DOI:
10.1039/D2RA07077D
(Paper)
RSC Adv., 2023,
13, 674-687
Phosphomolybdic acid hydrate encapsulated in MIL-53 (Fe): a novel heterogeneous heteropoly acid catalyst for ultrasound-assisted regioselective nitration of phenols†
Received
7th November 2022
, Accepted 5th December 2022
First published on 23rd December 2022
Abstract
In this study, a heterogeneous catalyst, PMA@MIL-53 (Fe) (MIL ⇒ Matériaux de l'Institut Lavoisier), has been used to replace the usual mineral acids such as sulfuric acid. A wide variety of nitration methods require the use of a mixture of acids such as concentrated nitric acid and sulfuric acid, which result in producing a large amount of acidic waste. During recent years, the use of the heterogeneous system for the nitration of aromatic compounds has been highly considered and used by chemists due to some specific advantages, i.e. easy separation of the product from the reaction mixture, the possibility of recycling and reusing the catalyst, etc. Herein, the catalyst was synthesized using a metal–organic framework and a heteropoly phosphomolybdic acid. The PMA@MIL-53 (Fe) was prepared using a similar method of MIL-53 (Fe) synthesis. Afterwards, FeCl3·6H2O and 1,4-benzene dicarboxylic acid (BDC) in a dimethylformamide solution were placed in an ultrasound bath and, then, HPA (heteropoly acid) was added to the reaction mixture. The PMA (phosphomolybdic acid) encapsulation in MIL-53 (Fe) was confirmed using various analysis. Under optimal conditions, the catalytic activity of PMA@MIL-53 (Fe) was evaluated in nitration of phenol under ultrasonic waves. Besides, the ratio of the two products of ortho and para was obtained using GC. Optimum conditions were reached after 15 minutes, in such a way that the loaded PMA was 0.02 g under optimal conditions, the efficiencies of ortho-nitrophenol and para nitrophenol were 54.98 and 45.01, respectively.
1. Introduction
The aromatic nitration reactions can be considered as one of the most industrially important organic reactions due to adding a nitrogen substituent to the aromatic rings without the double-nitration by-products under mild reaction conditions.1–3 In this sense, only a single nitro substitution has been added to the aromatic ring unless there is also a strong activating substituent.4,5
Nitrated phenols are one of the most important classes of the substituted aromatic compounds.6 Besides, they are important synthetic intermediates and essential starting materials in advanced organic material synthesis, i.e. their reduction was a useful route to the corresponding aminophenols and synthesis of phenol-containing natural products.7,8 Although there are several multistep total synthesis and catalytic methods to achieve nitrophenol compounds, the one-pot reaction between a very powerful nitration reagent (i.e. conc. HNO3 & H2SO4, N2O5 or NO2/O3 and metal nitrates) and phenol derivatives, in the presence of a solid acid as catalyst, is to a great extent accomplished as an intensively explored industrial method.4,9–13 The mechanistic studies show that these reactions proceed through a nitronium ion (NO2+) which is attacked by π electrons delocalized around the aromatic rings leading to breakage of one of the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds to avoid a five-valent nitrogen.14–16 Although the nitration of phenol is also very fast, these methods are not selective for the direct nitration of phenol, producing a mixture of ortho- and para-nitrophenols.1 Besides, dilute nitric acid must be used to avoid the oxidation and overoxidation of phenols to unspecified resinous products. Furthermore, because of the presence of strong activating OH groups on the starting arenes, overnitration (double nitration) to di-nitro aromatics is possible.17–19
O bonds to avoid a five-valent nitrogen.14–16 Although the nitration of phenol is also very fast, these methods are not selective for the direct nitration of phenol, producing a mixture of ortho- and para-nitrophenols.1 Besides, dilute nitric acid must be used to avoid the oxidation and overoxidation of phenols to unspecified resinous products. Furthermore, because of the presence of strong activating OH groups on the starting arenes, overnitration (double nitration) to di-nitro aromatics is possible.17–19
To address the above issues and to enable a practical nitration process, researchers have been trying to optimize the selectivity and efficiency of this reaction using catalytic methods.20–22 But, they need equivalents of strong acid-catalysis, operated at very high temperatures.12,23–26 Considerably, drastic reaction conditions would greatly limit the energy efficiency.7 In addition, the reaction progress requires very powerful reagents, leading to the formation of by-products and limiting the reaction yield and selectivity.27–29 Thus, the development of catalytic reactions under mild conditions using the ultrasound-assisted method as an ideal medium has attracted significant attention in the arena of nitration reactions, because the reactions can be carried out at room temperature, leading to a reduction in the by-products formation and increasing the efficiency of the main products.30–36
During recent years, metal–organic frameworks (MOFs) have been regarded as one of the most popular and useful catalysts for laboratory and commercial scale organic synthesis.37–47 On the ecological side, MOFs have been known as the low-cost and green solid supports, applied to immobilize the soluble catalysts due to their crucial role in lowering the toxicity, having heterogeneous48,49 high specific surface area, possessing porous nature and also structural diversity, being economically viable and also having safe and recyclable characteristics to synthesize organic molecules – leading to the synthesis of a wide variety of MOF catalysts,50,51 i.e. MIL-53 (Fe) which is an Fe-based MOF with high thermal stability and a very high surface area. In this sense it has greatly attracted researchers because of its exceptional tunability and functionality.52–57 Moreover, researchers have also aimed at designing effective functionalized MOF catalysts with organic and organometallic moieties.48,57–62 These methods which were applied to modify various types of catalysts,61 heteropoly acids and other transition metal-catalysts have been successfully encapsulated into the pores of MIL-53 (Fe). In addition, these composites signify the application of MOF-functionalized catalytic species in organic synthesis and functional group transformations.63 Thus, catalytic complex connection to the metal–organic frameworks (MOFs) as support is a new strategy to modify physiochemical properties, pore size, surface area and topology of the target porous material moieties.64–68
During recent decades, heteropoly acid catalysts, regarded as a member of Keggin structure crystals, have attracted great attention in catalysis science owing to a variety of important advantages.69–72 Therefore, researchers have focused on the synthesis of heterogenized heteropoly acids.73–75 Accordingly, we would introduce a novel phosphomolybdic acid hydrate (PMA) encapsulated @MIL-53 (Fe) MOF using the co-synthesis method as a novel functionalized MOF catalyst.
Considering our interest in the synthesis and application of MOF-based heterogeneous catalysts – using natural raw materials; herein, we aim at proposing the synthesis of PMA@MIL-53 (Fe) and its application in the direct nitration of phenol to the related nitro phenol derivatives in high yield and selectivity under ultrasonic waves.
2. Experimental section
2.1. Chemicals
Thin layer chromatography (TLC with silica gel plates 60F-254) was used to identify the desired compounds, improve the reactions, determine the reaction time and ensure the purity of the products and initial materials.
Solvents and other reagents were of analytical grade and utilized without further purification. In this study, all of the materials, reagents and solvents were obtained from Merck, Fluca, and Sigma Aldrich.
2.2. Instrumentation
GC model 7890B Agilent was equipped with column Rtx@5MS (length 30 m, internal diameter 0.25 mm). X-Ray powder diffraction spectrometer (diffraction type: D8 advance broker, λ = 1.5406 Å) was used to identify the solid sample. Thermal analysis of the samples was performed with Netzsch-TGA 209 F1. Fourier transformed infrared (FTIR) spectra were recorded using the FT-IR Spectrometer 470 Shimadzu using KBr pellets. The vibrational frequencies in the unit are wavelengths (cm−1). XPS spectra were recorded on a Bes Tek spectrometer with A1 Kα X-rays as photon source (1486.6 eV). N2 adsorption–desorption isotherm was used to determine the specific surface areas in Brunauer–Emmett–Teller (BET) at liquid nitrogen temperature (Micromeritics ASAP 2000). The weight percentage of hetero poly acid which was loaded in the pores and cavities was measured using a pair of induced plasma spectroscopy (ICP) and the Optima 8000 ICP-OES. The surface morphology of the composite was investigated applying scanning electron microscopy (SEM), (LEO 1430VP models) and high resolution transmission electron microscopy (HR-TEM) (Zeiss Libra 120 models), ultrasonic waves were generated using a Sonic 6mx ultrasonic bath with a pulse power of 37 kHz.
2.3. Synthesis of metal–organic framework MIL-53 (Fe)
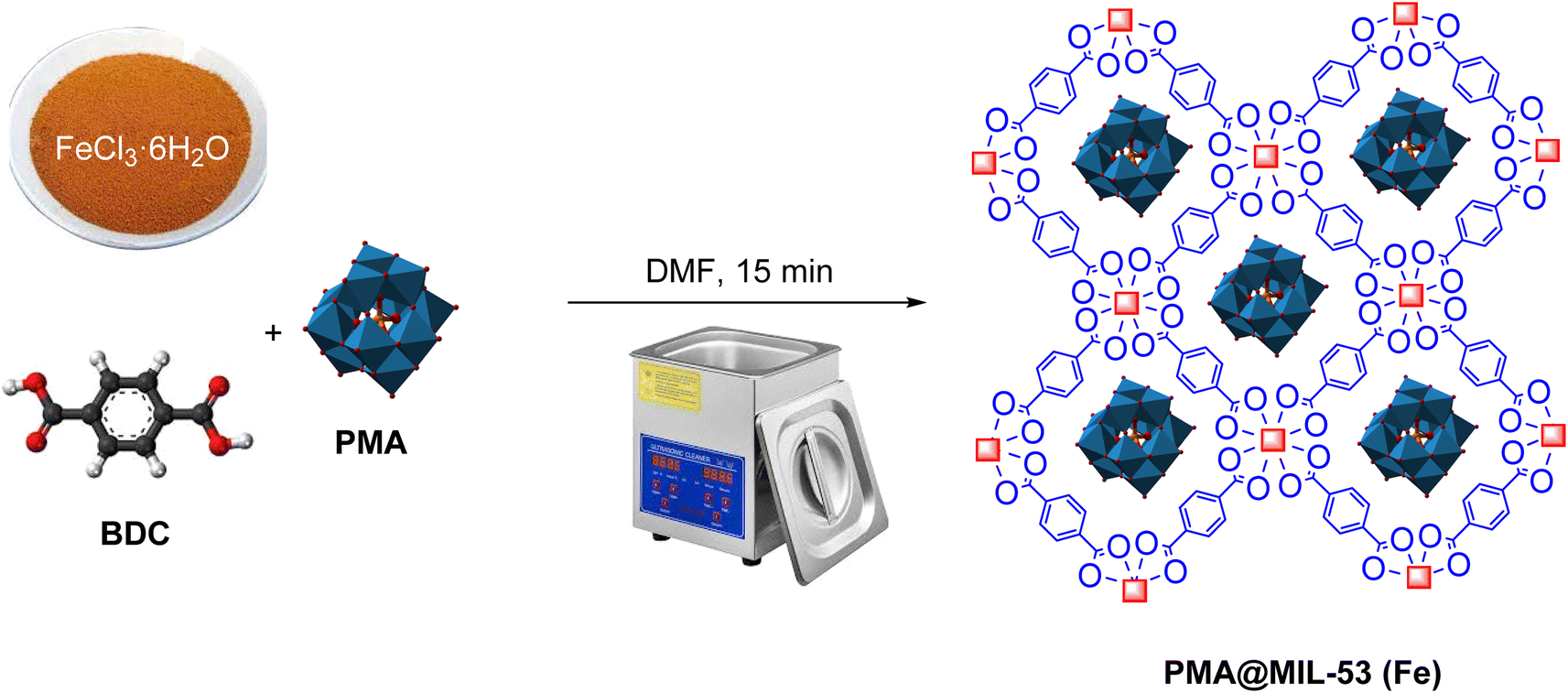
The synthesis of the metal–organic framework MIL-53 (Fe) using ultrasonic waves has been reported by Gordon et al.76 First, 1.27 g (1 mmol) of FeCl3·6H2O and 1.166 g (1 mmol) of 1,4-benzenedicarboxylate (BDC) were dissolved with 5 mL of DMF in a 51 mL flask. Afterwards, the container was irradiated in ultrasonic bath for 15 minutes. The resulting powder was separated from the solution using centrifuge and, then, heated to 150 °C for 24 hours.
2.4. Synthesis of PMA@MIL-53 (Fe) using ultrasonic waves
PMA@MIL-53 (Fe) is synthesized using a method similar to MIL-53 (Fe) synthesis. In this sense, heteropoly acid was added to the reaction medium. Moreover, capsulation was occurred in MIL using interaction of facial cavity walls. Primarily, 1.27 g (1 mmol) of FeCl3·6H2O and 1.166 g (1 mmol) of 1–4 benzene decarboxylic acid (BDC) with 5 mL of DMF were completely dissolved in several flasks. Subsequently, different amounts of phosphomolybdic acid hydrate (PMA) (0.01 and 0.02 g) were added to each container and, then, placed in the ambient temperature for 15 minutes under ultrasonic radiation. At the end of the reaction, the synthesized compound was separated using centrifuge and, then, heated for 15 hours at 150 °C (Scheme 1).
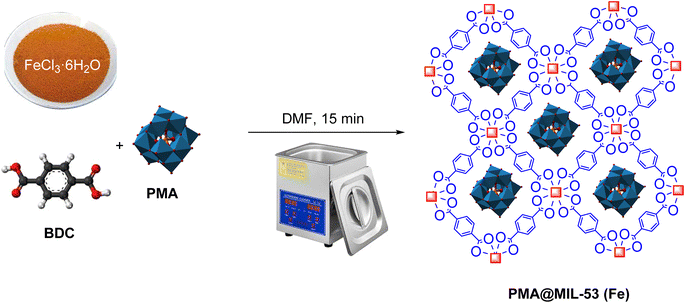 |
| | Scheme 1 Synthesis of PMA@MIL-53 (Fe) catalyst. | |
2.5. Nitration of phenol by PMA@MIL-53 (Fe) catalyst
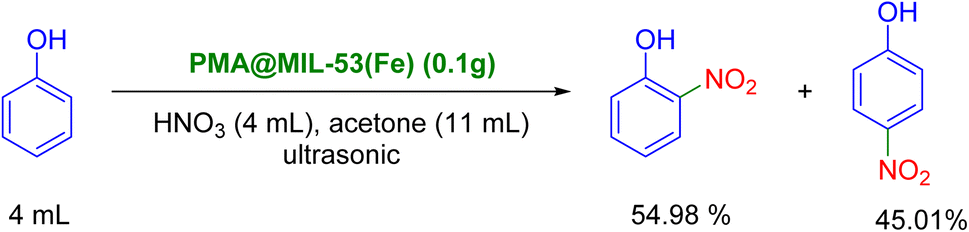
The catalytic activity of PMA@MIL-53 (Fe) was investigated using the phenolic nitration reaction under ultrasonic waves. In this sense, 11 mL acetone, 4 mL phenol, 4 mL nitric acid and 0.1 g of catalyst were added to the flask (25 mL). The reaction was performed in an ultrasonic bath under ultrasonic irradiation. It is worth mentioning that ultrasonic waves provide the energy for the reaction and the temperature was fixed at room temperature by pouring cold water in the bath. The progression of the reaction was investigated using TLC. Moreover, the catalyst was separated using centrifuge after the end of the reaction. The efficiency of the reaction and the percentage of isomers ortho and para were determined using GC as reported in ESI† (Scheme 2).
 |
| | Scheme 2 Nitration of phenol by PMA@MIL-53 (Fe) catalyst. | |
3. Results and discussion
3.1. Characterization of catalyst
In order to confirm the encapsulation of the PMA compound in MIL-53 (Fe), the FT-IR, XRD, XPS, TGA, EDS, ICP-OES, X-ray mapping, Fe-SEM, HR-TEM and nitrogen adsorption–desorption characterization techniques were used.
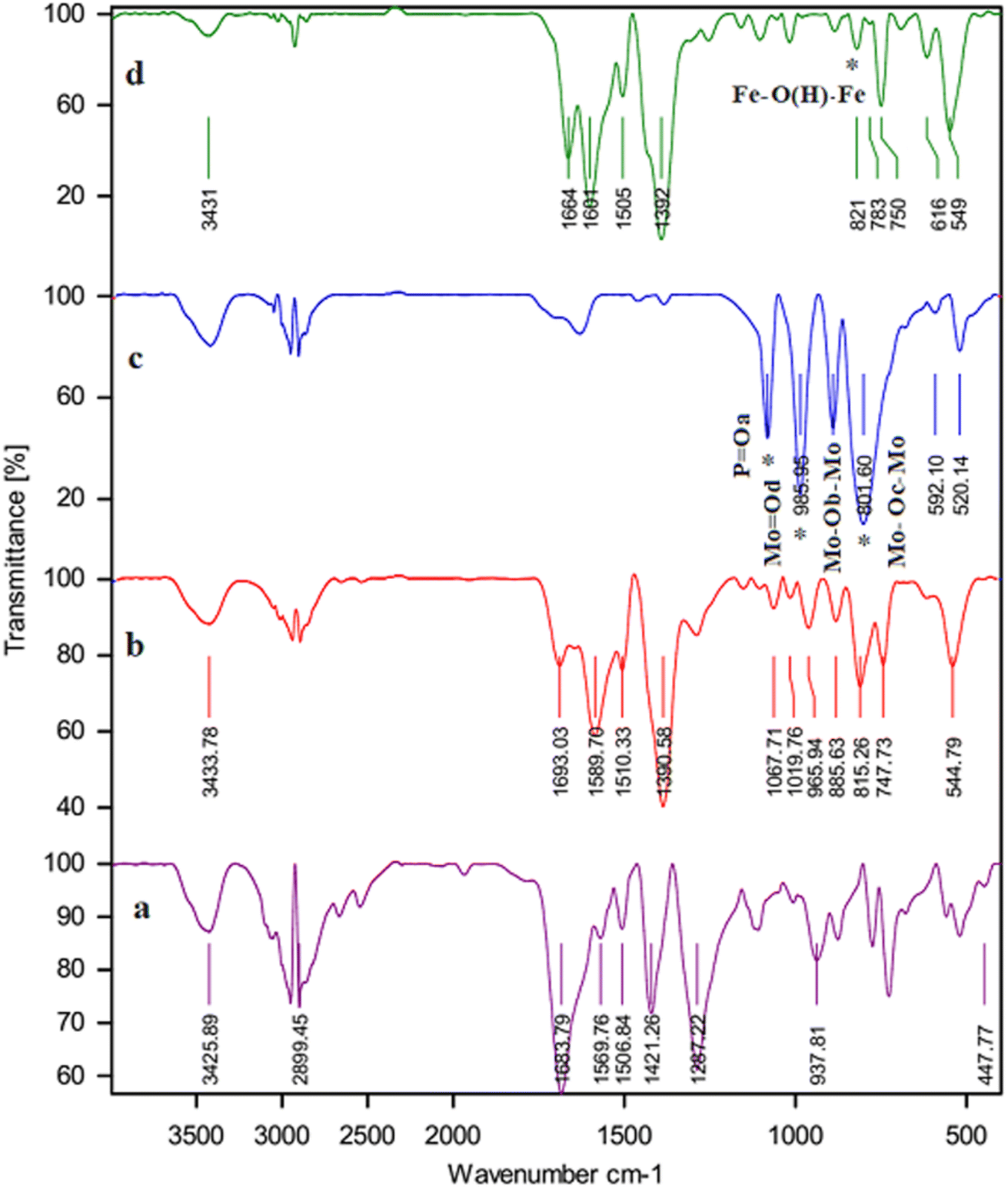
3.1.1. The results of FT-IR. FT-IR spectroscopy confirmed the addition of PMA to the framework structure, as shown in Fig. 1. The comparison of the two FT-IR spectra of the bed and the PMA@MIL-53 (Fe) show that the appearance of new peaks at 792, 889, 968, and 1063 cm−1 correspond to the PMA@MIL-53 (Fe). The peaks at 789![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 888, 969 and 1059 cm−1 are attributed to the vibrations of –P–O, Mo–Oe–Mo and Mo–Oc–Mo. Comparison of the spectra shows that heteropoly acid is well located in the composite structure. Moreover, the vibrational frequency has a slight change due to the interactions of heteropoly acid with the framework. Thus, it can be concluded that the capsule process is well performed.
888, 969 and 1059 cm−1 are attributed to the vibrations of –P–O, Mo–Oe–Mo and Mo–Oc–Mo. Comparison of the spectra shows that heteropoly acid is well located in the composite structure. Moreover, the vibrational frequency has a slight change due to the interactions of heteropoly acid with the framework. Thus, it can be concluded that the capsule process is well performed.
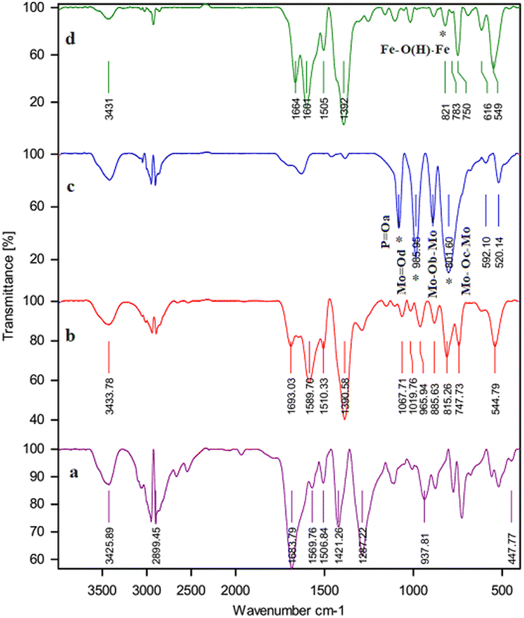 |
| | Fig. 1 FT-IR spectrum (a) H2BDC, (b) MIL-53 (Fe), (c) PMA and (d) PMA@MIL-53 (Fe). | |
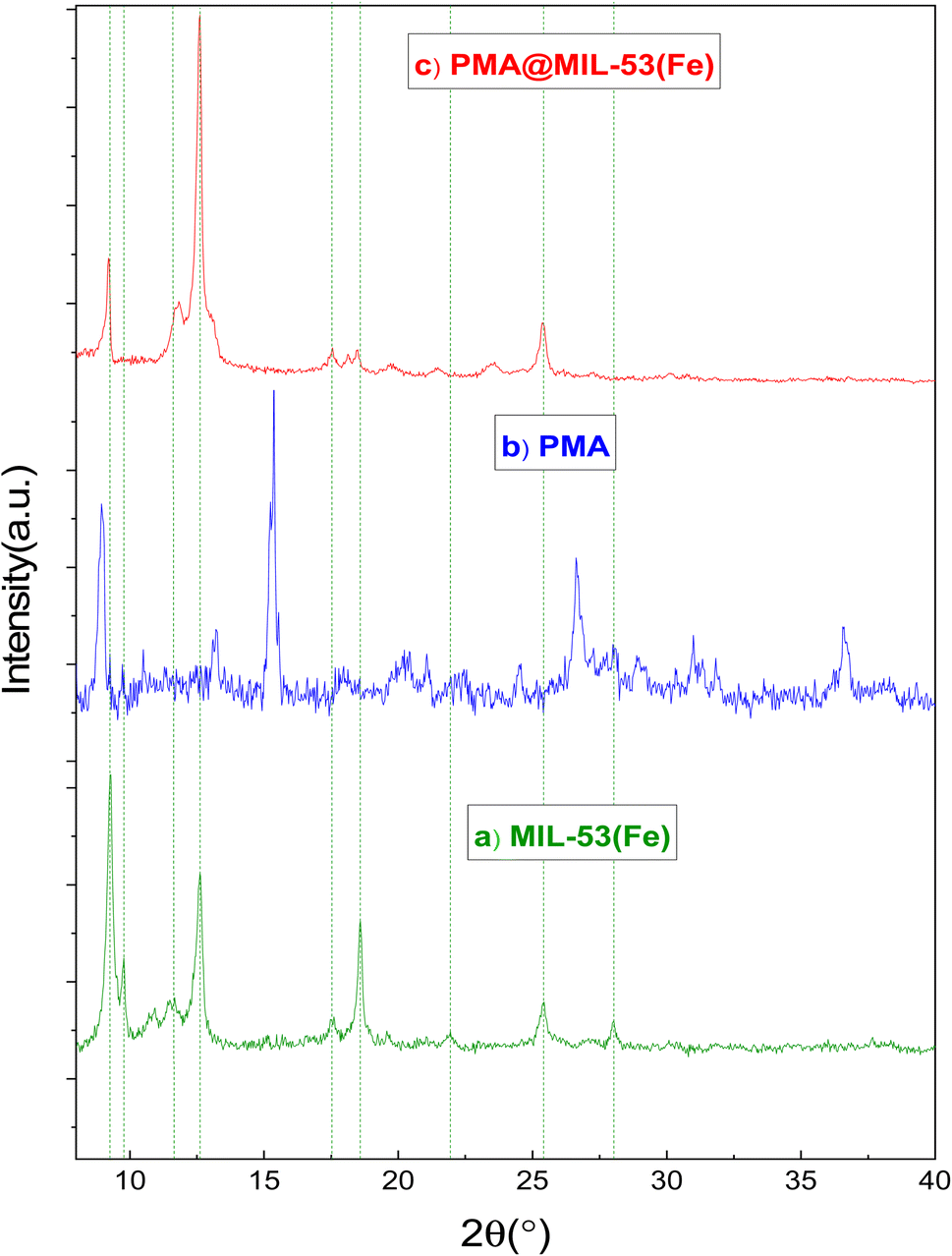
3.1.2. X-Ray diffraction (XRD). Powder XRD measurements were performed using a diffraction spectrometer (diffraction type: D8 advance broker, λ = 1.5406 Å). Fig. 2 shows that the XRD pattern for PMA, MIL-53 and PMA@MIL-53 are similar to previous reports. There are minor changes in the XRD pattern as compared to MIL-53 (Fe) due to capsulation heteropoly acid. These changes show that PMA is encapsulated in the framework. XRD patterns indicate that MIL-53 structure is not destroyed and structural characteristics are preserved during the process of encapsulating.
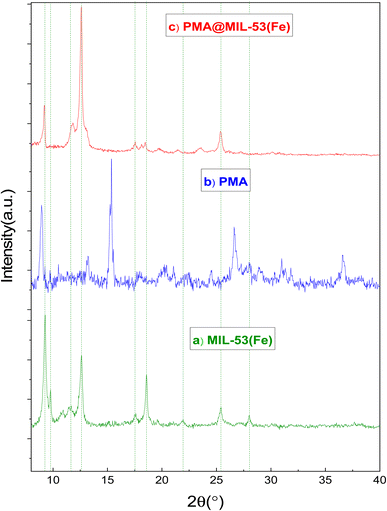 |
| | Fig. 2 XRD patterns (a) MIL-53 (Fe) and (b) PMA (c) PMA@MIL-53 (Fe). | |
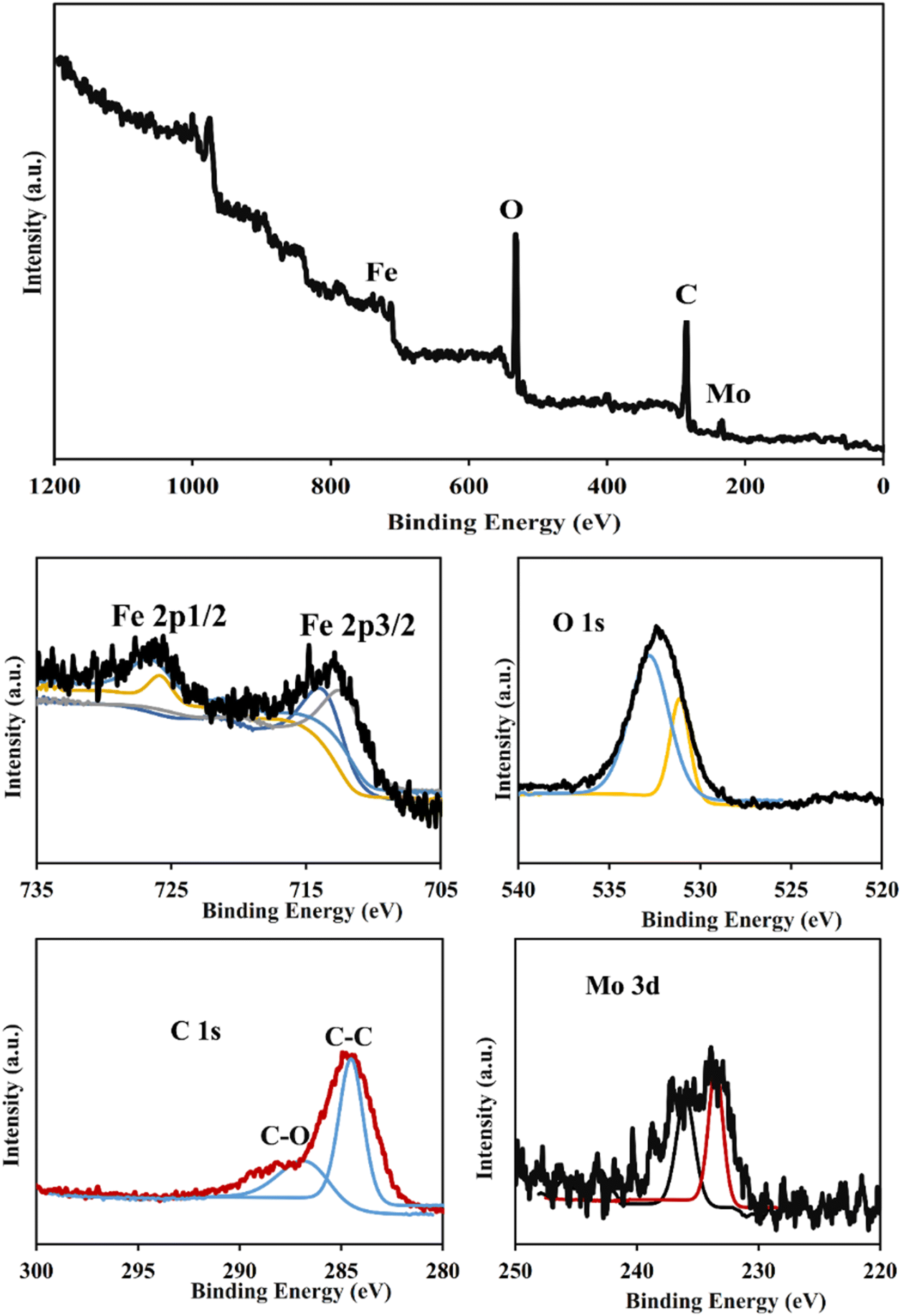
3.1.3. X-Ray photoelectron spectroscopy (XPS) analysis. XPS analysis was performed to study the elemental composition and chemical states of PMA@MIL-53 (Fe) MOF (Fig. 3). The XPS survey spectra show the presence of Fe, C, and O elements in the prepared sample that is in good agreement with the previous report on its patent MOF (MIL-53 (Fe)). The high-resolution XPS spectra show two peaks at around 713.1 eV and 726.3 eV binding energy which are attributed to the Fe 2p (Fe 2p3/2 and Fe 2p1/2 of Fe(III)), respectively. The comparison of the XPS spectra of the PMA@MIL-53 (Fe) MOF to its parent MIL-53 (Fe) shows a slight shift on Fe peaks along with the appearance of new peaks at 233.4 eV and 235.8 eV corresponding to the Mo due to the interactions of heteropoly acid with the framework. This result confirms the addition of PMA to the MIL-53 (Fe) MOF.
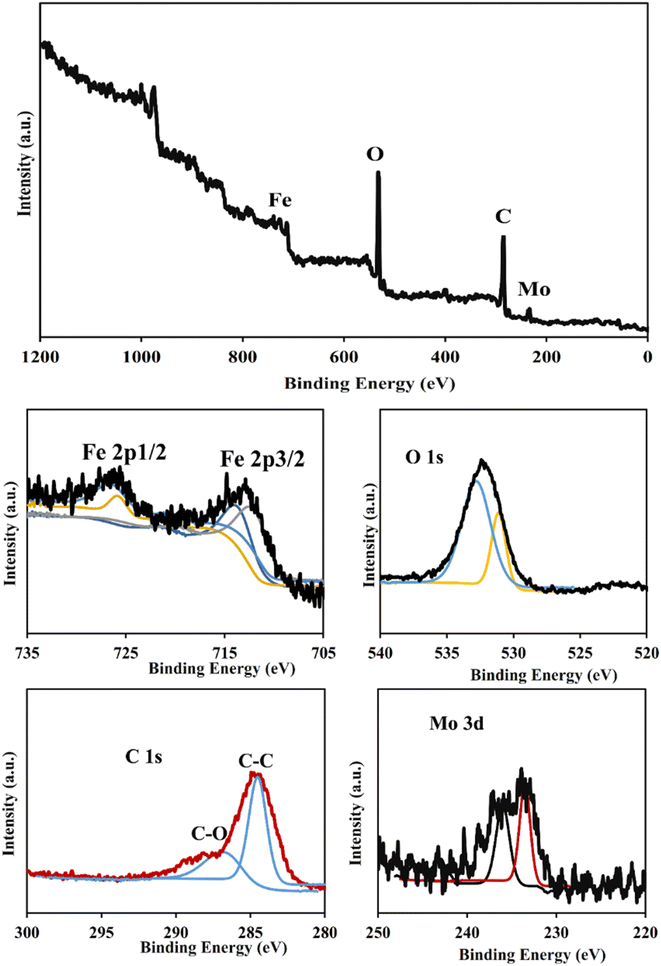 |
| | Fig. 3 XPS analysis of PMA@MIL-53 (Fe). | |
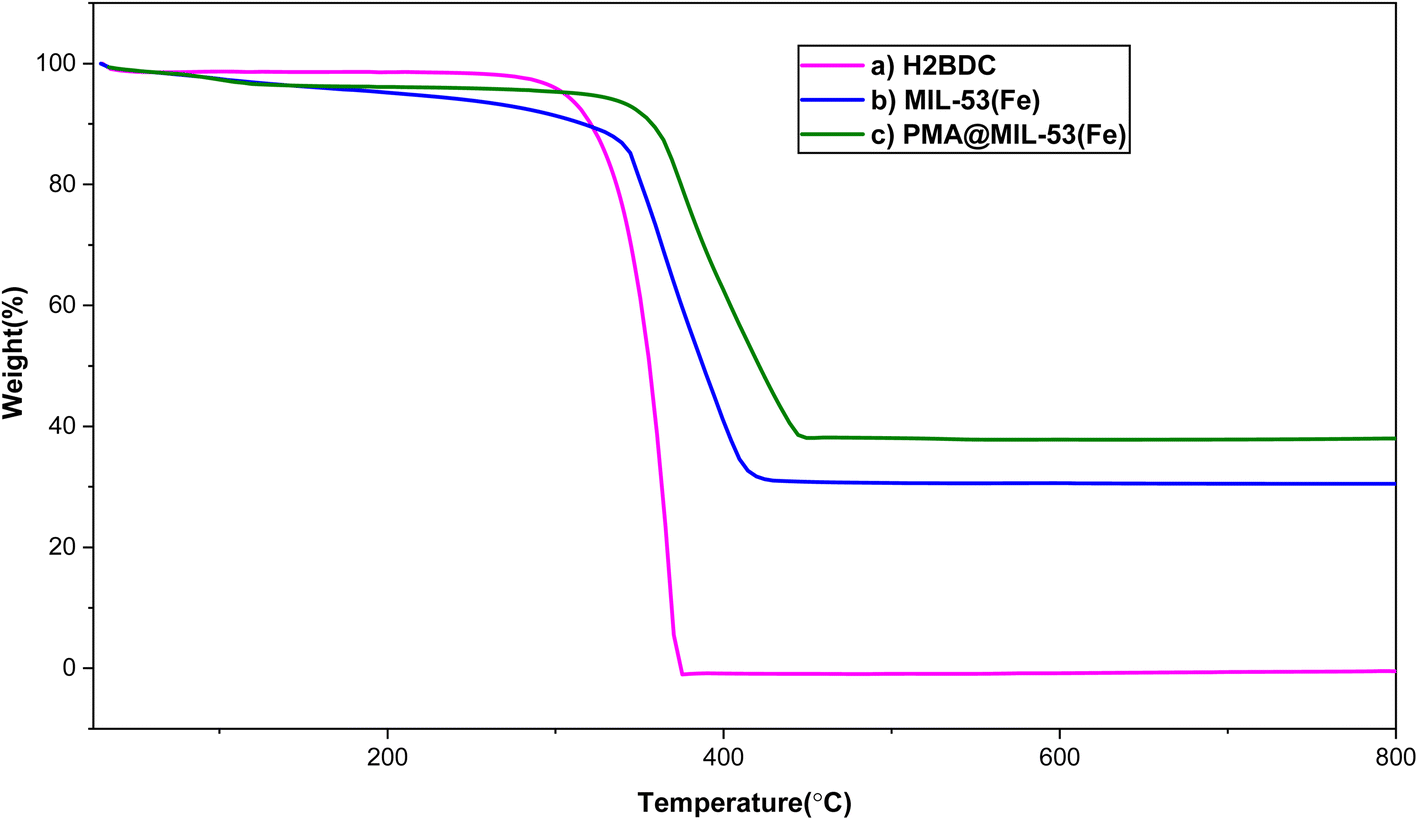
3.1.4. The results of thermal gravimetric analysis (TGA). Thermal behavior of H2BDC, MIL-53 (Fe) and PMA@MIL-53 was investigated using TGA analysis. Besides, the obtained data are presented in Fig. 4. Regarding the curves, two major weight losses were observed. The first stage is at temperature below 211 °C which can be due to the removal of some moisture in the samples and DMF in the MOFs cavities. In addition, the H2BDC is stable at up to 285 °C and, significantly, it was completely decomposed from 300 to 375 °C. In this sense, it is worth mentioning that it was selected as our reference for the decomposition of organic groups in the prepared samples. Regarding the TGA curves of MIL-53 and PMA@MIL-53 comparison, it is clear that the weight loss of PMA@MIL-53 is much less than that of H2BDC and MIL-53 (Fe) respectively, which is due to the heteropoly acid place in the cavities of the framework. It is well concluded that the encapsulation process of PMA heteropoly acid has been successfully performed. In addition, the thermal stability of the sample increased via PMA addition to the structure.
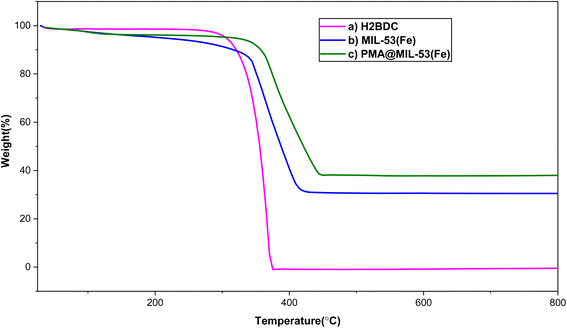 |
| | Fig. 4 TGA curves of (a) H2BDC, (b) MIL-53 (Fe) and (c) PMA@MIL-53. | |
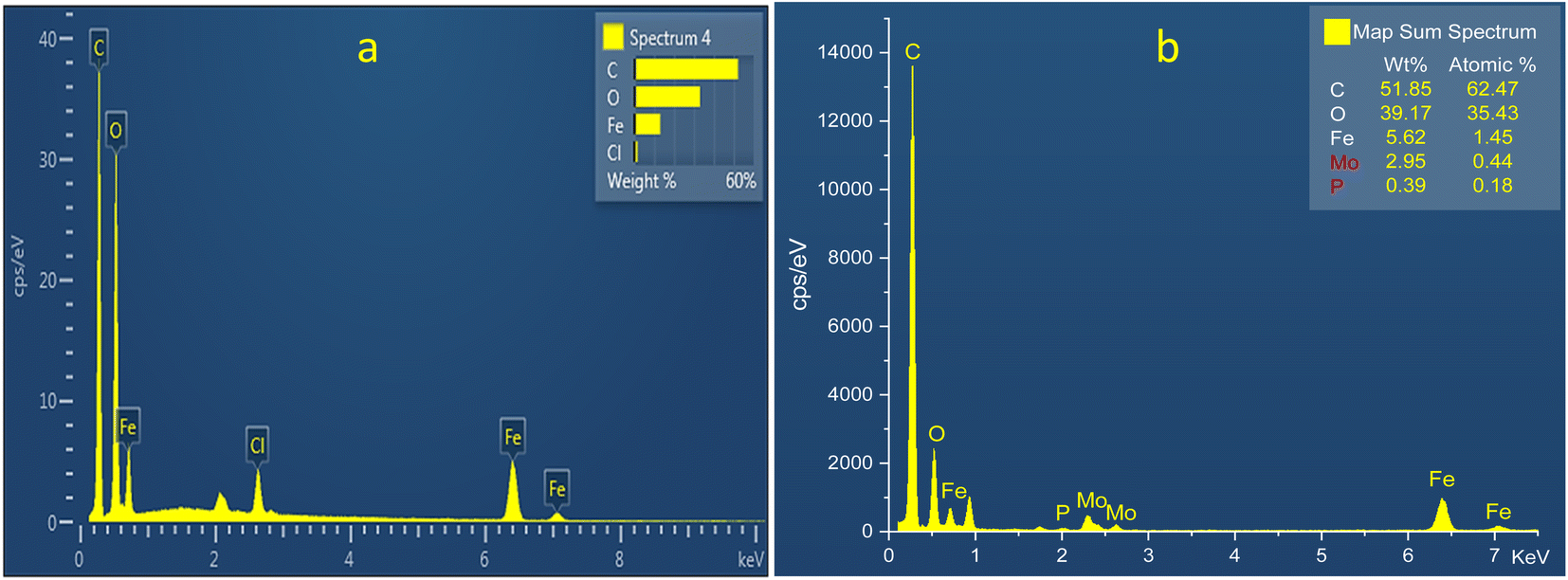
3.1.5. Energy-dispersive X-ray spectroscopy. The elemental composition of the synthesized MIL-53 (Fe) (b) PMA@MIL-53 (Fe) composites was carried out using EDX analysis (Fig. 5). The EDX measurements indicate that all of the required elements, including C (kα = 0.277 keV), O (kα = 0.525 keV) and Fe (Lα = 0.705, kα = 6.924 and kβ = 7.07 keV) species in MIL-53 (Fe), confirm the formation of the targeted MOF material. Additionally, the EDX measurements on PMA@MIL-53 (Fe) composites represent that, all of the X-ray signals of its parent MOF material, the position of a sharp peaks of Mo (Lα = 2.293 keV) and P (Kα = 2.013 keV) elements is attributed to PMA segments. These results confirm the encapsulation of PMA nanostructures into MIL-53 (Fe) metal–organic frameworks and the successful formation of the target heteropoly acid heterogeneous catalyst.
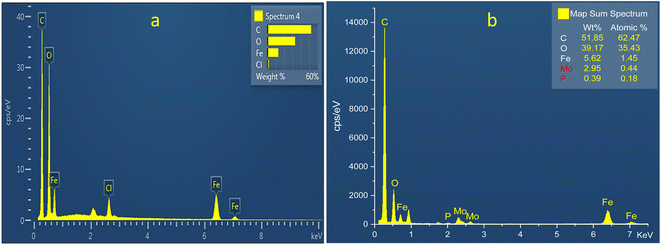 |
| | Fig. 5 EDS analysis of (a) MIL-53 (Fe) (b) PMA@MIL-53 (Fe). | |
3.1.6. Inductively coupled plasma mass spectrometry. Inductively coupled plasma analysis of PMA (10 wt%) @MIL-53 (Fe) indicates that the sample contains 60985 ppm of molybdenum, while molybdenum forms 63.075 wt% of phosphomolybdic acid (PMA). Regarding that 1 wt% = 10000 ppm, the amount of molybdenum is 6.0985 wt in this sense, the amount of molybdenum – as equivalent to phosphomolybdic acid – must be calculated. Therefore, by dividing 6.0985 (wt% Mo) by 63.075 and multiplying it by 100, the amount of PMA is obtained as 9.67 wt% (the actual value of the loaded PMA). It should be noted that there is often a difference between theoretical values and actual values of the loaded PMA in composite. As shown in Table 1, in higher values of PMA, the amount of this difference is greater. The reason for this difference is logical and predictable. Accordingly, two main reasons (in addition to minor systematic and individual errors) can be presented: (a) for many reasons that are beyond the scope of this discussion, the distribution of PMA in MIL-53 (Fe) cavities is random, and we cannot assume a uniform distribution for PMA in the MIL-53 (Fe) structure. For example, we cannot say that all holes of MIL-53 (Fe) are filled by PMA. (b) The second major reason is related to the sampling of the synthesized composite. Therefore, the difference of the samples in the analysis causes more influence of the factor, the non-uniformity of PMA distribution in MIL-53 (Fe) and as a result, it will be an influential factor in the observed error.77
Table 1 The characteristics of PMA@MIL-53 (Fe) composite
| Entry |
Sample |
Mo (ppm) |
Actual loading of PMA (wt%) |
| 1 |
MIL-53 (Fe) |
— |
— |
| 2 |
PMA (10 wt%)@MIL-53 (Fe) |
60985 |
9.67 |
| 3 |
PMA (20 wt%)@MIL-53 (Fe) |
121545 |
19.27 |
3.1.7. Elemental mapping analysis. In addition, the distribution of the elements was investigated using EDX-mapping analysis (Fig. 6). Elemental mapping images revealed the high density of C, O and Fe elements as the MOF support and also the high content of Mo and P elements as being uniformly distributed around the composite system, confirming the formation and excellent accessibility of the encapsulated heteropoly acid catalyst.
 |
| | Fig. 6 X-Ray mapping analysis of PMA@MIL-53 (Fe). | |
3.1.8. Scanning electron microscopy (SEM). The morphology of the catalyst was observed using scanning electron microscopy (SEM) (LEO 1430VP models). Fig. 7 shows the structure of MIL-53 (Fe) before and after the capsulation PMA. The results show that with the addition of hetero poly acid to the framework, the surface became rough and uneven, which indicates the presence of PMA. Based on the results, it is assured that the hetero poly acid was located in the pores of the framework.
 |
| | Fig. 7 SEM image of (a) MIL-53 (Fe) and (b) PMA@MIL-53 (Fe). | |
3.1.9. High resolution transmission electron microscopy (HR-TEM). The particle size and morphology of the PMA@MIL-53 (Fe) composite were measured using HR-TEM analysis (Fig. 8). The obtained high magnification TEM graphs illustrated that the size distribution of the MIL-53 (Fe) crystals is not uniform and crystal sizes ranged from 200–500 nm. The magnified TEM image reveals that very small black particles in different sizes (1–2 nm) were closely attached to the surface of the MIL-53 (Fe) crystals, which indicate the existence of PMA in the PMA@MIL-53 (Fe) composite. The above results show that the MIL-53 (Fe) was well prepared and PMA functionalities have successfully encapsulated onto its pores.
 |
| | Fig. 8 HR-TEM images PMA@MIL-53 (Fe). | |
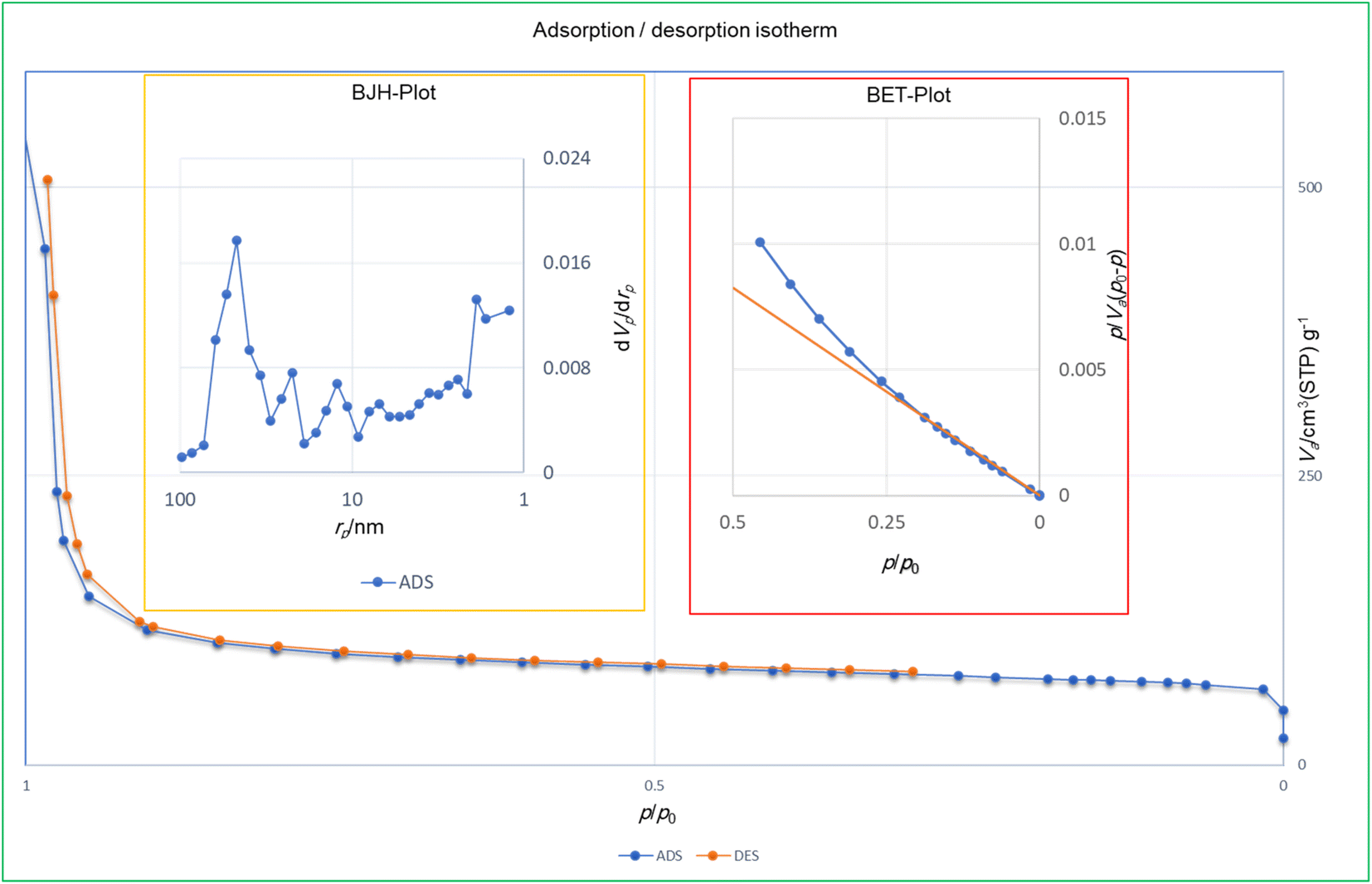
3.1.10. Nitrogen adsorption–desorption measurements. The porosity properties of the prepared PMA@MIL-53 (Fe) composite were measured using nitrogen adsorption–desorption analysis (Table 2). Surface areas, pores volumes and average pore sizes of the catalysts were measured using N2 physi-sorption and Micromeritics ASAP 2000. Fig. 5 shows the nitrogen adsorption–desorption isotherms of the PMA@MIL-53 (Fe) framework. The results show that, with the addition of PMA, the total pore volume (Vtotal) and mean pore diameter (Dmean) decreased from 0.067928 cm3 g−1 to 0.0215 cm3 g−1 and 18.23 to 16.784 nm, respectively. It is worth to mention that according to literature,78 MIL-53 (Fe) only opens its pores in the presence of guest molecules; therefore, unlike MIL-101 it does not have a high surface area. Additionally, in this work the surface area of pure MIL-53 (Fe) is significantly bigger than that of earlier reports on identical samples that were produced under optimal conditions.79 It can also be seen from Table 2 that the surface area decreases from 320 (m2 g−1) to 136.39 (m2 g−1) for MIL-53 (Fe) and PMA (20 wt%)@MIL-53 (Fe), due to the presence of heavy phosphomolybdic moieties.80 This evidence confirms the encapsulation of PMA and the formation of the composite (Fig. 9).
Table 2 Summarized characteristics of PMA@MIL-53 (Fe) composite and its parents
| Entry |
Sample |
SBETa (m2 g−1) |
| Specific surface area. |
| 1 |
MIL-53 (Fe) |
320 |
| 2 |
PMA (10 wt%)@MIL-53 (Fe) |
218.2 |
| 3 |
PMA (20 wt%)@MIL-53 (Fe) |
136.39 |
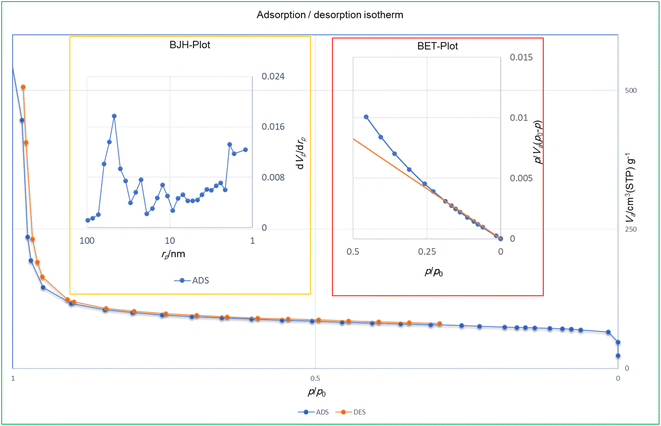 |
| | Fig. 9 Surface area, pore volume and pore size distribution curves of PMA@MIL-53 (Fe). | |
3.2. Catalytic study
3.2.1. Optimization the nitration of phenol. Regarding the significance of nitrated phenol derivatives, the possibility of phenol nitration was investigated using the high scale electrophilic aromatic substitution reactions of phenol with nitric acid. Besides, we also applied PMA@MIL-53 (Fe) MOF as an effective heterogeneous heteropoly acid catalyst. In this sense, the effect of various reaction parameters (i.e. catalyst loadings and reaction time) was investigated. Accordingly, the results are shown in Table 3.
Table 3 Screening the reaction parameters for the nitration of phenol under the catalysis of PMA@MIL-53 (Fe)

|
| Entry |
Catalyst |
Time (min) |
Yielda,b,c (%) |
| Total |
Ortho |
Para |
| GC yields. Conditions: phenol (4 mL), nitric acid (4 mL) PMA@MIL-53 (Fe) (100 mg) in acetone (10 mL) at room temperature under ultrasound irradiation. The reaction in the absence of HNO3. |
| 1 |
— |
5 |
NR |
— |
— |
| 2 |
MIL-53 (Fe) |
5 |
NR |
— |
— |
| 3 |
PMA (10 wt%)@MIL-53 (Fe) |
5 |
29.5 |
16.41 |
13.09 |
| 4 |
PMA (10 wt%)@MIL-53 (Fe) |
10 |
75.13 |
41.11 |
34.02 |
| 5 |
PMA (10 wt%)@MIL-53 (Fe) |
15 |
95.62 |
51.91 |
43.71 |
| 6 |
PMA (20 wt%)@MIL-53 (Fe) |
5 |
38.54 |
20.71 |
17.83 |
| 7 |
PMA (20 wt%)@MIL-53 (Fe) |
10 |
76.94 |
44.02 |
32.92 |
| 8 |
PMA (20 wt%)@MIL-53 (Fe) |
15 |
99.99 |
54.98 |
45.01 |
| 9 |
PMA (20 wt%)@MIL-53 (Fe) |
15 |
NRc |
— |
— |
The present study was initiated using the reaction of phenol (4 mmol) with nitric acid (4 mmol) as the model reaction in 10 mL of acetone as solvent and in presence of 0.1 g of PMA@MIL-53 (Fe) catalyst (Table 1) under ultrasonic irradiation for different reaction times. The transformation process with the reaction time for the nitration reaction was monitored using TLC and, then, the yields were calculated using gas chromatography (GC). It was observed that, while increasing the required reaction time, product yield was also increased. These results show that time plays a critical role in this catalytic reaction. In addition, a clear difference was found in the reaction selectivity and efficiency under different reaction times (entries 1–3, Table 3). Moreover, 95.62% yield of the ortho- and para-mononitrophenols mixture was concurrently obtained in 15 min (ortho/para ratio was 54:46). Afterwards, the effect of PMA loading on the reaction was evaluated (Table 3 entries 4–6). It was observed that when the concentration of PMA in the catalyst increased to 20 mg the time required for the completion of the reaction decreased. It was found out that the increase in PMA loading in MOF catalyst improves the reaction yield (up to 99.99%) and selectivity (ortho/para ratio was 55:45) which suggests that the phenol substrate is almost completely converted to its corresponding mononitrated ortho- and para-nitrophenols. These results show that the PMA plays a critical role in this catalytic reaction. Hence, the results also show that ortho/para-nitrophenols can be effectively prepared in presence of PMA@MIL-53 (Fe) (including 20 mg of PMA content) catalyst in acetone solvent system at room temperature, giving good yield in less reaction time (Table 3). Finally, the results of these catalytic investigations indicated that the ortho-nitro product was formed easier than its para-nitro isomer and provided the corresponding products in higher yields. Moreover, it is worth mentioning that the reaction involved a slight ortho-selectivity for all of the tested reactions. The exact explanation for this selectivity is due to the hydrogen bonding in its related intermediate, which causes stability and ease of formation. It is worth to mention that the reaction was evaluated in the absence of HNO3 or the absence of catalyst, in both cases the reaction process stalled.
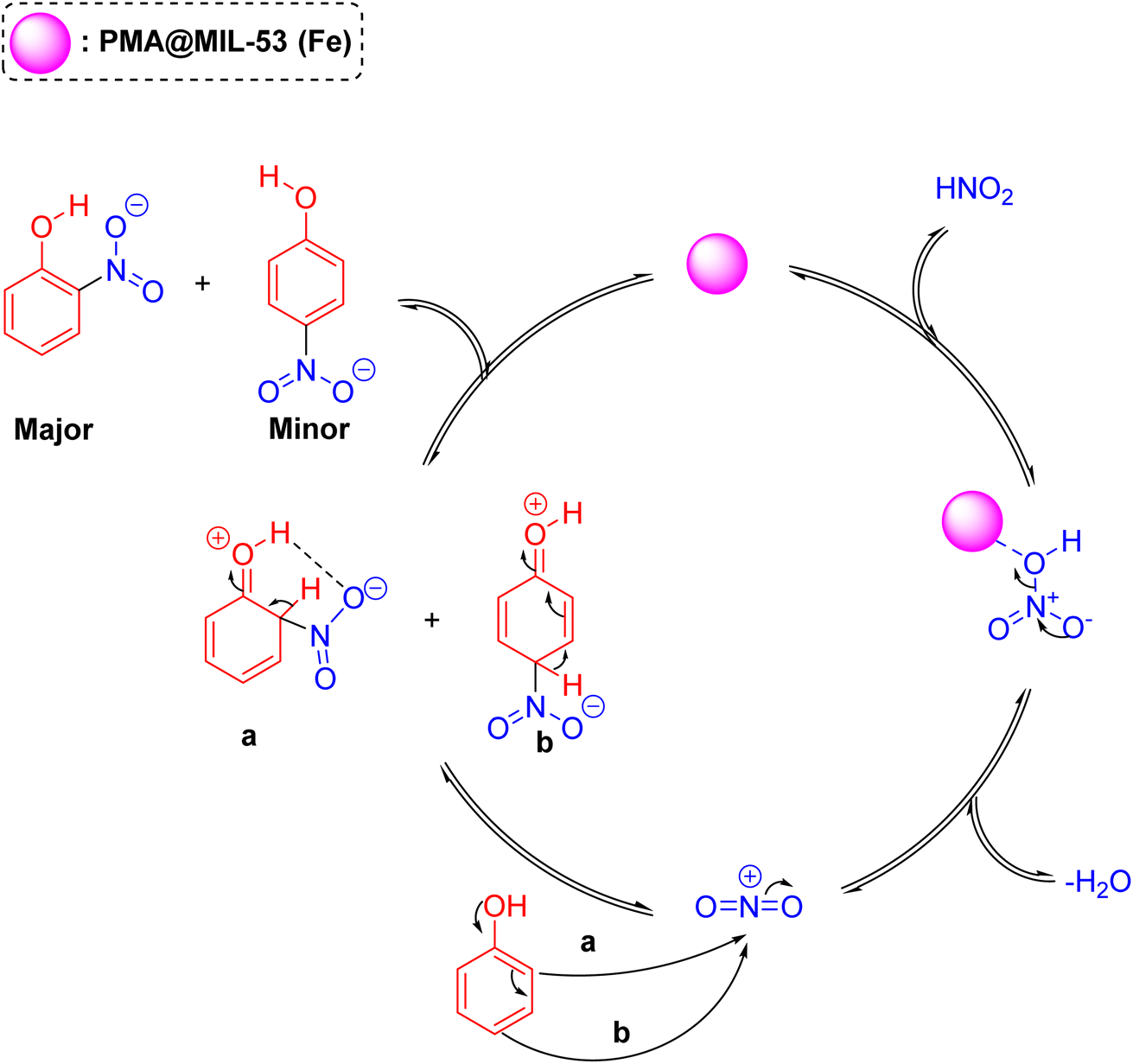
3.3. Mechanism
The plausible mechanism of the phenol nitration reaction in presence of the PMA@MIL-53 (Fe) acid-catalyst is shown in Scheme 3. It encompasses two-steps: first, generating nitronium ion (NO2+) via acid-catalyzed water elimination from nitric acid and, then, electrophilic substitution reaction. In the second step, the π electrons were delocalized around the aromatic rings, leading to break one of the NO bonds to avoid a five-valent nitrogen, finally the removal of a proton from the substituted carbon leads to the aromatization of products.
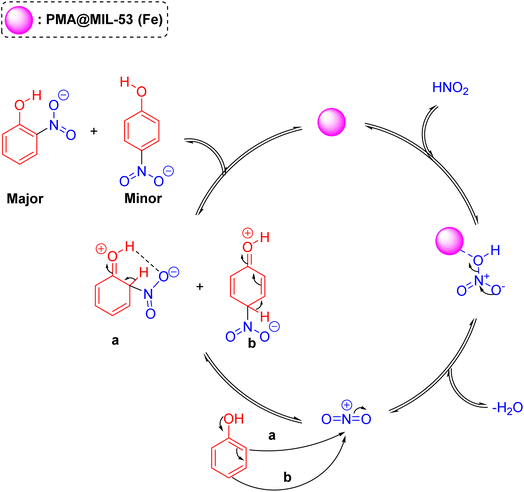 |
| | Scheme 3 Plausible mechanism for the nitration of phenol catalyzed by PMA@MIL-53 (Fe) solid acid. | |
3.4. Recovery and reusability
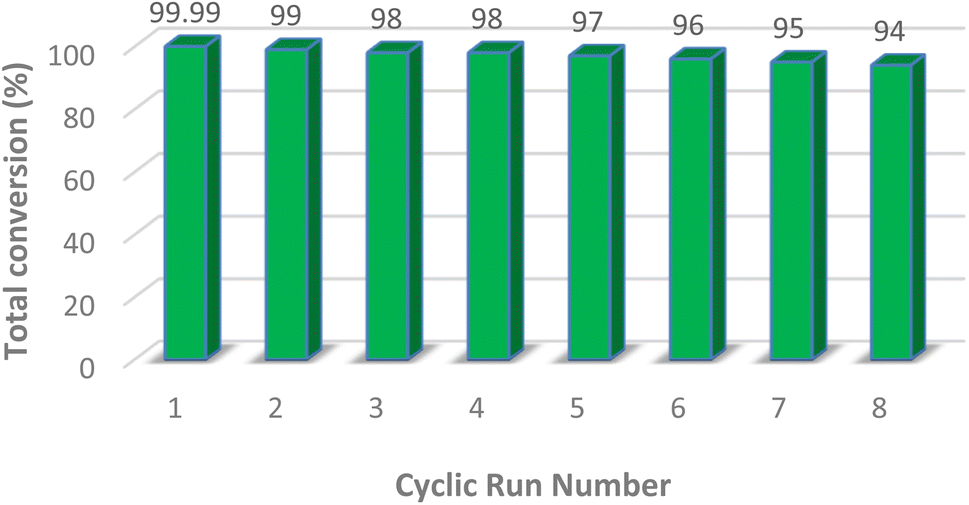
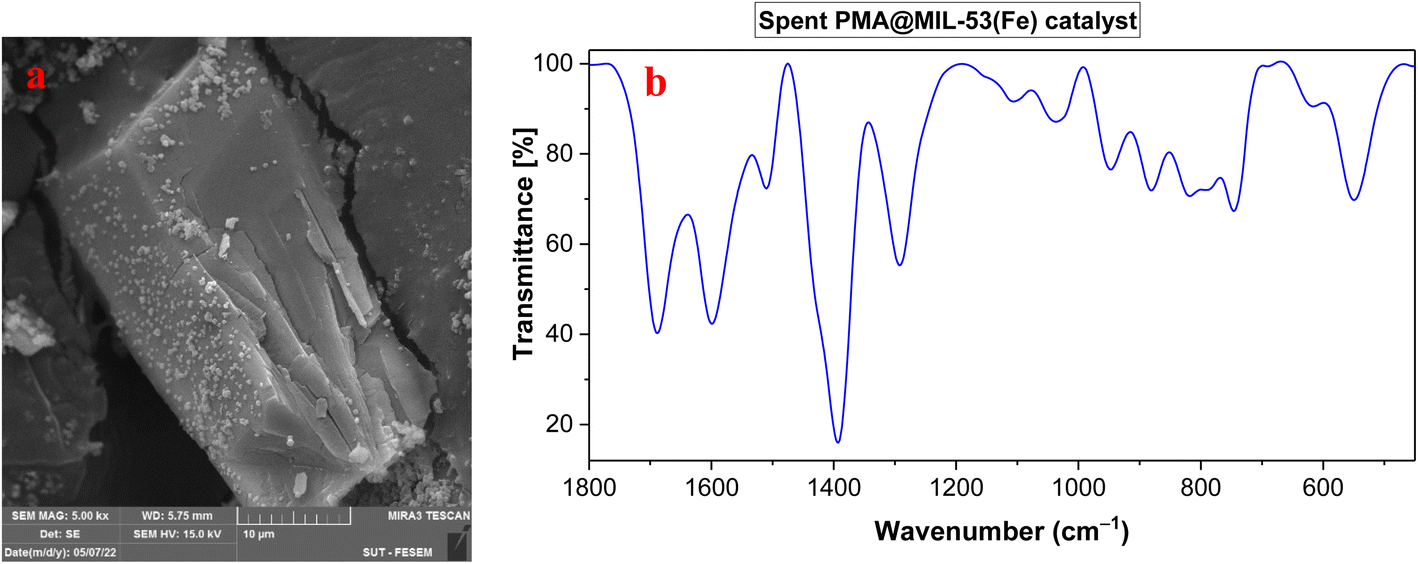
As an added benefit of PMA@MIL-53 (Fe), it can be recovered and reused for the same reaction several times. Herein, PMA@MIL-53 (Fe) was separated from the reaction mixture by centrifugation, rinsed multiple times with ethanol, dichloromethane and acetone, dried at 80 °C under vacuum and, further, reused straightforwardly for the fresh nitration reaction. Based on the result of its recyclable catalytic effectiveness, this became clear that the catalyst has such an excellent catalytic influence even after becoming reused for 8 attempts (Fig. 10). In addition, SEM and FT-IR spectra of the recovered catalyst were compared with the fresh one that showed no morphological, functionalities changes respectively (Fig. 11a and b), thus providing a strong evidence for excellent durability and stability of the catalyst.
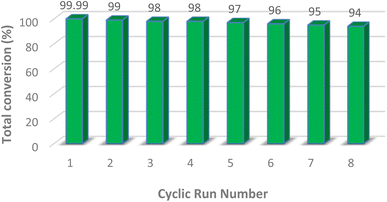 |
| | Fig. 10 Reusability of PMA@MIL-53 (Fe) catalyst. | |
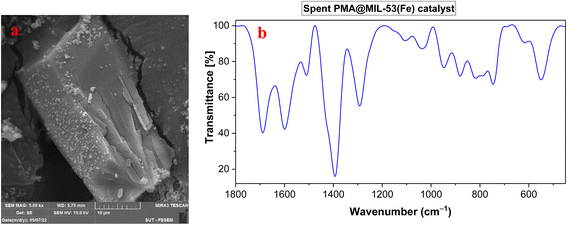 |
| | Fig. 11 (a) SEM image and (b) FT-IR spectrum of the recovered PMA@MIL-53 (Fe) catalyst. | |
3.5. Hot filtration test
The heterogeneous nature was investigated using a hot filtration study of the PMA@MIL-53 (Fe) catalyst under optimum operating conditions to determine whether PMA was leaching out or not from MIL-53 (Fe). This test reveals that the leaching of PMA did not take place after the reaction, clarifying that the PMA was still bound to the MIL-53 (Fe) framework. This provides strong evidence of its structural stability and gives good results even after its multiple reuses.
3.6. Comparison
We have also compared the efficiency of our catalyst with some literature reports. The catalyst is effective against the nitration reaction under milder conditions at shorter reaction time and higher yields with more regioselectivity and reusability (Table 4). It shows the uniqueness of the PMA@MIL-53 (Fe) heterogeneous catalyst over other catalysts.
Table 4 Comparison of the catalytic activity of PMA@MIL-53 (Fe) in the nitration of phenol with other reported catalyst systems in the literature
| Entry |
Catalyst |
Time (min) |
Yield (%) |
Ref |
| Total |
Ortho |
Para |
| 1 |
Montmorillonite KSF |
60 |
84 |
44 |
40 |
81 |
| 2 |
Tetrabutylammonium chromate |
2880 |
80 |
44 |
36 |
82 |
| 3 |
[KHSO4·2KHSO5·K2SO4] |
180 |
79 |
40 |
39 |
83 |
| 4 |
Cerium(IV) ammonium nitrate (CAN) |
1440 |
69 |
28 |
41 |
84 |
| 5 |
Silica sulfuric acid |
25 |
79 |
36 |
43 |
85 |
| 6 |
PMA@MIL-53 (Fe) |
15 |
99.99 |
54.98 |
45.01 |
This work |
4. Conclusion
We have developed a novel heteropoly acid encapsulated in MOF support and explored its efficiency as catalyst for phenol nitration reactions. The mentioned catalyst shows good to excellent yields in acetone at room temperature. Moreover, this catalyst can be effortlessly removed from the reaction mixture via simple filtration or centrifugation and reused eight times which shows its stability, recyclability and longevity.
Conflicts of interest
There are no conflicts to declare.
References
- K. A. Juárez-Ornelas, J. O. C. Jiménez-Halla, T. Kato, C. R. Solorio-Alvarado and K. Maruoka, Org. Lett., 2019, 21, 1315–1319 CrossRef PubMed.
- B. T. Ergan, M. Bayramoğlu and E. Şahin, Journal of Microwave Power and Electromagnetic Energy, 2017, 51, 314–324 Search PubMed.
- E. Bolzacchini, M. Bruschi, G. Galliani, J. Hjorth, M. Orlandi and B. Rindone, in ACS National Meeting Book of Abstracts, 2007 Search PubMed.
- Y. H. Sriram, T. Fatima, K. C. Rajanna, M. S. Kumar and R. M. Raju, Int. J. Chem. Kinet., 2017, 49, 622–632 CrossRef CAS.
- E. Bolzacchini, J. Hjorth, S. Meinardi, M. Orlandi, B. Rindone and E. Rosenbohm, in ACS National Meeting Book of Abstracts, 2000, vol. 40, pp. 395–397 Search PubMed.
- S. Mondal, J. Singh, S. Singh, S. Vishwakarma, K. Mitra, A. Kumari, R. Singh, S. K. Sen Gupta and B. Ray, New J. Chem., 2020, 44, 10878–10884 RSC.
- C. Zhang, J. Dong, M. Liu, W. Zhao and D. Fu, J. Hazard. Mater., 2019, 373, 547–557 CrossRef CAS PubMed.
- C.-C. Du, X.-F. Wang, S.-B. Zhou, D.-Z. Wang and D. Jia, CrystEngComm, 2017, 19, 6758–6777 RSC.
- C. A. Bunton, E. D. Hughes, G. J. Minkoff and R. I. Reed, Nature, 1946, 158, 514–515 CrossRef CAS.
- N. G. Tran, H. Kalyvas, K. M. Skodje, T. Hayashi, P. Moënne-Loccoz, P. E. Callan, J. Shearer, L. J. Kirschenbaum and E. Kim, J. Am. Chem. Soc., 2011, 133, 1184–1187 CrossRef CAS PubMed.
- T. C. Bruice, M. J. Gregory and S. L. Walters, J. Am. Chem. Soc., 1968, 90, 1612–1619 CrossRef CAS.
- M. A. Mondal, D. Mandal and K. Mitra, J. Chem. Sci., 2017, 129, 39–43 CrossRef CAS.
- M. R. Patil, P. H. Mohite, S. Shisodia and R. S. Keri, Lett. Org. Chem., 2015, 12, 129–135 CrossRef CAS.
- L. C. Raiford, J. Am. Chem. Soc., 1922, 44, 158–165 CrossRef CAS.
- M. Suresh, K. C. Rajanna, Y. H. Sriram and Y. R. Rao, SN Appl. Sci., 2019, 1, 485 CrossRef.
- C. M. R. Rocha, J. A. R. Rodrigues, P. J. S. Moran and R. Custodio, J. Mol. Model., 2014, 20, 2524 CrossRef PubMed.
- X. Cheng, Q. Chen, Y. Li, G. Huang, Y. Liu, S. Lu, Y. Zheng, W. Qiu, K. Lu, X. Qiu, F. Bianchi, C. Yan, B. Yuan, M. Shao, Z. Wang, M. R. Canagaratna, T. Zhu, Y. Wu and L. Zeng, Environ. Sci. Technol., 2021, 55, 4410–4419 CrossRef CAS PubMed.
- C. Wu, Q. Bian, T. Ding, M. Tang, W. Zhang, Y. Xu, B. Liu, H. Xu, H.-B. Li and H. Fu, ACS Catal., 2021, 11, 9561–9568 CrossRef CAS.
- R. S. Glass, J. Chem. Educ., 1983, 60, A283 CrossRef.
- W. Zhang, J. Zhang, S. Ren and Y. Liu, J. Org. Chem., 2014, 79, 11508–11516 CrossRef CAS PubMed.
- M. Mohammadi, M. Khodamorady, B. Tahmasbi, K. Bahrami and A. Ghorbani-Choghamarani, J. Ind. Eng. Chem., 2021, 97, 1–78 CrossRef CAS.
- M. Mohammadi and A. Ghorbani-Choghamarani, Appl. Organomet. Chem., 2022, 36, e6905 CrossRef CAS.
- B. B. Thummar, U. P. Tarpada and D. K. Raval, J. Heterocycl. Chem., 2014, 51, 1740–1746 CrossRef CAS.
- S. P. N. Sudhan, R. N. Ahmed, H. Kiyani and S. S. Mansoor, J. Saudi Chem. Soc., 2018, 22, 269–278 CrossRef CAS.
- Z. L. Shen, X. P. Xu and S. J. Ji, J. Org. Chem., 2010, 75, 1162–1167 CrossRef CAS PubMed.
- V. Kumar, S. Ghosh, A. K. Saini, S. M. Mobin and B. Mondal, Dalton Trans., 2015, 44, 19909–19917 RSC.
- A. Kalita, R. C. Deka and B. Mondal, Inorg. Chem., 2013, 52, 10897–10903 CrossRef CAS PubMed.
- B. Baghernejad, M. M. Heravi, H. A. Oskooie and F. F. Bamoharram, Bull. Chem. Soc. Ethiop., 2012, 26, 145–152 CrossRef CAS.
- Z. Tunalı, K. Sagdic, F. Inci and B. Ö. Öztürk, React. Chem. Eng., 2022, 7, 1617–1625 RSC.
- H. Yu, Q. Zhong, Y. Liu, Y. Guo, Y. Xie, W. Zhou and W. Yao, Ultrason. Sonochem., 2020, 64, 104844 CrossRef CAS PubMed.
- F. Penteado, B. Monti, L. Sancineto, G. Perin, R. G. Jacob, C. Santi and E. J. Lenardão, Asian J. Org. Chem., 2018, 7, 2368–2385 CrossRef CAS.
- Z. Zhao, Y. Xue, G. Xu, D. Chen, J. Zhou, P. Liu, S. Han and H. Lin, Int. J. Energy Res., 2017, 41, 1081–1095 CrossRef CAS.
- M. K. Elmkaddem, P. De Caro, S. Thiébaud-Roux, Z. Mouloungui and E. Vedrenne, OCL: Oilseeds Fats, Crops Lipids, 2016, 23(5), D507 CrossRef.
- A. Nikseresht, A. Daniyali, M. Ali-Mohammadi, A. Afzalinia and A. Mirzaie, Ultrason. Sonochem., 2017, 37, 203–207 CrossRef CAS PubMed.
- A. Afzalinia, A. Mirzaie, A. Nikseresht and T. Musabeygi, Ultrason. Sonochem., 2017, 34, 713–720 CrossRef CAS PubMed.
- H. Veisi, A. Nikseresht, A. Rostami and S. Hemmati, Res. Chem. Intermed., 2019, 45, 507–520 CrossRef CAS.
- W. Xue, D. Huang, X. Wen, S. Chen, M. Cheng, R. Deng, B. Li, Y. Yang and X. Liu, J. Hazard. Mater., 2020, 390, 122128 CrossRef CAS PubMed.
- F. Lorignon, A. Gossard and M. Carboni, Chem. Eng. J., 2020, 393, 124765 CrossRef CAS.
- N. Shekarlab, R. Ghorbani-Vaghei and S. Alavinia, J. Organomet. Chem., 2021, 949, 121971 CrossRef CAS.
- M. Younas, M. Rezakazemi, M. Daud, M. B. Wazir, S. Ahmad, N. Ullah, Inamuddin and S. Ramakrishna, Prog. Energy Combust. Sci., 2020, 80, 100849 CrossRef.
- L. Yang, H. Zhang, P. Tao, X. Lu, X. Li, C. Wang, B. Wang, F. Yue, D. Zhou and Q. Xia, ACS Appl. Mater. Interfaces, 2021, 13, 8474–8487 CrossRef CAS PubMed.
- A. Dhakshinamoorthy, A. M. Asiri and H. Garcia, ACS Catal., 2019, 9, 1081–1102 CrossRef CAS.
- M. Koolivand, M. Nikoorazm, A. Ghorbani-Choghamarani, M. Mohammadi, A. Ghorbani-Choghamarani and M. Mohammadi, Appl. Organomet. Chem., 2022, 36, e6656 CrossRef CAS.
- S. M. Ramish, A. Ghorbani-Choghamarani and M. Mohammadi, Sci. Rep., 2022, 12, 1479 CrossRef CAS PubMed.
- N. Hussain-Khil, A. Ghorbani-Choghamarani and M. Mohammadi, Sci. Rep., 2021, 11, 15657 CrossRef CAS PubMed.
- A. Nikseresht, S. Ghasemi and S. Parak, Polyhedron, 2018, 151, 112–117 CrossRef CAS.
- J. Babamoradi, R. Ghorbani-Vaghei and S. Alavinia, Can. J. Chem., 2022, 100, 412–421 CrossRef CAS.
- R. Oozeerally, D. L. Burnett, T. W. Chamberlain, R. J. Kashtiban, S. Huband, R. I. Walton and V. Degirmenci, ChemCatChem, 2021, 13, 2517–2529 CrossRef CAS.
- R. Oozeerally, S. D. K. Ramkhelawan, D. L. Burnett, C. H. L. Tempelman and V. Degirmenci, Catalysts, 2019, 9, 812 CrossRef CAS.
- R. Oozeerally, D. L. Burnett, T. W. Chamberlain, R. I. Walton and V. Degirmenci, ChemCatChem, 2018, 10, 706–709 CrossRef CAS PubMed.
- T. W. Chamberlain, V. Degirmenci and R. I. Walton, ChemCatChem, 2022, 14, e202200135 CrossRef CAS.
- H. Chen, Y. Liu, T. Cai, W. Dong, L. Tang, X. Xia, L. Wang and T. Li, ACS Appl. Mater. Interfaces, 2019, 11, 28791–28800 CrossRef CAS PubMed.
- H. Chen, W. Zeng, Y. Liu, W. Dong, T. Cai, L. Tang, J. Li and W. Li, ACS Appl. Mater. Interfaces, 2021, 13, 16364–16373 CrossRef CAS PubMed.
- J. Guo, J. Wu, Y. Guan, J. Wang, Q. Liu, X. Mao, X. Qi, P. He and H. Wang, Energy Fuels, 2021, 35, 3252–3265 CrossRef CAS.
- R. El Osta, A. Carlin-Sinclair, N. Guillou, R. I. Walton, F. Vermoortele, M. Maes, D. de Vos and F. Millange, Chem. Mater., 2012, 24, 2781–2791 CrossRef CAS.
- P. L. Llewellyn, P. Horcajada, G. Maurin, T. Devic, N. Rosenbach, S. Bourrelly, C. Serre, D. Vincent, S. Loera-Serna, Y. Filinchuk and G. Férey, J. Am. Chem. Soc., 2009, 131, 13002–13008 CrossRef CAS PubMed.
- S. Ghasemi, M. Yousefi, A. Nikseresht and H. Omidi, Process Biochem., 2021, 102, 92–101 CrossRef CAS.
- Y. Dou, H. Zhang, A. Zhou, F. Yang, L. Shu, Y. She and J.-R. Li, Ind. Eng. Chem. Res., 2018, 57, 8388–8395 CrossRef CAS.
- J. Chen, R. Liu, Y. Guo, L. Chen and H. Gao, ACS Catal., 2015, 5, 722–733 CrossRef CAS.
- K. Manna, P. Ji, F. X. Greene and W. Lin, J. Am. Chem. Soc., 2016, 138, 7488–7491 CrossRef CAS PubMed.
- F. Ghobakhloo, D. Azarifar, M. Mohammadi, H. Keypour and H. Zeynali, Inorg. Chem., 2022, 61, 4825–4841 CrossRef CAS PubMed.
- A. Ghorbani-Choghamarani, R. Sahraei, Z. Taherinia and M. Mohammadi, J. Iran. Chem. Soc., 2021, 18, 827–838 CrossRef CAS.
- Y. Zhang, V. Degirmenci, C. Li and E. J. M. Hensen, ChemSusChem, 2011, 4, 59–64 CrossRef CAS PubMed.
- G. Cai, P. Yan, L. Zhang, H.-C. Zhou and H.-L. Jiang, Chem. Rev., 2021, 121(20), 12278–12326 CrossRef CAS PubMed.
- Z. Zhuang and D. Liu, Nano-Micro Lett., 2020, 12, 132 CrossRef CAS PubMed.
- A. Valverde-Gonzalez, M. Iglesias and E. M. Maya, Chem. Mater., 2021, 33(17), 6616–6639 CrossRef CAS.
- A. Modak, A. Ghosh, A. R. Mankar, A. Pandey, M. Selvaraj, K. K. Pant, B. Chowdhury and A. Bhaumik, ACS Sustainable Chem. Eng., 2021, 9(37), 12431–12460 CrossRef CAS.
- H. Zhang, H. Yang, Z. Pan and Q. Cheng, React. Chem. Eng., 2022, 7, 1626–1639 RSC.
- L. Peng, X. Gao, Y. Liu, J. Zhang and L. He, Energy Fuels, 2021, 35, 4182–4190 CrossRef CAS.
- M. J. Hülsey, B. Zhang, Z. Ma, H. Asakura, D. A. Do, W. Chen, T. Tanaka, P. Zhang, Z. Wu and N. Yan, Nat. Commun., 2019, 10, 1330 CrossRef PubMed.
- S. Li, Z. Chen, X. Ling, J. Cao, Q. Wang, Y. Zhou and J. Wang, Appl. Surf. Sci., 2019, 496, 143650 CrossRef.
- Y. Shi, Z. Guo, Q. Wang, L. Zhang, J. Li, Y. Zhou and J. Wang, ChemCatChem, 2017, 9, 4426–4436 CrossRef CAS.
- S. Xiong, Y. Guan, C. Luo, L. Zhu and S. Wang, Energy Fuels, 2021, 35, 14462–14483 CrossRef CAS.
- R. Al-Faze, E. F. Kozhevnikova and I. V. Kozhevnikov, ACS Omega, 2021, 6, 9310–9318 CrossRef CAS PubMed.
- B. Zhang, H. Asakura, J. Zhang, J. Zhang, S. De and N. Yan, Angew. Chem., 2016, 128, 8459–8463 CrossRef.
- J. Gordon, H. Kazemian and S. Rohani, Microporous Mesoporous Mater., 2012, 162, 36–43 CrossRef CAS.
- A. Nikseresht and E. Aderang, Appl. Chem., 2021, 16, 25–38 Search PubMed.
- J. Gordon, H. Kazemian and S. Rohani, Mater. Sci. Eng., C, 2015, 47, 172–179 CrossRef CAS PubMed.
- P. Dinh Du and P. Ngoc Hoai, Adv. Mater. Sci. Eng., 2021, 2021, 1–13 CrossRef.
- E. Yılmaz, E. Sert and F. S. Atalay, J. Taiwan Inst. Chem. Eng., 2016, 65, 323–330 CrossRef.
- M. Shi, S.-C. Cui and W.-P. Yin, Eur. J. Org. Chem., 2005, 2005, 2379–2384 CrossRef.
- A. R. Pourali and A. Goli, J. Chem. Sci., 2011, 123, 63–67 CrossRef CAS.
- M. A. Zolfigol, E. Madrakian and E. Ghaemi, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2001, 40, 1191–1195 Search PubMed.
- W.-B. Pan, L.-M. Wei, L.-L. Wei, C.-C. Wu, F.-R. Chang and Y.-C. Wu, J. Chin. Chem. Soc., 2005, 52, 581–588 CrossRef CAS.
- A. Ghorbani-Choghamarani, H. Goudarziafshar, M. Nikoorazm and S. Yousefi, Can. J. Chem., 2009, 87, 1144–1147 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Rasoul Bagheriniaa,
Masoud Mohammadi
*a,
Rasoul Bagheriniaa,
Masoud Mohammadi