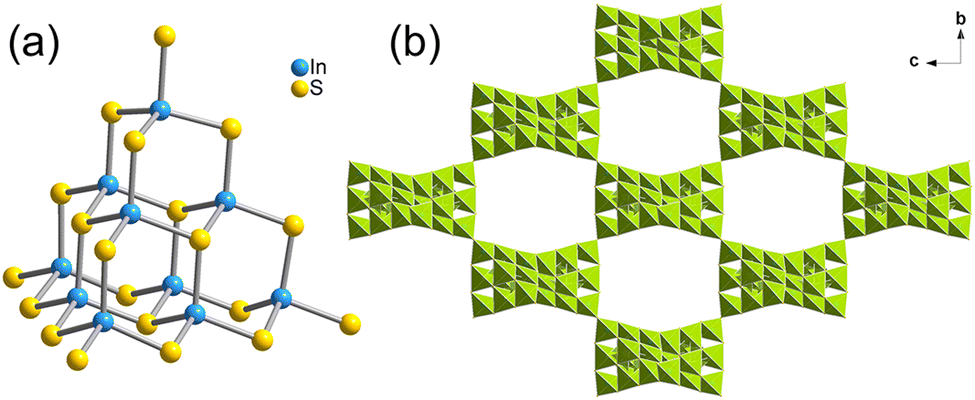DOI:
10.1039/D2QM00810F
(Research Article)
Mater. Chem. Front., 2023,
7, 128-136
Competitive coordination and Lewis-base interactions of amines in supertetrahedral cluster-based materials by a controllable naked T3-InS precursor method: from isolated clusters with various substituted ligands, chains to 3-D open frameworks†
Received
11th August 2022
, Accepted 4th November 2022
First published on 8th November 2022
Abstract
Investigation of structural evolution and assembly via inter-cluster transformation is highly effective to thoroughly understand the crystal growth process, which is crucial for the accurate synthesis of clusters and cluster-based materials. Herein, we use a ligand-free supertetrahedral (Tn) T3-InS cluster (T3-0) as the precursor to achieve diverse transformation products by tuning the composition of amines and other synthetic parameters, which include not only singly and doubly substituted T3 clusters (T3-1 and T3-2) and a double T3 cluster with six substituting groups (T3-3) but also T3 cluster based chains (T3-4) and three-dimensional cristobalite-topological open frameworks (T3-5). Importantly, we examine the role of amine in transformation processes using HRMS, comparative transformation experiments and DFT theoretical calculations. Notably, the results show that amines with high basicity are more likely to interact with the terminal metals of Tn clusters to produce low dimensional materials, and the competition between amines and cluster derived intermediates is crucial for accurate synthesis of Tn clusters. Then, a reasonable SN1-like mechanism is proposed for the surface reaction of cluster transformation. This work is significant not only for understanding the role of amines in the nucleation processes of metal chalcogenide Tn clusters but also for providing a novel method for the accurate synthesis of a series of cluster based materials with specific Tn skeletons.
Introduction
Clusters are considered as bridges not just between phases of matter but also across chemical disciplines, and have greatly promoted the overall development of chemistry.1 The ability to regulate the evolution of a cluster nucleus in a solution system is essential for getting the desired crystalline clusters. At the beginning of the 20th century, classical nucleation theory proposed by Volmer and Weber stated that molecules or atoms directly form a stable crystal phase through the crystallization process in a system.2–6 With the advancement of science and technology and theoretical studies, researchers have discovered that the nucleation and growth of clusters in solution are governed by more variable laws.7–9 Metal chalcogenide supertetrahedral clusters (MCSCs) possess the characteristics of lucid structures and adjustable composition and have tremendous potential to solve various problems involving the surface and structural science of metal chalcogenide semiconductor materials.10–12 Notably, MCSCs as building units can be constructed into open frameworks through a variety of connection techniques rather than merely existing as isolated clusters. In recent years, these open frameworks have received considerable attention because of their fascinating structures and potential applications, such as gas adsorption,13,14 ion exchange15–19 and catalysis.20–24 However, a pressing issue in the synthesis of these materials is that minor adjustments to the synthesis settings often produce products with glaring structural variations. This sensitive response seems to indicate that there is a fierce competition among various active substances during the synthesis of these compounds, which results in high stochasticity in the synthesis.
To realize the precise synthesis of MCSCs, researchers have made a lot of efforts and proposed many beneficial synthesis methodologies. For example, the synthesis strategies of “multivalent metal complementarity” and “superbase-assisted crystallization” proposed by Feng and co-workers offer significant theoretical guidance for the efficient preparation of MCSCs.25–27 In last several years, Li,28–31 Huang,32–34 Stefanie Dehnen35–37 and others have used ionic liquids to develop many unique MCSCs that are difficult to obtain from solvothermal synthesis, making thermal synthesis of ionic liquids one of the important means to obtain MCSCs. Both new synthesis strategies and new ionothermal synthesis methods have greatly promoted the development of MCSCs. However, the problem of allergic reaction of products to parameter fluctuations in solvothermal synthesis, which is a traditional and important synthesis method, has not been well solved. The reason is the lack of a further understanding of the factors influencing formation and assembly of clusters during the solvothermal synthesis of MCSCs, which plays a crucial role in improving the controllability of synthesis. In the process of tackling this problem, an insurmountable challenge lies in the requirement of high temperature, high pressure and closed synthesis conditions for solvothermal synthesis. These characteristics make it challenging for many advanced technical means to monitor the changing process of solvothermal synthesis of MCSCs, such as low temperature transmission electron microscopy (cryo-TEM),38,39 atomic electron tomography (AET)40 and atomic force microscopy (AFM).4
Currently, one of the efficient methods to investigate the formation process of clusters is to use inter-cluster transformation to analyze the structural evolution of compounds. This method has been widely used in the accurate synthesis of a variety of clusters, such as III–V semiconductor nanocrystals,41,42 noble metal clusters43–47 and sulfide nanocrystals.48,49 Note that the surface initiation reaction of clusters is the key to transformation synthesis, which opens up more possibilities for cluster precursor transformation and assembly. Previously, isolated MCSCs were sporadically reported to be used as reactants to build new open frameworks or construct new metal doped isolated MCSCs.50–52 However, the use of MCSC transformation to explore the formation and assembly of MCSCs has been ignored by researchers, which may be the simplest and most efficient way to comprehend the content related to solvothermal synthesis of MCSCs at present.
At present, protonated amines are widely employed as cationic templates to regulate the formation of MCSCs. Due to the negative charge of the MCSC skeleton, cationic species can regulate the formation and assembly of clusters through electrostatic interactions. More significantly, amines involved in the assembly of MCSCs are being increasingly reported, including the synthesis of multidimensional compounds, such as [Ga10S16(NC6H7)2(N2C12H12)]2− and SCIF-X (X = 1, 2,…,9).53,54 Moreover, amines also participate in the construction of isolated clusters, such as ISC-X (X = 1, 2,…,9) and [Ga10S16(NC7H9)4].25,55 The structural diversity of these compounds further indicates that amines may play multiple important roles in the structural regulation of MCSCs. For example, they may act as neutral ligands to reduce the skeletal charge of clusters and as counterions to stabilize clusters after being protonated.25 However, the role of organic amines in regulating the assembly dimension of Tn clusters is still unclear.
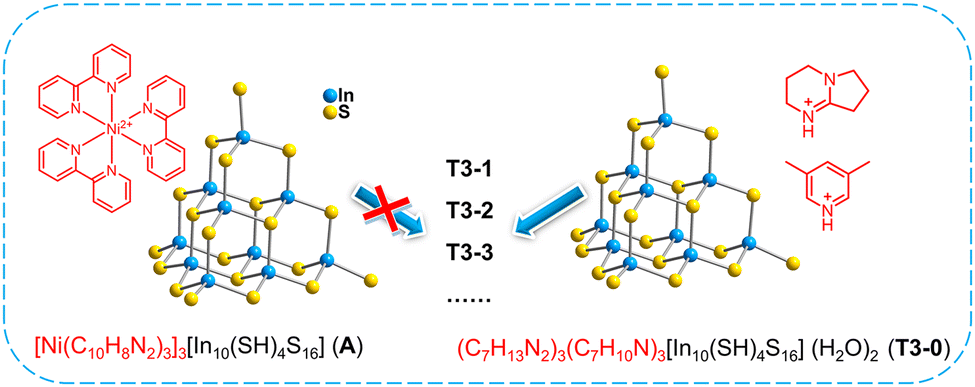
Herein, by adjusting the composition of amines, we realized diversified transformation with a free T3-InS cluster (HDBN)3(C7H10N)3[In10S16(SH)4](H2O)2 (T3-0) as the precursor. Isolated T3-InS clusters with various substituted ligands were obtained, namely, (HDBN)4.5(C7H10N)0.5 [In10S16(SH)3(DBN)] (T3-1), (HDBN)4[In10S16(SH)2(DBN)2] (T3-2) and (HDBN)6 [In20S33(DBN)6] (T3-3). Then, 1D chains (HDBN2)2(CH3NH3)2 [In10S17(DBN)2] (T3-4) and 3D open frameworks (HDBN)1.5(C2H8NO)4.5[In10S18] (T3-5) constructed from T3-InS clusters were transformed. The existing types of amines in the series of transformation products were confirmed by HRMS, and theoretical computation was used to confirm the energy difference in cluster transformation. Importantly, the possible reaction mechanism on the surface of the T3-InS cluster at the beginning of transformation and the effect of the alkalinity of amines on the adjustment of the cluster structure were proposed.
Results and discussion
The phase purity of a series of compounds was demonstrated by PXRD (Fig. S1–S7, ESI†). The EDS (Fig. S8, ESI†) and FTIR (Fig. S9, ESI†) were used to analyze the composition of compounds, and the stability was characterized by TGA (Fig. S10, ESI†).
Single crystal structure analysis
Crystal diffraction data analysis demonstrates that the outcomes of cluster transformation vary according to synthetic conditions. The analysis of crystal diffraction data shows that the naked T3-0 cluster can be used as a precursor to produce a variety of transformation products with different numbers of substituted ligands or different cluster assembly methods under various synthesis conditions. Single crystal structure information of a series of compounds is listed in the ESI† (Tables S1–S3).
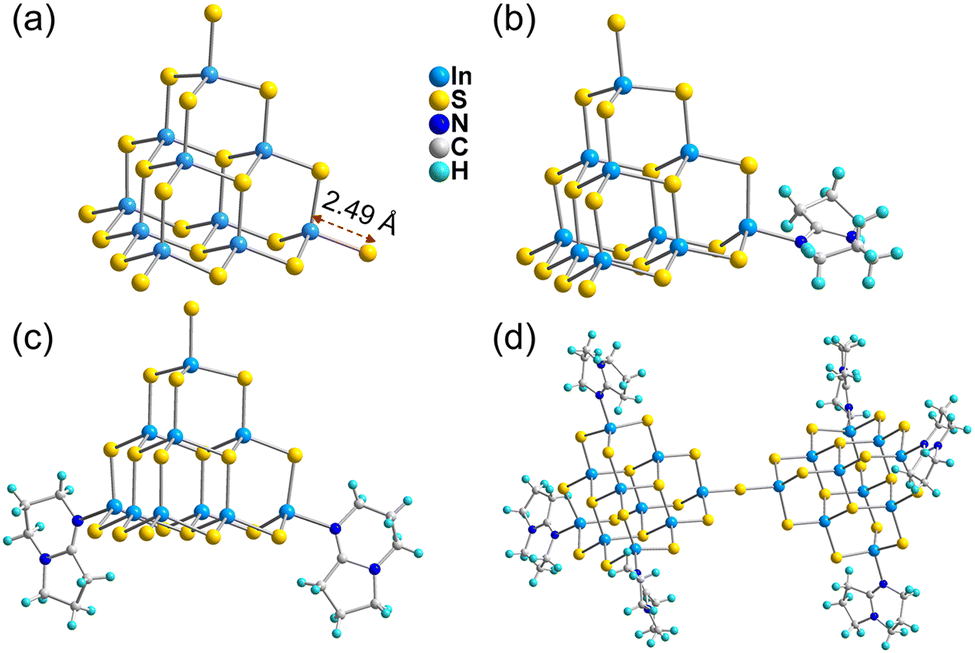
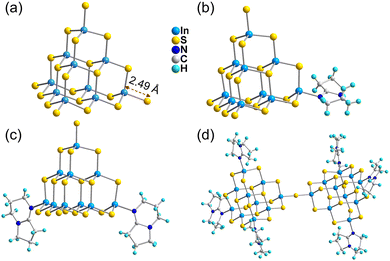
The ligand-free protected T3-InS cluster, i.e., T3-0, crystallizes in the Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group of the cubic system. As shown in Fig. 1(a), T3-0 is a classical T3 supertetrahedral cluster, which is composed of indium atoms coordinated tetrahedrally with sulfur atoms. And the S atoms can coordinate in a number of modes, such as end groups, double coordination, and triple coordination. Notably, the length of the terminal In–S bond of T3-0 is as high as 2.491 Å, which indicates that the terminal S atom is likely to be protonated. This is beneficial for reducing the negative charge and improving the stability of clusters, and the anionic formula of T3-0 is [In10S16(SH)4]6−.
m space group of the cubic system. As shown in Fig. 1(a), T3-0 is a classical T3 supertetrahedral cluster, which is composed of indium atoms coordinated tetrahedrally with sulfur atoms. And the S atoms can coordinate in a number of modes, such as end groups, double coordination, and triple coordination. Notably, the length of the terminal In–S bond of T3-0 is as high as 2.491 Å, which indicates that the terminal S atom is likely to be protonated. This is beneficial for reducing the negative charge and improving the stability of clusters, and the anionic formula of T3-0 is [In10S16(SH)4]6−.
 |
| | Fig. 1 Supertetrahedral T3 isolated cluster in T3-0 (a), T3-1 (b) T3-2 (c) and T3-3 (d). | |
The conversion product T3-1 crystallizes in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group of the triclinic system. As shown in Fig. 1(b), the T3-1 cluster possesses an anion skeleton, which is highly similar to that of the T3-0 cluster. The difference is that a terminal S–In bond in the T3-1 cluster is broken, and the exposed In atom is combined with N atom at the 5-position of 1,5-diazabicyclo[4.3.0]-5-nonene (DBN) to form a single DBN substituted T3-InS cluster. Additionally, the other terminal S–In bonds in the T3-1 cluster are significantly shorter than those in the T3-0 cluster, and the bond lengths are 2.434, 2.455 and 2.456 Å. The formula of the anionic T3-1 cluster is [In10S16(SH)3(DBN)]5−.
space group of the triclinic system. As shown in Fig. 1(b), the T3-1 cluster possesses an anion skeleton, which is highly similar to that of the T3-0 cluster. The difference is that a terminal S–In bond in the T3-1 cluster is broken, and the exposed In atom is combined with N atom at the 5-position of 1,5-diazabicyclo[4.3.0]-5-nonene (DBN) to form a single DBN substituted T3-InS cluster. Additionally, the other terminal S–In bonds in the T3-1 cluster are significantly shorter than those in the T3-0 cluster, and the bond lengths are 2.434, 2.455 and 2.456 Å. The formula of the anionic T3-1 cluster is [In10S16(SH)3(DBN)]5−.
T3-2 crystallized in the I2/m space group of the monoclinic system. As shown in Fig. 1(c), in addition to maintaining the basic skeleton of the T3-InS cluster, it is noteworthy that the two terminal S atoms of the T3-InS cluster in T3-2 are replaced with DBN molecules, exhibiting the gradual substitution trend of ligands for T3-0 and T3-1 during the transformation process. The cluster formula of T3-2 is [In10S16(SH)2(DBN)2]4−. Notably, T3-0 cluster transformation produced T3-1 and T3-2 clusters, which are metastable phase crystals. After increasing the conversion temperature, the most stable isolated double T3 cluster T3-3 [In20S33(DBN)6]6− can be obtained, which has been previously reported by us56 and others.57 As shown in Fig. 1(d), T3-3 is composed of two T3-InS clusters assembled by corner-sharing, and the other six terminal S atoms are replaced with DBN molecules through N–In bonds.
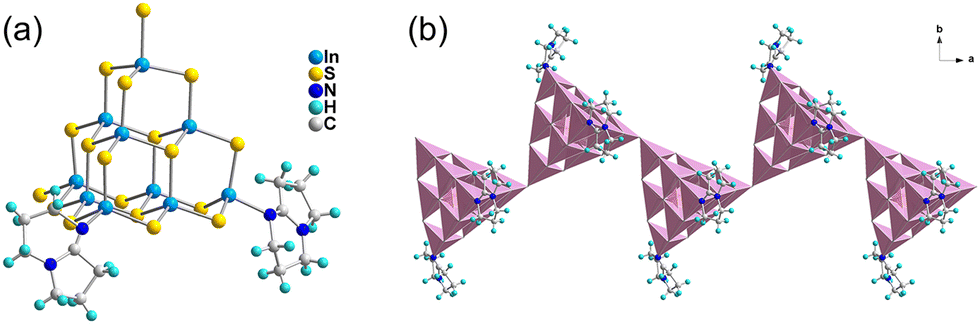
T3-4 crystallizes in the I2/a space group of the monoclinic system, which is a 1D chain compound. As shown in Fig. 2(a), the building block of T3-4 is the T3-InS cluster, in which two terminal S atoms are replaced with DBN molecules. Then, adjacent building blocks are assembled by corner-sharing to obtain a Z-shaped 1D chain compound [In10S17(DBN)2]4− extending along the a-axis (Fig. 2(b)). It is worth noting that the S–In bond length of T3-4 is 2.544 Å and the In–S–In bond angle of T3-4 is 116.74°, which are significantly larger than the bond length and angle obtained from similar assembly methods. This result may be attributed to the steric hindrance effect of DBN molecules on the same side of the skeleton. According to Fig. S11 (ESI†), the adjacent chain structures are staggered along the [100] direction, and the protonated DBN and methylamine are distributed between the adjacent chain structures as counterions. Methylamine is too disordered to be solved by X-ray crystallographic analysis, and its existence is judged by TGA and EA.
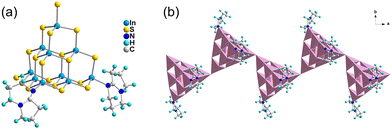 |
| | Fig. 2 Supertetrahedral T3 cluster in T3-4 (a) and the 1D chain structure of T3-4 along the a-axis (b). | |
T3-5 crystallized in the I41/acd space group of the tetragonal system. As shown in Fig. 3(a), the building block of T3-5 is a classical T3-InS supertetrahedral cluster. Adjacent T3-InS clusters assemble to form a 3D cristobalite topology through corner-sharing of the clusters. A single framework possesses a rectangular channel of 11.91 Å × 19.38 Å along the a-axis. The overall structure is formed by double interpenetration of two frameworks, and the structure presents a rectangular channel in the [111] direction (Fig. S12, ESI†). The anionic framework formula of T3-5 is [In10S18]6−.
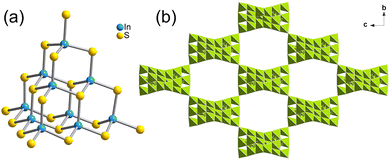 |
| | Fig. 3 (a) Supertetrahedral T3 cluster in T3-5 and (b) 3D framework of T3-5 viewed from the a-axis. | |
Countercation analysis
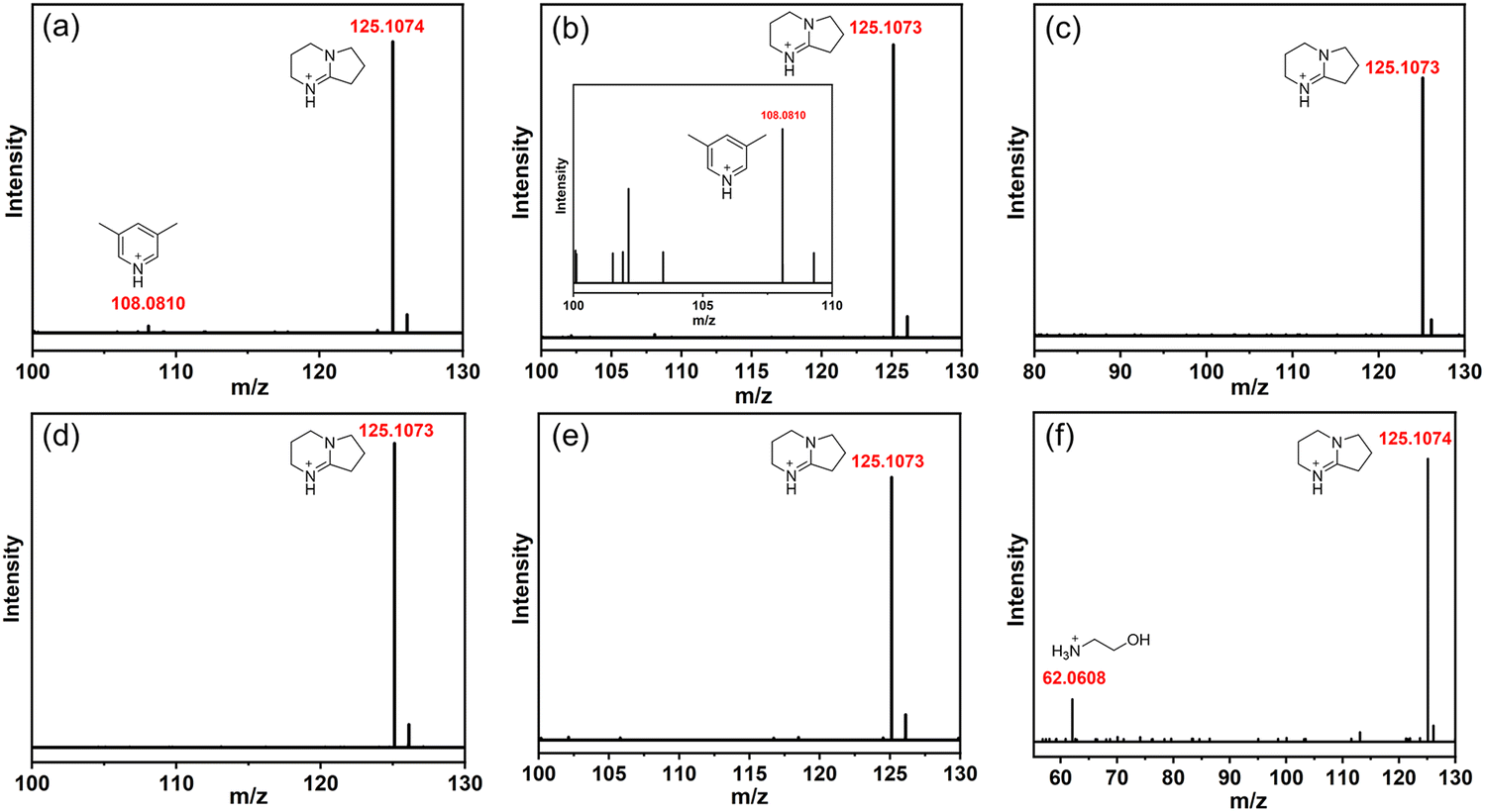
A series of transformation results show that the adjustment of the amine has a great influence on the outcome of the cluster transformation. Therefore, we analyzed the protonated amine composition of a series of compounds through high-resolution mass spectrometry (HRMS, ESI, cationic mode). As shown in Fig. 4(a), there are two types of protonated amines in T3-0, namely, protonated 3,5-dimethylpyridine (m/z = 108.0810) and DBN (m/z = 125.1074). The two types of protonated amines remained in the conversion product T3-1 (Fig. 4(b)). However, when T3-0 was converted to T3-2 and T3-3, the countercation was only [HDBN]+ (Fig. 4(c) and (d)). The m/z of [CH3NH3]+ in the T3-4 structure is beyond the testing range of HRMS (Fig. 4(e)), but its existence can be confirmed by TGA (Fig. S10, ESI†). Then, the mass spectrum proved that T3-5 contained [NH3CH2CH2OH]+ and [HDBN]+ (Fig. 4(f)), of which [HDBN]+ comes from T3-0. These results demonstrate the competitiveness of protonated amines as counterions of conversion products, with [HDBN]+ clearly outperforming its rivals.
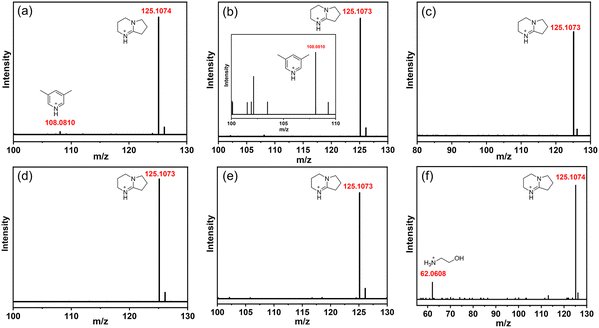 |
| | Fig. 4 HRMS spectra of counterions in compounds T3-0–T3-5 (corresponding to a–f, respectively). | |
Analysis of transformation conditions
A series of synthesis studies were carried out using cluster T3-0 as a precursor (Fig. S1, ESI†). First of all, we adjusted the synthesis time to understand its effect on clustering transformation. The relevant synthesis results are summarized in Table S4 (ESI†). When the cluster T3-0 is transformed at 180 °C, a mixture of clusters T3-1 or T3-2 and T3-3 can be obtained with the extension of reaction time (Table S4, entries 1 and 2, ESI†). Until the reaction time was extended to 5 days, the clusters were all transformed into cluster T3-3, and on further extending the reaction time to 12 days, the products did not change (Table S4, entries 3 and 4, ESI†). An interesting phenomenon is that T3-1 and T3-2 are occasionally obtained alternately during the early stage of the reaction when they are converted at 180 °C. These results indicate that cluster T3-3 is a stable transformation product of cluster T3-0, while clusters T3-1 and T3-2 are metastable products in this transformation process.
Then, the difference in conversion results is more obvious after the temperature of the reaction system is adjusted. The relevant synthesis results are summarized in Table S5 (ESI†). At the beginning, cluster T3-0 did not transform at 100 °C, and there were only many tiny T3-0 crystals in the system (Table S5, entry 1, ESI†). When the conversion temperature was raised to 120 °C, the T3-0 cluster exhibited transition to T3-1 (Table S5, entry 2, ESI†), and the purity of the product was verified by PXRD (Fig. S2, ESI†). On further increasing the conversion temperature to 140–160 °C, cluster T3-2 was obtained (Table S5, entries 3, 4 and Fig. S3, ESI†), which showed better thermodynamic stability than T3-1. Notably, the cluster T3-3 was obtained at a conversion temperature of 180 °C, and the product did not change when the temperature was further increased to 200 °C (Table S5, entries 5, 6 and Fig. S4, ESI†). These results indicate that T3-3 has the highest thermodynamic stability in this series of molecular clusters, which is consistent with the previous results of conversion time control experiments (Table S4, ESI†).
Finally, the amine environment in the conversion synthesis was adjusted (Table S6, ESI†). When DBN and methylamine (volume ratio = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1; Table S6, entry 1, ESI†) were used to form the amine environment for conversion reaction, the conversion result was not affected and cluster T3-3 was still obtained. However, after adjusting the amount of DBN and methylamine (volume ratio = 1:3; Table S6, entry 2, ESI†), the structure of the product changed dramatically and the 1D chain compound T3-4 (Fig. S5, ESI†) was obtained. It cannot be ignored that the dosage ratio of amines must be strictly controlled during the conversion reaction to T3-4, otherwise T3-3 may be generated. However, after completely changing the transformation environment of cluster T3-0 with ethanolamine (Table S6, entry 3, ESI†), compound T3-5 was obtained (Fig. S6, ESI†), which has a pure inorganic anion skeleton rather than an organic–inorganic skeleton like other conversion products. These results show that DBN is more likely to participate in the construction of an anionic skeleton in the synthesis of MCSCs than other amines. In addition, other amines such as pyridine, piperidine, imidazole and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) were also used for conversion, but only a yellow green powder was obtained. These results again show that suitable amines play a crucial role in obtaining supertetrahedral clusters.
:1; Table S6, entry 1, ESI†) were used to form the amine environment for conversion reaction, the conversion result was not affected and cluster T3-3 was still obtained. However, after adjusting the amount of DBN and methylamine (volume ratio = 1:3; Table S6, entry 2, ESI†), the structure of the product changed dramatically and the 1D chain compound T3-4 (Fig. S5, ESI†) was obtained. It cannot be ignored that the dosage ratio of amines must be strictly controlled during the conversion reaction to T3-4, otherwise T3-3 may be generated. However, after completely changing the transformation environment of cluster T3-0 with ethanolamine (Table S6, entry 3, ESI†), compound T3-5 was obtained (Fig. S6, ESI†), which has a pure inorganic anion skeleton rather than an organic–inorganic skeleton like other conversion products. These results show that DBN is more likely to participate in the construction of an anionic skeleton in the synthesis of MCSCs than other amines. In addition, other amines such as pyridine, piperidine, imidazole and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) were also used for conversion, but only a yellow green powder was obtained. These results again show that suitable amines play a crucial role in obtaining supertetrahedral clusters.
Transformation control experiment
In order to understand the initiation process of surface reaction during cluster transformation, we synthesized cluster A with exactly the same anionic skeleton as that of T3-0 according to the literature.58 Compared with T3-0, the countercations of cluster A are composed of metal complexes [Ni(C10H8N2)3]2+. Under the same conditions, we performed transformation control experiments using compound A as the precursor. Regrettably, neither isolated clusters nor multidimensional compounds were obtained, and only a large amount of yellow precipitate was left in the reaction system. The experiment shows that the counterions, which can provide protons, may play a non-negligible role in the initiation of cluster surface reactions in the transformation and assembly of T3-InS clusters (Fig. 5).
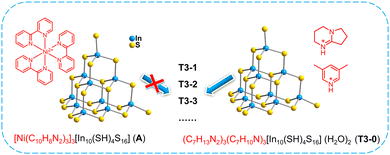 |
| | Fig. 5 Comparative experiment of cluster transformation of A and T3-0. | |
Theoretical calculation and analysis
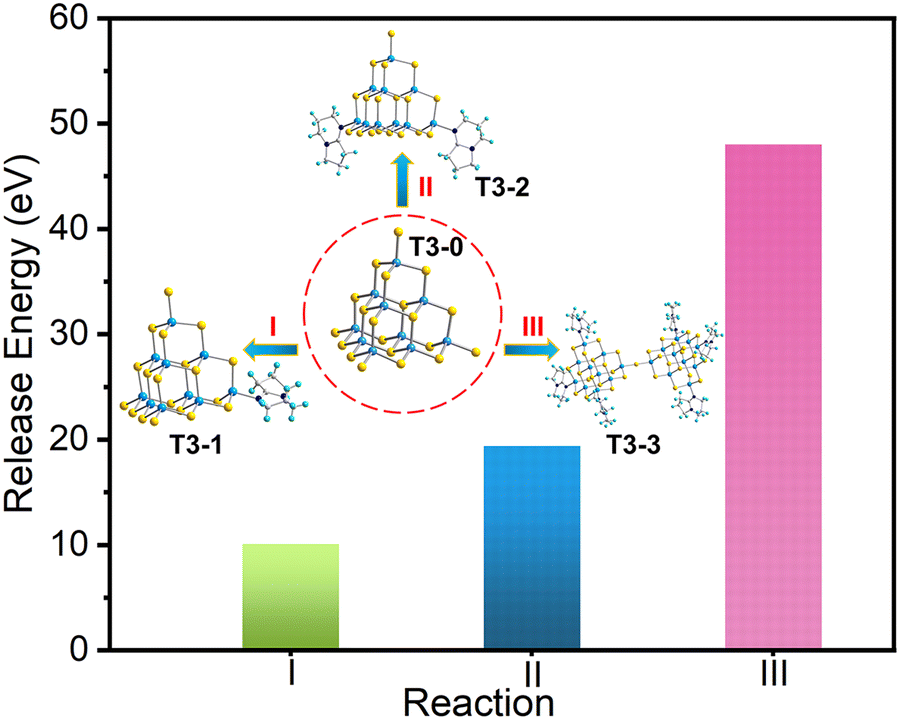
To further analyze the series conversion results with T3-0 as the precursor, we calculated the electronic energy of a series of isolated T3 clusters, and the relevant results are listed in Table S7 (ESI†). Based on the conservation of matter and electrons, we list the corresponding conversion equations and calculate the binding energies based on the known electron energy. First, compounds T3-1, T3-2 and T3-3 are obtained from T3-0 as the precursor, and the conversion equations are as follow:| | | [In10S16(SH)4]6− + [HDBN]+ → [In10S16(SH)3(DBN)]5− + H2S | (I) |
| | | [In10S16(SH)4]6− + 2[HDBN]+ → [In10S16(SH)2(DBN)2]4− + 2H2S | (II) |
| | | 2[In10S16(SH)4]6− + 6[HDBN]+ → [In20S33(DBN)6]6− + 7H2S | (III) |
As shown in Fig. 6, when the T3-0 cluster was transformed into compounds T3-1, T3-2 and T3-3, the energy of the system decreased to 10.106, 19.431 and 48.041 eV, respectively. The analysis of energy stability displays that compound T3-0 as a precursor shows a thermodynamic trend of transforming into other isolated clusters. Moreover, the stability of compounds T3-1, T3-2 and T3-3 boosts gradually, which is consistent with the experimental results. Notably, this stability law maintains a negative correlation with the number of –SH groups at the cluster terminal. This may be attributed to the higher reactivity of the terminal –SH group compared to other S sites in the cluster.59
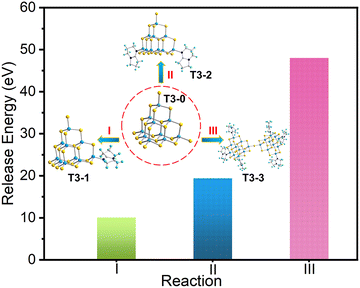 |
| | Fig. 6 Released energy of conversion reaction of compounds T3-1, T3-2 and T3-3. | |
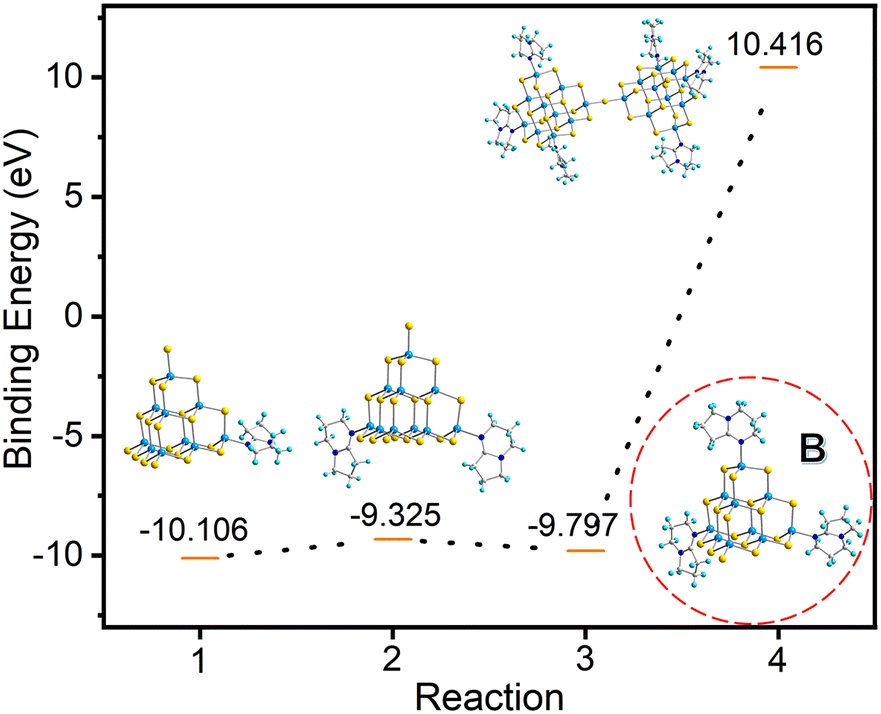
In order to understand the formation process of compound T3-3, we hypothesize that the trisubstituted intermediate B (Fig. 7) might be formed during the transformation. The structure of B is characterized using the T3-InS cluster in which three terminal S atoms are replaced with DBN molecules. Then, this structure is optimized and electronic energy is concomitantly calculated (Table S7, ESI†). Based on our hypothetical route, compound T3-0 is gradually transformed into double supertetrahedral T3-3 through metastable phase clusters T3-1, T3-2 and intermediate B. The corresponding step-by-step equation is as follows:
| | | [In10S16(SH)4]6− + [HDBN]+ → [In10S16(SH)3(DBN)]5−+ H2S | (1) |
| | | [In10S16(SH)3(DBN)]5− + [HDBN]+ → [In10S16(SH)2(DBN)2]4− + H2S | (2) |
| | | [In10S16(SH)2(DBN)2]4− + [HDBN]+ → [In10S16(SH)(DBN)3]3− + H2S | (3) |
| | | 2[In10S16(SH)(DBN)3]3− → [In20S33(DBN)6]6− + H2S | (4) |
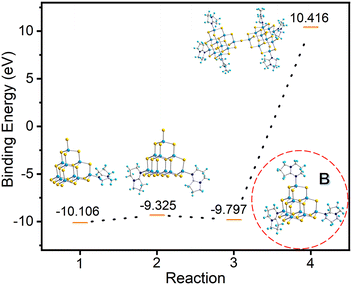 |
| | Fig. 7 Binding energies for the stepwise transformation of compound T3-0via compounds T3-1 and T3-2 to compound T3-3. | |
As shown in Fig. 7, the gradual transformation from compound T3-0 to T3-1 and T3-2 reduces the system energy. Assuming that compound T3-2 is converted to intermediate B, the system energy continues to decrease. However, when intermediate B is converted to compound T3-3, it needs to absorb energy in the conversion process and the system energy increases. Based on these results, if intermediate B does exist, theoretically, product B can be easily obtained in the synthesis process, but this is contrary to the actual results. Then, we consider the transformation process of T3-1 and T3-2 to T3-3. The transformation equations are listed as follows:
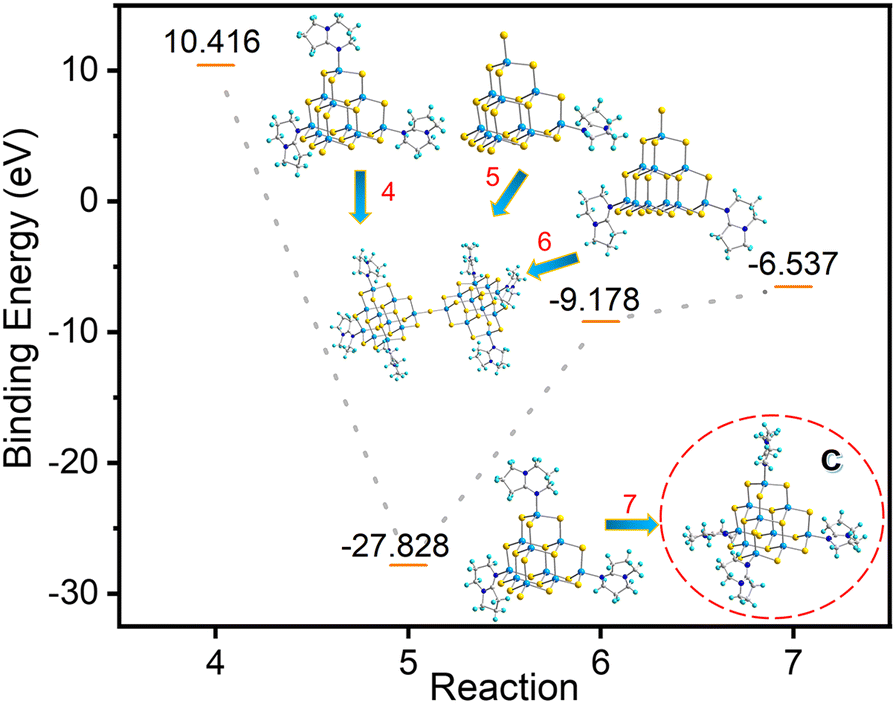
| | | 2[In10S16(SH)3(DBN)]5− + 4[HDBN]+ → [In20S33(DBN)6]6− + 5H2S | (5) |
| | | 2[In10S16(SH)2(DBN)2]4− + 2[HDBN]+ → [In20S33(DBN)6]6− + 3H2S | (6) |
| | | [In10S16(SH)(DBN)3]3− + [HDBN]+ → [In10S16(DBN)4]2− + H2S | (7) |
As shown in Fig. 8, the system energy decreases when both T3-1 and T3-2 form T3-3, with the decrease in system energy in the case of T3-1 being more dramatic. Compared with compound T3-2, the presence of protonated 3,5-dimethylpyridine in compound T3-1 may be more conducive to cluster transformation. In addition, according to binding energy analysis of eqn (7), the trisubstituted intermediate B possesses a strong tendency to transform into the reported tetrasubstituted cluster C (Fig. 8).24 However, no trace of C clusters was found in a large number of transformation experiments. Therefore, we further believe that intermediate B was not generated during the conversion from compound T3-1 to compound T3-3. The cluster T3-3 mainly comes from the transformation products of T3-0 through the metastable cluster T3-1.
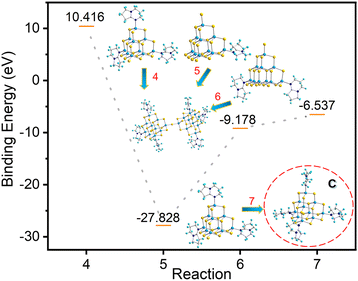 |
| | Fig. 8 Binding energy of conversion from compounds T3-1 and T3-2 and possible intermediate B to compound T3-3. | |
Speculation on the transformation mechanism of T3 clusters
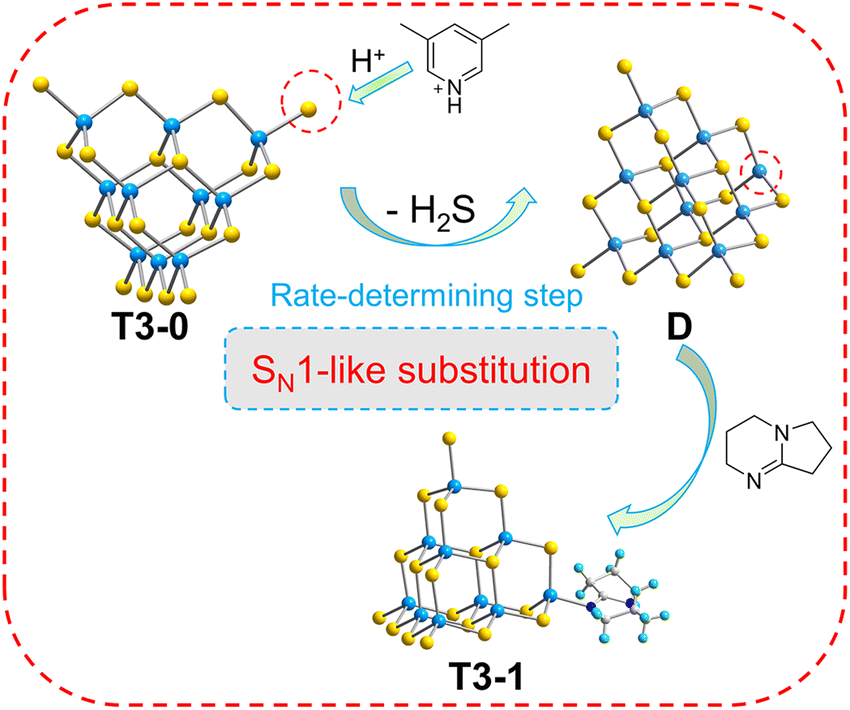
Based on the above experimental and theoretical calculation results, the transformation mechanism of T3 clusters is speculated using the transformation process of T3-0 to T3-1 (Fig. 9). Significantly, T3-0 contains protonated 3,5-dimethylpyridine and DBN with pKa values of 13.960 and 23.79,61 respectively, which means that protonated 3,5-dimethylpyridine is strongly acidic and easily loses protons. This causes the protonated 3,5-dimethylpyridine to lose the H+ of the N atom under the electrostatic attraction of the T3-InS anion cluster. Then, the proton is transferred to the –SH group of the T3-InS cluster, allowing it to escape in the form of H2S. The exposed and unsaturated In atom in intermediate D immediately coordinates with DBN molecules, which occurs by a reaction similar to SN1 substitution in organic chemistry. According to the mechanism, the reduction of the negative charge of the cluster skeleton will make it more difficult to capture the protons from counterions. Therefore, the initiation of the transformation process requires higher energy, which is compatible with the change rule of experimental temperature for the transformation of different isolated clusters.
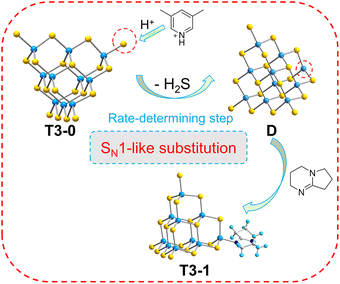 |
| | Fig. 9 Conversion mechanism of T3-0 to T3-1. | |
The structure of the products clearly differed after adjusting the types of amines in the conversion process. When amines with low basicity are used, the conversion product of T3-0 is no longer isolated clusters but the Z-shaped 1D chain T3-4 and the 3D open framework T3-5 are formed. Notably, half of the terminal S atoms of the T3-InS cluster in compound T3-4 are replaced with DBN, and the other half is involved in the inter-cluster assembly. This result attributed to the difference in the charge density of nitrogen atoms in amines. The pKa value of DBN is as high as 23.79, and electron density on N atom in it is much higher than that in CH3NH2 (pKa = 18.4),60 so it preferentially combines with the In atom of intermediate. For the preparation of T3-5, we speculate that although there is a large amount of ethanolamine in the synthetic environment, it has a lower charge density on the N atom (pKa = 17.5)60 and cannot form a stable interaction with the exposed In atom of intermediate D, due to which D eventually forms T3-5 through self-assembly.
Conclusions
By using the ligand-free T3-InS isolated cluster T3-0 as the precursor, we realized its transformation into a variety of gradually substituted isolated clusters, 1D chains and 3D open frameworks employing T3-InS clusters as a building unit. Importantly, in combination with HRMS analysis, theoretical calculation and comparative transformation experiments, a reasonable transformation mechanism of the T3-InS cluster is proposed. A key driving force for the surface reaction in cluster transformation is the capture of protons of protonated amine by the anion skeleton of the cluster. Of special note the coordination ability of nitrogen atoms of amines is closely related to their basicity, which can significantly affect the diversity of growth of cluster nuclei and assembly modes. This work is of great significance for understanding the controllable synthesis of MCSCs and their open frameworks, and provides a new path to prepare a series of materials with the same size of Tn clusters.
Author contributions
L. S. designed the experiments and wrote the paper. L. S. and H.-Y. Z. carried out most of the experiments. F.-Q. Z. provided theoretical calculations for research. L. W. revised the paper and supplemented experimental data. X.-M. Z. revised the paper and directed the research.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by NSFC (21871167, 22266034, and 91961201), 1331 project of Shanxi Province.
Notes and references
- Z. Luo, A. W. Castleman, Jr. and S. N. Khanna, Reactivity of metal clusters, Chem. Rev., 2016, 116, 14456–14492 CrossRef CAS.
- G. C. Sosso, J. Chen, S. J. Cox, M. Fitzner, P. Pedevilla, A. Zen and A. Michaelides, Crystal nucleation in liquids: Open questions and future challenges in molecular dynamics simulations, Chem. Rev., 2016, 116, 7078–7116 CrossRef CAS PubMed.
- M. Sleutel, J. Lutsko, A. E. Van Driessche, M. A. Duran-Olivencia and D. Maes, Observing classical nucleation theory at work by monitoring phase transitions with molecular precision, Nat. Commun., 2014, 5, 5598 CrossRef CAS.
- J. Chen, E. Zhu, J. Liu, S. Zhang, Z. Lin, X. Duan, H. Heinz, Y. Huang and J. J. D. Yoreo, Building two-dimensional materials one row at a time: Avoiding the nucleation barrier, Science, 2018, 362, 1135–1139 CrossRef CAS PubMed.
- K. Z. Takahashi, T. Aoyagi and J.-I. Fukuda, Multistep nucleation of anisotropic molecules, Nat. Commun., 2021, 12, 5278 CrossRef CAS PubMed.
- S. Jeon, T. Heo, S.-Y. Hwang, J. Ciston, K. C. Bustillo, B. W. Reed, J. Ham, S. Kang, S. Kim, J. Lim, K. Lim, J. S. Kim, M.-H. Kang, R. S. Bloom, S. Hong, K. Kim, A. Zettl, W. Y. Kim, P. Ercius, J. Park and W. C. Lee, Reversible disorder–order transitions in atomic crystal nucleation, Science, 2021, 371, 498–503 CrossRef CAS.
- J. F. Lutsko, How crystals form: A theory of nucleation pathways, Sci. Adv., 2019, 5, eaav7399 CrossRef CAS PubMed.
- Y. Y. Zhang, D. S. Zhang, T. Li, M. Kurmoo and M. H. Zeng, In situ metal-assisted ligand modification induces Mn4 cluster-to-cluster transformation: A crystallography, mass spectrometry, and DFT study, Chem. – Eur. J., 2020, 26, 721–728 CrossRef CAS PubMed.
- X. Kang, Y. Li, M. Zhu and R. Jin, Atomically precise alloy nanoclusters: Syntheses, structures, and properties, Chem. Soc. Rev., 2020, 49, 6443–6514 RSC.
- B. Silva-Gaspar, R. Martinez-Franco, G. Pirngruber, A. Fécant, U. Diaz and A. Corma, Open-framework chalcogenide materials-from isolated clusters to highly ordered structures- and their photocalytic applications, Coord. Chem. Rev., 2022, 453, 214243 CrossRef CAS.
- Y. Liu, J. Zhang, B. Han, X. Wang, Z. Wang, C. Xue, G. Bian, D. Hu, R. Zhou, D.-S. Li, W. Zhenxing, O. Zhongwen, L. Mingde and W. Tao, New insights into Mn–Mn coupling interaction-directed photoluminescence quenching mechanism in Mn2+-Doped semiconductors, J. Am. Chem. Soc., 2020, 142, 6649–6660 CrossRef CAS.
- C. Xue, X. Fan, J. Zhang, D. Hu, X.-L. Wang, X. Wang, R. Zhou, H. Lin, Y. Li, D.-S. Li, X. Wei, D. Zheng, Y. Yang, K. Han and T. Wu, Direct observation of charge transfer between molecular heterojunctions based on inorganic semiconductor clusters, Chem. Sci., 2020, 11, 4085–4096 RSC.
- K. Sasan, Q. Lin, C. Mao and P. Feng, Open framework metal chalcogenides as efficient photocatalysts for reduction of CO2 into rrenewable hydrocarbonfuel, Nanoscale, 2016, 8, 10913–10916 RSC.
- X. Chen, X. Bu, Y. Wang, Q. Lin and P. Feng, Charge- and size- complementary
multimetal-induced morphology and phase control in zeolite-type metal chalcogenides, Chem. – Eur. J., 2018, 24, 10812–10819 CrossRef CAS PubMed.
- X. M. Zhang, D. Sarma, Y. Q. Wu, L. Wang, Z. X. Ning, F. Q. Zhang and M. G. Kanatzidis, Open-framework oxysulfide based on the supertetrahedral [In4Sn16O10S34]12− cluster and efficient sequestration of heavy metals, J. Am. Chem. Soc., 2016, 138, 5543–5546 CrossRef CAS PubMed.
- H. Yang, M. Luo, L. Luo, H. Wang, D. Hu, J. Lin, X. Wang, Y. Wang, S. Wang, X. Bu, P. Feng and T. Wu, Highly selective and rapid uptake of radionuclide cesium based on robust zeolitic chalcogenide via stepwise ion-exchange strategy, Chem. Mater., 2016, 28, 8774–8780 CrossRef CAS.
- L. Wang, H. Pei, D. Sarma, X.-M. Zhang, K. MacRenaris, C. D. Malliakas and M. G. Kanatzidis, Highly selective radioactive 137Cs+ capture in an open-framework oxysulfide based on supertetrahedral cluster, Chem. Mater., 2019, 31, 1628–1634 CrossRef CAS.
- B. Zhang, J. Li, D. N. Wang, M. L. Feng and X. Y. Huang, Fast and effective decontamination of aqueous mercury by a highly stable zeolitic-like chalcogenide, Inorg. Chem., 2019, 58, 4103–4109 CrossRef CAS.
- W.-A. Li, Y.-C. Peng, W. Ma, X.-Y. Huang and M.-L. Feng, Rapid and selective removal of Cs+ and Sr2+ ions by two zeolite-type sulfides via ion exchange method, Chem. Eng. J., 2022, 442, 136377 CrossRef CAS.
- D. Liu, Y. Liu, P. Huang, C. Zhu, Z. Kang, J. Shu, M. Chen, X. Zhu, J. Guo, L. Zhuge, X. Bu, P. Feng and T. Wu, Highly tunable heterojunctions from multimetallic sulfide nanoparticles and silver nanowires, Angew. Chem., Int. Ed., 2018, 57, 5374–5378 CrossRef CAS PubMed.
- M. Hao, Q. Hu, Y. Zhang, M. Luo, Y. Wang, B. Hu, J. Li and X. Huang, Soluble supertetrahedral chalcogenido T4 clusters: High stability and enhanced hydrogen evolution activities, Inorg. Chem., 2019, 58, 5126–5133 CrossRef CAS PubMed.
- D. Hu, X. Wang, X. Chen, Y. Wang, A. N. Hong, J. Zhong, X. Bu, P. Feng and T. Wu, S-Doped Ni(OH)2 nano-electrocatalyst confined in semiconductor zeolite with enhanced oxygen evolution activity, J. Mater. Chem. A, 2020, 8, 11255–11260 RSC.
- R. Hu, X. L. Wang, J. Zhang, D. Hu, J. Wu, R. Zhou, L. Li, M. Li, D. S. Li and T. Wu, Multi-metal nanocluster assisted Cu–Ga–Sn tri-doping for enhanced photoelectrochemical water splitting of BiVO4 film, Adv. Mater. Interfaces, 2020, 7, 2000016 CrossRef CAS.
- Q. Lin, X. Bu, C. Mao, X. Zhao, K. Sasan and P. Feng, Mimicking high-silica zeolites: Highly stable germanium- and tin-rich zeolite-type chalcogenides, J. Am. Chem. Soc., 2015, 137, 6184–6187 CrossRef CAS PubMed.
- T. Wu, X. Bu, P. Liao, L. Wang, S.-T. Zheng, R. Ma and P. Feng, Superbase route to supertetrahedral chalcogenide clusters, J. Am. Chem. Soc., 2012, 134, 3619–3622 CrossRef CAS PubMed.
- J. Zhang, X. Bu, P. Feng and T. Wu, Metal chalcogenide supertetrahedral clusters: Synthetic control over assembly, dispersibility, and their functional applications, Acc. Chem. Res., 2020, 53, 2261–2272 CrossRef CAS PubMed.
- J. Zhang, P. Feng, X. Bu and T. Wu, Atomically precise metal chalcogenide supertetrahedral clusters: Frameworks to molecules, and structure to function, Natl. Sci. Rev., 2021, 9, nwab076 CrossRef PubMed.
- Y. Peng, Q. Hu, Y. Liu, J. Li and X. Huang, Discrete supertetrahedral tn chalcogenido clusters synthesized in ionic liquids: Crystal structures and photocatalytic activity, ChemPlusChem, 2020, 85, 2487–2498 CrossRef CAS.
- Y. Wang, Z. Zhu, Z. Sun, Q. Hu, J. Li, J. Jiang and X. Huang, Discrete supertetrahedral T5 selenide clusters and their Se/S solid solutions: Ionic-liquid-assisted precursor route syntheses and photocatalytic properties, Chem. – Eur. J., 2020, 26, 1624–1632 CrossRef CAS.
- C. F. Du, J. R. Li, B. Zhang, N. N. Shen and X. Y. Huang, From T2,2@bmmim to alkali @T2,2@bmmim ivory ball-like clusters: Ionothermal syntheses, precise doping, and photocatalytic properties, Inorg. Chem., 2015, 54, 5874–5878 CrossRef CAS PubMed.
- D.-D. Yang, W. Li, W.-W. Xiong, J.-R. Li and X.-Y. Huang, ionothermal synthesis of discrete supertetrahedral Tn (n = 4, 5) clusters with tunable components, band gaps, and fluorescence properties, Dalton Trans., 2018, 47, 5977–5984 RSC.
- J. R. Li, Z. L. Xie, X. W. He, L. H. Li and X. Y. Huang, Crystalline open-framework selenidostannates synthesized in ionic liquids, Angew. Chem., Int. Ed., 2011, 50, 11395–11399 CrossRef CAS PubMed.
- Y. Liu, Y. Wang, Y. Peng, Q. Hu, J. Li and X. Huang, Ionic-Liquid-assisted precursor route syntheses of highly dispersed supertetrahedral T3 In–S/Se clusters for photocatalytic hydrogen production, ChemistrySelect, 2022, 7, e202200585 CAS.
- W.-W. Xiong, J.-R. Li, B. Hu, B. Tan, R.-F. Li and X.-Y. Huang, Largest discrete supertetrahedral clusters synthesized in ionic liquids, Chem. Sci., 2012, 3, 1200–1204 RSC.
- S. Santner, J. Heine and S. Dehnen, Synthesis of crystalline chalcogenides in ionic liquids, Angew. Chem., Int. Ed., 2016, 55, 876–893 CrossRef CAS PubMed.
- Z. Wu, I. Nussbruch, S. Nier and S. Dehnen, Ionothermal access to defined oligomers of supertetrahedral selenido germanate clusters, JACS Au, 2022, 2, 204–213 CrossRef CAS PubMed.
- B. Peters, S. Santner, C. Donsbach, P. Vöpel, B. Smarsly and S. Dehnen, Ionic liquid cations as methylation agent for extremely weak chalcogenido metalate nucleophiles, Chem. Sci., 2019, 10, 5211–5217 RSC.
- R. E. Schreiber, L. Houben, S. G. Wolf, G. Leitus, Z. L. Lang, J. J. Carbo, J. M. Poblet and R. Neumann, Real-time molecular scale observation of crystal formation, Nat. Chem., 2017, 9, 369–373 CrossRef CAS.
- A. E. S. Van Driessche, N. Van Gerven, P. H. H. Bomans, R. R. M. Joosten, H. Friedrich, D. Gil-Carton, N. Sommerdijk and M. Sleutel, Molecular nucleation mechanisms and control strategies for crystal polymorph selection, Nature, 2018, 556, 89–94 CrossRef CAS PubMed.
-
J. Zhou, Y. Yang, Y. Yang, D. S. Kim, A. Yuan, X. Tian, C. Ophus, F. Sun, A. Schmid, M. Nathanson, H. Heinz, Q. An, H. Zeng, P. Ercius and J. Miao, Capturing Nucleation at 4D Atomic Resolution, Conference: APS March Meeting 2019, Boston, MA, March 4–8, 2019; Related Information: 2019, https://arxivorg/abs/180710709, 2018, Medium: ED, Size: 40 p.
- B. M. Cossairt, Shining light on indium phosphide quantum dots: Understanding the interplay among precursor conversion, nucleation, and growth, Chem. Mater., 2016, 28, 7181–7189 CrossRef CAS.
- M. R. Friedfeld, J. L. Stein, A. Ritchhart and B. M. Cossairt, Conversion reactions of atomically precise semiconductor clusters, Acc. Chem. Res., 2018, 51, 2803–2810 CrossRef CAS.
- Y. Kamei, Y. Shichibu and K. Konishi, Generation of small gold clusters with unique geometries through cluster-to-cluster transformations: Octanuclear clusters with edge-sharing gold tetrahedron motifs, Angew. Chem., Int. Ed., 2011, 50, 7442–7445 CrossRef CAS PubMed.
- L. Y. Yao, F. K. Hau and V. W. Yam, addition reaction-induced cluster-to-cluster rransformation: Controlled self-assembly of luminescent polynuclear gold(I) μ3-sulfido clusters, J. Am. Chem. Soc., 2014, 136, 10801–10806 CrossRef CAS PubMed.
- L. Y. Yao and V. W. Yam, Photoinduced isomerization-driven structural transformation between decanuclear and octadecanuclear gold(I) sulfido clusters, J. Am. Chem. Soc., 2015, 137, 3506–3509 CrossRef CAS PubMed.
- G. T. Xu, L. L. Wu, X. Y. Chang, T. W. H. Ang, W. Y. Wong, J. S. Huang and C. M. Che, Solvent-induced cluster-to-cluster transformation of homoleptic gold(I) thiolates between catenane and ring-in-ring structures, Angew. Chem., Int. Ed., 2019, 58, 16297–16306 CrossRef CAS.
- L. L. Yan, L. Y. Yao and V. W. Yam, Concentration- and solvation-induced reversible structural transformation and assembly of polynuclear gold(I) sulfido complexes, J. Am. Chem. Soc., 2020, 142, 11560–11568 CrossRef CAS.
- R. García-Rodríguez, M. P. Hendricks, B. M. Cossairt, H. Liu and J. S. Owen, Conversion reactions of cadmium chalcogenide nanocrystal precursors, Chem. Mater., 2013, 25, 1233–1249 CrossRef.
- Y. Yang, Y. Li, C. Luan, N. Rowell, S. Wang, C. Zhang, W. Huang, X. Chen and K. Yu, Transformation pathways in colloidal CdTeSe magic-size clusters, Angew. Chem., Int. Ed., 2021, 61, e202114551 Search PubMed.
- O. Yaghi, Z. Sun, D. E. Richardson and T. Groy, Directed transformation of molecules to solids: Synthesis of a microporous sulfide from molecular germanium sulfide cages, J. Am. Chem. Soc., 1994, 116, 807–808 CrossRef CAS.
- C. L. Bowes, W. U. Huynh, S. J. Kirkby, A. Malek, G. A. Ozin, S. Petrov, M. Twardowski and D. Young, Dimetal linked open frameworks: [(CH3)4N]2(Ag2,Cu2)Ge4S10, Chem. Mater., 1996, 8, 2147–2152 CrossRef CAS.
- J. Lin, Q. Zhang, L. Wang, X. Liu, W. Yan, T. Wu, X. Bu and P. Feng, Atomically precise doping of monomanganese ion into coreless supertetrahedral chalcogenide nanocluster inducing unusual red shift in Mn2+ emission, J. Am. Chem. Soc., 2014, 136, 4769–4779 CrossRef CAS PubMed.
- P. Vaqueiro and M. L. Romero, Gallium– sulfide supertetrahedral clusters as building blocks of covalent organic–inorganic networks, J. Am. Chem. Soc., 2008, 130, 9630–9631 CrossRef CAS.
- T. Wu, R. Khazhakyan, L. Wang, X. Bu, S. T. Zheng, V. Chau and P. Feng, Three-dimensional covalent co-assembly between inorganic supertetrahedral clusters and imidazolates, Angew. Chem., Int. Ed., 2011, 50, 2536–2539 CrossRef CAS.
- P. Vaqueiro and M. L. Romero, [Ga10S16(NC7H9)4]2−: A hybrid supertetrahedral nanocluster, Chem. Commun., 2007, 3282–3284 RSC.
- L. Sun, H. Y. Zhang, J. Zhang, Y. J. Jia, Y. Z. Yu, J. J. Hou, Y. X. Wang and X. M. Zhang, A quasi-D3-symmetrical metal chalcogenide cluster constructed by the corner-sharing of two T3 supertetrahedra, Dalton Trans., 2020, 49, 13958–13961 RSC.
- J. Wu, B. Jin, X. Wang, Y. Ding, X.-L. Wang, D. Tang, X. Li, J. Shu, D.-S. Li, Q. Lin, Y.-B. Wu and T. Wu, Breakdown of valence shell electron pair repulsion theory in an H-bond-stabilized linear sp-hybridized sulfur, CCS Chem., 2021, 3, 2584–2590 CrossRef CAS.
- Y.-P. Zhang, X. Zhang, W.-Q. Mu, W. Luo, G.-Q. Bian, Q.-Y. Zhu and J. Dai, Indium sulfide clusters integrated with 2,2′-bipyridine complexes, Dalton Trans., 2011, 40, 9746–9751 RSC.
- T. Wu, Q. Zhang, Y. Hou, L. Wang, C. Mao, S. T. Zheng, X. Bu and P. Feng, Monocopper doping in Cd–In–S supertetrahedral nanocluster via two-step strategy and enhanced photoelectric response, J. Am. Chem. Soc., 2013, 135, 10250–10253 CrossRef CAS PubMed.
- J.-N. Li, Y. Fu, L. Liu and Q.-X. Guo, First-principle predictions of basicity of organic amines and phosphines in acetonitrile, Tetrahedron, 2006, 62, 11801–11813 CrossRef CAS.
-
D. Margetic, Superbases for Organic Synthesis: Guanidines, Amidines, Phosphazenes and Related Organocatalysts, ed. T. Ishikawa, John Wiley & Sons, Ltd, 2009, ch. 2, p. 21 Search PubMed.
|
| This journal is © the Partner Organisations 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *c and
Xian-Ming
Zhang
*c and
Xian-Ming
Zhang