
Experimental and computational studies on rhodium-catalyzed C4(5)aryl–H activation/annulation of imidazoles with alkynes: facile synthesis of six types of N-heterocycles†
Ya-Nan
Tian
,
Shihai
Lv
,
Lingyu
Huang
,
Chaoying
Wen
,
Yanyan
Yang
,
Xiangfei
Kong
,
Qiping
Zhu
 * and
Shiqing
Li
*
* and
Shiqing
Li
*
Guangxi Key Laboratory of Electrochemical and Magneto-Chemical Functional Materials, College of Chemistry and Bioengineering, Guilin University of Technology, Guilin 541004, China. E-mail: lisq@glut.edu.cn
First published on 10th November 2022
Abstract
C–H annulations at N- and C2-aryls of an imidazole have been researched well, while the annulation on C4(5)-aryls especially the reactivity and site-selectivity among these aryls remains unknown. Herein, a molecular engineering strategy involving six reaction modes based on the rhodium-catalyzed C4(5)aryl–H activation/annulation of imidazoles with alkynes has been developed, giving diverse neutral/cationic N-heterocycles with broad scope (>60 examples) and high selectivity. More importantly, through a series of intramolecular competition experiments and DFT calculations, the reactivity of the peripheral C–H bonds has been studied and ranked for the first time: C2aryl–H > C4(5)aryl–H > C4(5)styryl–H/C2styryl–H > residual C4(5)aryl–H/C4(5)styryl–H. Furthermore, the remote bulky C2-subsituent is found to have a big influence on the regioselectivity of C4(5)aryl–H activation.
Introduction
Transition metal-catalyzed site-selective C–H bond activation that selectively provides a single product among other possible isomers/products is a holy grail in chemistry,1 and it is one of the ultimate pursuits in synthetic chemistry.2,3 Insight into the reactivity and selectivity among multiple reactive C–H bonds of a substrate can help in precisely designing more complex molecules by sequential C–H functionalization. One route depends on the innate electron density/pKa of different C–H bonds,4 with examples of molecular engineering of sequential C–H arylation of thiophene,5 thiazole,6 imidazole,7 and pyrazole8 for the programmed synthesis of multi-aryl substituted heterocycles. Another route to realizing site-selectivity depends on the introduction of a directing group (DG).9 However, the installation and removal of nonessential DGs are trivial and lead to poor atom and step economy. Recently, using transient DGs10 has been regarded as a step-economical way to assemble functional molecules without pre-installing and dismantling the DGs. Besides, C–H activation mediated by intrinsic DGs, which are already existing in the parent molecules, is also considered as an appealing route to functionalize the parent molecules at a late stage.11 The site-selectivity of more complex systems, such as a substrate containing two or more DGs with multiple competitive C–H bonds, is attractive and challenging but less reported.12Among the diverse N-heterocyclic intrinsic DGs such as pyridine, imidazole, pyrazole, and indole, imidazole is very interesting as it is an innate DG core with all the five atoms (N1, C2, N3, C4 and C5) prone to associate with a metal. For example, the C–H annulations of N1/3-aryl13 and C2-aryl14 imidazoles have been well developed (Fig. 1A, left). Particularly, lots of cationic C–H annulation reactions of N-aryl imidazolium salts have been disclosed by Choudhury15 and Wang.16 However, all these reports only discussed the mono- or/and di-annulation of the same benzene ring (without other competitive aryls) located at the N1, N3, or C2 position, just giving C2-substituted products, such as imidazo[2,1-a]isoquinolines. To the best of our knowledge, the C–H activation/annulation of C4(5)-aryl of an imidazole (Fig. 1A, right) has remained unknown, although it might confer complementary reactivity such as giving imidazo[5,1-a]isoquinolines, which has been widely applied to drugs,17 DNA affinity,18 nitrogen heterocyclic carbenes (NHCs),19 and fluorescent dyes20 (Fig. 1B). Furthermore, the site- and regioselectivity among the competitive peripheral C–H bonds in a more complicated imidazole-centred system, e.g. 1,4,5-trisubstituted imidazole, are interesting, yet they have not been disclosed. Herein, we unlock and rank the reactivity and site-selectivity of different C–H bonds on the C2-, C4-, and C5-groups of an imidazole based on the combination of experiments and density functional theory (DFT) calculations. We also find that the steric factor of a C2-subsituent has a large influence on the regioselectivity of unsymmetric C4(5)-aryls. By employing the site-selective groups and adjusting the ring-junction group (N1 or C2 blocks), derivatization and diversification of the imidazole core are presented and six reaction modes have been disclosed (Fig. 1C).
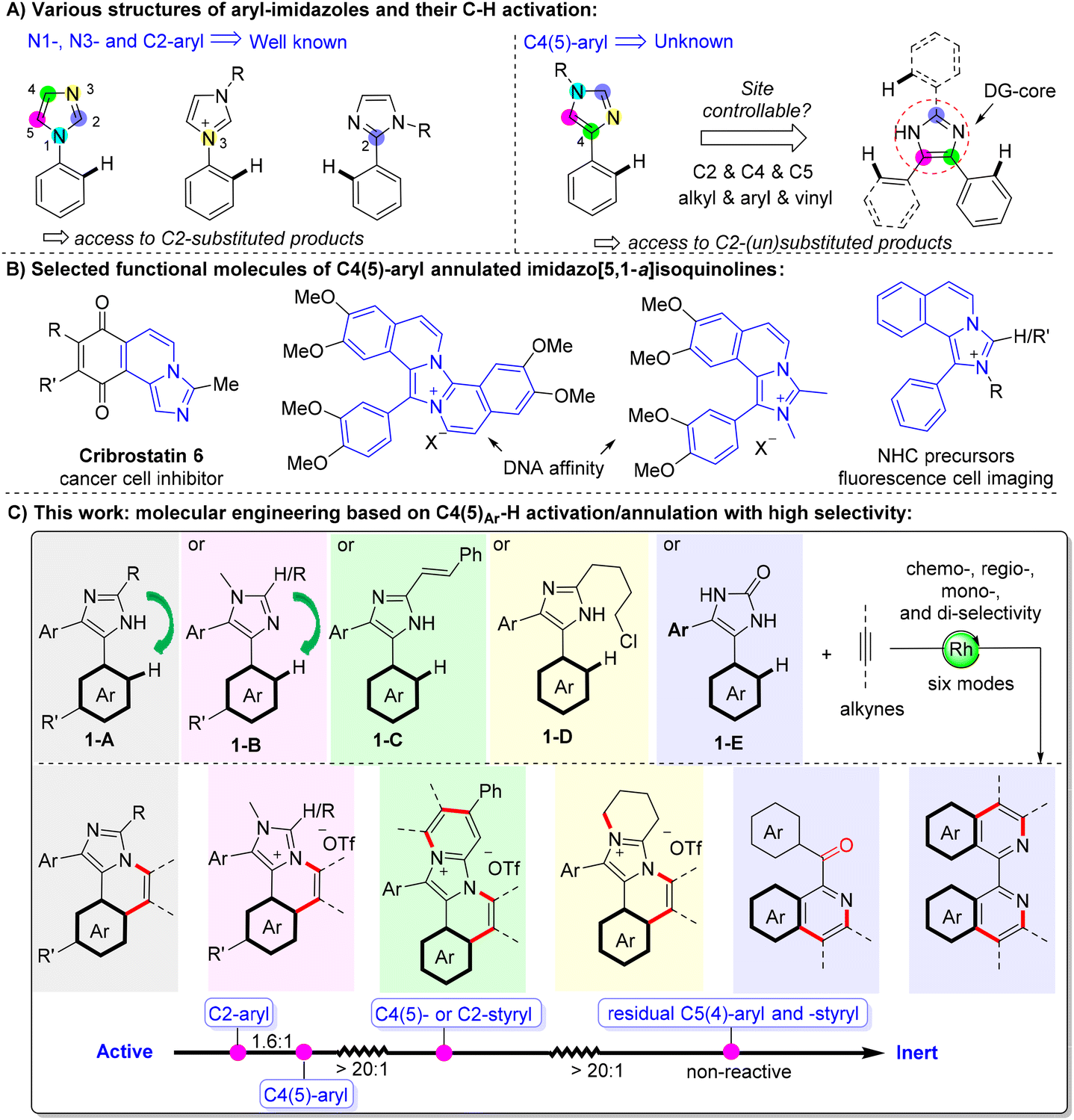 | ||
| Fig. 1 C–H activation/annulation of aryl imidazole. (A) Various structures of aryl imidazoles and their C–H activation. (B) Selected functional molecules of C4(5)-aryl annulated imidazo[5,1-a]isoquinolines. (C) This work: molecular engineering based on C4(5)Ar–H activation/annulation with high selectivity. | ||
Results and discussion
Initially, site-selectivity between C2alkyl–H and C4(5)aryl–H was firstly investigated by using 2-isopropyl-4,5-diphenyl-1H-imidazole (1a) and diphenylacetylene (2a) as standard reagents. After a systematical screening of the reaction conditions (see Table S1 in the ESI†), imidazo[5,1-a]isoquinoline 3a was exclusively generated in 93% yield in the presence of [Cp*RhCl2]2 (2.5 mol%) and Cu(OTf)2 (1 equiv.) in MeOH at 100 °C for 10 h under air (Scheme 1), without generating [5,6]-bicyclic 3a′,213a′′ and diannulated 3a′′′. The result did not change even if 2 equiv. of alkynes were used (Table S1,† entry 12), implying that C4- or C5-aryl is more reactive than C2-alkyl because of the acidity difference {pKa = 55.5 for C2alkyl–H and 51.9 for C4(5)aryl–H}. Meanwhile, tautomeric equilibrium in N–H imidazoles made the C4- and C5-positions equivalent, but the residual C4/5-aryl became very inert when N–H was annulated, which may be attributed to the steric hindrance of the two internal C–H bonds (bay position).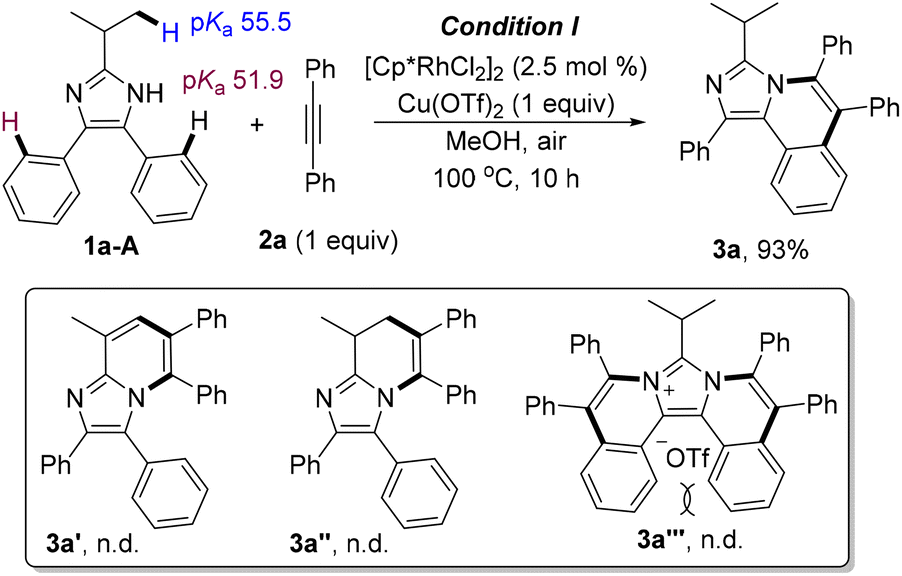 | ||
| Scheme 1 C4(5)–H annulation of 2-isopropyl-4,5-diphenyl-1H-imidazole. | ||
Subsequently, the scope of the annulation of 4,5-diaryl imidazoles 1-A and alkynes was explored (Scheme 2, upper). Both alkyl and aryl alkynes were compatible, giving imidazo[5,1-a]isoquinoline derivatives 3a–g in good to high yields. Thienyl-substituted alkyne was also tolerated albeit with a low yield of 35% (3h). The symmetric substrates with two same substituents respectively at the C4 and C5 phenyl positions underwent the C–H annulation smoothly, giving 3i–l in 59–88% yields. 4,5-Dithiophenyl imidazole was well tolerated to generate 3m in 82% yield. The C2 groups with examples of n-propyl (3n), cyclohexyl (3o) and pentafluorophenyl (3p) were untouched in this protocol. The structure of 3o was further determined by single-crystal X-ray diffraction.22 The reaction of 2-isopropyl-4-methyl-5-phenyl-1H-imidazole 1j-A and 2a gave product 3q in a moderate yield by using Cu(OAc)2·H2O as the oxidant and acetone as the solvent. Treatment of unsymmetric imidazole 1k-A with 2a afforded two isolatable isomers 3r and 3r′ in 40% and 48% yields, respectively. Imidazole 1l-A with two meta-OMe reacted with 2a to give a couple of isomers (3s/3s′ = 10/1) in 95% yield with a good selectivity, where the rhodium catalyst was prone to attack the less hindered C–H bond. The major structure of 3s was determined by single-crystal X-ray diffraction.22 The annulation of 2-isopropyl-1H-phenanthro[9,10-d]imidazole and 2a gave a large-size rigid molecule 3t in 90% yield.
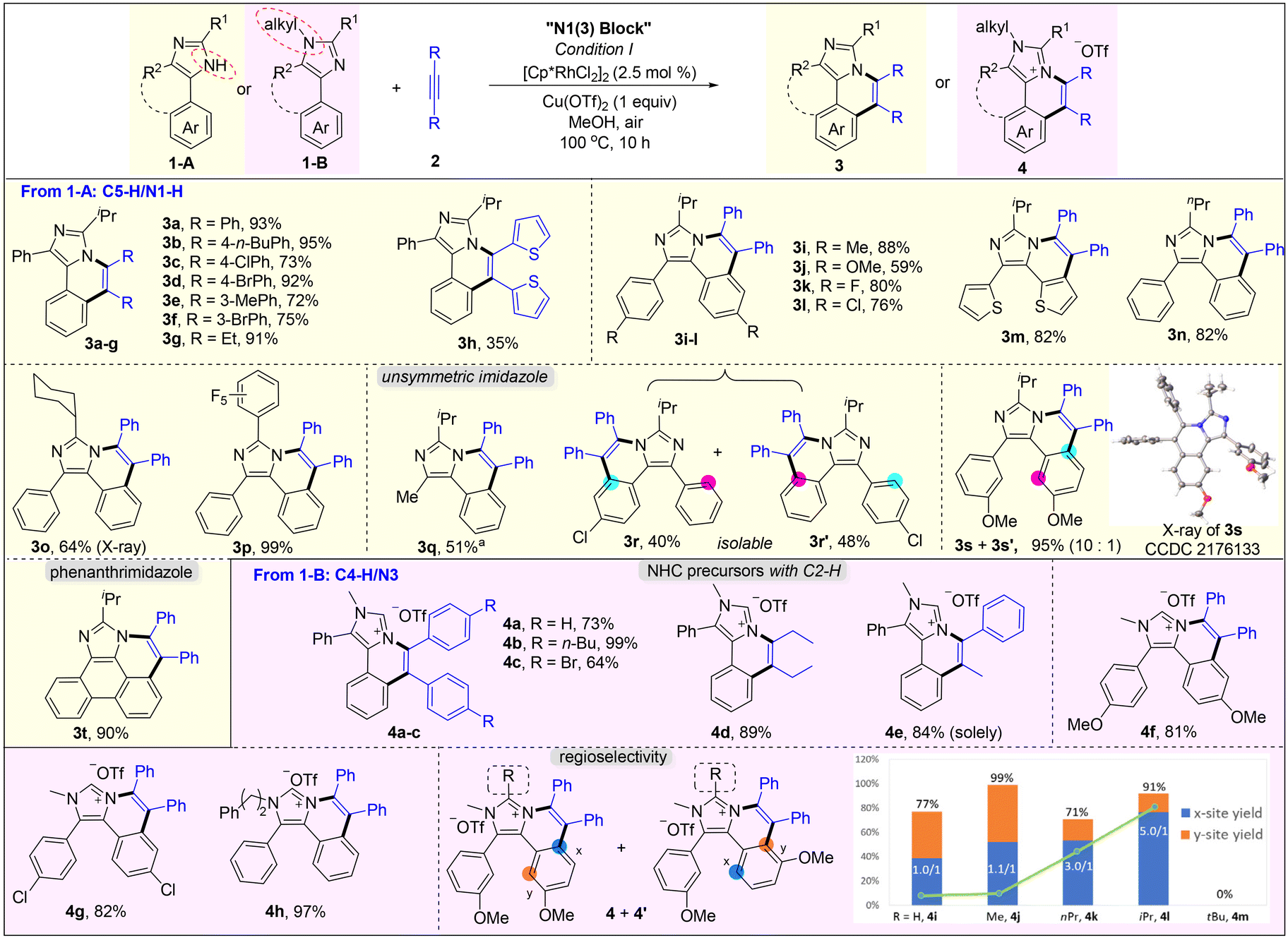 | ||
Scheme 2 N1 block. Conditions I: imidazole 1 (0.1 mmol), alkyne 2 (0.1 mmol), [RhCp*Cl2]2 (2.5 mol%), and Cu(OTf)2 (1 equiv.) in MeOH (1 mL) at 100 °C for 10 h under air, isolated yield. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Cu(OAc)2·H2O (2.2 equiv.) as the oxidant and acetone (1 mL) as the solvent under a N2 atmosphere. Cu(OAc)2·H2O (2.2 equiv.) as the oxidant and acetone (1 mL) as the solvent under a N2 atmosphere. | ||
When using N-substituted 4,5-diaryl imidazoles 1-B to react with alkynes, the C–H annulation occurred on C4-aryl, giving cationic imidazo[5,1-a]isoquinolines (Scheme 2, lower). All aryl–aryl, alkyl–alkyl, and aryl–alkyl internal alkynes were compatible and furnished the corresponding salts 4a–e in good to quantitative yields. 1-Phenyl-1-propyne reacted with 2a selectively to offer 4e as a sole product in 84% yield. Varying the substituent at the C4/C5 aryl and the N1 positions delivered products 4f–h in 81–97% yields. Notably, our method provides a significantly simple route to novel non-symmetric N-heterocyclic carbene (NHC) precursors (4a–h) with naked C2–H, which cannot be obtained from previous imidazole-based C–H annulations.13–16 Also, they are potential ligands that can bond with DNA.18 The unsatisfied selectivity between the two ortho positions in unsymmetric meta-substituted reagents has been the long-standing problem of rhodium-catalyzed C–H annulation and the solutions are unknown. Kanai and Matsunaga et al. handled this problem well through Co-catalysis.23 Herein, we found that the regioselectivity was influenced by the remote bulky C2-substituent of imidazole. Imidazole 1e-B bearing methoxy at the meta-position reacted with 2a to give a mixture of 4i and 4i′ in 77% yield with a ratio of 1.0. When changing the C2 group from H to Me, nPr, and iPr, the regio-selective ratio increased to 1.1, 3.0 and 5.0, respectively. Upon further enhancing the steric hindrance to tBu, however, the C–H annulations ceased which is perhaps due to the major obstacle in the process of alkyne insertion.
Next, some imidazoles (1-C and 1-D) with additional reactive sites on C2 groups have been designed and prepared to expand the synthetic utility of this methodology (Scheme 3). The reaction of 2-(4-chlorobutyl)-4,5-diaryl-1H-imidazoles (1-C) and alkynes afforded the title imidazo[5,1-a]isoquinolinium salts 5a–e in 33–77% yields, through Rh(III)-catalyzed C5aryl–H activation/annulation and subsequent intramolecular N-quaternization. Upon using a styryl (1-D) instead of chlorobutyl, C4(5)aryl–H activation/annulation and sequential C2vinyl–H activation/annulation proceeded smoothly to obtain ladder-type polycyclic aromatic hydrocarbons (PHAs) 6a–h in moderate to high yields with a broad substrate scope. When employing 4,5-diphenyl-1,3-dihydro-2H-imidazol-2-one (1-E) as the substrate, interestingly, a novel ring-opening C–H annulation reaction occurred to give isoquinolines (7a and 7b) in moderate yields under conditions III, in which the carbonyl may be formed from the hydrolysis of imine.24 Gratifyingly, by treatment of 1-E with 2 molecules of alkyne, 1,1′-bisisoquinolines 8a and 8b could be efficiently obtained in fair yields through an unprecedented ring-opening, decarbonylative double C–H annulation, which serves as a complementary method to our previous work.25 These results revealed that both the C4 and C5 aryls can be activated when the imidazole ring is destroyed.
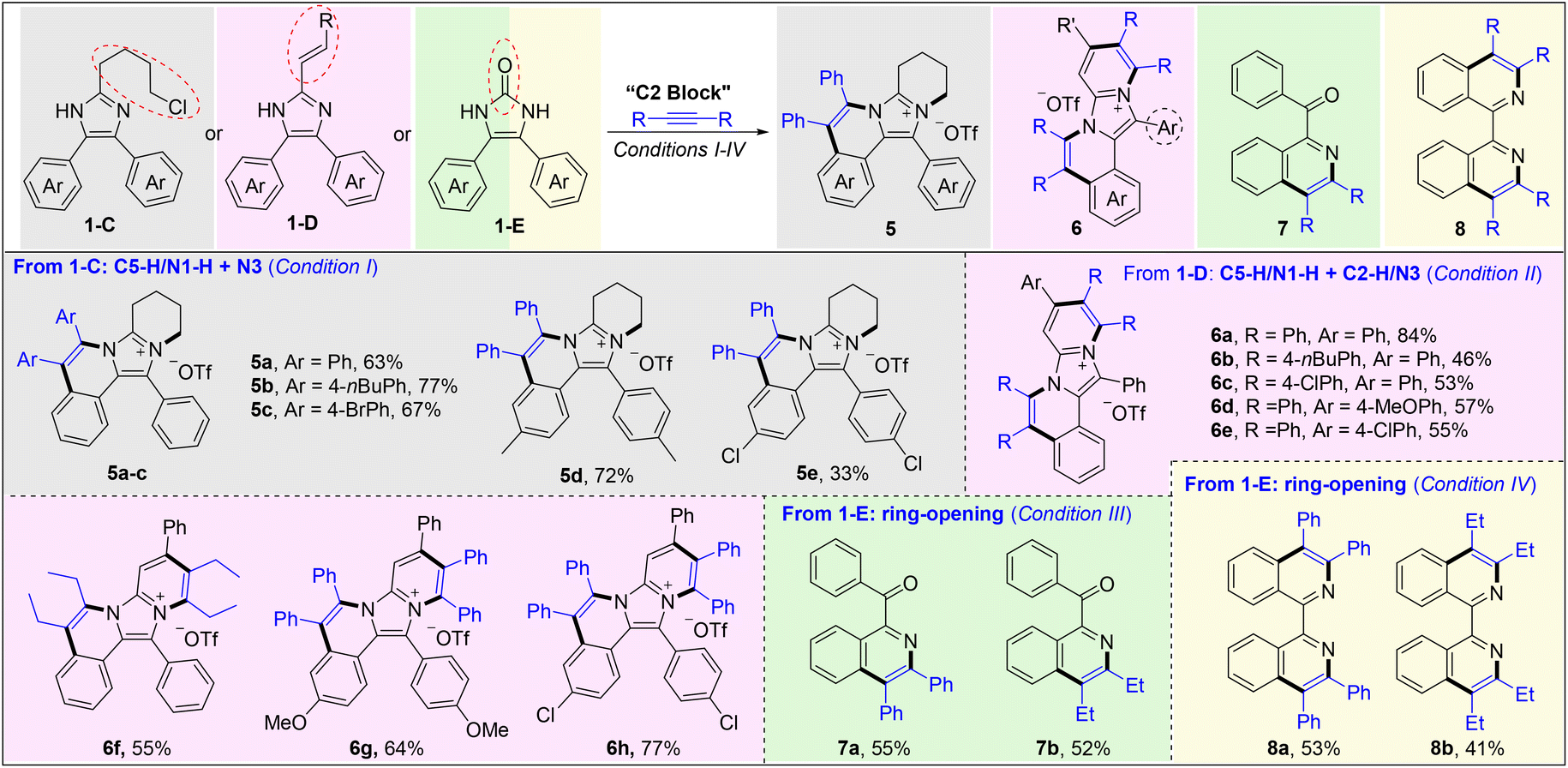 | ||
| Scheme 3 C2 block. Conditions I: imidazole 1 (0.1 mmol), alkyne 2 (0.1 mmol), [RhCp*Cl2]2 (2.5 mol%), and Cu(OTf)2 (1 equiv.) in MeOH (1 mL) at 100 °C for 10 h under air, isolated yield. Conditions II: imidazole 1-D (0.1 mmol), alkyne 2 (0.2 mmol), [RhCp*Cl2]2 (2.5 mol%), and Cu(OTf)2 (2 equiv.) in MeOH (1 mL) at 100 °C for 10 h under air. Conditions III: imidazole 1-E (0.1 mmol), alkyne 2 (0.1 mmol), [RhCp*Cl2]2 (2.5 mol%), and Cu(OAc)2·H2O (2.2 equiv.) in acetone (0.5 mL) at 60 °C for 12 h under air. Conditions IV: imidazole 1-E (0.1 mmol), alkyne 2 (0.2 mmol), [RhCp*Cl2]2 (2.5 mol%), Cu(OAc)2·H2O (1 equiv.) and MesCOOH (0.6 equiv.) in toluene (0.5 mL) at 120 °C for 12 h under air. | ||
To gain insights into the reaction mechanism, we conducted some deuterium-labeling experiments for all the five types of substrates 1-A to 1-E. Low H/D scrambling (<7% D) at the ortho-positions of 1a-A/B/C indicates that the C–H activation may be irreversible for these three types of reactions (Fig. 2A). A significant H/D scrambling, 54% D for C4(5)-aryl & 27% D for C2-vinyl, was observed on 1a-D suggesting that the initial C4(5)aryl–H and C2vinyl–H activation are reversible. While no H/D scrambling was occurred on compound 10, indicating that the second C4(5)aryl–H or C2vinyl–H activation is irreversible (Fig. 2B). The ortho-positions of 1-E showed 37% and 75% deuterium incorporation, respectively, under conditions III and IV, revealing a reversible C–H activation process in the imidazole ring-opening reactions. The kinetic isotope effect (KIE) values of 4/1 and 2/1 were measured for competition and parallel competition experiments, respectively, by using substrates 1a-A and/or d10-1a-A with 2a under conditions I, implying that the C–H cleavage is the rate-determining step (Fig. 2C). To our delight, a cyclorhodium intermediate Rh-NH with a free N–H bond which is obviously observed by 1H NMR with a chemical shift at 13.93 ppm was easily obtained through filtration in the reaction of 1a-D and [Cp*RhCl2]2 (Fig. 2D), while complex Rh-N was not detected. A satisfactory yield was obtained in the reaction of 1a-D and 2a by using Rh-NH as the catalyst (Fig. 2E). This result suggested that the C–H activation proceeded prior to N–H activation, which is contrary to the previous work.14d,26
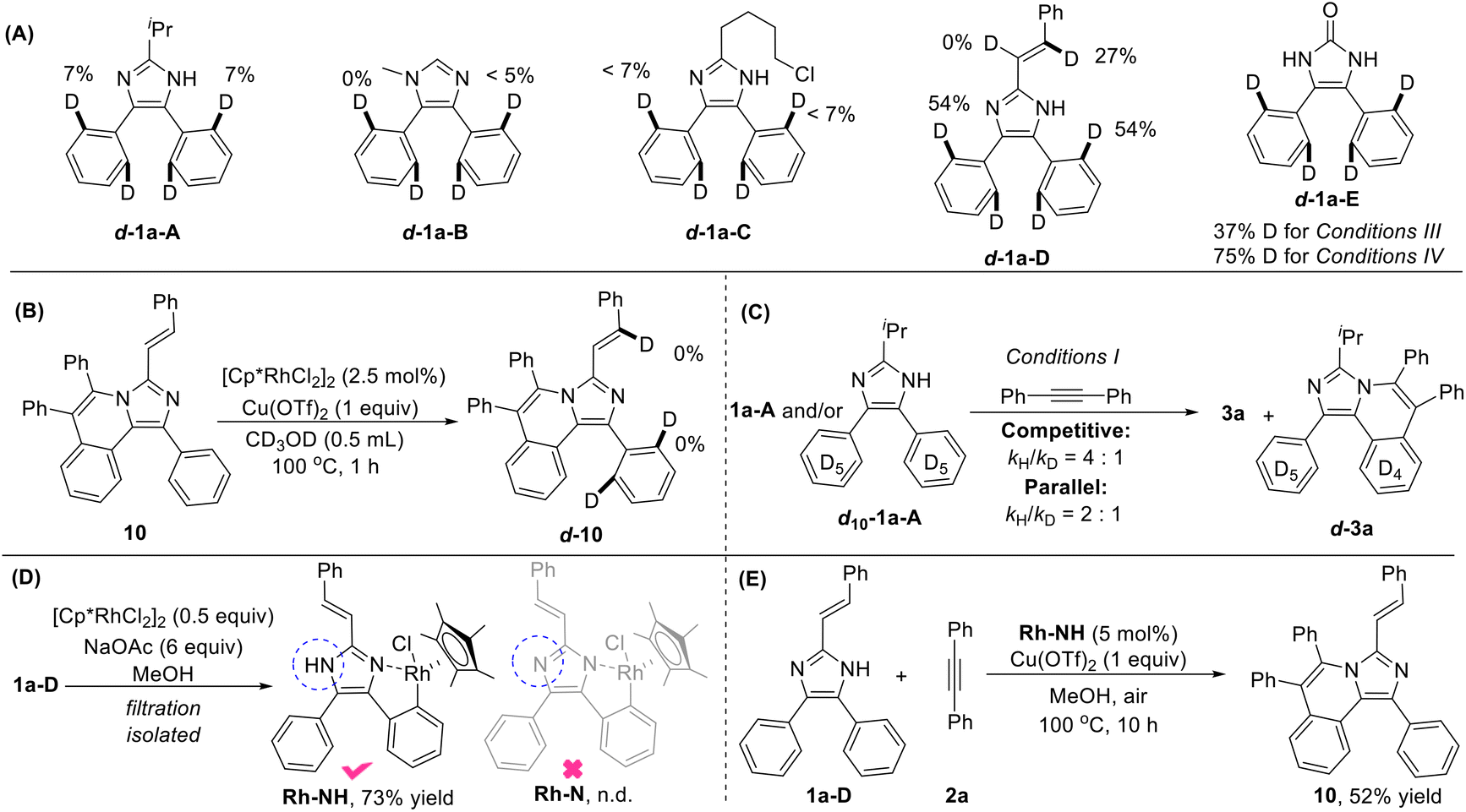 | ||
| Fig. 2 (A) H/D exchange experiments of substrates 1a-A to E. (B) H/D exchange experiment of 10. (C) Kinetic isotope effect (KIE). (D) Synthesis of complex Rh-NH. (E) Reaction of 1a-D and 1a using Rh-NH as the catalyst. | ||
Finally, we turned our attention to unlock the reactivity and selectivity among the multiple C–H bonds located at the C2, C4, and C5 positions through intramolecular competition experiments (Fig. 3). The reactivity between C4(5)-aryl and C2-vinyl was initially investigated by using 4,5-diphenyl-2-styryl imidazole 1a-D and 1 equiv. of 2a under conditions I, giving C4-annulated 10 as the single product in 76% yield without C2-annulated 10′ being detected (Fig. 3A). The reaction of compound 10 and hex-3-yne generated 11 solely in 73% yield, indicating that the reactivity of C2styryl–H is much higher than that of residual C4(5)aryl–H (Fig. 3B). The reaction of 2,4,5-triaryl imidazole 12 with 1 equiv. of 2a afforded C4(5)-annulated 13 and C2-annulated 13′ in 25% and 40% yields, respectively, showing a bit higher reactivity of C2-aryl than that of C4(5)-aryl with a ratio of 1/1.6 (Fig. 3C). Interestingly, 13 and 13′ exhibit significantly different fluorescence properties, with photoluminescence peaking at 498 and 447 nm, respectively. Upon increasing 2a to 2 equiv., both 13 and 13′ could transform into a diannulated salt 14 which was an analogue of the DNA affinity agent.18 The competition experiment of C4-vinyl and C5-aryl in imidazole 15 generated compound 16 as the sole product with a yield of 50%, showing a higher reactivity of C4(5)-aryl than that of C4(5)-vinyl (Fig. 3D). Based on the above results, a comprehensive ranking of the reactivity of the peripheral C–H bonds is C2aryl–H > C4(5)aryl–H > C4(5)styryl–H/C2styryl–H > residual C4(5)aryl–H/C4(5)styryl–H. It is noted that the residual C4(5) aryl and vinyl became very inert (Fig. 3E).
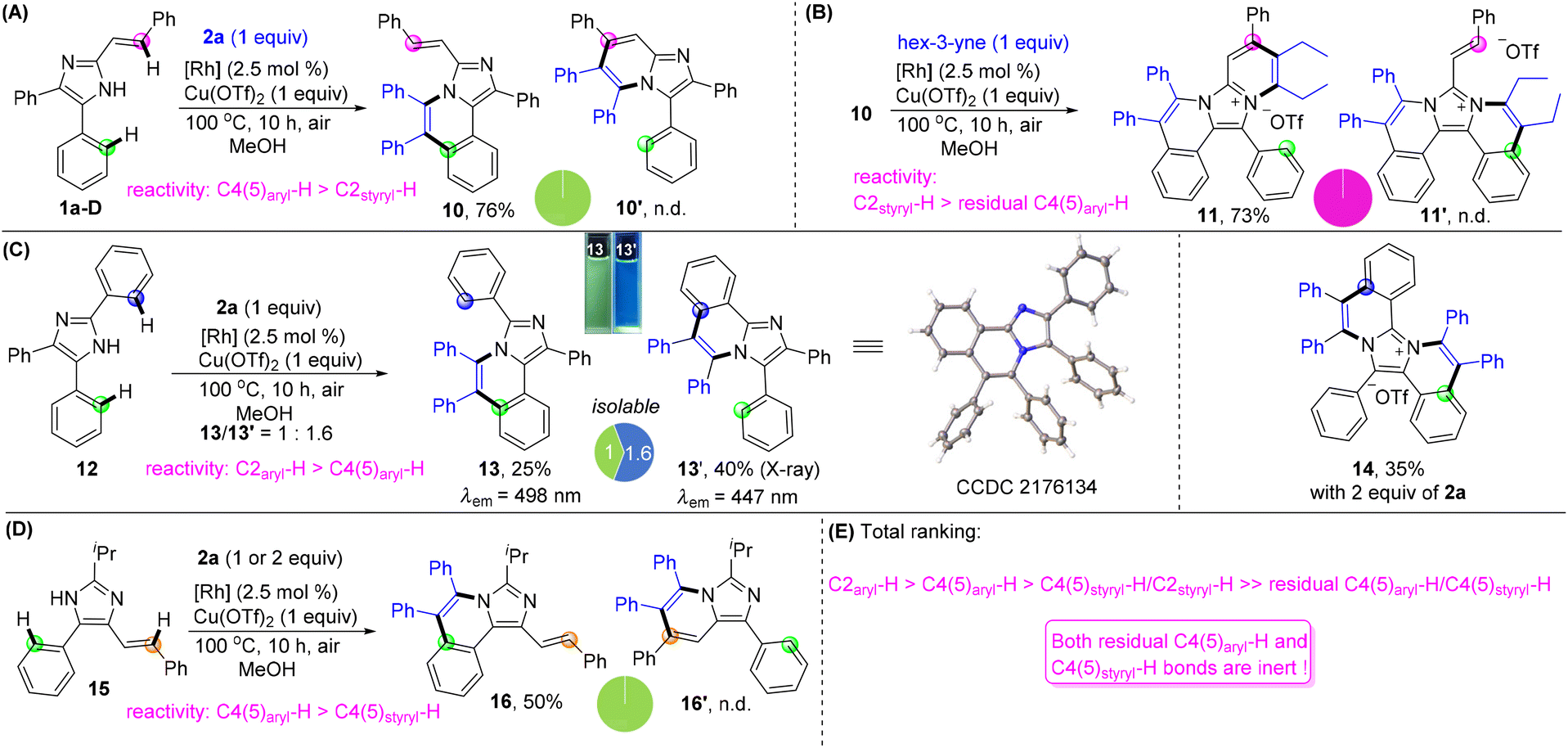 | ||
| Fig. 3 Studies on reactivity and selectivity. (A) C4(5)-aryl versus C2-styryl. (B) C2-styryl versus residual C4(5)-aryl. (C) C4(5)-aryl versus C2-aryl. Inset: Fluorescence photos of 13 and 13′ in CH2Cl2 under a UV lamp (365 nm). (D) C4(5)-aryl versus C4(5)-styryl. (E) Total reactive ranking of peripheral C–H bonds. | ||
To shed light on the origin of different C–H activities, the pKa value of the C–H bond for substrates 1a-D, 10, 12 and 15 is calculated (Fig. 4A) based on density functional theory (DFT). For the substituted group connected to the second position of imidazole (1a-D, 10 and 12), the pKa value of the C2–H bond is 44.6, 47.1 and 48.0 for 1a-D, 10 and 12, respectively, and is lower than the pKa value of the C4(5)–H bond (52.1, 54.7 and 51.6 for 1a-D, 10 and 12, respectively). This difference can be attributed to the stronger inductive effect since the C2 carbon atom is directly connected to two nitrogen atoms of imidazole, leading to a lower pKa value. For 15, the inductive effect from imidazole on the generation of phenyl and styryl groups is similar, and the pKa values are 49.4 and 48.1 for the C4(5)aryl–H bond and C4(5)styryl–H bond, respectively. This is due to the fact that styrene and imidazole are coplanar, which enhances the transmission of the inductive effect and results in lower pKa of the C–H bond in the styryl group compared with the C–H bond in the aryl group. Thus, compounds 10 and 12 exhibited pKa-driven activation behavior, while 1a-D and 15 did not.
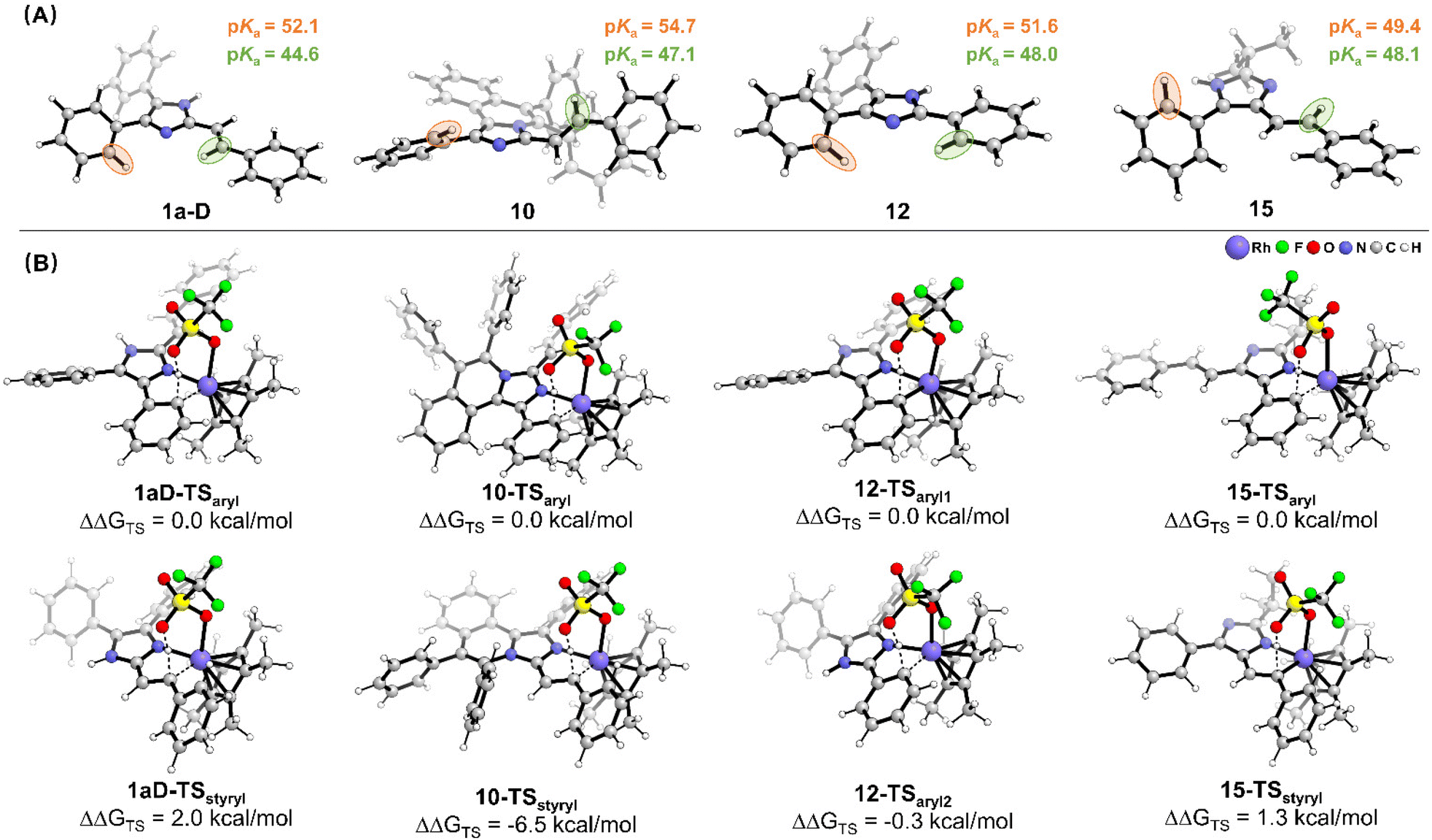 | ||
| Fig. 4 (A) The DFT optimized structures of the substrate with the corresponding pKa value of key C–H bonds. (B) The C–H activation transition states with the corresponding relative free energy. | ||
Considering that the interaction between the substrate and the catalyst during C–H activation will bring about a deformation of the substrate molecular structure, it will affect the kinetics of the reaction. Therefore, the C–H activation transition states and corresponding relative energies were calculated. As shown in Fig. 4B, the relative energy of Cstyryl–H activation transition states is 2.0 kcal mol−1 and 1.3 kcal mol−1 higher than that of Caryl–H for 1a-D and 15, respectively, which is in good agreement with the experimental observed reactivity. Despite the lower pKa value of the C2styryl–H bond, the imidazole ligand and the styryl group form a stable conjugated system, which requires overcoming a higher activation energy barrier for C–H activation. In this case, the chemoselectivity is dominated by the conjugation effect. For 10, due to the repulsion of adjacent phenyl groups, the styryl group and the imidazole cannot form a stable conjugate system. Thus, the C–H activation prefers to occur at the lower pKa C2styryl–H bond with a significant difference in the relative energy (10-TSstyryl: ΔΔGTS = −6.5 kcal mol−1). In this case, the chemoselectivity is dominated by the inductive effect. As for 12, although the phenyl group at the second position is conjugated to the imidazole, the slight single bond rotation of the phenyl group in the C–H activation transition state does not result in a higher energy barrier. Judging from the relative energies of the two transition states (12-TSaryl2: ΔΔGTS = −0.3 kcal mol−1), the reaction chemoselectivity continues to be dominated by the inductive effect.
Conclusions
In conclusion, we have developed a molecular engineering strategy based on unprecedented rhodium-catalyzed C4(5)aryl–H activation/annulation to construct six types of N-heterocycles with broad substrate scope (>60 examples) just by simply altering the N1- and C2-substituents. The systematic competition experiments and DFT calculations indicate, for the first time, the order of the reactivity of peripheral C–H bonds: C2aryl–H > C4(5)aryl–H > C4(5)styryl–H/C2styryl–H > residual C4(5)aryl–H/C4(5)styryl–H. The reactivity and site-selectivity are determined by a combination of inductive and conjugation effects. The residual C4(5)-aryl can be activated only if the imidazole ring is destroyed, which is exemplified as an efficient way to assemble 1,1′-bisisoquinolines via uncommon decarbonylative double C–H annulation. Furthermore, we find that the remote bulky C2-group has a significant effect on the regioselectivity of C4(5)aryl–H activation. The intrinsic reactivity and selectivity revealed in this paper can provide guidance in designing and synthesizing novel and more complicated imidazole-based optoelectronic materials and drugs, expanding the current imidazole chemistry. Detailed mechanistic studies and further application of these imidazole molecules are currently ongoing in our laboratory.Author contributions
Conceptualization: S. L. Methodology: Y.-N. T. Mechanistic research: S. L. and L. H. Fluorescence test: Y. Y. and C. W. DFT calculations: X. K. and Q. Z. Writing: Y.-N. T. and S. L.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (22001049, and 22261013), the Guangxi Natural Science Foundation (2020GXNSFBA297003, AD22035990, and AD19245095), and the Key Laboratory of Electrochemical and Magneto-Chemical Functional Materials (EMFM20181108).Notes and references
- J. F. Hartwig, Catalyst-Controlled Site-Selective Bond Activation, Acc. Chem. Res., 2017, 50, 549–555 CrossRef CAS PubMed.
- Z. Huang and G. Dong, Site-Selectivity Control in Organic Reactions: A Quest to Differentiate Reactivity among the Same Kind of Functional Groups, Acc. Chem. Res., 2017, 50, 465–471 CrossRef CAS PubMed.
- K. I. Goldberg and A. S. Goldman, Large-Scale Selective Functionalization of Alkanes, Acc. Chem. Res., 2017, 50, 620–626 CrossRef CAS.
- J. C. Lewis, R. G. Bergman and J. A. Ellman, Direct Functionalization of Nitrogen Heterocycles via Rh-Catalyzed C–H Bond Activation, Acc. Chem. Res., 2008, 41, 1013–1025 CrossRef CAS.
- S. Yanagisawa, K. Ueda, H. Sekizawa and K. Itami, Programmed Synthesis of Tetraarylthiophenes through Sequential C-H Arylation, J. Am. Chem. Soc., 2009, 131, 14622–14623 CrossRef CAS PubMed.
- (a) L.-C. Campeau, M. Bertrand-Laperle, J.-P. Leclerc, E. Villemure, S. Gorelsky and K. Fagnou, C2, C5, and C4 Azole N-Oxide Direct Arylation Including Room-Temperature Reactions, J. Am. Chem. Soc., 2008, 130, 3276–3277 CrossRef CAS; (b) L.-C. Campeau, D. R. Stuart, J.-P. Leclerc, M. Bertrand-Laperle, E. Villemure, H.-Y. Sun, S. Lasserre, N. Guimond, M. Lecavallier and K. Fagnou, Palladium-Catalyzed Direct Arylation of Azine and Azole N-Oxides: Reaction Development, Scope and Applications in Synthesis, J. Am. Chem. Soc., 2009, 131, 3291–3306 CrossRef CAS.
- J. M. Joo, B. B. Touré and D. Sames, C–H Bonds as Ubiquitous Functionality: A General Approach to Complex Arylated Imidazoles via Regioselective Sequential Arylation of All Three C-H Bonds and Regioselective N-Alkylation Enabled by SEM-Group Transposition, J. Org. Chem., 2010, 75, 4911–4920 CrossRef CAS PubMed.
- R. Goikhman, T. L. Jacques and D. Sames, C–H Bonds as Ubiquitous Functionality: A General Approach to Complex Arylated Pyrazoles via Sequential Regioselective C-Arylation and N-Alkylation Enabled by SEM-Group Transposition, J. Am. Chem. Soc., 2009, 131, 3042–3048 CrossRef CAS.
- (a) J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Towards Mild Metal-Catalyzed C–H Bond Activation, Chem. Soc. Rev., 2011, 40, 4740–4761 RSC; (b) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Ruthenium(II)-Catalyzed C–H Bond Activation and Functionalization, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS PubMed; (c) J. Lv, Q. Zhang, M. Cai, Y. Han and S. Luo, Aromatic Aminocatalysis, Chem. – Asian J., 2018, 13, 740–753 CrossRef CAS PubMed; (d) C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, A Comprehensive Overview of Directing Groups Applied in Metal-Catalysed C–H Functionalization Chemistry, Chem. Soc. Rev., 2018, 47, 6603–6743 RSC; (e) O. T. O'Sullivan and M. J. Zdilla, Properties and Promise of Catenated Nitrogen Systems As High-Energy-Density Materials, Chem. Rev., 2020, 120, 5682–5744 CrossRef.
- (a) H. Sun, N. Guimond and Y. Huang, Advances in the Development of Catalytic Tethering Directing Groups for C–H Functionalization Reactions, Org. Biomol. Chem., 2016, 14, 8389–8397 RSC; (b) Q. Zhao, T. Poisson, X. Pannecoucke and T. Besset, The Transient Directing Group Strategy: A New Trend in Transition-Metal-Catalyzed C–H Bond Functionalization, Synthesis, 2017, 4808–4826 CAS; (c) P. Gandeepan and L. Ackermann, Transient Directing Groups for Transformative C–H Activation by Synergistic Metal Catalysis, Chem, 2018, 4, 199–222 CrossRef CAS; (d) S. S. John-Campbell and J. A. Bull, Transient Imines as ‘Next Generation’ Directing Groups for the Catalytic Functionalisation of C–H Bonds in a Single Operation, Org. Biomol. Chem., 2018, 16, 4582–4595 RSC; (e) J. I. Higham and J. A. Bull, Transient Imine Directing Groups for the C–H Functionalisation of Aldehydes, Ketones and Amines: An Update 2018–2020, Org. Biomol. Chem., 2020, 18, 7291–7315 RSC; (f) Y. Wu and B. Shi, Transition Metal-Catalyzed C-H Activation via Imine-Based Transient Directing Group Strategy, Chin. J. Org. Chem., 2020, 40, 3517–3535 CrossRef CAS; (g) G. Liao, T. Zhang, Z.-K. Lin and B. F. Shi, Transition Metal-Catalyzed Enantioselective C–H Functionalization via Chiral Transient Directing Group Strategies, Angew. Chem., Int. Ed., 2020, 59, 19773–19786 CrossRef CAS; (h) C. Jacob, B. U. W. Maes and G. Evano, Transient Directing Groups in Metal-Organic Cooperative Catalysis, Chem. – Eur. J., 2021, 27, 13899–13952 CrossRef CAS PubMed.
- L. Zheng and R. Hua, C–H Activation and Alkyne Annulation via Automatic or Intrinsic Directing Groups: Towards High Step Economy, Chem. Rec., 2018, 18, 556–569 CrossRef CAS PubMed.
- (a) H.-X. Dai, A. F. Stepan, M. S. Plummer, Y.-H. Zhang and J.-Q. Yu, Divergent C–H Functionalizations Directed by Sulfonamide Pharmacophores: Late-Stage Diversification as a Tool for Drug Discovery, J. Am. Chem. Soc., 2011, 133, 7222–7228 CrossRef CAS PubMed; (b) Y.-J. Liu, H. Xu, W.-J. Kong, M. Shang, H.-X. Dai and J.-Q. Yu, Overcoming the limitations of Directed C–H Functionalizations of Heterocycles, Nature, 2014, 515, 389–393 CrossRef CAS PubMed; (c) H. Wang, M. M. Lorion and L. Ackermann, Overcoming the Limitations of C–H Activation with Strongly Coordinating N-Heterocycles by Cobalt Catalysis, Angew. Chem., Int. Ed., 2016, 55, 10386–10390 CrossRef CAS PubMed.
- (a) J.-R. Huang, L. Dong, B. Han, C. Peng and Y.-C. Chen, Synthesis of Aza-Fused Polycyclic Quinolines via Double C–H Bond Activation, Chem. – Eur. J., 2012, 18, 8896–8900 CrossRef CAS PubMed; (b) J.-R. Huang, Q.-R. Zhang, C.-H. Qu, X.-H. Sun, L. Dong and Y.-C. Chen, Rhodium(III)-Catalyzed Direct Selective C(5)–H Oxidative Annulations of 2-Substituted Imidazoles and Alkynes by Double C–H Activation, Org. Lett., 2013, 15, 1878–1881 CrossRef CAS; (c) Q. Ge, B. Lia and B. Wang, Synthesis of Substituted Benzo[ij]Imidazo[2,1,5-de]-Quinolizine by Rhodium(III)-Catalyzed Multiple C–H Activation and Annulations, Org. Biomol. Chem., 2016, 14, 1814–1821 RSC; (d) Z. Liu, S. Guo, P. Wang, Z. Yan and T. Mu, Oxidative Annulations via Double C–H Bond Cleavages: Approach to Quinoline Derivatives, Appl. Organomet. Chem., 2021, 35, e6156 CAS.
- (a) L. Zheng and R. Hua, Modular Assembly of Ring-Fused and π-Extended Phenanthroimidazoles via C–H Activation and Alkyne Annulation, J. Org. Chem., 2014, 79, 3930–3936 CrossRef CAS PubMed; (b) S. Karthik, J. Ajantha, C. M. Nagaraja, S. Easwaramoorthi and T. Gandhi, Synthesis and Photophysics of Extended π-Conjugated Systems of Substituted 10-Aryl-Pyrenoimidazoles, Org. Biomol. Chem., 2016, 14, 10255–10266 RSC; (c) J. M. Villar, J. Suárez, J. A. Varela and C. Saá, N-Doped Cationic PAHs by Rh(III)-Catalyzed Double C–H Activation and Annulation of 2-Arylbenzimidazoles with Alkynes, Org. Lett., 2017, 19, 1702–1705 CrossRef CAS; (d) P. K. Dutta and S. Sen, (Benz)Imidazole-Directed Cobalt(III)-Catalyzed C–H Activation of Arenes: A Facile Strategy to Access Polyheteroarenes by Oxidative Annulation, Eur. J. Org. Chem., 2018, 5512–5519 CrossRef CAS.
- (a) D. Ghorai and J. Choudhury, Exploring a Unique Reactivity of N-Heterocyclic Carbenes (NHC) in Rhodium(III)-Catalyzed Intermolecular C–H Activation/Annulation, Chem. Commun., 2014, 50, 15159–15162 RSC; (b) D. Ghorai and J. Choudhury, Rhodium(III)–N-Heterocyclic Carbene-Driven Cascade C–H Activation Catalysis, ACS Catal., 2015, 5, 2692–2696 CrossRef CAS; (c) R. Thenarukandiyil and J. Choudhury, Rhodium(III)-Catalyzed Activation and Functionalization of Pyridine C–H Bond by Exploring a Unique Double Role of “N-Heterocyclic Carbene–Pyridyl” Ligand Platform, Organometallics, 2015, 34, 1890–1897 CrossRef CAS; (d) D. Ghorai, C. Dutta and J. Choudhury, Switching of “Rollover Pathway” in Rhodium(III)-Catalyzed C–H Activation of Chelating Molecules, ACS Catal., 2016, 6, 709–713 CrossRef CAS; (e) R. Thenarukandiyil, H. Thrikkykkal and J. Choudhury, Rhodium(III)-Catalyzed Nonaromatic sp2 C-H Activation/Annulation Using NHC as a Directing and Functionalizable Group, Organometallics, 2016, 35, 3007–3013 CrossRef CAS; (f) R. Thenarukandiyil, C. Dutta and J. Choudhury, Switching of Reaction Pathway from C–C Rollover to C-N Ring-Extension Annulation, Chem. – Eur. J., 2017, 23, 15529–15533 CrossRef CAS; (g) C. Dutta, S. S. Rana and J. Choudhury, Leveraging Metallotropism-Enabled Substrate Activation in Cobalt-Catalyzed Annulation Chemistry: Protic NHC Template Is the Key, ACS Catal., 2019, 9, 10674–10679 CrossRef CAS; (h) P. Karak, C. Dutta, T. Dutta, A. L. Koner and J. Choudhury, Orchestrated Catalytic Double Rollover Annulation: Rapid Access to N-Enriched Cationic and Neutral PAHs, Chem. Commun., 2019, 55, 6791–6794 RSC.
- (a) Q. Ge, B. Li, H. Song and B. Wang, Rhodium(III)-Catalyzed Cascade Oxidative Annulation Reactions of Aryl Imidazolium Salts with Alkynes Involving Multiple C–H Bond Activation, Org. Biomol. Chem., 2015, 13, 7695–7710 RSC; (b) R. Li, Y. Hu, R. Liu, R. Hu, B. Li and B. Wang, Ruthenium(II)-Catalyzed Oxidative Annulation Reactions of Arylimidazolium Salts via N-Heterocyclic Carbene-Directed C-H Activation, Adv. Synth. Catal., 2015, 357, 3885–3892 CrossRef CAS.
- (a) M. T. Hoyt, R. Palchaudhuri and P. J. Hergenrother, Cribrostatin 6 Induces Death in Cancer Cells Through a Reactive Oxygen Species (ROS)-Mediated Mechanism, Invest. New Drugs, 2011, 29, 562–573 CrossRef CAS PubMed; (b) D. J. Asby, M. G. Radigois, D. C. Wilson, F. Cuda, C. L. L. Chai, A. Chen, A. S. Bienemann, M. E. Light, D. C. Harrowven and A. Tavassoli, Triggering Apoptosis in Cancer Cells with an Analogue of Cribrostatin 6 that Elevates Intracellular ROS, Org. Biomol. Chem., 2016, 14, 9322–9330 RSC.
- (a) B. Juskowiak, E. Galezowska, N. Koczorowska and T. W. Hermann, Aggregation and G-Quadruplex DNA-Binding Study of 6a,12a-Diazadibenzo-[a,g]Fluorenylium Derivative, Bioorg. Med. Chem. Lett., 2004, 14, 3627–3630 CrossRef CAS; (b) L. Madry, E. Galezowska, A. Gluszynska, T. W. Hermann, M. Zabel and B. Juskowiak, Interactions of the Oxidation Products of Papaverine with Guanine Quadruplexes, Pol. J. Chem., 2006, 80, 921–929 CAS; (c) B. Rubis, M. Kaczmarek, N. Szymanowska, E. Galezowska, A. Czyrski, B. Juskowiak, T. Hermann and M. Rybczynska, The Biological Activity of G-Quadruplex DNA Binding Papaverine-Derived Ligand in Breast Cancer Cells, Invest. New Drugs, 2009, 27, 289–296 CrossRef CAS PubMed; (d) E. Galezowska, J. Kosman, A. Stepien, B. Rubis, M. Rybczynska and B. Juskowiak, Bioactive Papaverine Derivatives Bind G-Quadruplexes Selectively, Chem. Pap., 2012, 66, 79–84 CAS; (e) D. N. Osinnikova, E. B. Moroshkina and D. M. Glushkina, Thermodynamics of Interaction and Structure of DNA Complexes with Phenacylimidazo[5,1-a]Isoquinoline Derivatives, Conf. Ser., 2015, 661, 012020 CrossRef.
- M. Alcarazo, S. J. Roseblade, A. R. Cowley, R. Fernández, J. M. Brown and J. M. Lassaletta, Imidazo[1,5-a]pyridine: A Versatile Architecture for Stable N-Heterocyclic Carbenes, J. Am. Chem. Soc., 2005, 127, 3290–3291 CrossRef CAS PubMed.
- (a) G. Volpia, B. Laceb, C. Garinoa, E. Priolaa, E. Artusoa, P. Cerreia Vioglioc, C. Baroloa, A. Find, A. Genree and C. Prandi, New Substituted Imidazo[1,5-a]Pyridine and Imidazo[5,1-a]Isoquinoline Derivatives and their Application in Fluorescence Cell Imaging, Dyes Pigm., 2018, 157, 298–304 CrossRef; (b) S. Mahata, A. Bhattacharya, J. P. Kumar, B. B. Mandal and V. Manivannan, Naked-Eye Detection of Pd2+ Ion Using a Highly Selective Fluorescent Heterocyclic Probe by “Turn-off” Response and in-vitro Live Cell Imaging, J. Photochem. Photobiol., A, 2020, 394, 112441 CrossRef CAS; (c) S. Mahata, S. Dey, B. B. Mandal and V. Manivannan, 3-(2-Hydroxyphenyl)imidazo[5,1-a]Isoquinoline as Cu(II) Sensor, Its Cu(II) Complex for Selective Detection of CN− Ion and Biological Compatibility, J. Photochem. Photobiol., A, 2022, 427, 113795 CrossRef CAS.
- C. Luo, P. Gandeepan, Y.-C. Wu, C.-H. Tsai and C.-H. Cheng, Cooperative C(sp3)–H and C(sp2)–H Activation of 2-Ethylpyridines by Copper and Rhodium: A Route toward Quinolizinium Salts, ACS Catal., 2015, 5, 4837–4841 CrossRef CAS.
- CCDC 2192426 (3o), 2176133 (3s), and 2176134 (13′) contain the supplementary crystallographic data for this paper.†.
- B. Sun, T. Yoshino, M. Kanai and S. Matsunaga, Cp*CoIII Catalyzed Site-Selective C-H Activation of Unsymmetrical O-Acyl Oximes: Synthesis of Multisubstituted Isoquinolines from Terminal and Internal Alkynes, Angew. Chem., Int. Ed., 2015, 54, 12968–12972 CrossRef CAS.
- X.-L. Wu and L. Dong, Synthesis of α-Ketone-isoquinoline Derivatives via Tandem Ruthenium(II)-Catalyzed C–H Activation and Annulation, Org. Lett., 2018, 20, 6990–6993 CrossRef CAS.
- S. Li, H. Lv, R. Xie, Y. Yu, X. Ye and X. Kong, The C–H Activation/Bidirecting Group Strategy for Selective Direct Synthesis of Diverse 1,1′-Biisoquinolines, Org. Lett., 2020, 22, 4207–4212 CrossRef CAS PubMed.
- Y. Xu, L. Zhang, M. Liu, X. Zhang, X. Zhang and X. Fan, Synthesis of Benzoazepine Derivatives via Rh(III)-Catalyzed inert C(sp2)–H Functionalization and [4 + 3] Annulation, Org. Biomol. Chem., 2019, 17, 8706–8710 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and NMR data. CCDC 2192426 (3o), 2176133 (3s) and 2176134 (13′). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2qo01390h |
| This journal is © the Partner Organisations 2023 |