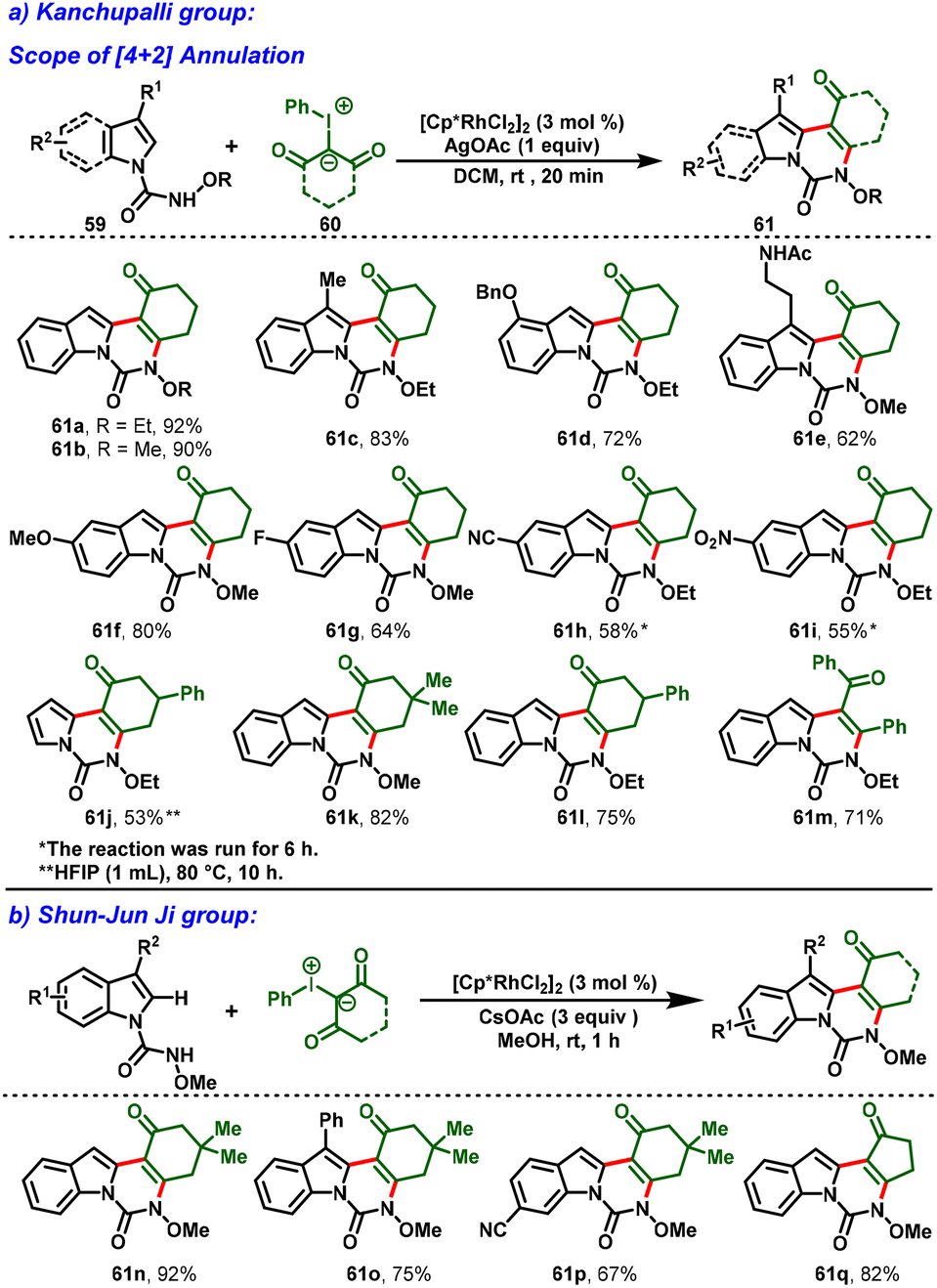
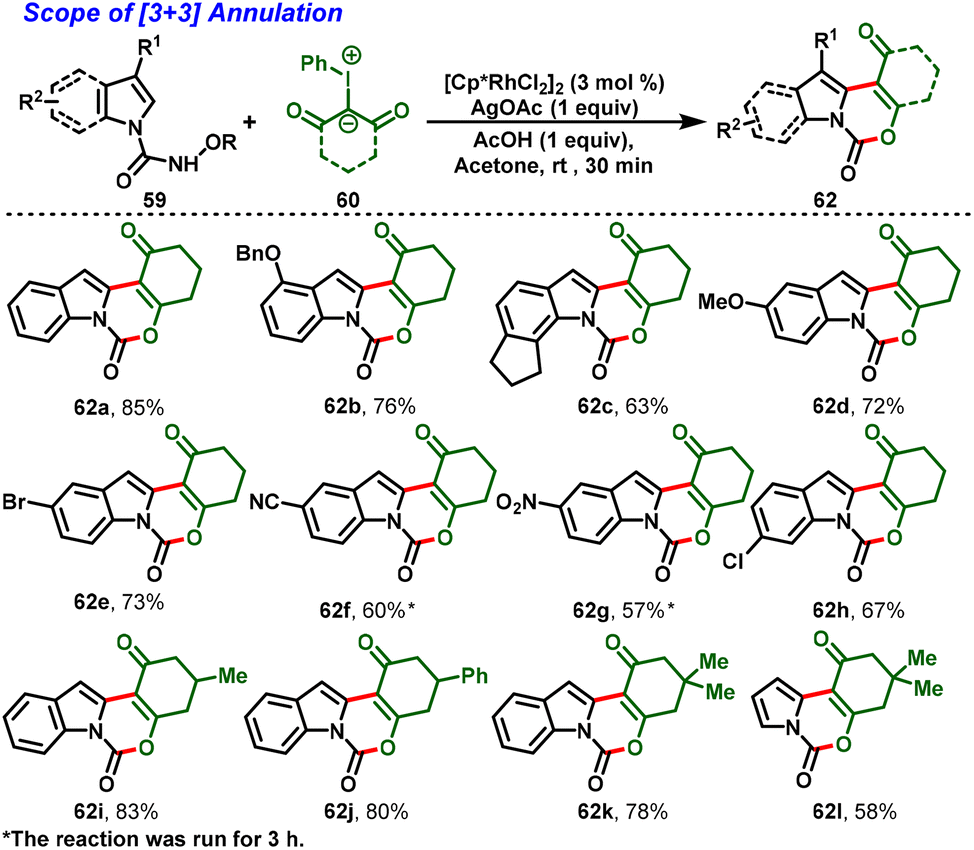
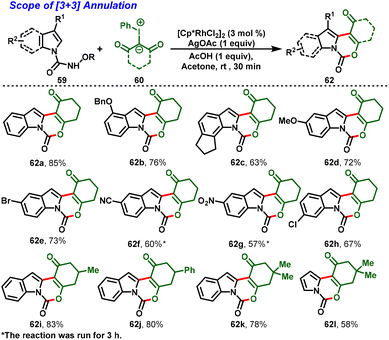
In 2021, Kanchupalli and his group developed an acid-switchable chemodivergent protocol for the synthesis of synthetically attractive 3,4-dihydroindolo[1,2-c]quinazoline-1,6(2H,5H)-dione (61) and 1H-[1,3]oxazino[3,4-a]indol-1-one compounds (62) via Rh(III)-catalyzed [4 + 2] and [3 + 3] annulation reactions. In this protocol, N-alkoxycarbamoyl indoles (59) are used as directing groups and iodonium ylides (60) act as carbene precursors to enable tri- and tetracyclic N-heterocycles under mild conditions. Interestingly, in the presence of [Cp*RhCl2]2, AgOAc and DCM as a solvent, the desired [4 + 2] annulation was achieved (Scheme 16a). In contrast, a complete switch in selectivity was observed using a catalytic system comprising [Cp*RhCl2]2, AgOAc, AcOH, and acetone as the solvent, which delivered the [3 + 3] annulation product in excellent yields (Scheme 17). Further, this method features a wide substrate scope with great functional group tolerance and moderate-to-excellent yields. In addition, one-pot synthesis and gram-scale synthesis were demonstrated, and the annulated products were further extended to various synthetic transformations.28
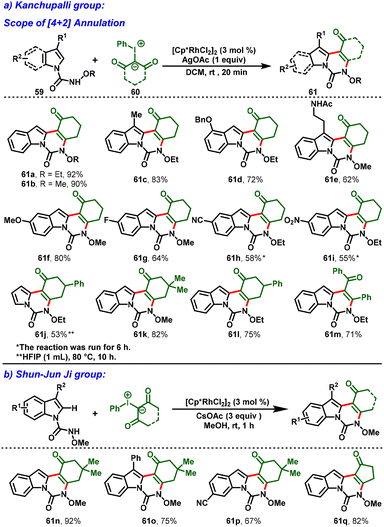 |
| | Scheme 16 Rh(III)-catalyzed [4 + 2]-annulations of indole N-carbomylindoles with iodonium ylides. | |
 |
| | Scheme 17 Rh(III)-catalyzed [3 + 3]-annulations of indole N-carbomylindoles with iodonium ylides. | |
Similarly, in 2021, Shun-Jun Ji and his group reported an Rh(III)-enabled C–H functionalization/cyclization cascade process for the synthesis of indoloquinazolinone scaffolds using hypervalent iodonium ylides and N-alkoxy-1H-indole-1-carboxamides. The strategy shows wide functional group tolerance, excellent yields, and mild conditions. Interestingly, the indoloquinazolinones were obtained simply by filtration without tedious column chromatography. Moreover, the catalytic system can be recycled at least ten times, which can be useful for industry applications (Scheme 16b).29
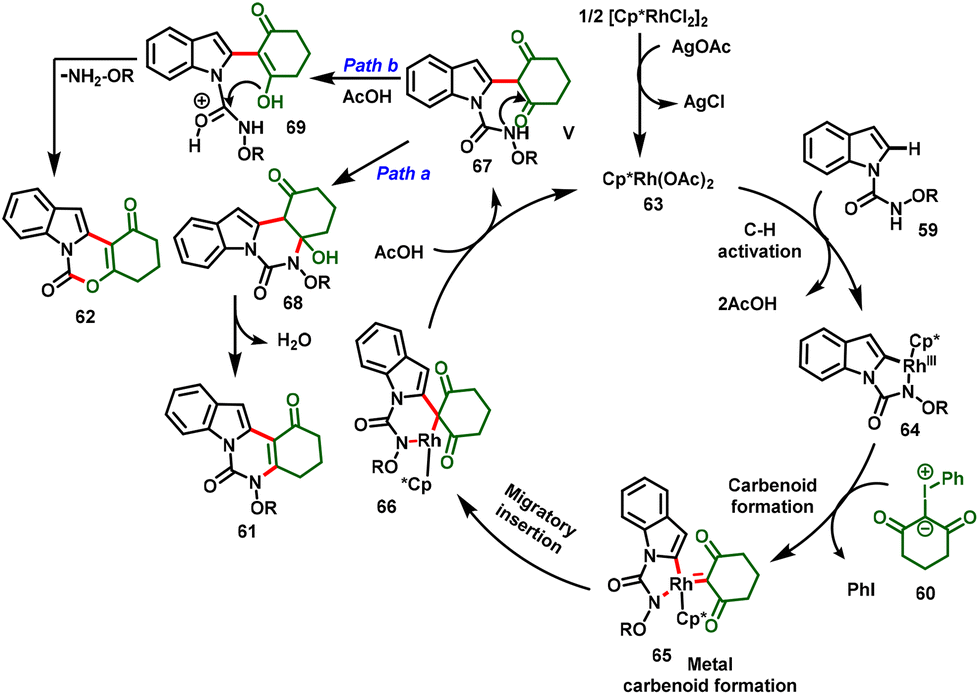
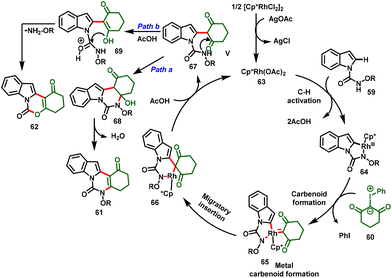
The proposed mechanism initiated with the C–H bond activation of N-alkoxycarbamoyl indole at the C-2 position with the active catalyst 63 generating the five-membered cyclometallated intermediate 64. Simultaneously, coordination of iodonium ylide followed by elimination of PhI leads to the formation of metal–carbenoid 65. Further migratory insertion affords intermediate 66. Then, protodemetallation of intermediate 66 leads to the formation of alkylated intermediate 67 and regenerates the active Rh catalyst (63). Further, intermediate 67 follows two different annulation pathways for the desired products. In the case of Path a, the highly nucleophilic amide –NH group undergoes nucleophilic addition to the ketone carbonyl, followed by dehydration to favor the [4 + 2] annulated product (61). In contrast, Path b proceeding through an excess of acetic acid protonates the amidic carbonyl and activates more nucleophilicity on the –OH group to attack the electrophilic carbon, and elimination of alkoxyl-amine furnishes the [3 + 3] annulation product (62) (Scheme 18).
 |
| | Scheme 18 Proposed catalytic cycle for acid-controlled chemodivergent reaction. | |
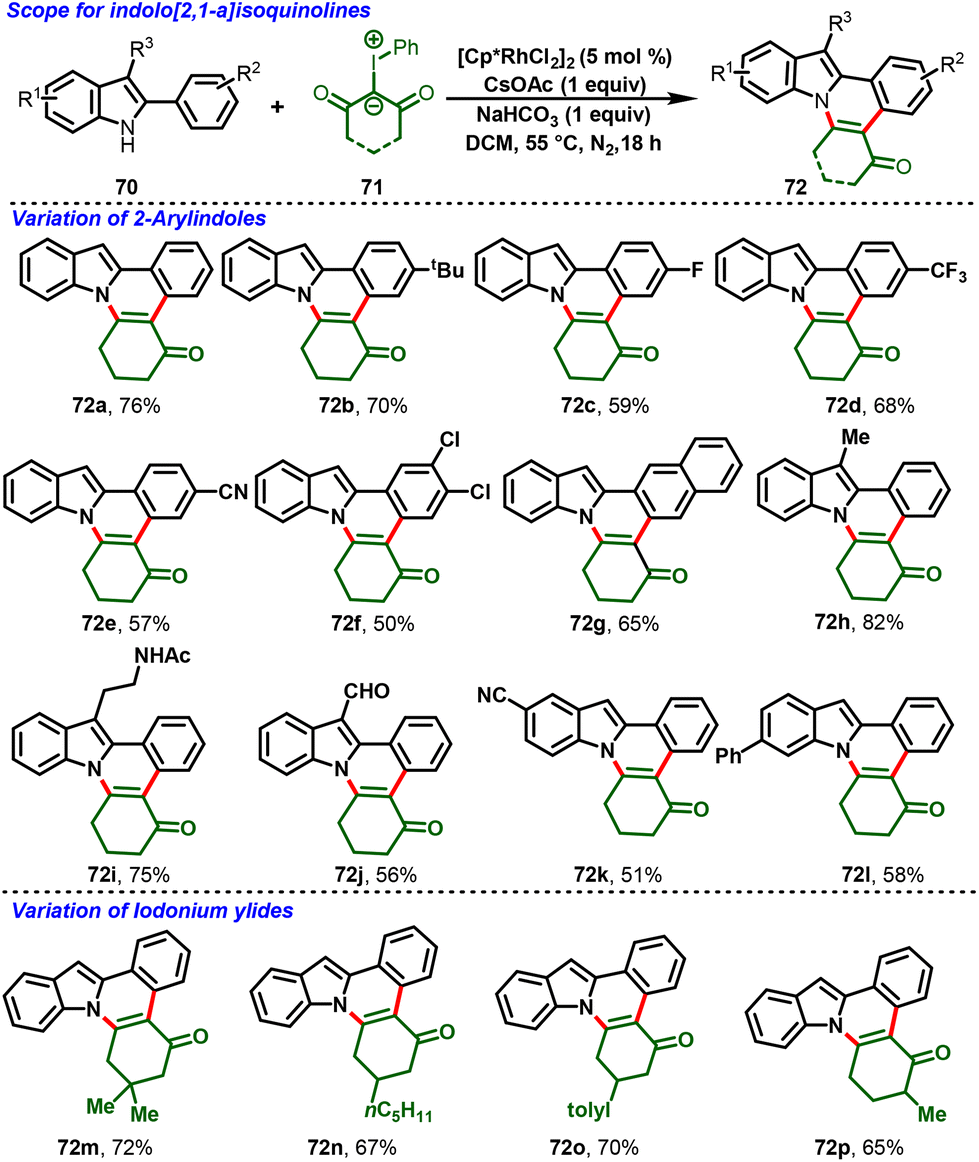
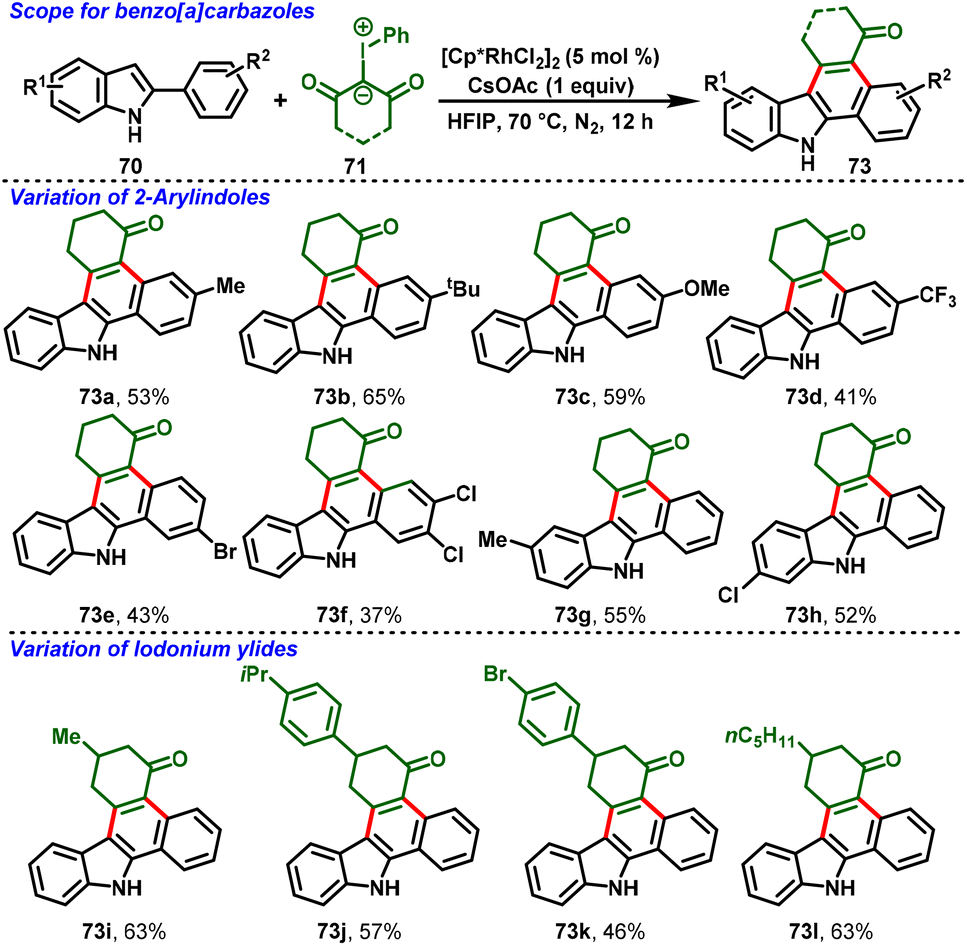
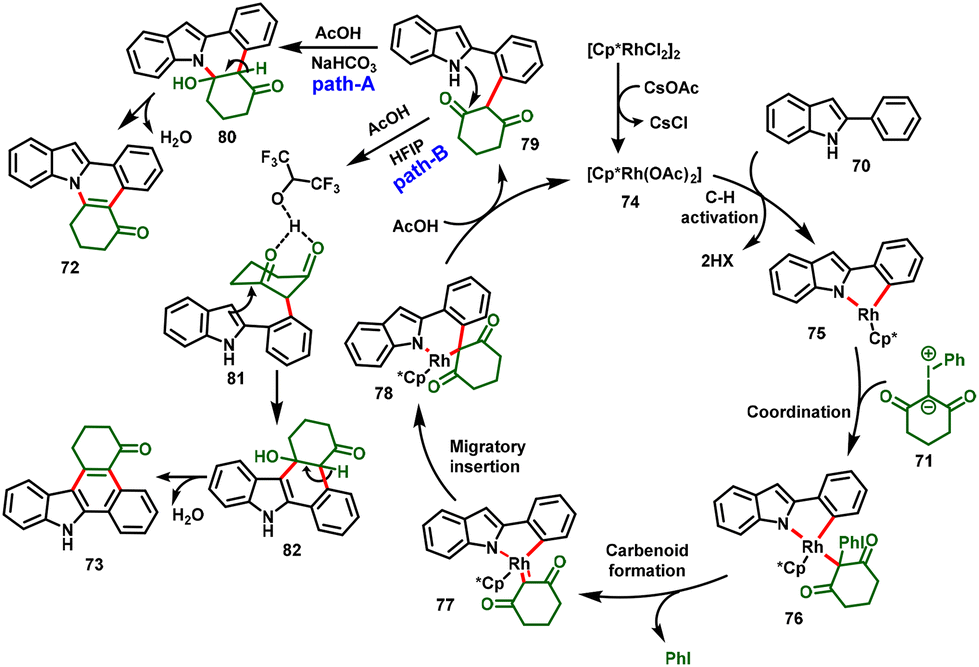
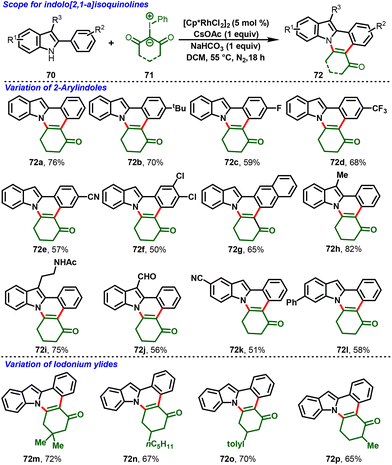
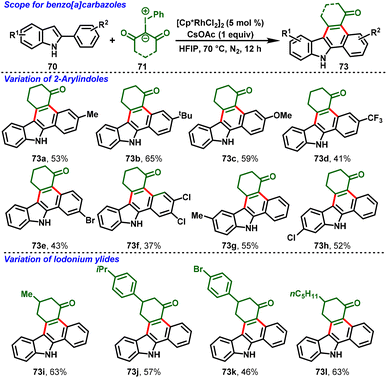
Very recently, a breakthrough work was reported by Kanchupalli and his group. The group disclosed a switchable Rh(III)-catalyzed C–H functionalization/[4 + 2]-annulation reaction of 2-arylindoles (70) and highly reactive iodonium ylides (71). Notably, the solvent controlled the regioselectivity of the annulation; using DCM, indolo[2,1-a]isoquinoline cores (72) were delivered exclusively (Scheme 19), whereas using polar HFIP as the magic solvent predominately enabled benzo[a]carbazole scaffolds (73) (Scheme 20). The reaction features excellent regioselectivity and good functional group tolerance under simple conditions, which selectively afforded indole fused polycyclic compounds. The annulated product further underwent synthetically attractive transformations, viz. Suzuki, Heck, dehydrogenative Heck couplings, reduction, and 1,6-conjugate addition.30
 |
| | Scheme 19 Synthesis of indolo[2,1-a]isoquinoline derivatives. | |
 |
| | Scheme 20 Synthesis of benzo[a]carbazole scaffolds. | |
The proposed mechanism is depicted in Scheme 21. The ligand exchange with CsOAc forms an active catalytic system [Cp*Rh(OAc)2] (74), and ortho C–H bond activation of the aryl group in 2-phenylindole follows, leading to the formation of metallocycle intermediate 75. Then, iodonium ylide coordination followed by extrusion of iodobenzene forms the five-membered Rh-carbenoid intermediate 77. Further migratory insertion to generate six-membered Rh-cyclic complex 78 followed by protonolysis affords the indole C2-alkylated product (79) along with regeneration of the active catalyst 74. Intermediate 79 follows two different pathways: in path A, the NaHCO3 base abstracts the proton from the indole NH group, which is followed by immediate N-centered intramolecular nucleophilic addition to the carbonyl group with subsequent dehydration, favoring 7,8-dihydroindolo[1,2-f]phenanthridin-5(6H)-ones (72). On the other hand, in Path B, the magic solvent HFIP forms a hydrogen bond by coordination with the 1,3-dicarbonyl group, and transfers nucleophilicity from N1 to C3 of 2-penylindole, followed by indole C3-position nucleophilic addition to the carbonyl center, and subsequent dehydration delivers 1,2,3,9-tetrahydro-4H-dibenzo[a,c]carbazol-4-one derivatives (73).
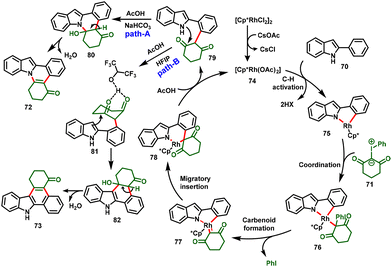 |
| | Scheme 21 Plausible catalytic cycle. | |
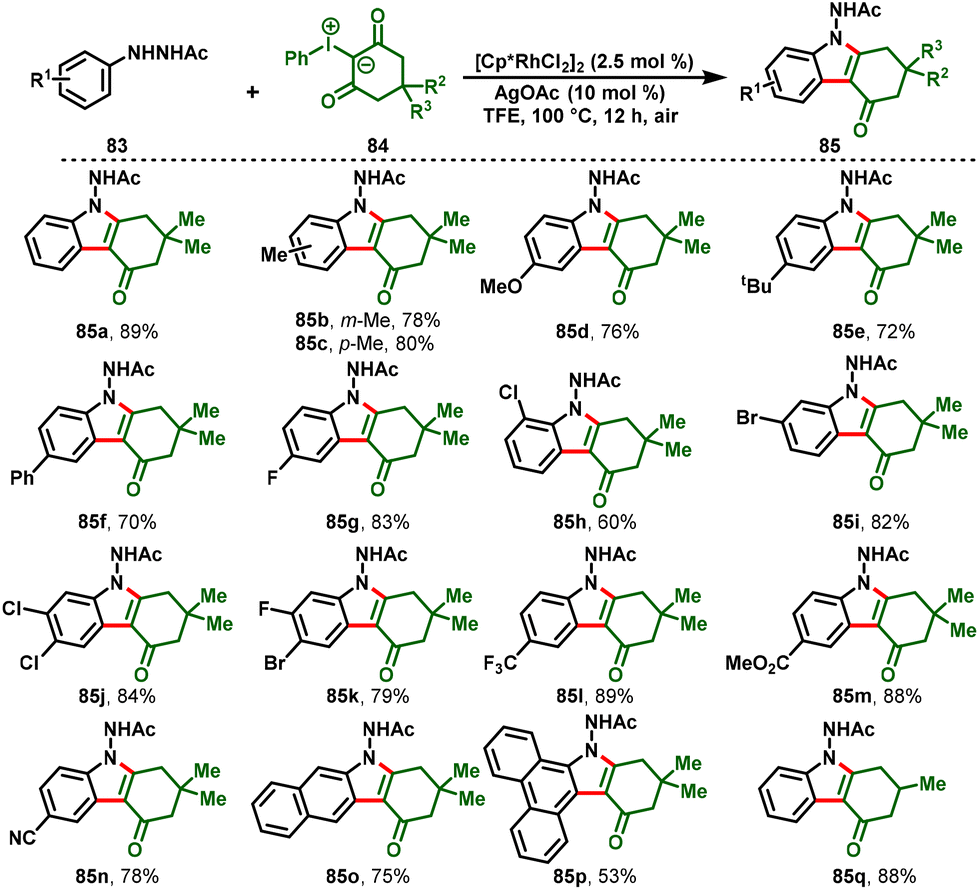
He Li and his co-workers developed a new protocol that achieved tetrahydrocarbazol-4-ones (85) via Rh(III)-catalyzed intramolecular [3 + 2]-annulation reactions between arylhydrazines (83) as directing groups and iodonium ylides (84) as carbene precursors. The synthetic protocol covered a wide substrate scope with broad functional group tolerance in moderate-to-excellent yields. Additionally, the gram-scale reaction and synthetic transformations of the desired product demonstrated the synthetic practicality and utilization of this method (Scheme 22).31
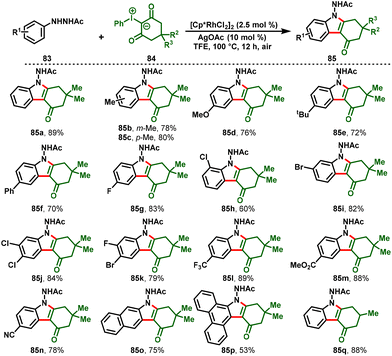 |
| | Scheme 22 Synthesis of tetrahydrocarbazol-4-ones via Rh(III)-catalyzed C–H activation/annulation of arylhydrazines with iodonium ylides. | |
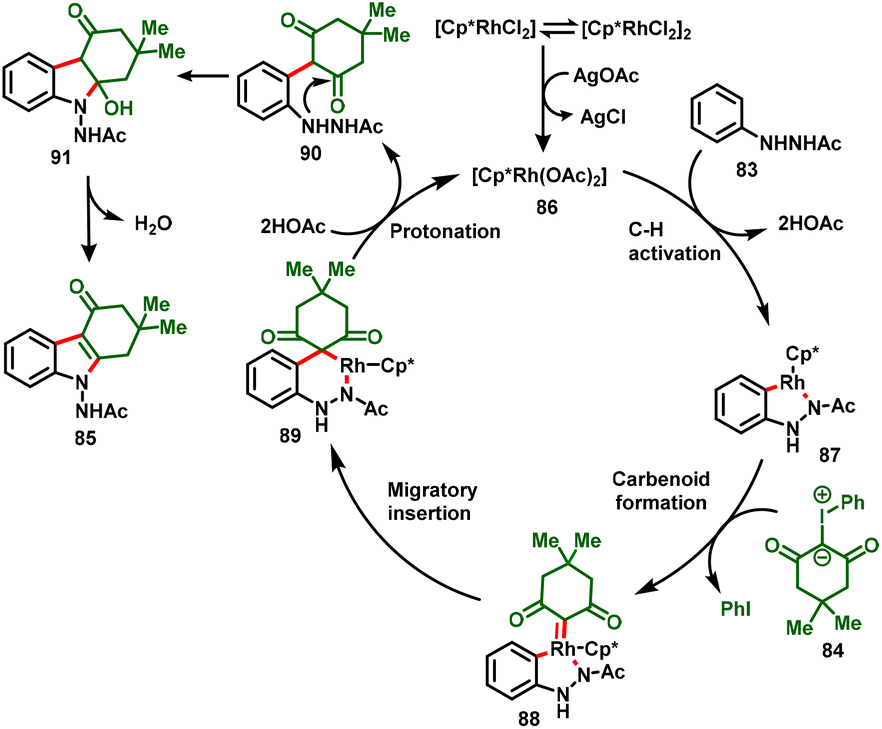
The authors also conveyed a possible reaction mechanism for the reaction, which is depicted in Scheme 23. Initially, the precatalyst [Cp*RhCl2]2 forms an active catalyst [Cp*Rh(OAc)2] (86) with treatment with the additive AgOAc. The active catalyst 86 undergoes coordination with N’-phenylacetohydrazide to afford five-membered rhodacycle 87, which results in irreversible cyclometallation. Further, the addition of iodonium ylides to the Rh center followed by the elimination of PhI gives carbenoid intermediate 88. Migratory insertion then delivers the six-membered rhodacycle 89, which upon protonation through in situ generated HOAc affords intermediate 90 and regenerates the active catalyst (86). Finally, intermediate 90 undergoes an intramolecular nucleophilic addition to deliver the desired product (85) with the release of water.
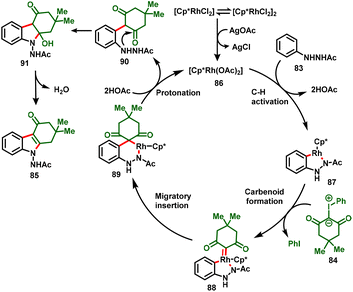 |
| | Scheme 23 Proposed mechanism. | |
5.2 Dihydrophenanthridine derivatives
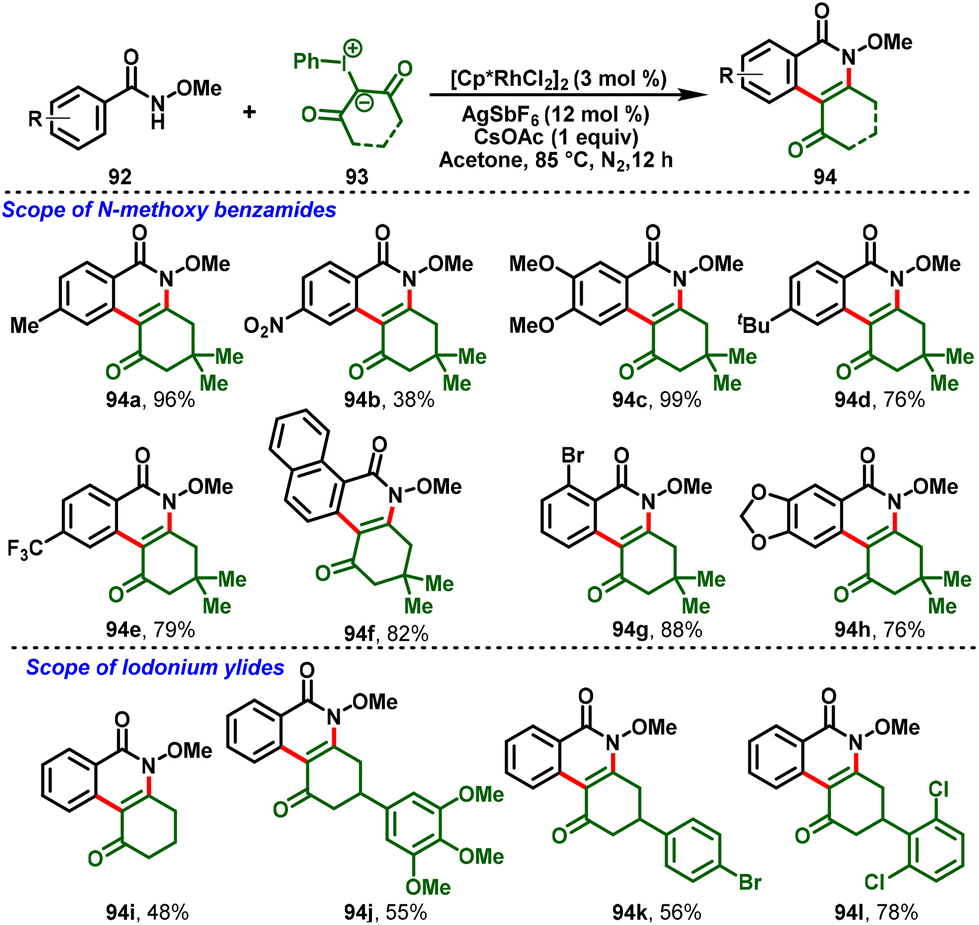
Uma Maheswari and co-workers disclosed the Rh(III)-catalyzed C–H activation of N-methoxybenzamide (92) with hypervalent iodonium ylides (93) which proceeds via domino intermolecular C–H functionalization followed by intramolecular condensation to access dihydrophenanthridine cores (94). A detailed mechanistic investigation was conducted using KIE experiments, deuterium labelling studies, competitive studies, and DFT calculations, and indicated that the C–H activation process is not a rate-limiting step. This method was extended to the derivatization of dihydrophenanthridines with alkynes via Rh(III)-catalyzed peri-C–H/O–H activation/annulation processes to obtain the fluorescent pyranoisocoumarins, which were further studied for their photo-physical properties (Scheme 24).32
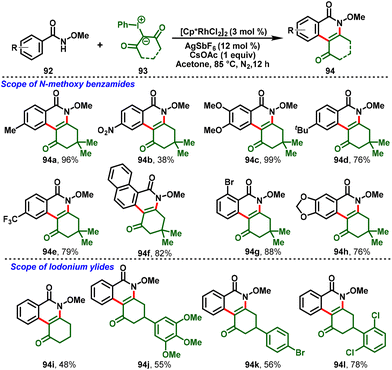 |
| | Scheme 24 Synthesis of dihydrophenanthridines via Rh(III)-catalyzed C–H activation of N-methoxybenzamide with iodonium ylides. | |
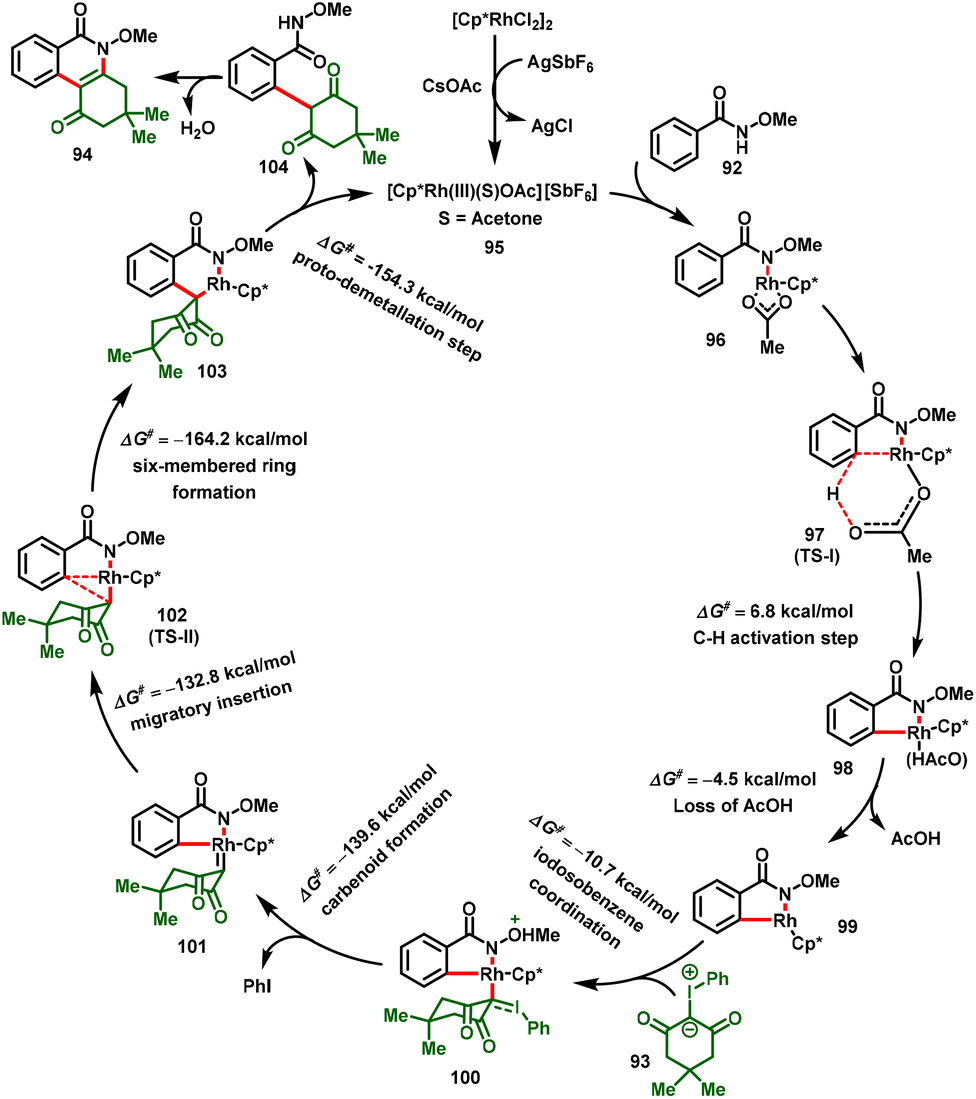
Based on DFT calculations, the authors proposed a catalytic cycle for the reaction, and the plausible mechanism is illustrated in Scheme 25. The catalytic cycle starts with the formation of Int-96 by coordination with the NH of N-methoxybenzamide. Further, Int-96 undergoes C–H bond activation via concerted metalation-deprotonation (CMD) to form the five-membered rhodacycle Int-98, and loss of AcOH provides Int-99. According to DFT calculations, these two steps require relatively low energies (6.8 kcal mol−1 and 11.3 kcal mol−1), indicating that the C–H activation step is not the rate-limiting step. Furthermore, Int-99 coordination of the iodosocarbene followed by deiodination affords the Rh–carbene intermediate Int-101. Int-101 then undergoes through migratory insertion to furnish Int-103viaTS-II. This step is located at −132.8 kcal mol−1 with a barrier of 6.8 kcal mol−1 from the carbenoid intermediate. Subsequently, protonation of the C–Rh bond of Int-103 with acetic acid delivers the alkylated product 104 and regenerates the active catalyst 95. Finally, intermediate 104, tandemly intramolecular nucleophilic cyclization and dehydration deliver corresponding dihydrophenanthridine-1,6(2H,5H)-dione skeleton (94).
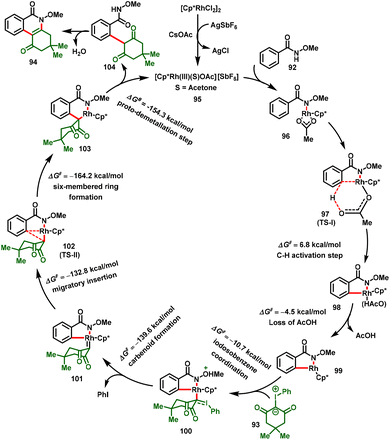 |
| | Scheme 25 Plausible mechanism. | |
5.3 2H-imidazo-fused polycyclic derivatives
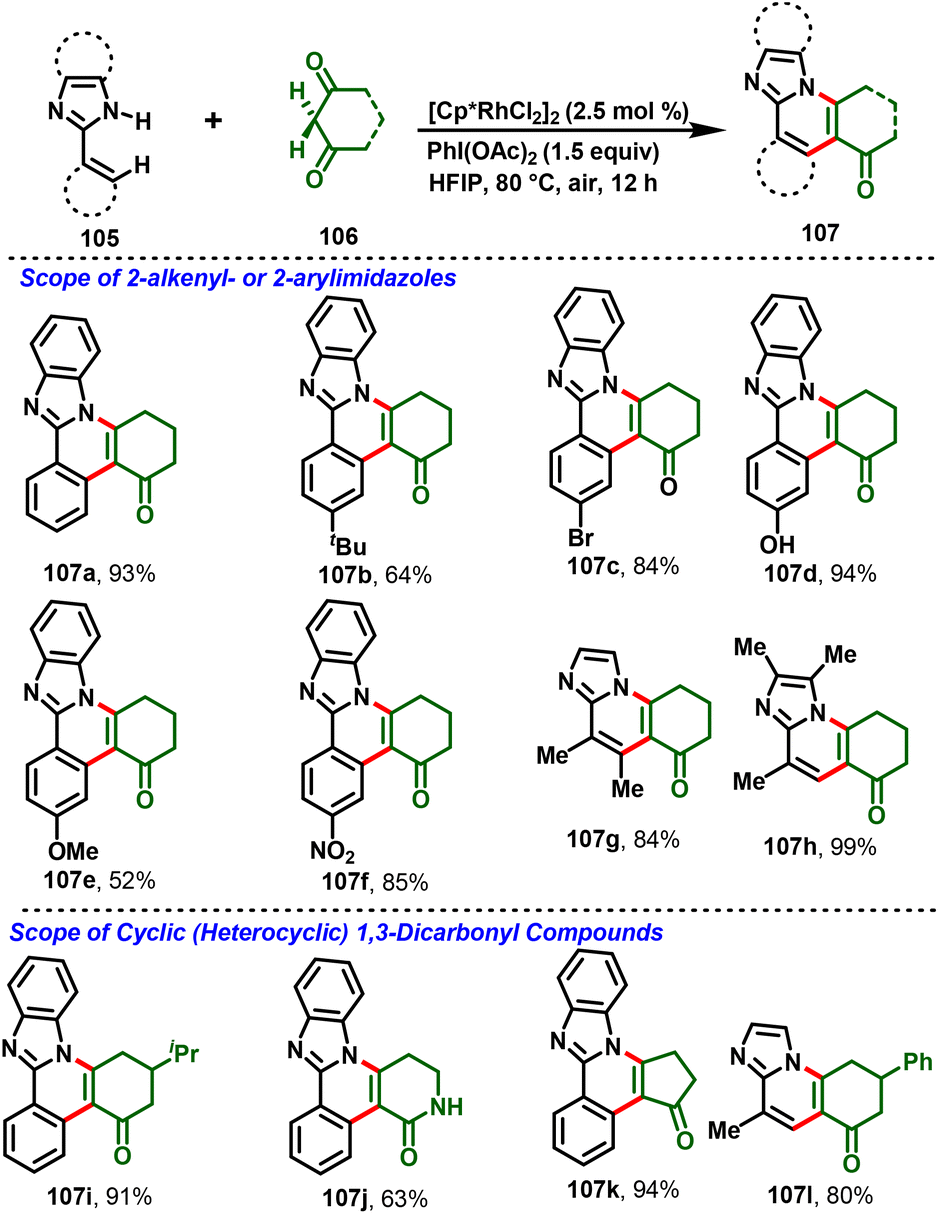
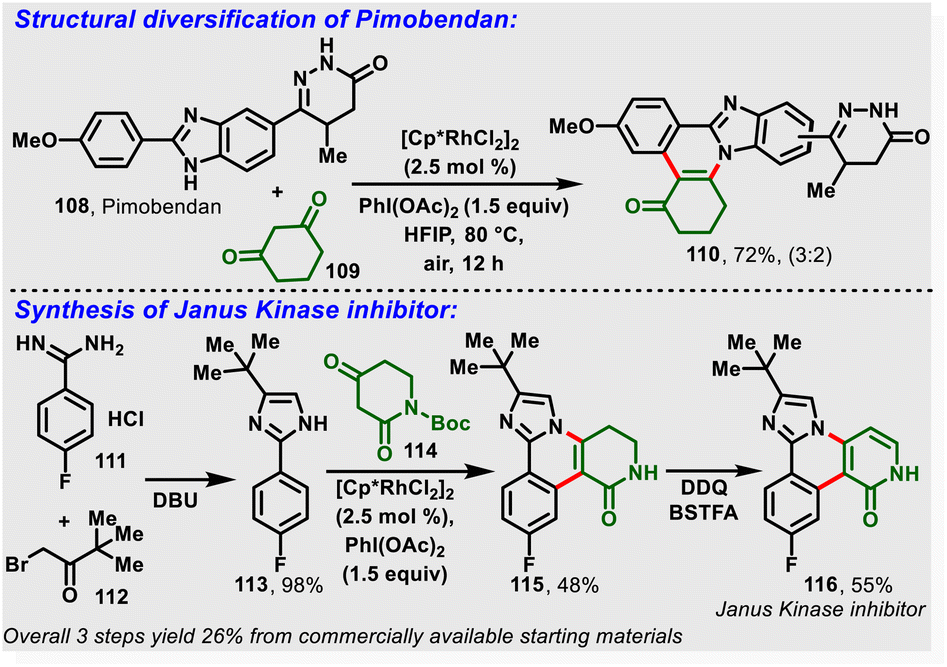
Most recently, the nitrogen-assisted, Rh(III)-catalyzed [4 + 2]-annulation of 2-arylimidazoles/benzimidazole (105) with (hetero)cyclic 1,3-dicarbonyl substrates (106) was reported. Notably, the reaction proceeds via in situ generation of iodosocarbene with a PhI(OAc)2/HFIP system and gives the desired substituted imidazo-fused polycyclic compounds (107) in moderate-to-high yields (Scheme 26). Moreover, the method enabled structural diversification of the drug pimobendan and three-step synthesis of a Janus kinase inhibitor (116) with 26% overall yield (Scheme 27).33a Similarly, Kanchupalli and co-workers disclosed the synthesis of substituted tetracyclic and pentacyclic bridgehead N-heterocycles via Ru-catalyzed C–H functionalization of 2-arylbenzimidazoles/2-arylimidazoles with iodonium ylides.33b
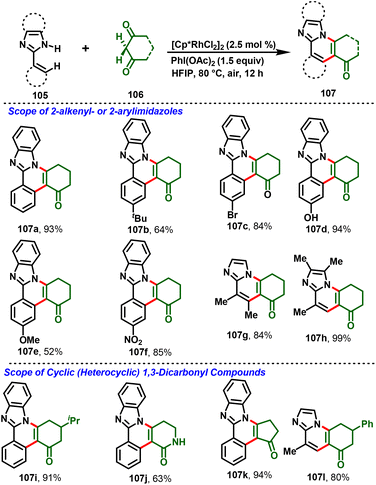 |
| | Scheme 26 Synthesis of 2H-indazole derivatives. | |
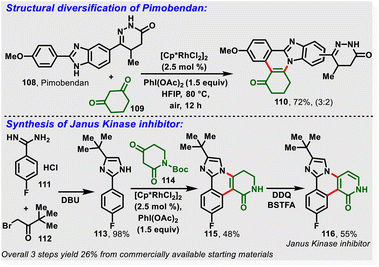 |
| | Scheme 27 Synthetic diversification of pimobendan and synthesis of Janus kinase inhibitor. | |
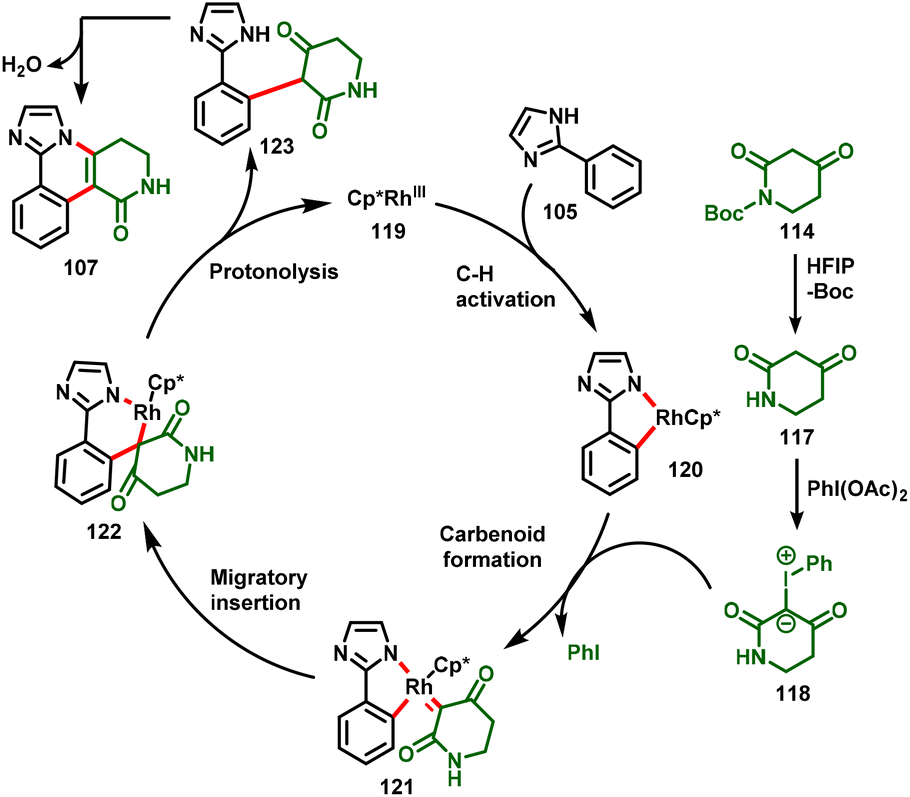
The mechanism for the annulation of 2-arylimidazole/benzimidazole with 1,3-dicarbonyl compounds is shown in Scheme 28, in which the reaction is proposed to proceed through the in situ formation of carbene intermediate (118). Initially, the N-Boc protected piperidine-2,4-dione (114) is deprotected under HFIP solvent to give the piperidine-2,4-dione (117), which reacts with PhI(OAc)2 to form the piperidine-derived iodonium ylide (118). Additionally, cyclometallated Rh-complex 120 is achieved by the concerted metalation–deprotonation (CMD) of 2-arylimidazole with the active catalyst (119), which undergoes subsequent coordination with the iodonium ylide and affords the metalcarbenoid intermediate 121via loss of iodobenzene. Further migratory insertion of intermediate 121 provides intermediate 122, which further undergoes proto-demetallation to afford the alkylated product (123) along with the regeneration of the RhIII species (119). Finally, intramolecular nucleophilic addition followed by dehydration of the alkylated product (123) occurs to give the desired annulated product (107).
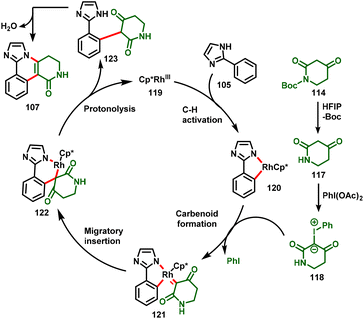 |
| | Scheme 28 Proposed mechanism. | |
5.4 1,2-Benzothiazines and their derivatives
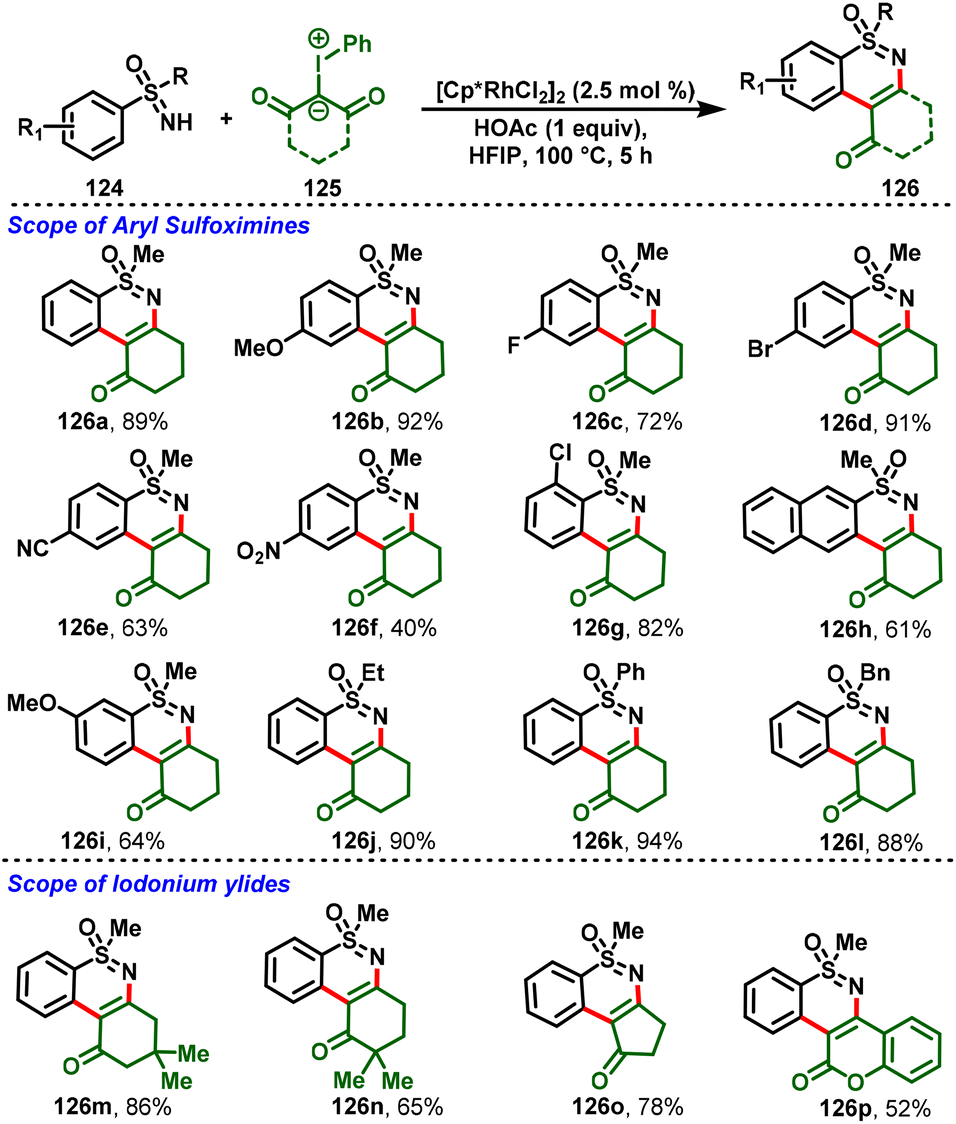
Pan and co-workers showed that S-aryl sulfoximines (124) are also good candidates for the C–H activation by converting them into the corresponding 1,2-benzothiazine derivatives (126) through the rhodium-catalyzed C–H activation/cyclization of S-aryl sulfoximines (124) with iodonium ylides (125). The authors found that a series of tricyclic and tetracyclic sulfoximine derivatives could be obtained via C–H and N–H bond functionalization. Moreover, the reactions feature good substrate scope and functional group tolerance, along with moderate-to-high yields (Scheme 29).34 Later, Yafei Ji et al., and Chen et al., individually reported Rh-catalyzed cascade C–H functionalization/annulation of sulfoximines with iodonium ylides for the synthesis of 1,2-benzothiazine derivatives.35,36
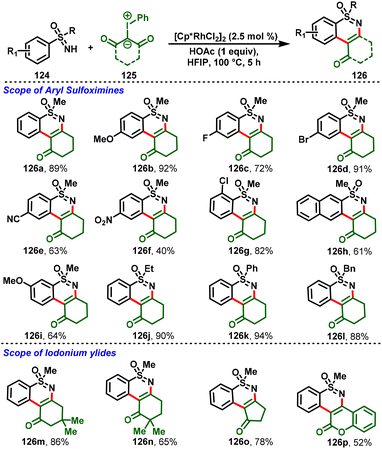 |
| | Scheme 29 Synthesis of polycyclic 1,2-benzothiazines. | |
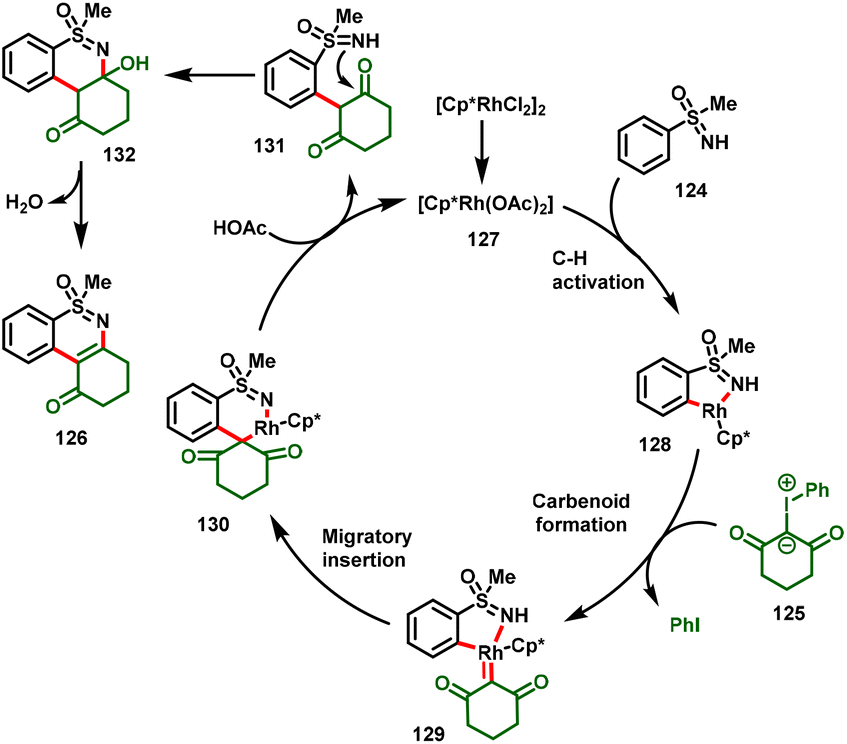
A plausible reaction mechanism is presented in Scheme 30. The catalytic cycle begins with the generation of active catalyst 127, which undergoes a concerted metalation–deprotonation (CMD) process with the S-aryl sulfoximines to give intermediate 128. Furthermore, intermediate 128 forms the metal–carbenoid system 129via the coordination of iodonium ylide with the loss of iodobenzene, in which the by-product PhI was detected in the reaction system to validate the mechanism. Further migratory insertion of the Rh–aryl bond into the carbene center leads to the formation of six-membered rhodacycle complex 130, and subsequent intramolecular nucleophilic attack of the sulfoximine –NH to the carbonyl delivers intermediate 131. Subsequently, elimination of water provides the corresponding desired product (126).
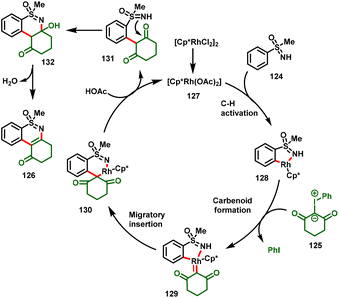 |
| | Scheme 30 Proposed mechanism. | |
5.5 Cinnoline and its derivatives
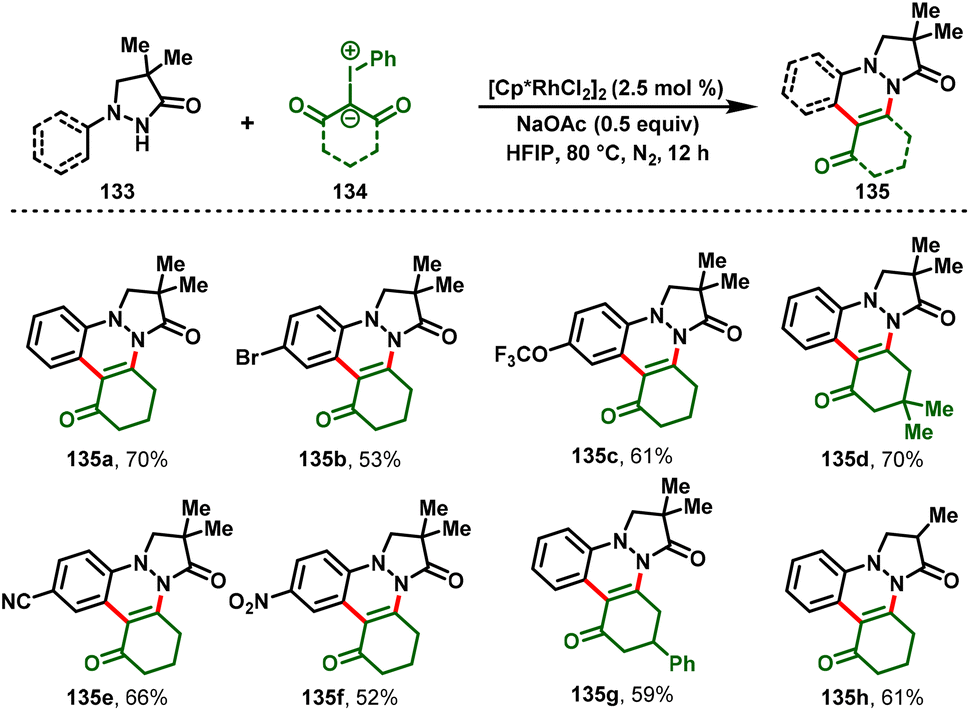
Yaqian Li and co-workers recently disclosed rhodium(III)-catalyzed cascade annulations to obtain complex pyrazolo[1,2-a]cinnoline derivatives. A range of pyrazolidiones (133) and iodonium ylides (134) underwent Rh(III)-catalyzed C–H functionalization smoothly and afforded the substituted cinnolines (135) in moderate-to-excellent yields with good functional group tolerance (Scheme 31).37
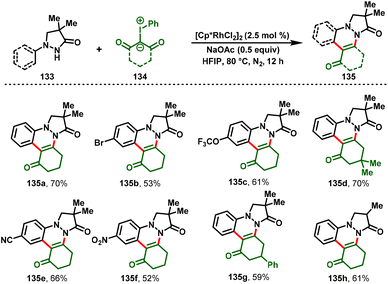 |
| | Scheme 31 Synthesis of pyrazolo[1,2-a]cinnoline via Rh(III)-catalyzed annulation of pyrazolidiones and iodonium ylides. | |
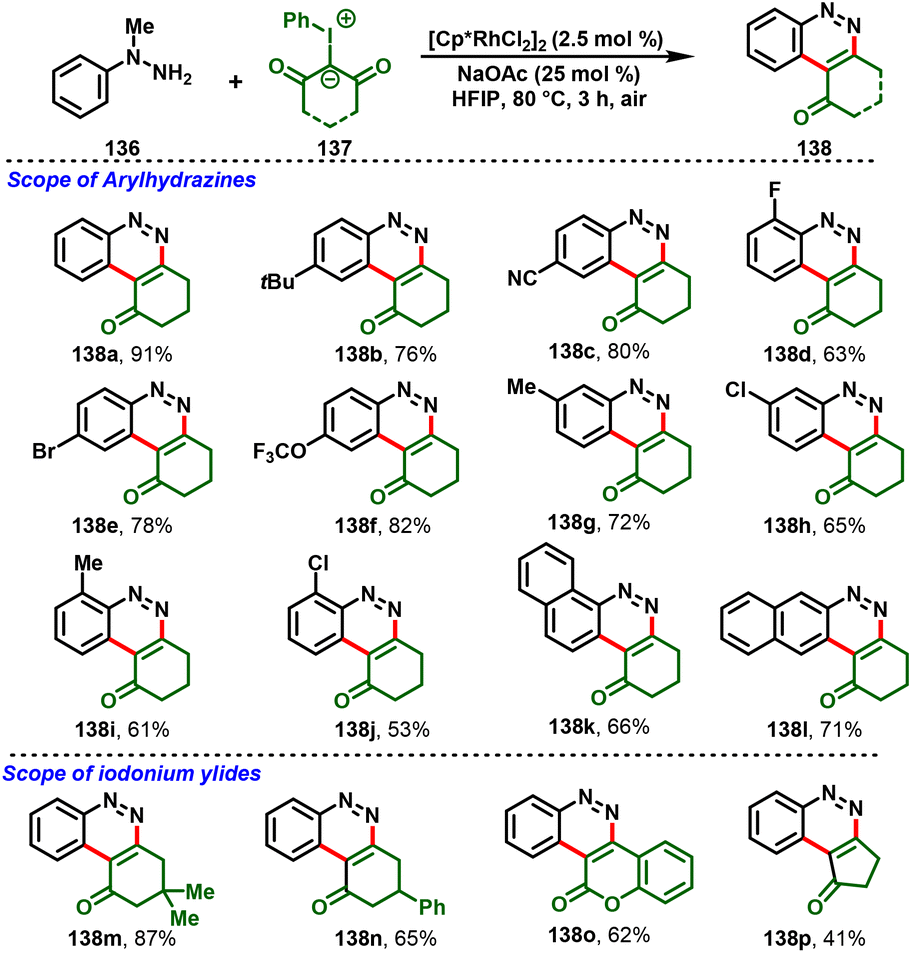
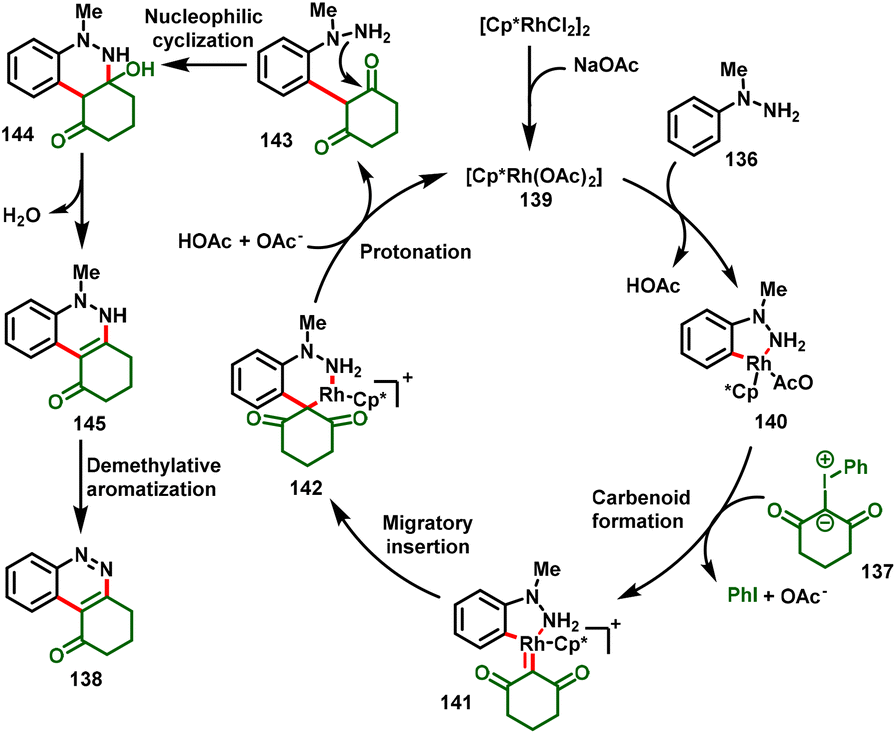
Similarly, Yu et al., developed the synthesis of ring-fused cinnolines (138) via Rh(III)-catalyzed annulation of N-methyl arylhydrazines (136) with iodonium ylides (137). This strategy proceeds via a cascade process, such as C–H activation/annulation, dehydration and demethylative aromatization, under simple reaction conditions. A variety of N-methyl arylhydrazines could be cyclized under the optimized conditions to afford the corresponding oxycycloalkyl-fused cinnolines (138) in good yields (Scheme 32).38
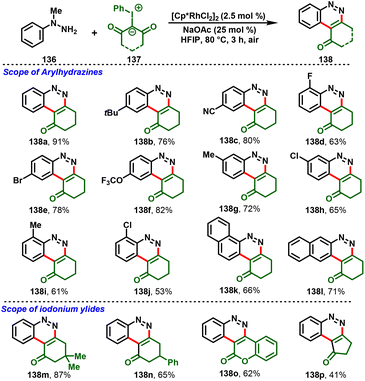 |
| | Scheme 32 Rh(III)-catalyzed C–H activation/annulation of N-methyl arylhydrazines with iodonium ylides. | |
The authors performed various mechanistic studies, and control studies suggested that the catalytic cycle was initiated via C–H activation of N-methyl arylhydrazine with the active catalyst 139 to form the cyclorhodium intermediate 140. Coordination of iodonium ylide provides the metal–carbenoid intermediate 141 by elimination of PhI. Subsequently, intermediate 141 undergoes migratory insertion to achieve six-membered rhodium complex 142, which further undergoes protonolysis with in situ formed HOAc to deliver the species 143. Finally, intramolecular nucleophilic addition results in tandem cyclization–dehydration followed by demethylative aromatization to furnish the desired cinnoline frameworks (138) (Scheme 33).
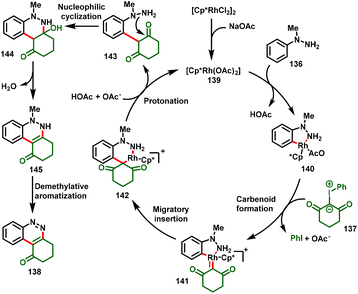 |
| | Scheme 33 Proposed mechanism. | |
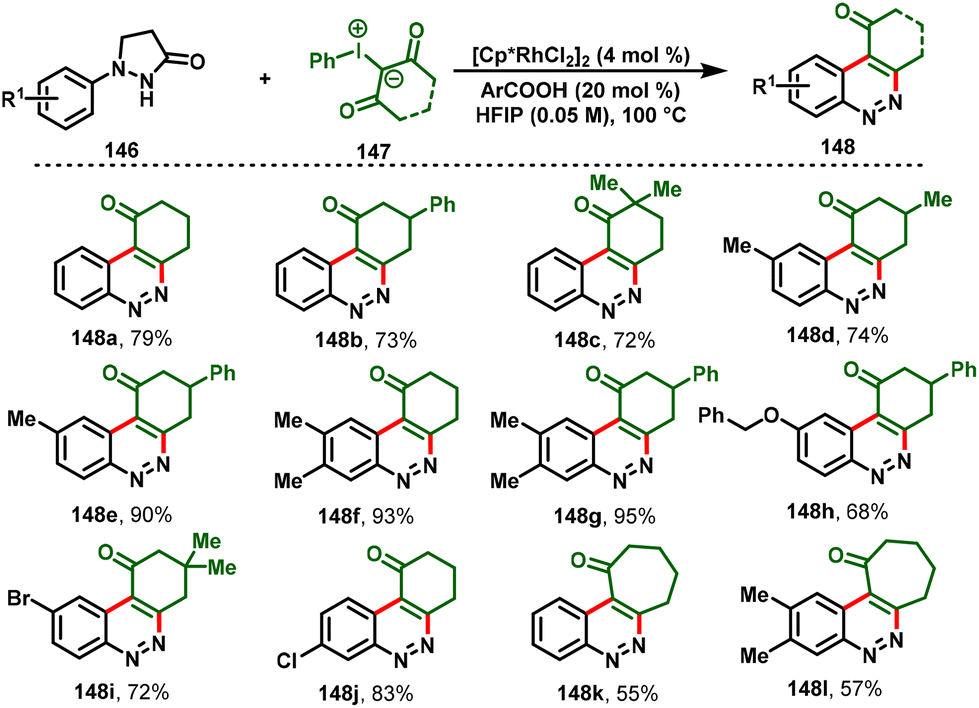
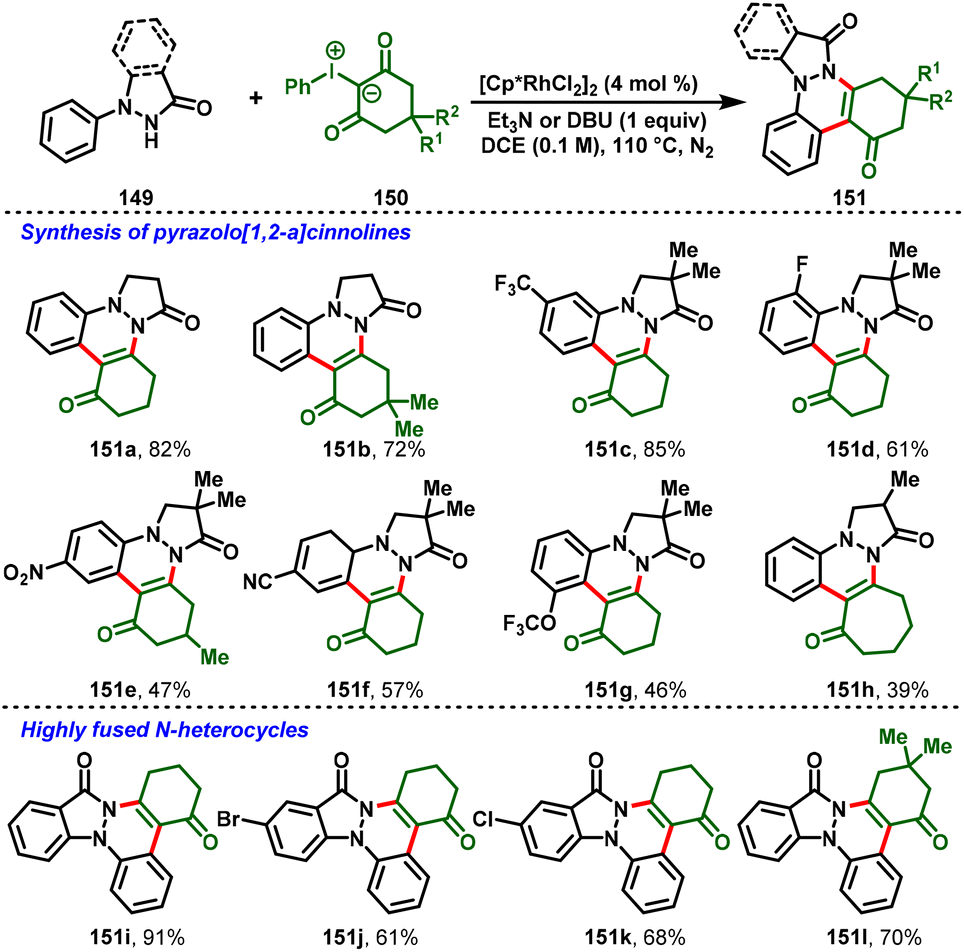
Later, the group of Xiao-Qiang Hu disclosed a switchable approach for cinnolines (148) and pyrazolo[1,2-a]cinnoline scaffolds (151) via Rh(III) C–H functionalization of pyrazolidiones with iodonium ylides. Notably, the use of a Rh(III)/HFIP solvent system allowed the elimination process to give cinnoline derivatives (148) in excellent yields (Scheme 34). In contrast, the combination of Rh(III)/organic base and the solvent DCE afforded tetra- and pentacyclic cinnolines (151) exclusively (Scheme 35). Moreover, the protocol showed attractive advantages, such as mild conditions, a broad substrate scope, and tunable fused N-heterocyclic frameworks. Furthermore, the synthetic utility was expanded by scale-up of the cinnoline derivative reaction and diverse synthetic transformations.39
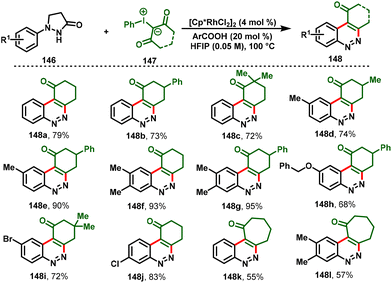 |
| | Scheme 34 Synthesis of 1,2-benzothiazine derivatives. | |
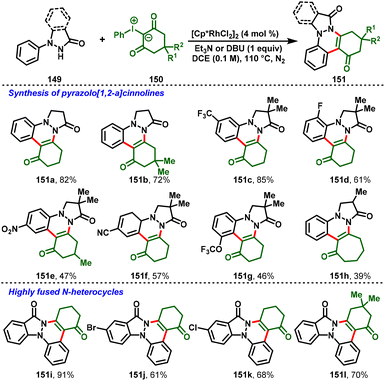 |
| | Scheme 35 Synthesis of 1,2-benzothiazine derivatives. | |
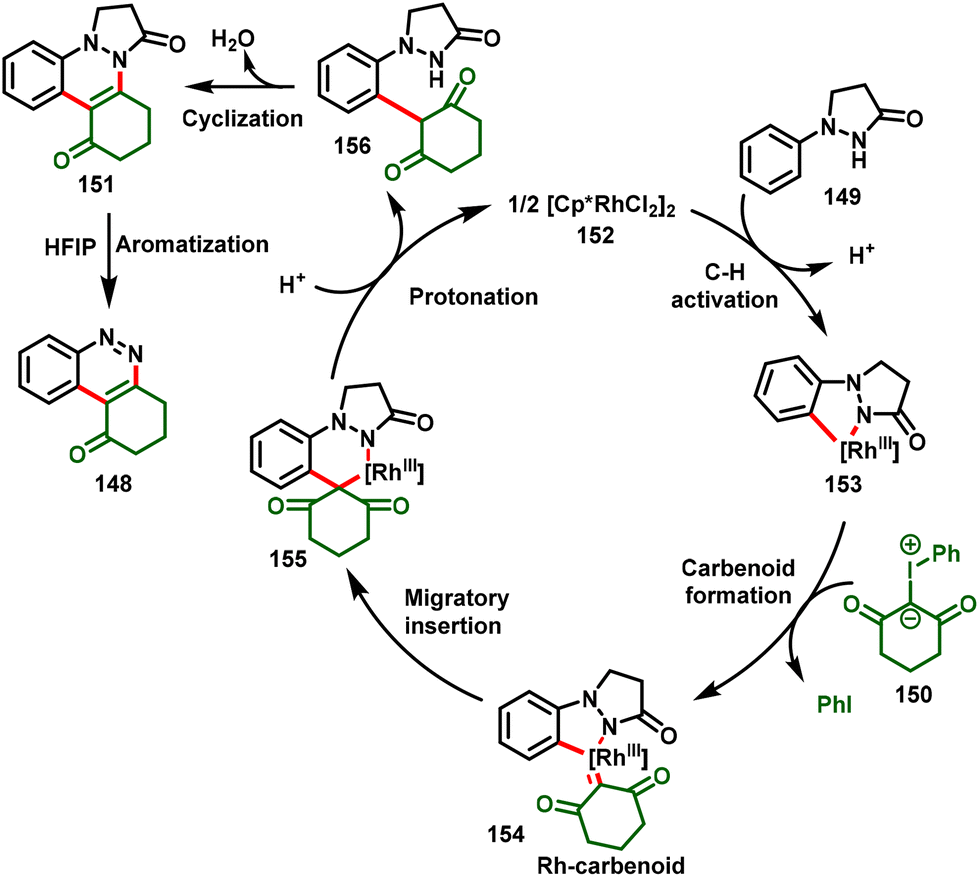
Based on the preliminary mechanistic study and previous reports, the authors proposed a possible mechanism, as depicted in Scheme 36. Initially, the active Rh catalyst (152) activates the C–H bond of 1-phenylpyrazolidinone via a concerted metalation–deprotonation cascade process to form rhodacycle intermediate 153, followed by the addition of iodonium ylide to access Rh(III)-carbenoid intermediate 154 by subsequent loss of iodobenzene. Further, highly reactive intermediate 154 undergoes migratory insertion followed by subsequent protonation and demetallation to furnish the intermediate 156 and regenerate the active catalyst 152. Eventually, species 156 stimulates the nucleophilic cyclization–dehydration to afford cinnoline 151 in the solvent DCE. In the case of HFIP, cinnoline 151 further undergoes the aromatization process, which favors the aromatized product (148).
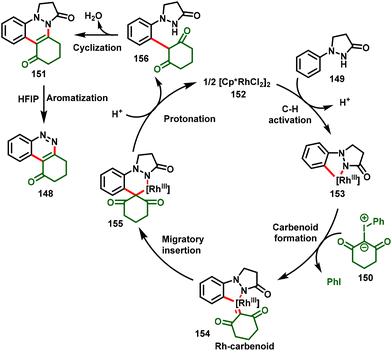 |
| | Scheme 36 Plausible mechanism. | |
Conclusions
As demonstrated by the reactions summarized in this review, iodonium ylides exhibit unique and diverse chemical reactivity in transition-metal-catalyzed C–H functionalizations. This allows for the construction of diverse carbon–carbon and carbon–heteroatom bonds. Notably, many functional groups, including aldehydes, carboxylic acids, amides, sulfoxonium ylides, weakly directing amines, etc., were employed as coupling partners with iodonium ylides, leading to the delivery of the diverse carbo/heterocyclizations. Interestingly, most of these strategies provide tri-, tetra- and pentacyclic scaffolds, which are difficult to synthesize using other methods. Moreover, several chemo-divergent annulations were also examined. Hence, iodonium ylides are a perfect alternative to carbene precursors, which is desirable to offer new opportunities for organic synthesis.
Despite this remarkable progress, several challenges still need to be addressed. Therefore, we detail some key issues that will benefit further developments. (1) As of the above results, the iodonium ylides that have been investigated are relatively limited. Additionally, most of the iodonium ylides are derived from cyclic 1,3-dione scaffolds. In the future, the development of acyclic-derived iodonium ylides would be highly desirable. (2) To date, the enantioselective versions of the annulations have not yet been demonstrated; these would lead to new types of asymmetric transformations and are also highly desirable. Moreover, the great influence of the structure of the iodonium ylides and their properties on the catalytic system should be investigated. Such investigations would be helpful in the refinement and further improvement of the field of catalysis. (3) In particular, rhodium and ruthenium catalysts are highly utilized in catalytic approaches; hence, introducing catalysts based on inexpensive and earth-abundant metals, such as Co, Fe and Ni, would be highly advantageous. We hope that this review will inspire chemists and might become an interesting research direction in the near future.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
The authors S. K., V. B., M. M., and S. N. would like to thank DoP, India and National Institute of Pharmaceutical Education and Research (NIPER), Hyderabad for the fellowship. V. K. thanks the DST-Inspire faculty. The project was supported by the Department of Science & Technology-Inspire Faculty (Grant No. DST/INSPIRE/04/2018/001660) and National Institute of Pharmaceutical Education and Research (NIPER), Hyderabad.NIPERH Research Communication No.NIPERH/2022/112.
References
-
(a) R. G. Bergman, Nature, 2007, 446, 391–393 CrossRef CAS PubMed
 ;
(b) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624–655 CrossRef CAS PubMed ;
(c) J. Grover, G. Prakash, N. Goswami and D. Maiti, Nat. Commun., 2022, 13, 1085 CrossRef CAS PubMed ;
(d) S. K. Sinha, S. Guin, S. Maiti, J. P. Biswas, S. Porey and D. Maiti, Chem. Rev., 2022, 122, 5682–5841 CrossRef CAS PubMed ;
(e)
N. Goswami, R. Mohan and D. Maiti, Comprehensive Organometallic Chemistry IV, Elsevier, 2021 Search PubMed ;
(f) N. Goswami, T. Bhattacharya and D. Maiti, Nat. Rev. Chem., 2021, 5, 646 CrossRef CAS ;
(g) R. H. Crabtree and A. Lei, Chem. Rev., 2017, 117, 8481–8482 CrossRef CAS .
;
(b) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624–655 CrossRef CAS PubMed ;
(c) J. Grover, G. Prakash, N. Goswami and D. Maiti, Nat. Commun., 2022, 13, 1085 CrossRef CAS PubMed ;
(d) S. K. Sinha, S. Guin, S. Maiti, J. P. Biswas, S. Porey and D. Maiti, Chem. Rev., 2022, 122, 5682–5841 CrossRef CAS PubMed ;
(e)
N. Goswami, R. Mohan and D. Maiti, Comprehensive Organometallic Chemistry IV, Elsevier, 2021 Search PubMed ;
(f) N. Goswami, T. Bhattacharya and D. Maiti, Nat. Rev. Chem., 2021, 5, 646 CrossRef CAS ;
(g) R. H. Crabtree and A. Lei, Chem. Rev., 2017, 117, 8481–8482 CrossRef CAS .
-
(a) A. Biffis, P. Centomo, A. D. Zotto and M. Zecca, Chem. Rev., 2018, 118, 2249–2295 CrossRef CAS ;
(b) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS ;
(c) E. Negishi, Angew. Chem., Int. Ed., 2011, 50, 6738–6764 CrossRef CAS ;
(d) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442–4489 CrossRef CAS ;
(e) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417–1492 CrossRef CAS ;
(f) L. C. Campeau and N. Hazari, Organometallics, 2019, 38, 3–35 CrossRef CAS ;
(g) D. G. Brown and J. Bostrom, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS .
-
(a) D. L. Davies, S. A. Macgregor and C. L. McMullin, Chem. Rev., 2017, 117, 8649–8709 CrossRef CAS PubMed ;
(b) X.-S. Xue, P. Ji, B. Zhou and J.-P. T. Cheng, Chem. Rev., 2017, 117, 8622–8648 CrossRef CAS PubMed ;
(c) S. R. Sahoo, S. Dutta, S. A. Al-Thabaiti, M. Mokhtar and D. Maiti, Chem. Commun., 2021, 57, 11885–11903 RSC .
- For selected reviews on C–H activation, see:
(a) L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS ;
(b) T. Newhouse and P.S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362–3374 CrossRef CAS PubMed ;
(c) J.W. Delord and F. Glorius, Nat. Chem., 2013, 5, 369–375 CrossRef PubMed ;
(d) P. Gandeepan and C.H. Cheng, Chem. – Asian J., 2016, 11, 448–460 CrossRef CAS ;
(e) T. Iwai and M. Sawamura, ACS Catal., 2015, 5, 5031–5040 CrossRef CAS ;
(f) B. Liu, F. Hu and B.F. Shi, ACS Catal., 2015, 5, 1863–1881 CrossRef CAS ;
(g) Y. Ping, Q. Ding and Y. Peng, ACS Catal., 2016, 6, 5989–6005 CrossRef CAS ;
(h) R.Y. Zhu, E.M. Farmer, Y.Q. Chen and J.Q. Yu, Angew. Chem., Int. Ed., 2016, 55, 10578–10599 CrossRef CAS ;
(i) G.C. Newton, S.G. Wang, C.C. Oliveira and N. Cramer, Chem. Rev., 2017, 117, 8908–8976 CrossRef ;
(j) R. Shang, L. Ilies and E. Nakamura, Chem. Rev., 2017, 117, 9086–9139 CrossRef CAS ;
(k) K. Murakami, S. Yamada, T. Kaneda and K. Itami, Chem. Rev., 2017, 117, 9302–9332 CrossRef CAS ;
(l) H.F.T. Klare, ACS Catal., 2017, 7, 6999–7002 CrossRef CAS ;
(m) Y. Wei, P. Hu, M. Zhang and W. Su, Chem. Rev., 2017, 117, 8864–8907 CrossRef CAS ;
(n) H.M.L. Davies and D. Morton, ACS Cent. Sci., 2017, 3, 936–943 CrossRef CAS PubMed ;
(o) Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787–8863 CrossRef CAS ;
(p) J. R. Hummel, J.A. Boerth and J.A. Ellman, Chem. Rev., 2017, 117, 9163–9227 CrossRef CAS .
-
(a) P. de Frémont, N. Marion and S. P. Nolan, Coord. Chem. Rev., 2009, 253, 862–892 CrossRef ;
(b) F. K. Zinn, M. S. Viciu and S. P. Nolan, Annu. Rep. Prog. Chem., Sect. B: Org. Chem., 2004, 100, 231–249 RSC ;
(c) D. Zhu, L. Chen, H. Fan, Q. Yao and S. Zhu, Chem. Soc. Rev., 2020, 49, 908–950 RSC ;
(d) B. Chattopadhyay and V. Gevorgyan, Angew. Chem., Int. Ed., 2012, 51, 862–872 CrossRef CAS ;
(e) A. V. Gulevich and V. Gevorgyan, Angew. Chem., Int. Ed., 2013, 52, 1371–1373 CrossRef CAS PubMed ;
(f) H. M. L. Davies and J. S. Alford, Chem. Soc. Rev., 2014, 43, 5151–5162 RSC ;
(g) P. Anbarasan, D. Yadagiri and S. Rajasekar, Synthesis, 2014, 46, 3004–3023 CrossRef CAS ;
(h) D. Kaiser, I. Klose, R. Oost, J. Neuhaus and N. Maulide, Chem. Rev., 2019, 119, 8701–8780 CrossRef CAS PubMed ;
(i) A. Yoshimura, M. S. Yusubov and V. V. Zhdankin, Org. Biomol. Chem., 2016, 14, 4771–4781 RSC ;
(j) A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328–3435 CrossRef CAS PubMed ;
(k) M. Jia and S. Ma, Angew. Chem., Int. Ed., 2016, 55, 9134–9166 CrossRef CAS PubMed ;
(l) S. G. Dawande, V. Kanchupalli, J. Kalepu, H. Chennamsetti, B. S. Lad and S. Katukojvala, Angew. Chem., Int. Ed., 2014, 53, 4076–4080 CrossRef CAS ;
(m) V. Kanchupalli, D. Joseph and S. Katukojvala, Org. Lett., 2015, 17, 5878–5881 CrossRef CAS PubMed ;
(n) V. Kanchupalli, L. A. Thorbole, J. Kalepu, D. Joseph, M. Arshad and S. Katukojvala, Org. Lett., 2022, 24, 3850–3854 CrossRef CAS PubMed ;
(o) S.-F. Zhu and Q.-L. Zhou, Acc. Chem. Res., 2012, 45, 1365–1377 CrossRef CAS PubMed ;
(p) Y. Xia, D. Qiu and J. Wang, Chem. Rev., 2017, 117, 13810–13889 CrossRef CAS ;
(q) K. Wang and J. Wang, Synlett, 2019, 30, 542–551 CrossRef CAS .
-
(a) S. Nunewar, S. Kumar, S. Talakola, S. Nanduri and V. Kanchupalli, Chem. - Asian J., 2021, 16, 443–459 CrossRef CAS ;
(b) S. Kumar, S. Nunewar, S. Oluguttula, S. Nanduri and V. Kanchupalli, Org. Biomol. Chem., 2021, 19, 1438–1458 RSC .
-
(a) F. Hu, Y. Xia, C. Ma, Y. Zhang and J. Wang, Chem. Commun., 2015, 51, 7986–7995 RSC ;
(b) Y. Xiang, C. Wang, Q. Ding and Y. Peng, Adv. Synth. Catal., 2019, 361, 919–944 CrossRef CAS ;
(c) X. Wu, S. Sun, J.-T. Yu and J. Cheng, Synlett, 2019, 30, 21–29 CrossRef CAS ;
(d) J. Vaitla and A. Bayer, Synthesis, 2019, 51, 612–628 CrossRef CAS ;
(e) N. Jha, N. K. Khot and M. Kapur, Chem. Rec., 2021, 21, 4088–4122 CrossRef CAS PubMed ;
(f) S. Bera, A. Biswas and R. Samanta, Chem. Rec., 2021, 51, 3411–3428 CrossRef .
-
(a) R.-Y. Yang and L.-X. Dai, Chin. J. Org. Chem., 1994, 14, 113–130 CAS ;
(b) V. V. Zhdankin and P. J. Stang, Chem. Rev., 2008, 108, 5299–5358 CrossRef CAS PubMed ;
(c) A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328–3435 CrossRef CAS PubMed .
-
(a) M. S. Yusubov, A. Yoshimura and V. V. Zhdankin, Arkivoc, 2016, 9(i), 342–374 Search PubMed ;
(b) P. Muller, Acc. Chem. Res., 2004, 37, 243–251 CrossRef ;
(c) E. Malamidou-Xenikaki and S. Spyroudis, Synlett, 2008, 18, 2725–2740 Search PubMed ;
(d) M. Tong, X. Zhang, Y. Wang and Z. Wang, Chin. J. Org. Chem., 2021, 41, 126–143 CrossRef .
- E. Gudriniece, O. Neiland and G. Zh Vanags, Obshch. Khim., 1957, 27, 2737–2740 CAS .
- Y. Hayasi, T. Okada and M. Kawanisi, Bull. Chem. Soc. Jpn., 1970, 43, 2506–2511 CrossRef CAS .
- G. F. Koser and S. M. Yu, J. Org. Chem., 1975, 40, 1166–1168 CrossRef CAS .
-
(a) R. M. Moriarty, B. R. Bailey, O. Prakash and I. Prakash, J. Am. Chem. Soc., 1985, 107, 1375–1378 CrossRef CAS ;
(b) K. Schank and C. Lick, Synthesis, 1983, 1983, 392–395 CrossRef ;
(c) A. Asouti and L. P. Hadjiarapoglou, Tetrahedron Lett., 1998, 39, 9073–9076 CrossRef CAS ;
(d) R. M. Moriarty, S. Tyagi, D. Ivanov and M. Constantinescu, J. Am. Chem. Soc., 2008, 130, 7564–7565 CrossRef CAS ;
(e) A. Antos, Y. Elemes, A. Michaelides, J. A. Nyxas, S. Skoulika and L. P. Hadjiarapoglou, J. Org. Chem., 2012, 77, 10949–10954 CrossRef CAS PubMed ;
(f) D. Kalpogiannaki, C. I. Martini, A. Nikopoulou, J. A. Nyxas, V. Pantazi and L. P. Hadjiarapoglou, Tetrahedron, 2013, 69, 1566–1575 CrossRef CAS ;
(g) J. Tao, C. D. Estrada and G. K. Murphy, Chem. Commun., 2017, 53, 9004–9007 RSC ;
(h) A. Antos, Y. Elemes, A. Michaelides, J. A. Nyxas, S. Skoulika and L. P. Hadjiarapoglou, J. Org. Chem., 2012, 77, 10949–10954 CrossRef CAS .
- C. Tejo, H. Q. Yeo and P. W. H. Chan, Synlett, 2014, 25, 201–204 CAS .
-
(a)
J. Cossy and A. Guerinot, in Advances in Heterocyclic Chemistry, Elsevier., 2016, 119, 107–142 Search PubMed ;
(b) N. Kaur, P. Grewal and K. Poonia, Synth. Commun., 2021, 51, 2423–2444 CrossRef CAS .
-
(a) A. Deiters and S. F. Martin, Chem. Rev., 2004, 104, 2199–2238 CrossRef CAS ;
(b) Y. F. Liang and N. Jiao, Acc. Chem. Res., 2017, 50, 1640–1653 CrossRef CAS PubMed ;
(c) B. Desai, M. Patel, B. Z. Dholakiya, S. Rana and T. Naveen, Chem. Commun., 2021, 57, 8699–8725 RSC .
- Y. Jiang, P. Li, J. Zhao, B. Liu and X. Li, Org. Lett., 2020, 22, 7475–7479 CrossRef CAS PubMed .
- Z. Dong, P. Li, X. Li and B. Liu, Chin. J. Chem., 2021, 39, 2489–2494 CrossRef CAS .
- S. Kumar, S. Nunewar and V. Kanchupalli, Asian J. Org. Chem., 2022, 11, e202100689 CAS .
-
(a) P. Nealmongkol, K. Tangdenpaisal, S. Sitthimonchai, S. Ruchirawat and N. Thasana, Tetrahedron, 2013, 69, 9277–9283 CrossRef CAS ;
(b) V. R. L. J. Bloemendal, J. C. M. V. Hest and F. P. J. T. Rutjes, Org. Biomol. Chem., 2020, 18, 3203–3215 RSC .
- C. Yin, L. Li and C. Yu, Org. Biomol. Chem., 2022, 20, 1112–1116 RSC .
- Y. Dai, X. Li and B. Liu, Chin. J. Org. Chem., 2021, 41, 4476–4483 CrossRef .
- X. Li, Y. Shen, G. Zhang, X. Zheng, Q. Zhao and Z. Song, Org. Lett., 2022, 24, 5281–5286 CrossRef CAS .
- P. Xie, H. Gao, X. Li, Y. Jiang and B. Liu, Org. Chem. Front., 2022, 9, 3823–3827 RSC .
- F. Wu, L. Xiao, H. Xie, S. Y. Chen, J. L. Song, Y. C. Zheng, Y. Z. Liu and S. S. Zhang, Org. Biomol. Chem., 2022, 20, 5055–5059 RSC .
-
(a) S. Takano and K. Ogasawara, Alkaloids, 1989, 36, 225–252 CAS ;
(b) E. Vitaku, D. T. Smith and J. T. Njardason, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS ;
(c) A. P. Taylor, R. P. Robinson, Y. M. Fobian, D. C. Blakemore, L. H. Jones and O. Fadeyi, Org. Biomol. Chem., 2016, 14, 6611–6634 RSC ;
(d) A. J. Kochanowska-Karamyan and M. T. Hamann, Chem. Rev., 2010, 110, 4489–4497 CrossRef CAS PubMed .
-
(a) B. Nie, W. Wu, Y. Zhang, H. Jiang and J. I. Zhang, Org. Chem. Front., 2020, 7, 3067–3099 RSC ;
(b) T. Naveen, Tetrahedron, 2021, 84, 132025 CrossRef CAS ;
(c) J. C. Lewis, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2008, 41, 1013–1025 CrossRef CAS .
- S. Nunewar, S. Kumar, H. Pandhare, S. Nanduri and V. Kanchupalli, Org. Lett., 2021, 23, 4233–4238 CrossRef CAS PubMed .
- Z. P. Han, M. M. Xu, R. Y. Zhang, X. P. Xu and S. J. Ji, Green Chem., 2021, 23, 6337–6340 RSC .
- S. Nunewar, S. Kumar, P. Priyanka, P. Girase and V. Kanchupalli, Chem. Commun., 2022, 58, 6140–6143 RSC .
- H. Li, H. Gu, Y. Lu, N. Xu, N. Han, J. Li, J. Liu and J. Liu, J. Org. Chem., 2022, 87, 8142–8150 CrossRef CAS PubMed .
- S. Mayakrishnan, M. Tamizmani and N. U. Maheswari, Chem. Commun., 2020, 56, 15462–15465 RSC .
-
(a) Z. Zhong, M. Liang, Z. Zhang, H. Cui, N. Wang, S. Mai and H. Tao, Org. Lett., 2022, 24, 4850–4854 CrossRef CAS ;
(b) S. Nunewar, S. Kumar, A. W. Meshram and V. Kanchupalli, J. Org. Chem., 2022 DOI:10.1021/acs.joc.2c01429 .
- G. Huang, Y. Shan, J. T. Yu and C. Pan, Org. Biomol. Chem., 2021, 19, 10085–10089 RSC .
- L. Chen, Z. Wang, Y. Wang, L. Hao, X. Xu, G. Wu and Y. Ji, Org. Biomol. Chem., 2022, 20, 887–894 RSC .
- J. Huang, F. Liu, F. Du, L. Zeng and Z. Chen, Green Synth. Catal. DOI:10.1016/j.gresc.2022.06.002 .
- Z. Yang, Y. Zhou, H. Li, J. Lei, P. Bing, B. He and Y. Li, Asian J. Org. Chem., 2022, 11, e202100656 CAS .
- C. Pan, C. Yuan, D. Chen, Y. Chen and J. T. Yu, Asian J. Org. Chem., 2022, 11, e202100809 CAS .
- R. Li, Y. X. Hou, J. H. Xu, Y. Gao and X. Q. Hu, Org. Chem. Front., 2022, 9, 2181–2186 RSC .
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ,
Mohd.
Mujahid
,
Saiprasad
Nunewar
and
Vinaykumar
Kanchupalli
,
Mohd.
Mujahid
,
Saiprasad
Nunewar
and
Vinaykumar
Kanchupalli