
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVitrimer ionogels towards sustainable solid-state electrolytes†
Fengdi Li,
Giao T. M. Nguyen,
Cédric Vancaeyzeele ,
Frédéric Vidal and
Cédric Plesse*
,
Frédéric Vidal and
Cédric Plesse*
Laboratory of Physicochemistry of Polymers and Interfaces, CY Cergy Paris University, 5 Mail Gay Lussac, 95000 Neuville sur Oise, France. E-mail: cedric.plesse@cyu.fr
First published on 27th February 2023
Abstract
The growing demand for flexible, stretchable, and wearable devices has boosted the development of ionogels used as polymer electrolytes. Developing healable ionogels based on vitrimer chemistry is a promising approach to improve their lifetimes as these materials are usually subjected to repeated deformation during functioning and are susceptible to damage. In this work, we reported in the first place the preparation of polythioether vitrimer networks based on the not extensively studied associative S-transalkylation exchange reaction using thiol–ene Michael addition. Thanks to the exchange reaction of sulfonium salts with thioether nucleophiles, these materials demonstrated vitrimer properties such as healing and stress relaxation. The fabrication of dynamic polythioether ionogels was then demonstrated by loading 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide or 1-ethyl-3-methylimidazolium trifluoromethanesulfonate (EMIM triflate) within the polymer network. The resulting ionogels exhibited Young's modulus of 0.9 MPa and ionic conductivities in the order of 10−4 S cm−1 at room temperature. It has been found that adding ionic liquids (ILs) changes the dynamic properties of the systems, most likely due to a dilution effect of the dynamic functions by the IL but also due to a screening effect of the alkyl sulfonium OBrs-couple by the ions of the IL itself. To the best of our knowledge, these are the first vitrimer ionogels based on an S-transalkylation exchange reaction. While the addition of ILs resulted in less efficient dynamic healing at a given temperature, these ionogels can provide materials with more dimensional stability at application temperatures and can potentially pave the way for the development of tunable dynamic ionogels for flexible electronics with a longer lifespan.
Introduction
The growing demand for flexible, stretchable, and wearable electronic devices has emphasized a need for stretchable electronic and ionically conducting materials. Among these materials, the recent development in ionically conducting materials has boosted the development of electrochemical devices (e.g., batteries and fuel cells, supercapacitors, electrochromic devices) and has opened the door to flexible and wearable sensors, intronics, etc.1–8 These ionically conducting materials are gel-like materials that make it possible to combine the mechanical properties of the polymer network with the ionic conductivity of the liquid electrolyte.3 Depending on the nature of the liquid phase, different types of gels are described: hydrogels (salt + water), organogels (salt + organic solvent), or more recently ionogels (ionic liquid).9 Ionogels are termed as a class of ionically conducting materials when the ionic liquid (IL) is percolated through a polymer network. Such materials are more precisely achieved by in situ gel network formation in an IL or by swelling a polymer gel network with ILs.1,2 Ionogels combine the mechanical properties of the crosslinked polymer networks with the ionic conductivity, non-volatility and non-flammability of ILs.1,2,7–9In the frame of their applications and especially in the rising field of flexible, stretchable, and wearable devices, ionogels are usually subjected to repeated deformation during functioning, making them susceptible to damage. Thus, developing a simple and effective approach to improve their durability and life span becomes crucial. Imparting ionogels with self-healing ability seems likely a promising strategy because of their capabilities to repair mechanically induced damages. Healable ionogels based on hydrogen bonds, ionic bonds or metal–ligand coordination have been widely studied.10–13 Despite that ionic and hydrogen bonds are known to achieve efficient self-healing performances, these are generally strongly impacted by heat, the presence of water and any other polar solvents, which might be limiting in regard to their practical applications. Another strategy to impart materials with healing functionality is to introduce reversible reactions including covalent bonds. Fabrication of dynamic reversible polymer networks has become a popular strategy, particularly by introducing exchangeable chemical bonds into polymer networks, which are known as covalent adaptable networks (CANs). Based on the intrinsic mechanism of bond exchange reaction, CANs are further divided into dissociative and associative mechanisms.14 In dissociative CANs, bonds are first broken and then reformed in response to external stimuli, such as heat or light.14–16 Such dissociative bond exchange reactions have been used to prepare healable ionogels. For example, ionogels composed of chemically crosslinked poly(furfuryl methacrylate-co-methyl methacrylate) (P(FMA-co-MMA)) and physically crosslinked poly(vinylidene fluoride-co-hexafluoropropylene) (P(VDF-co-HFP)) networks with 80 wt% of 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl) imide (EMIM TFSI) loading have been prepared.17 The furan-maleimide crosslinker used in the network endowed the ionogel with a fast thermal healing efficiency thanks to the dissociative reversible Diels–Alder exchange reaction while the IL content provided the material with an ionic conductivity of 3.3 mS cm−1 at room temperature. Li et al. designed ionic skins by impregnating 1,2-dimethyl-3-ethoxyethyl-imidazolium bis(trifluoromethanesulfonyl)imide (DEIM TFSI) into a mechanically robust poly(urea-urethane) network with an ionic conductivity of 1.2 mS cm−1.18 The dissociative dynamic hindered urea bonds and hydrogen bonds between the amide groups endowed ionogel with healing ability. However, dissociative CANs allow topology rearrangements due to uncrosslinking of the material associated with a sudden drop of viscosity. This feature is a drawback in applications that require dimensional stability, easy shaping, and welding process. On the contrary, CANs that rely on an associative bond exchange reaction are characterized by a constant crosslink density.19 The bond cleavage is accompanied by the simultaneous formation of a new crosslink, as a result, such systems can change their topology with no loss of connectivity, making such networks permanent and insoluble. More specifically, in 2011, the term ‘vitrimers’ was introduced by Leibler et al. for thermally triggered associative CANs.20 Some groups reported vitrimer ionogels. Healable and reprocessable gelatin ionogels based on reversible exchange of imine bonds have been designed for flexible supercapacitors.21 These dynamic ionogels demonstrated an ionic conductivity higher than 1 mS cm−1 at room temperature thanks to the use of 1-ethyl-3-methylimidazolium acetate (EMIM Ac). Xu et al. reported polyurethane (PU) ionogels demonstrating an ionic conductivity of about 14 mS cm−1.22 The cracked PU ionogels can be readily healed at room temperature and restore their original performance owing to the dynamic boronic ester crosslinker used in the polymer network. Another team also showcased healable and recyclable boronic ester-based ionogels using 1-butyl-3-methylimidazolium tetrafluoro-borate. However, imine bonds and boronic ester are sensitive to water and can undergo dissociative hydrolysis, which impacts the stability of the materials and limit their applications.23
Recently, Zhong et al. reported polythioether (PTE)/EMIM TFSI ionogels with tailorable surface and mechanical properties using thiol–ene Michael addition.1,2 The nearly ideal 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric reactivity of thiol–ene Michael addition enabled fine-tuning of surface and mechanical properties of ionogels. While these materials are initially permanent and non-dynamic networks, Hendriks et al. reported turning PTE networks into catalyst-free vitrimer networks by covalently incorporating an alkylation agent (butyl brosylate BuOBrs) through thermal treatment.24 The resulting poly(thioether-sulfonium) materials can be recycled and remolded without loss of mechanical properties at elevated temperature through the S-transalkylation reaction of sulfonium salts with thioether nucleophiles. Moreover, this exchange reaction is not water sensitive. Combining tailorable PTE/EMIM TFSI ionogels with S-transalkylation seems very promising to develop healable ionogels based on associative dynamic covalent bonds.
:1 stoichiometric reactivity of thiol–ene Michael addition enabled fine-tuning of surface and mechanical properties of ionogels. While these materials are initially permanent and non-dynamic networks, Hendriks et al. reported turning PTE networks into catalyst-free vitrimer networks by covalently incorporating an alkylation agent (butyl brosylate BuOBrs) through thermal treatment.24 The resulting poly(thioether-sulfonium) materials can be recycled and remolded without loss of mechanical properties at elevated temperature through the S-transalkylation reaction of sulfonium salts with thioether nucleophiles. Moreover, this exchange reaction is not water sensitive. Combining tailorable PTE/EMIM TFSI ionogels with S-transalkylation seems very promising to develop healable ionogels based on associative dynamic covalent bonds.
Herein, S-transalkylation polythioether networks were first prepared to study the dynamic properties of this not extensively studied chemistry. These networks were prepared through the thiol–ene Michael addition between multifunctional thiols and diacrylates using a photobase generator (PBG). The fabrication of dynamic polythioether ionogels based on this reaction was then demonstrated by loading 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl) imide (EMIM TFSI) or 1-ethyl-3-methylimidazolium trifluoromethanesulfonate (EMIM Triflate). They were chosen as ionic liquid in these ionogels owing to their high ionic conductivity. Thermal properties, mechanical properties, and the ionically conducting behavior of these ionogels were studied. Dynamic properties of these materials were examined with healing and stress relaxation experiments. To the best of our knowledge, these are the first vitrimer ionogels based on S-transalkylation exchange reaction. The design concept illustrated in this work could potentially open a new path for the development of flexible electrochemical-based electronics with a longer lifetime.
Experimental section
Materials
The photobase generator (PBG) 2-(9-oxoxanthen-2-yl)propionic acid 1,5,7-triazabicyclo[4.4.0]dec-5-ene salt and 1,4-butanediol bis(thioglycolate) (dithiol, DT) were purchased from TCI Chemicals. Poly(ethylene glycol) diacrylate (PEGDA, Mn = 700 g mol−1), trimethylolpropane tris(3-mercoptopropianate) (trithiol, TT), triethylamine, 1-ethyl-3-methylimidazolium trifluoromethanesulfonate (EMIM Triflate), and sodium bicarbonate (NaHCO3) were purchased from Sigma–Aldrich. 4-Bromobenzenesulfonyl chloride was purchased from Alfa Aesar. Magnesium sulfate heptahydrate (MgSO4·7H2O) was obtained from Acros Organics. 1-Ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide (EMIM TFSI) was purchased from Solvionic. Finally, hydrochloric acid 37% (HCl) and dichloromethane (DCM) were obtained from VMR Chemicals.Synthetic procedures
Methods and techniques
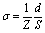 | (1) |
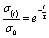 | (2) |
For vitrimers, relaxation times reflect associative exchange reactions and their temperature dependence can be fitted to the Arrhenius equation (eqn (3)):26,27
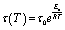 | (3) |
The values of τ were then plotted as a function of temperature to determine the activation energy Ea of the viscous flow induced by the associative exchange reaction. The topology freezing temperature Tv is another key characteristic for vitrimer materials. Conventionally, the hypothetical Tv is chosen as the temperature at which the viscosity equals 1012 Pa s as this value describes the liquid-to-solid transition of a glass-forming liquid.14,20 The relation between the viscosity η and the characteristic relaxation time τ can be expressed with the Maxwell relation (eqn (4)):28
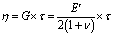 | (4) |
Results and discussion
Firstly, dynamic polythioether networks PTE-BuOBrs were prepared, i.e., without ionic liquid, to investigate their different properties and to select the optimal alkylating agent content for the future dynamic ionogels. Our group have reported the preparation and characterization of PTE-based ionogels using photocatalyzed thiol–ene Michael addition between a mixture of different thiol and acrylate functional groups.2 The resulting soft and stretchable ionogels with high ionic conductivity were obtained from poly(ethylene glycol) diacrylate (PEGDA), 1,4-butanediol bis(thioglycolate) (dithiol, DT) as chain extender trimethylolpropane tris(3-mercoptopropianate) (trithiol, TT) as crosslinker in the presence of 50 wt% ionic liquid relative to the total mixture weight. Therefore, the same composition of ionogel was used as starting point for the preparation of PTE-based polymer network in this work. PTE vitrimer was first synthesized by introducing BuOBr acting as alkylation agent into the polymer network. Ionogel vitrimer was then obtained by the in situ synthesis in the presence of ionic liquid i.e., EMITFSI or EMI OTf. The chemical structures of all chemicals, illustration of dynamic ionogels PTE-BuOBrs-IL are shown in Scheme 1.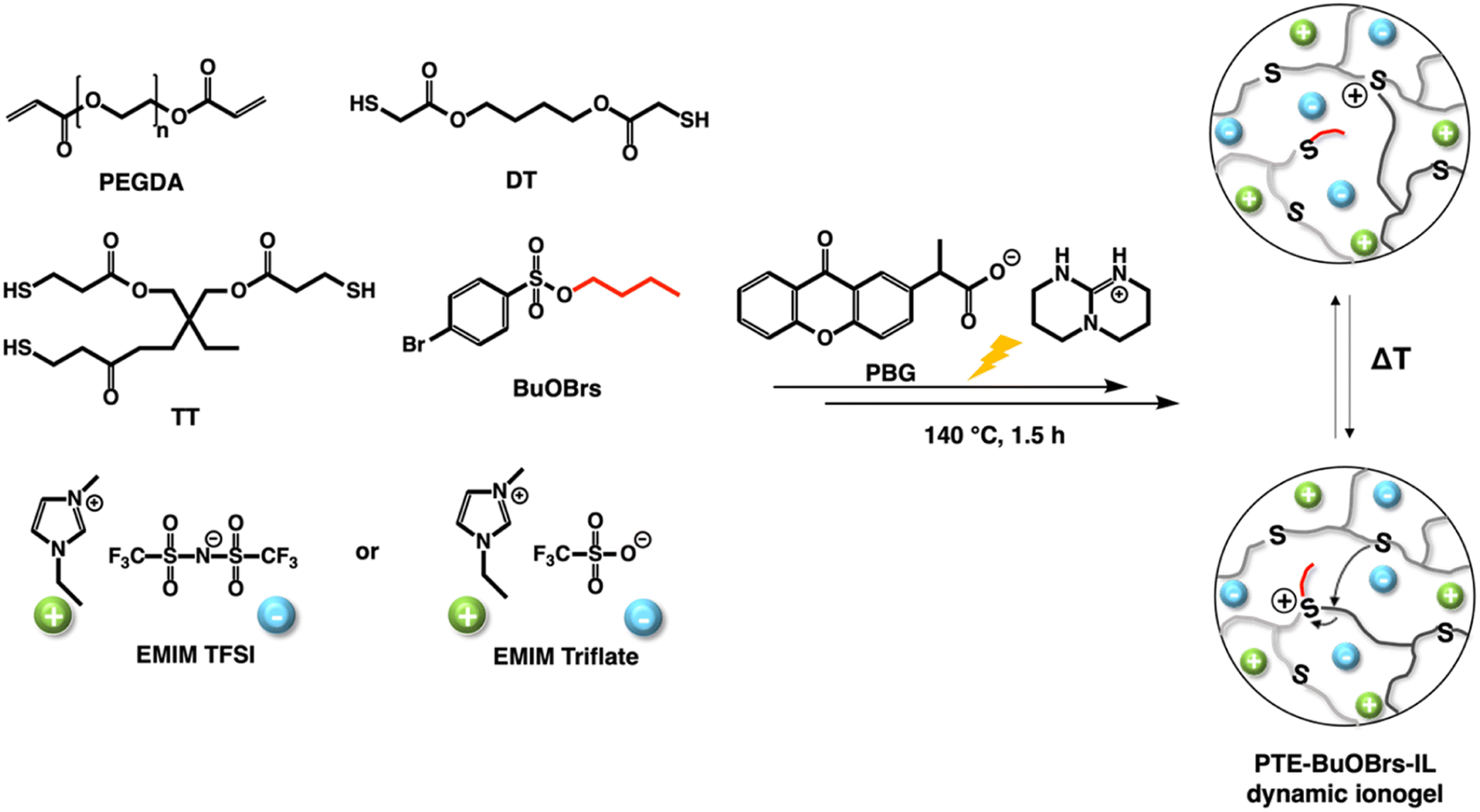 | ||
| Scheme 1 Chemical structures of the chemicals and illustrations of polythioether vitrimer PTE-BuOBrs and dynamic ionogels PTE-BuOBrs-IL. | ||
Synthesis and characterization of PTE-BuOBrs polythioether vitrimer networks
The PTE-BuOBrs polythioether vitrimer networks were first synthesized with the introduction of different quantities of BuOBrs (1, 2, 5 mol% vs. acrylate functional groups) with thiol and acrylate precursors in the presence of 1 wt% PBG (vs. the total weight of precursor mixture). Free-standing PTE-BuOBrs films were obtained by photopolymerization. A thermal activation at 140 °C for 1.5 h is necessary to covalently incorporate the alkylation agent into the PTE network.29 As a blank, a network without BuOBrs was prepared. The compositions and properties of all samples are illustrated in Table 1.:50:50 and various amounts of alkylation agent BuOBrs. Extractable contents of all samples before and after thermal treatment at 140 °C for 1.5 h are listed. Thermal and mechanical properties of thermal-treated PTE-BuOBrs networks and their reference PTE samples
| Sample | BUOBRS (mol%) | Extractable content before thermal treatment (wt%) | Extractable content after thermal treatment (wt%) | Tg (DSC) (°C) | Tα (DMA) (°C) | Young's modulus (MPa) | Elongation at break (%) |
|---|---|---|---|---|---|---|---|
| PTE | 0 | 3.9 | 3.6 | −44.7 | −30.0 | 1.7 ± 0.1 | 90 ± 11 |
| PTE-BuOBrs1 | 1 | 7 | 3.8 | −44.5 | −25.6 | 1.3 ± 0.1 | 71 ± 19 |
| PTE-BuOBrs2 | 2 | 6.7 | 7.7 | −44.6 | −24.3 | 1.2 ± 0.1 | 71 ± 1 |
| PTE-BuOBrs5 | 5 | 7.7 | 14.3 | −45.0 | −21.0 | 1.1 ± 0.2 | 53 ± 20 |
Rheological studies were carried out on PTE-BuOBrs5 (sample containing 5 mol% of alkylation agent) and its corresponding control sample (PTE) by following the in situ photopolymerization of the precursor mixtures at 30 °C. Storage modulus (G′) and loss modulus (G′′) were recorded as a function of time (Fig. 1a). The initial viscosities of the PTE-BuOBrs5 and PTE precursor mixtures were low (about 0.1 Pa s), which enable possible coating applications. The gel points, where G′ and G′′ curves intersected, were reached in less than 20 s, indicating the fast polymerization kinetics of thiol–ene Michael addition under catalytic activation of the photobase. The two samples reached a similar G′ plateau of about 300 kPa within 5 min. These results indicate the successful formation of polymer network through UV-activated thiol–ene Michael addition and that the presence of BuOBrs does not hinder the polymerization.
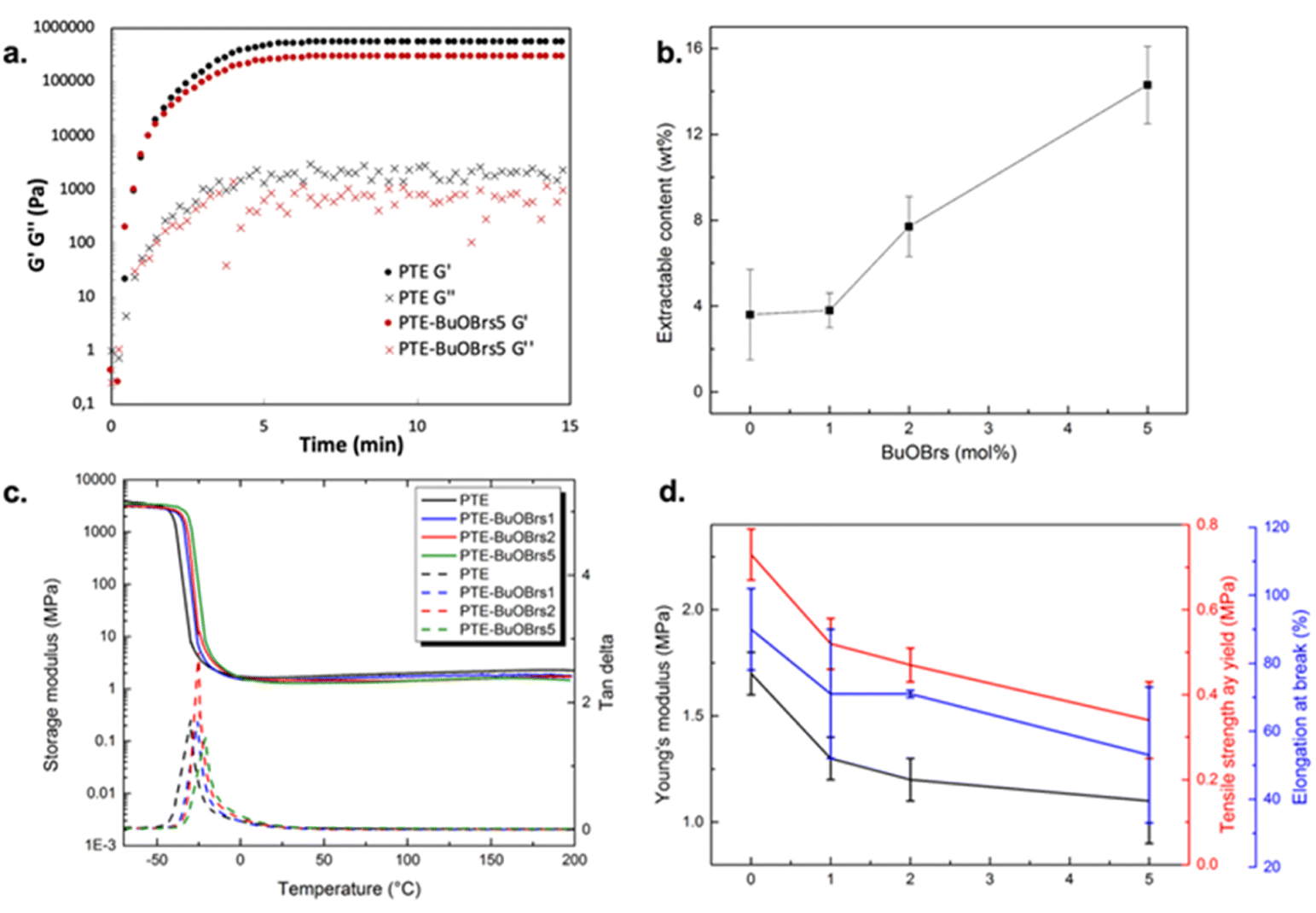 | ||
| Fig. 1 (a) Rheological properties of PTE-BuOBrs5 and PTE precursor mixtures as a function of time during photopolymerization; (b) soluble fractions of PTE-BuOBrs samples after thermal treatment at 140 °C for 1.5 h as a function of alkylation agent content; (c) DMA curves of PTE-BuOBrs series samples and PTE control sample; (d) Young's modulus, tensile strength at yield and elongation at break extracted from the stress–strain test as a function of BuOBrs mol% of PTE-BuOBrs samples. | ||
After verifying that the addition of alkylation agent does not disturb the formation of the polythioether networks, characterizations were carried out on PTE-BuOBrs samples that underwent a thermal activation at 140 °C for 1.5 h to covalently incorporate BuOBrs into the polymer network through alkylation of the thioether functions. The same procedure was also applied to the reference PTE sample. To verify the successful formation of polymer networks, soluble fractions of all UV-cured networks before and after thermal treatment were measured by Soxhlet extraction in DCM at 60 °C under 100 bar. Table 1 listed the soluble fractions of all materials. All samples except PTE-BuOBrs5 demonstrated similar and low extractable contents before and after the thermal treatment. This result indicates that the polymerization conditions are suitable for the formation of polymer network. To better understand the increase in soluble fraction of PTE-BuOBrs5 sample after thermal treatment, extractable contents of PTE and PTE-BuOBrs5 samples before and after thermal treatment were examined by 1H NMR. The NMR spectrum of PTE before and thermal treatment can be found in Fig. S2 and S3,† while those of PTE-BuOBrs5 before and thermal treatment can be found in Fig. S4 and S5.† The Soxhlet residues contained a large amount of oligo- or polythioether and traces of PEGDA, DT, and TT precursors. For PTE-BuOBrs5 sample, the alkylation agent could only be observed before thermal treatment, implying that the BuOBrs was successfully incorporated into the PTE networks after the thermal treatment. Concerning samples after applying thermal treatment of 1.5 h, an increase in soluble fractions has been observed with increasing alkylation agent content (Fig. 1b). This same tendency has been reported by Hendriks et al.,24 which could probably be explained by the cleavage of network chain ends by exchange to a butyl group, thus being removed from the bulk material. Minimum amounts of acrylate bonds originating from PEGDA were only extracted from thermally treated PTE and PTE-BuOBrs5 samples. Integrations of peaks originating from precursors were examined and the ratio between PEGDA, DT, and TT was calculated. In the case of stoichiometric reactivity, the theoretical ratio of PEGDA/DT/TT extracted should be 6/3/2, as samples were prepared using PEGDA, DT and TT precursors with a functional group ratio of 100:50:50. Based on the calculation, residues of all samples after extraction were found to demonstrate ratios close to 6/3/2, which confirmed the stoichiometric reactivity of thiol Michael addition. Moreover, ratios of PEGDA, DT and TT extracted before and after the thermal treatment remained unchanged. These results indicate that despite the increase in sol fractions of PTE-BuOBrs5 sample, thermally treated PTE-BuOBrs and PTE networks still consisted of the same compositions as pristine samples.
After thermal treatment, PTE-BuOBrs sample should demonstrate an ionically conducting behavior thanks to the formation of alkyl sulfonium on the PTE network and 4-bromobenzenesulfonate counter-ion during the thermal activation, as depicted in Scheme 1. Thus, the ionically conducting behaviors of PTE-BuOBrs2 sample before and after thermal treatment have been studied. The non-treated material showed no conducting behavior, while the samples after thermal treatment demonstrated a low ionic conductivity of 1.8 × 10−7 S cm−1 at 60 °C. These results confirmed the presence of sulfonate counter-ions and consequently the successful alkylation of thioether groups through thermal treatment. After verifying the well-formed nature of PTE-BuOBrs and the incorporation of alkylation agent through thermal treatment, different characterizations were carried out on these materials.
Fig. 1c shows the storage modulus and tanδ versus temperature of crosslinked PTE-BuOBrs networks with different loads of alkylation agent BuOBrs. At low temperatures, the storage modulus of glassy states of all samples were about 2 GPa, whereas the storage modulus decreased remarkably when the samples went through an evident and narrow α relaxation. All curves exhibit only one transition, which is in line with the results obtained by DSC. The onset temperature of the alpha transition slightly increased with the addition of BuOBrs. Furthermore, the storage moduli of rubbery states were similar. The relaxation temperature Tα of PTE-BuOBrs samples are resumed in Table 1. The slight increase in relaxation temperature with the BuOBrs load might be due to the increasing ionic interactions occurring after alkylation.
Tensile tests were performed on PTE-BuOBrs series samples to study the influence of alkylation agent content on the mechanical properties of the materials. Values of Young's modulus and elongation at break extracted from tensile tests are listed in Table 1 and are plotted as a function of BuOBrs content in Fig. 1d. It is clear that the modulus and stretchability of samples decrease with increasing alkylation agent content. This plasticizing-like effect can be explained by the presence of small uncrosslinked fractions evidenced by increasing extractable contents with BuOBrs content as well as the plasticizing role of 4-bromobenzenesulfonate.
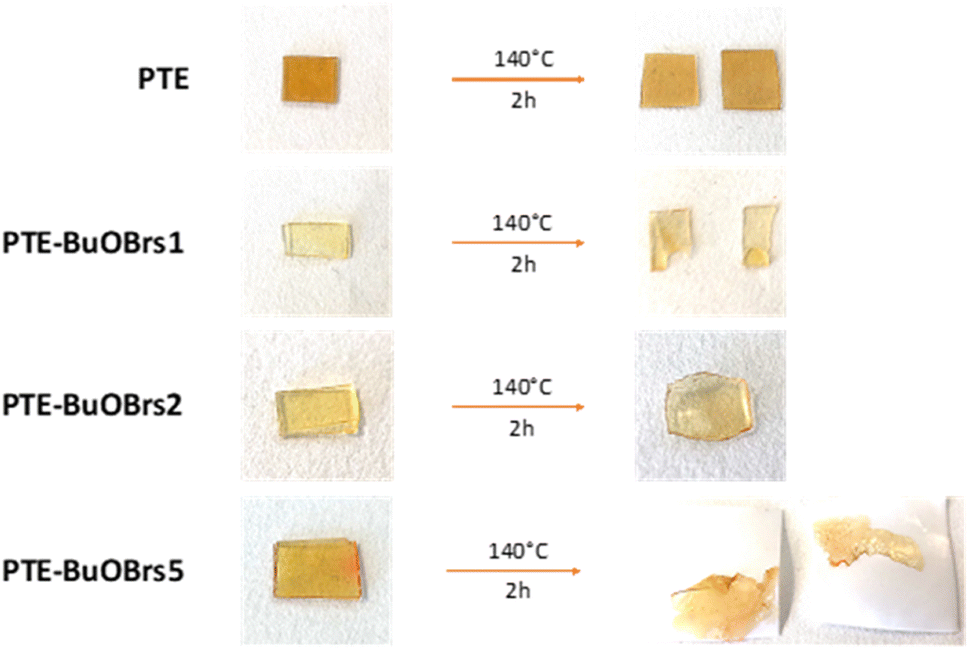 | ||
| Fig. 2 Healing behavior of PTE-BuOBrs samples and PTE control sample at 140 °C during 2 h. | ||
Fig. 3a compares the relaxation curves of samples containing 0, 2 and 5 mol% of alkylation agent at 140 °C, where the relative stress percentage is plotted as a function of time. Both PTE-BuOBrs2 and PTE-BuOBrs5 completely relaxed the applied stress after 86 min and 15 min respectively, whereas limited viscoelastic relaxation could be observed with the PTE control sample. These results provide convincing evidence that the alkylation agent BuOBrs plays a key role in network rearrangement in PTE-BuOBrs samples. Moreover, the full stress relaxation also indicates a swift exchange of thioether and sulfonium linkages and implies the dynamic nature of PTE-BuOBrs samples. Fig. 3b and c compare the stress relaxation behavior of PTE-BuOBrs2 and PTE-BuOBrs5 samples at different temperatures respectively. At 30 °C, after applying a constant deformation of 3% for 1 h, no significant stress relaxation could be seen in both samples. This result indicates that at low temperature, the transalkylation exchange reaction is inactivated and the network rearrangement is ‘frozen’, consequently these vitrimer materials behave like a classical crosslinked material. On the other hand, the relaxation rate increases with temperature, proving the temperature-dependent nature of the dynamic exchange reaction.
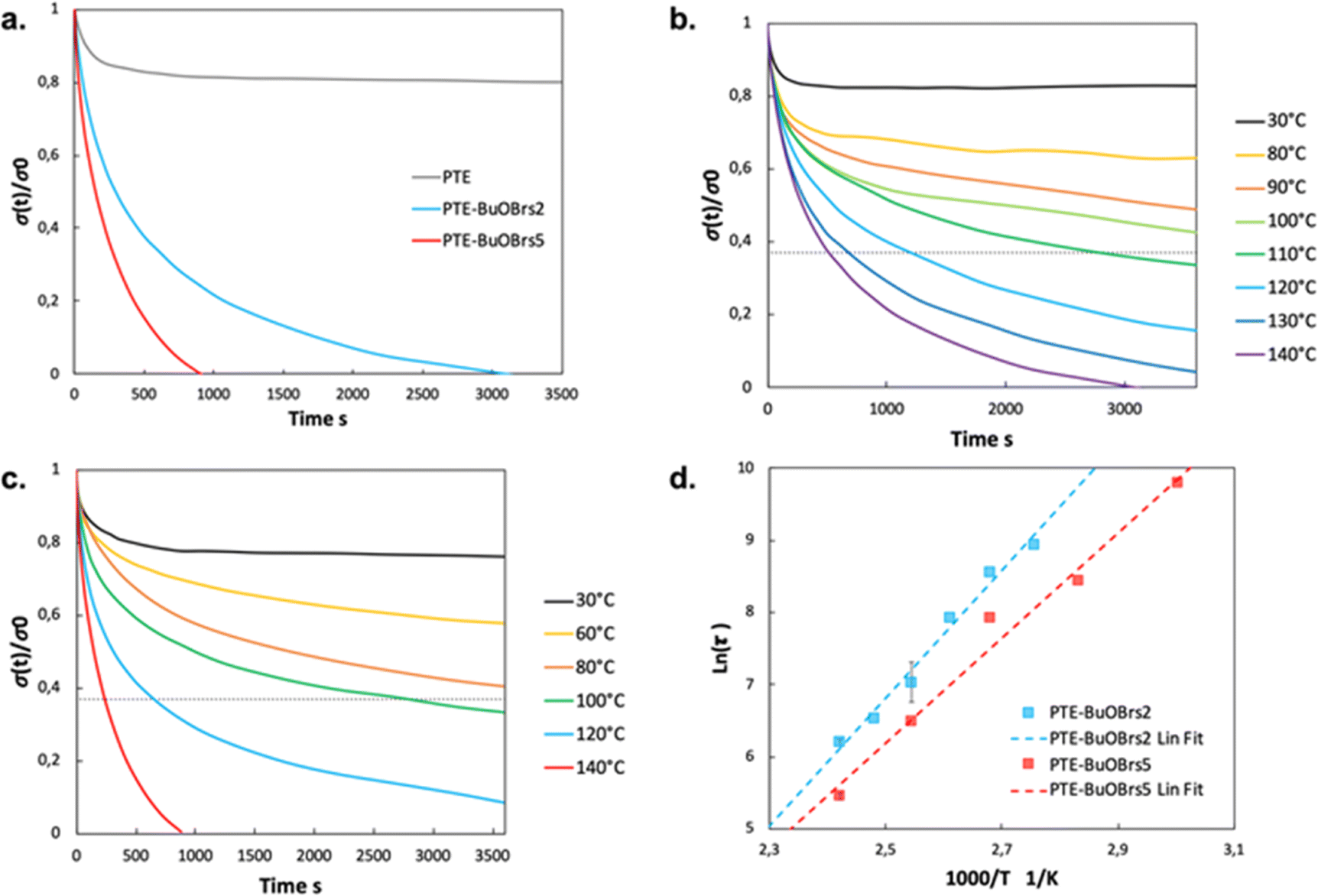 | ||
| Fig. 3 (a) Stress relaxation behaviors of PTE-BuOBrs samples with 2 and 5 mol% of alkylation agent content and their control sample PTE at 140 °C; (b) stress relaxation curves of PTE-BuOBrs2 sample at different temperatures (c) stress relaxation curves of PTE-BuOBrs5 sample at different temperatures; (d) Arrhenius linear fit of relaxation times of PTE-BuOBrs2 and PTE-BuOBrs5 samples plotted as a function of 1000/T. | ||
Relaxation time is defined as the time required to relax to 37% (1/e according to the Maxwell model for viscoelastic fluids) of initial stress.25 By comparing the stress relaxations curves shown in Fig. 3b and c, PTE-BuOBrs samples with higher BuOBrs content exhibit a lower relaxation time at the same temperature. Indeed, a faster network reorganization is expected with an increasing number of associative exchangeable bonds. One important feature of vitrimers is that they demonstrate an Arrhenius-like gradual decrease of viscosity at higher temperatures. To confirm the associative exchange mechanism of transalkylation reaction, relaxation times were plotted as a function of temperature. As shown in Fig. 3d, the relaxation times indeed followed the Arrhenius law, and an activation energy of 73.6 kJ mol−1 and 60.6 kJ mol−1 were calculated from the slope of Arrhenius linear fit of PTE-BuOBrs2 and PTE-BuOBrs5, respectively. These values are lower than those reported in the litterature,29,35,36 probably because of the polar environment of our polymer matrix which favors the transalkylation reaction that takes place between ionic reactants. The more available alkyl sulfonium existing in PTE-BuOBrs5 network resulted in faster exchange kinetics and consequently lower activation energies of the viscous flow. The topology freezing temperature Tv is another characteristic for vitrimer materials. Tv values were extrapolated based on the method described in the experimental section (Table 2). The hypothetical Tv of PTE-BuOBrs2 and PTE-BuOBrs5 samples were calculated to be 23 °C and 1 °C respectively. These values of Tv seems to indicate that the transalkylation reaction can start to take place at a such low temperature, while stress-relaxation experiments state a behavior similar to that of PTE network. Indeed, Du Prez et al. pointed out that extrapolation could result in significant errors when determining Tv in low Tg vitrimer materials,37 as in our case. The hypothetical Tv can be considered more as an estimation of relative importance for the exchange mechanism than a crucial characteristic of the materials. The fact that Tv value decreases with the increase in BuOBrs content is in line with the increase in dynamic site for exchange reaction, leading to a faster achievement of liquid-to-solid transition.
| Sample | TV (°C) | Ea (kJ mol−1) |
|---|---|---|
| PTE-BuOBrs2 | 23 | 73.6 |
| PTE-BuOBrs5 | 1 | 60.6 |
| Sample | IL type | IL content (wt%) | Extractable content after TC (wt%) | Tg (DSC) (°C) | Tα (DMA) (°C) | Young's modulus (MPa) | Elongation at break (%) |
|---|---|---|---|---|---|---|---|
| PTE-BUOBRS2 | — | — | 7.7 | −44.6 | −24.3 | 1.2 ± 0.1 | 71 ± 1 |
| PTE-BUOBRS2-TFSI50 | EMIM TFSI | 50 | 47.8 | −51.2 | −29.5 | 0.9 ± 0 | 55 ± 1 |
| PTE-BUOBRS2-Trif50 | EMIM triflate | 50 | 49.2 | −55.8 | −29.5 | 0.9 ± 0.1 | 33 ± 13 |
The rheological properties of the PTE-BuOBrs2-TFSI50 and PTE-BuOBrs2-Trif50 precursor mixtures were monitored during photopolymerization (Fig. S7†). The initial viscosities of the two precursor mixtures were 0.17 and 0.11 Pa s, respectively. Gel points were reached within 20 s. A G′ plateau of about 250 kPa and a G′′ plateau of 680 Pa were reached within 5 min, demonstrating similar polymerization kinetics to PTE-BuOBrs samples. Soluble fractions of PTE-BuOBrs2-TFSI50 and PTE-BuOBrs2-Trif50 in DCM were 47.8% and 49.2% respectively. Bearing in mind that 50 wt% of ILs were incorporated into ionogels, these results demonstrated the well-formed nature of the polymer networks.
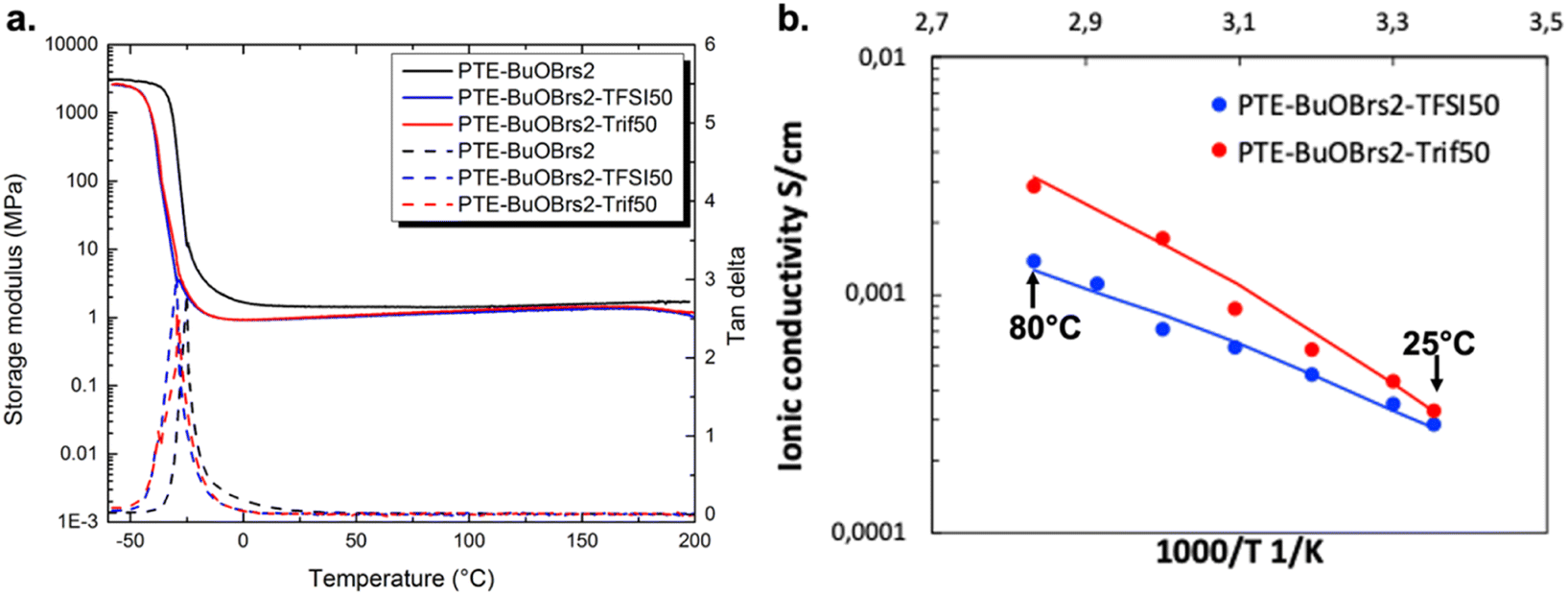 | ||
| Fig. 4 (a) DMA tests of PTE-BuOBrs-IL ionogels and PTE-BuOBrs2 sample; (b) ionic conductivity of PTE-BuOBrs2-TFSI50 and PTE-BuOBrs2-Trif50 samples at different temperatures. Solid lines: calculation according to VTF equation. | ||
where A is a temperature-independent constant related to the number of charge carriers, Ea is the pseudo-activation energy related to polymer segmental motion and R stands for the gas constant. T0 is a reference temperature usually correlated with the ideal glass transition temperature at which free volume disappears or at which the configurational entropy of the polymer chain becomes zero. T0 is usually 35 to 50 K below Tg in either case.38–40 The VTF behaviors of all PTE-BuOBrs-IL samples were studied with T0 set to Tg – 50 K and all parameters extrapolated are listed in Table 4. The values of R2 of the VTF linear fit are about 0.99, which shows that the VTF model is suitable for describing the ionic behavior of these materials, indicating that the movements of the charge carrier are correlated with the polymer chain movements. The parameter A was found to be 1.0 S K1/2 cm−1 for PTE-BuOBrs2-TFSI50 sample and 12.4 S K1/2 cm−1 for PTE-BuOBrs2-Trif50, while the Ea of ionic conduction of 5.6 kJ mol−1 and 8.5 kJ mol−1 was assigned to each material. The higher A parameter which is correlated to the number of charge carriers can be explained by the higher cation and anion concentration of EMIM triflate (5.3 mol L−1) than EMIM TFSI (3.9 mol L−1).41 Moreover, compared to the hydrophobic EMIM TFSI, EMIM triflate is known to be more hydrophilic,42 thus more susceptible to absorb ambient humidity, resulting in a higher conductivity in open air condition with better ion dissociation.
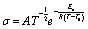
| Sample | Tg (°C) | Ionic conductivity at 30 °C (S cm−1) | A (VTF) (S K1/2·cm−1) | Ea (VTF) (kJ mol−1) | R2 (VTF) |
|---|---|---|---|---|---|
| PTE-BuOBrs2-TFSI50 | −51.2 | 2.8 × 10−4 | 1.0 | 5.6 | 0.9838 |
| PTE-BuOBrs2-Trif50 | −55.8 | 3.2 × 10−4 | 12.4 | 8.5 | 0.9818 |
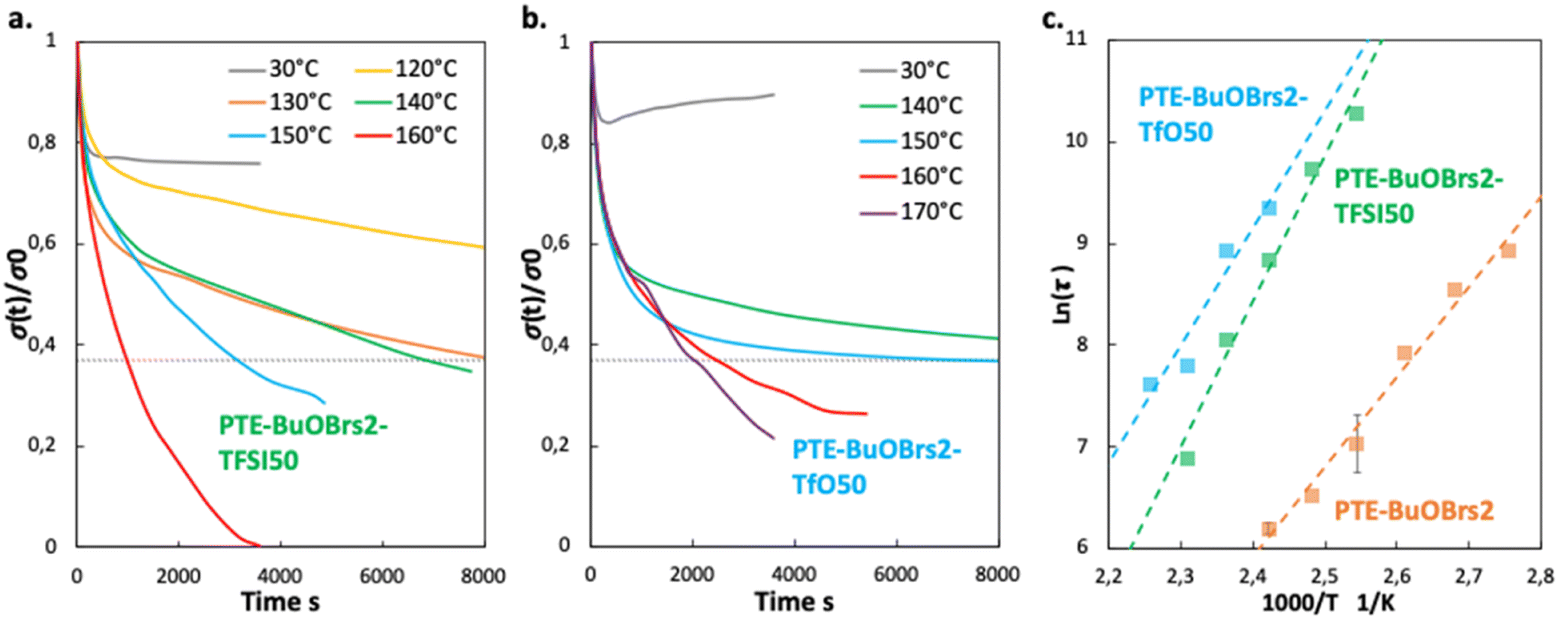 | ||
| Fig. 5 (a) Stress relaxation tests of PTE-BuOBrs2-TFSI50 sample at various temperatures; (b) stress relaxation tests of PTE-BuOBrs2-Trif50 sample at various temperatures; (c) Arrhenius linear plot extracted from relaxation times of PTE-BuOBrs2-TFSI50, PTE-BuOBrs2-TFSI50 and PTE-BuOBrs2 samples at different temperatures. | ||
Pictures of PTE-BuOBrs2-TFSI50 and PTE-BuOBrs2-Trif50 samples before and after stacked healing tests can be found in Fig. 6a and b respectively. For both samples, the two films couldn't be separated after the healing process, demonstrating that co-bonding took place at the interface thanks to the dynamic bond exchanges. However, the two pieces are not fused together as it was observed for the PTE-BuOBrs2 without IL, demonstrating either a lower healing efficiency or incomplete healing due to the much slower dynamic kinetics. These observations are consistent with the results obtained from stress relaxation experiments, which is the addition of ILs results in slower network rearrangement. The stacked healed samples showed similar ionically conducting behaviors as pristine samples (Fig. 6a and b), indicating that the healing process was able to ensure the ionic conduction between the two films and recover the pristine ionic conducting behavior. Fig. 6c and d present tensile test experiments as well as pictures of PTE-BuOBrs2-TFSI50 and PTE-BuOBrs2-Trif50 films after the edge-to-edge healing test respectively. After 4 h at 140 °C, the fractured surfaces fused together, and the cracks were almost invisible to the eye. Tensile testing of the two samples showed that the healed samples demonstrated very similar Young's modulus as pristine samples (about 0.8 MPa), indicating that the topological rearrangement enabled by transalkylation seems to have retrieved the pristine mechanical properties within the elastic domain. The elongation at break reduced sharply (down to 55%) with breaks localized randomly and not especially on the healing area. Healing conditions of 4 h at 140 °C allowed fractured samples to weld together but it seems that long time exposure to high temperature reduced the elasticity of the materials. After healing, scares are not visible with the eyes but residual traces of it can be guessed on the image of the cross-section of stacked healed ionogels, especially on the EMIM Triflate based one (red arrows in Fig. 6e and f).
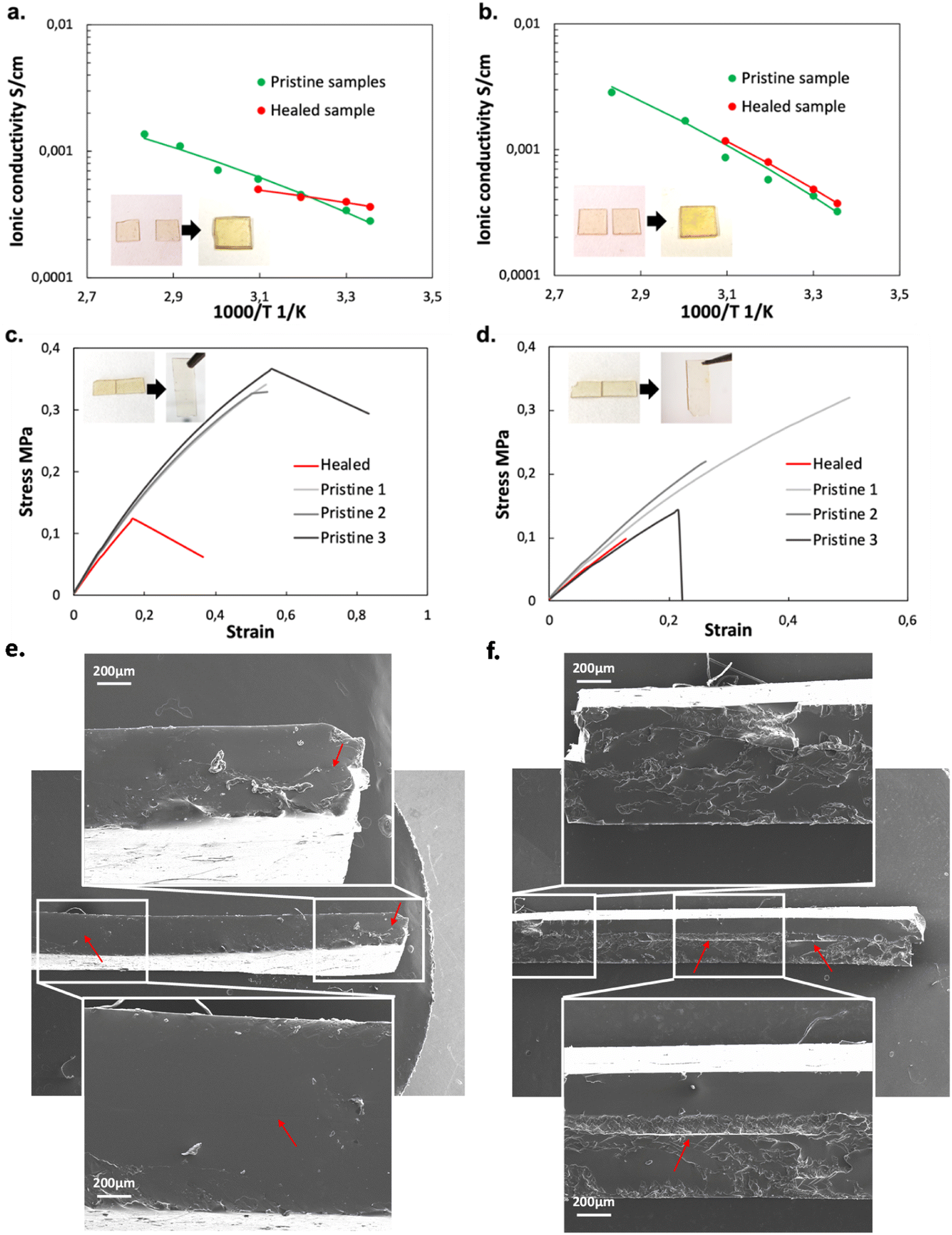 | ||
| Fig. 6 (a) Pictures and the ionic conductivity behavior of PTE-BuOBrs2-TFSI50 before and after stacked healing; (b) pictures and the ionic conductivity behavior of PTE-BuOBrs2-Trif50 before and after stacked healing; (c) pictures and the tensile testing curves of PTE-BuOBrs2-TFSI50 before and after interfacial healing; (d) pictures and the tensile testing curves of PTE-BuOBrs2-Trif50 before and after interfacial healing; SEM image of the cross-section of two pieces of (e) PTE-BuOBrs2-TFSI50 after stacked healing and of (f) PTE-BuOBrs2-Trif50 after stacked healing. | ||
Conclusions
In this work, PTE-BuOBrs vitrimers were synthesized through the thiol–ene Michael addition between multifunctional thiol and diacrylate using a photobase generator. These PTE-BuOBrs networks exhibited vitrimer properties after thermal activation treatment, allowing the formation of alkylsulfonium salts. Healing and stress relaxation at elevated temperatures were demonstrated thanks to the S-transalkylation exchange reaction of sulfonium salts with thioether nucleophiles. These materials could completely relax stress and demonstrated viscous flow energies ranging from 60.6 kJ mol−1 and 73.6 kJ mol−1. Vitrimer ionogels were then developed by synthesizing the PTE network in the presence of EMIM TFSI or EMIM triflate. These ionogels demonstrated Young's modulus of 0.9 MPa and ionic conductivities in the order of 10−4 S cm−1 at room temperature. Dynamic properties of these materials were examined with healing and stress relaxation experiments. It has been found that the addition of ILs modify the dynamic properties of the systems, probably because of a dilution effect of the dynamic functions by the IL but also because of a screening effect of the alkyl sulfonium OBrs− couple by the ions of the IL itself. A slowing down of the dynamic exchange kinetics and an increase of both their activation energy and Tv is observed. While it is resulting in a less efficient dynamic healing at a given temperature, it can provide also more dimensionally stable networks at application temperatures. To the best of our knowledge, this is the first work reporting vitrimer ionogels based on S-transalkylation exchange reaction. The ionogels developed herein are using mostly commercially available products, thus providing a simple and effective approach to developed tunable dynamic ionogels for flexible electronics with a longer lifespan.Abbreviations
| PEGDA | Poly(ethylene glycol) diacrylate |
| DT | 1,4-Butanediol bis(thioglycolate) |
| TT | Trimethylolpropane tris(3-mercoptopropianate) |
| PBG | 2-(9-Oxoxanthen-2-yl)propionic acid 1,5,7-triazabicyclo[4.4.0]dec-5-ene salt |
| IL | Ionic liquid |
| PTE | Polythioether |
| BuOBrs | Alkylating agent |
| EMIM TFSI | 1-Ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide |
| EMIM Triflate | 1-Ethyl-3-methylimidazolium trifluoromethanesulfonate |
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 Research and Innovation Program under grant agreement No. 825232 “WEAFING”.References
- Y. Zhong, G. T. M. Nguyen, C. Plesse, F. Vidal and E. W. H. Jager, ACS Appl. Mater. Interfaces, 2018, 10, 21601–21611 CrossRef CAS PubMed.
- Y. Zhong, G. T. M. Nguyen, C. Plesse, F. Vidal and E. W. H. Jager, J. Mater. Chem. C, 2019, 7, 256–266 RSC.
- C. H. Yang, B. Chen, J. J. Lu, J. H. Yang, J. Zhou, Y. M. Chen and Z. Suo, Extreme Mech. Lett., 2015, 3, 59–65 CrossRef.
- G. Gu, H. Xu, S. Peng, L. Li, S. Chen, T. Lu and X. Guo, Soft Robot., 2019, 6, 368–376 CrossRef PubMed.
- Z. Deng, H. Wang, P. X. Ma and B. Guo, Nanoscale, 2020, 1224–1246 RSC.
- C. Yang and Z. Suo, Nat. Rev. Mater., 2018, 3, 125–142 CrossRef CAS.
- Y. Ren, J. Guo, Z. Liu, Z. Sun, Y. Wu, L. Liu and F. Yan, Sci. Adv., 2019, 5, eaax0648 CrossRef CAS PubMed.
- J. Le Bideau, L. Viau and A. Vioux, Chem. Soc. Rev., 2011, 40, 907–925 RSC.
- E. Andrzejewska, A. Marcinkowska and A. Zgrzeba, Polymers, 2017, 62, 344–352 CAS.
- S. Xiang, F. Zheng, S. Chen and Q. Lu, ACS Appl. Mater. Interfaces, 2021, 13, 20653–20661 CrossRef CAS PubMed.
- D. Weng, F. Xu, X. Li, S. Li, Y. Li and J. Sun, ACS Appl. Mater. Interfaces, 2020, 12, 57477–57485 CrossRef CAS PubMed.
- Y. Shi, Y. Wang, Y. Gu, L. Zheng, S. Ma and X. Xu, Chem. Eng. J., 2020, 392, 123645 CrossRef CAS.
- X. Qu, W. Niu, R. Wang, Z. Li, Y. Guo, X. Liu and J. Sun, Mater. Horizons, 2020, 7, 2994–3004 RSC.
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2016, 7, 30–38 RSC.
- A. M. Wemyss, C. Bowen, C. Plesse, C. Vancaeyzeele, G. T. M. Nguyen, F. Vidal and C. Wan, Mater. Sci. Eng., R, 2020, 141, 100561 CrossRef.
- N. J. Van Zee and R. Nicolaÿ, Prog. Polym. Sci., 2020, 104, 101233 CrossRef CAS.
- Z. Tang, X. Lyu, A. Xiao, Z. Shen and X. Fan, Chem. Mater., 2018, 30, 7752–7759 CrossRef CAS.
- T. Li, Y. Wang, S. Li, X. Liu and J. Sun, Adv. Mater., 2020, 32, 2002706 CrossRef PubMed.
- J. M. Winne, L. Leibler and F. E. Du Prez, Polym. Chem., 2019, 10, 6091–6108 RSC.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- J. Liu, H. Song, Z. Wang, J. Zhang, J. Zhang and X. Ba, J. Mater. Sci., 2020, 55, 3991–4004 CrossRef CAS.
- J. Xu, H. Wang, X. Du, X. Cheng, Z. Du and H. Wang, Chem. Eng. J., 2021, 426, 130724 CrossRef CAS.
- J. Tang, J. Yang, H. Yang, R. Miao, R. Wen, K. Liu, J. Peng and Y. Fang, J. Mater. Chem. C, 2018, 6, 12493 RSC.
- Q. L. Lei, X. Xia, J. Yang, M. P. Ciamarra, R. Ni, M. Pica Ciamarra and R. Ni, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 27111–27115 CrossRef CAS PubMed.
- T. Wright, T. Tomkovic, S. G. Hatzikiriakos and M. O. Wolf, Macromolecules, 2019, 52, 36–42 CrossRef CAS.
- W. Denissen, G. Rivero, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Adv. Funct. Mater., 2015, 25, 2451–2457 CrossRef CAS.
- Y. Yang, S. Zhang, X. Zhang, L. Gao, Y. Wei and Y. Ji, Nat. Commun., 2019, 10, 1–8 CrossRef PubMed.
- C. He, S. Shi, D. Wang, B. A. Helms and T. P. Russell, J. Am. Chem. Soc., 2019, 141, 13753–13757 CrossRef CAS PubMed.
- B. Hendriks, J. Waelkens, J. M. Winne and F. E. Du Prez, ACS Macro Lett., 2017, 6, 930–934 CrossRef CAS PubMed.
- M. Röttger, T. Domenech, R. Van Der Weegen, A. Breuillac, R. Nicolaÿ and L. Leibler, Science, 2017, 356, 62–65 CrossRef PubMed.
- A. Breuillac, A. Kassalias and R. Nicolaÿ, Macromolecules, 2019, 52, 7102–7113 CrossRef CAS.
- D. J. Fortman, J. P. Brutman, G. X. De Hoe, R. L. Snyder, W. R. Dichtel and M. A. Hillmyer, ACS Sustain. Chem. Eng., 2018, 6, 11145–11159 CrossRef CAS.
- M. Chen, L. Zhou, Y. Wu, X. Zhao and Y. Zhang, ACS Macro Lett., 2019, 8, 255–260 CrossRef CAS PubMed.
- J. Han, T. Liu, C. Hao, S. Zhang, B. Guo and J. Zhang, Macromolecules, 2018, 51, 6789–6799 CrossRef CAS.
- Z. Tang, Y. Liu, Q. Huang, J. Zhao, B. Guo and L. Zhang, Green Chem., 2018, 20, 5454–5458 RSC.
- W. Xu, W. Yu, X. Chen, S. Liao and M. Luo, J. Appl. Polym. Sci., 2021, 51182 CrossRef CAS.
- M. Guerre, C. Taplan, J. M. Winne and F. E. Du Prez, Chem. Sci., 2020, 11, 4855–4870 RSC.
- G. Adam and J. H. Gibbs, J. Chem. Phys., 1965, 43, 139–146 CrossRef CAS.
- C. A. Angell, Solid State Ionics, 1983, 10, 3–16 CrossRef.
- M. C. Wintersgill and J. J. Fontanella, Polymer Electrolyte Reviews, Elsevier, London and New York, 1989, vol. 2 Search PubMed.
- P. Bonhôte, A.-P. Dias, N. Papageorgiou, K. Kalyanasundaram and M. Grätzel, Inorg. Chem., 1996, 35, 1168–1178 CrossRef PubMed.
- G. García-Miaja, J. Troncoso and L. Romaní, J. Chem. Thermodyn., 2009, 41, 161–166 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experiments discussed here including 1H NMR spectra, and characterizations by DSC and rheometer. See DOI: https://doi.org/10.1039/d2ra06829j |
| This journal is © The Royal Society of Chemistry 2023 |