
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInduced abundant oxygen vacancies in Sc2VO5−δ/g-C3N4 heterojunctions for enhanced photocatalytic degradation of levofloxacin†
Jian Feng *,
Liyao Zu,
Hongrong Yang,
Yuanyuan Zheng,
Ziying Chen,
Wei Song,
Ran Zhao,
Li Wang,
Xia Ran and
Bo Xiao
*,
Liyao Zu,
Hongrong Yang,
Yuanyuan Zheng,
Ziying Chen,
Wei Song,
Ran Zhao,
Li Wang,
Xia Ran and
Bo Xiao
Engineering Research Center for Molecular Medicine, School of Basic Medical Science, Guizhou Medical University, Guiyang 550025, China. E-mail: jfeng@gmc.edu.cn; Tel: +86 851 88174017
First published on 23rd December 2022
Abstract
Sc2VO5−δ/g-C3N4 heterojunctions (SVCs) with abundant oxygen vacancies (OVs) were synthesized by ultrasonic exfoliation combined with the thermal etching method. The structures, OVs and spatial separation of the photogenerated carriers were systematically characterized. The results manifested that the SVCs were successfully constructed via the strong interaction between g-C3N4 (CN) and Sc2VO5−δ (SV). The SVCs possessed a higher concentration of OVs than that of pristine CN and SV. The formation of the SVC heterostructures and the optimization of the OVs were the two major factors to accelerate the separation of the charge carriers and finally to improve the photocatalysis performance. The as-prepared 10%SVC (containing 10 wt% of SV) catalyst exhibited the highest OV concentration and the best photocatalytic performance. The levofloxacin (LVX) photodegradation activity showed a positive correlation with the OV concentration. The photocatalytic degradation efficiencies were 89.1, 98.8 and 99.0% on 10%SVC for LVX, methylene blue (MB) and rhodamine B (RhB), respectively. These photodegradation processes followed the pseudo first order kinetic equation. The apparent rate constant (kapp) of LVX degradation on 10%SVC was 11.0 and 7.5 times that of CN and SV. The h+, ˙OH and ˙O2− were the major reactive species in the photodegradation process.
1. Introduction
The development of modern industry and the consumption of fossil fuel have caused serious environmental pollution, which is severely harmful to human health and the sustainable development of society.1 Traditional techniques have been utilized to remove organic pollutants, including dyes and antibiotics.2–4 However, these traditional methods suffer from drawbacks, such as not being suitable for low concentration pollutants, high operation costs, low removal efficiency and secondary pollution.In the recent few decades, semiconductor based photocatalysis has been regarded as the most promising technology for the elimination of low concentration contaminants.5–7 The photocatalytic technology has mild reaction conditions, low cost, superior recoverability and less secondary pollution and etc. Therefore, photocatalysis technology has been extensively investigated for the removal of dyes and antibiotics.8–11 The significant progress has been made in this field recently.12 Various semiconductor materials with superior photocatalytic performance in wastewater treatment have aroused extensively research interests.13–15 Nevertheless, the low quantum efficiency and the narrow visible light response range have always been the major obstacles for the practical application of the photocatalytic technology.
Rare earth vanadates are a type of semiconductor materials with novel magnetic, optical, electronic and catalytic performances,16 which have been widely applied as laser hosts, phosphors, polarizers and catalysts.17 Especially, the electron on 3d orbital of V can be excited by the visible light.18 Rare earth vanadates are consequently considered as the promising candidates for highly efficient visible-light photocatalysts. Several rare earth vanadates, including CeVO4,19 DyVO4,20 EuVO4,21 GdVO4,17 HoVO4,22 LaVO4,18 NdVO4,23 PrVO4,24 SmVO4,25 TmVO4,21 YVO4,26 were successfully prepared and the visible light driven photocatalytic activities were investigated. Nevertheless, the low visible light utilization and the high charge recombination are still the key factors of low photocatalytic efficiency of rare earth vanadates.27 Doping,28 metal-substitution,29 morphology control,19 introduction of surface OVs30,31 and construction of heterojunction18,24,25,27 have been adopted to meliorate its photocatalytic quantum efficiency. Among them, surface OVs engineering can effectively regulate the electronic structure and charge migration, produce the active surface phases, and ameliorate the adsorption of the contaminants and intermediates in the photocatalytic degradation process.32 As a contrast, the bulk OVs are the deep charge traps, which can expedite the recombination of the photogenerated electrons and holes. The photocatalytic performance is hence reduced by the bulk OVs. Accurate introduction of surface OVs is crucial to improve the photocatalytic activity. According to previous literature, the control of synthesis temperature, substitution, doping, chemical etching and surface reduction are the effectual approaches to tune the OVs of the photocatalyst.33 Thereinto, the control of calcination temperature is a convenient method to engineer the OVs concentration on the surface or at the interface of the binary or ternary oxides catalysts during the post treatment process. It is believed that the calcination temperature influences the crystallinity, morphology, surface area and OVs concentration. In general, the calcination temperature range between 350 to 500 °C is advantageous to achieve the optimal surface OVs concentration of the oxides.33
The construction of heterojunction is another feasible and effective way to facilitate the spatial separation and the recombination rate of photogenerated charge carriers.34–37 Many rare earth vanadates based heterojunctions have been established to promote its photocatalytic degradation of contaminants. For instance, LaVO4/g-C3N4 heterojunction was fabricated and the visible light driven photocatalytic performance was assessed. 99% of trichloroethylene was mineralized on 3% LaVO4/g-C3N4, which were 3.89 and 4.67 times of LaVO4 and g-C3N4, respectively.18 In another work, the integration of UiO-66-NH2 with SmVO4 enhanced the light absorption, carrier lifetime and specific surface area of the composite. SmVO4/UiO-66-NH2 eliminated 50% of tetracycline in 90 min under solar light.25 La2O3/CeVO4@halloysite nanotubes composites also successfully fabricated by hydrothermal method. The construction of heterojunction between La2O3 and CeVO4 inhibited the recombination of photogenerated charge carriers.27 87.1% of tetracycline in 60 min under visible light irradiation on the optimized composite, which was 3.89 times higher than that of raw CeVO4. LaVO4/BiOBr heterojunction was also prepared by hydrothermal method. The introduction of LaVO4 heightened the solar energy harvesting and reduced the transfer resistance of the photogenerated charge carriers. 95.4% and 87.1% of acetone and toluene degraded under visible light irradiation over 3% LaVO4/BiOBr, which were 2.6 and 5.3 times higher than that of pristine BiOBr.38 As mentioned above, the spatial separation of the photogenerated charge and the photocatalytic activity can be effectively enhanced through constructing rare earth vanadates based heterojunction.
In this study, Sc2VO5−δ with abundant OVs was loaded on the ultrathin g-C3N4 nanosheets to form the Sc2VO5−δ/g-C3N4 heterojunctions. Transmission electron microscopy (TEM), high resolution TEM (HRTEM), scanning electron microscopy (SEM), Fourier-transform infrared spectra (FTIR) and X-ray photoelectron spectroscopy (XPS) results clarified the successful formation of SVCs and its ultrathin structure. Ultraviolet-visible diffuse reflectance spectra (DRS) and XPS VB spectra revealed it was type II heterojunction. Electron paramagnetic resonance (EPR) confirmed the existence of OVs and XPS O1s spectra determined the OVs concentration. Transient photocurrent response (TPC), electrochemical impedance spectroscopy (EIS) and photoluminescence spectra (PL) assessed the promotion of the separation efficiency and the inhibition of the recombination of the photogenerated charge carriers in SVCs. The influence of OVs and SV content on the photocatalytic degradation efficiency of LVX, MB and RhB was investigated. Due to the introduction of surface OVs and the formation of heterojunction, the as-prepared SVCs manifested highly enhanced photocatalytic degradation efficiency. This study supplied a new paradigm for further exploring the synergistic effect of surface OVs and heterostructure on the photocatalytic activity.
2. Experiment
2.1 Synthesis of SVCs
2.2 Characterization
X-ray diffraction (XRD) patterns were tested on Rigaku SmartLab diffractometer. FTIR spectra were obtained on Nicolet NEXUS 470 spectrometer. TEM and HRTEM images were recorded on JEOL-2100F transmission electron microscope. SEM, energy dispersion spectrum (EDS) and elemental mapping images were measured on JSM-4800F scanning electron microscope. XPS spectra were measured on Thermo ESCALAB 250XI system. EPR spectra were recorded on Bruker E500 spectrometer. The N2 adsorption desorption isotherms and BET specific surface area were analyzed by ASAP 2460 Micrometrics instrument. DRS spectra were determined on Shimadzu UV-2401 spectrophotometer. UV-vis spectra were conducted on Varian Cary 50 UV-vis spectrophotometer.2.3 Photoelectrochemical measurement
The photoelectrochemical experiments of CN, SVCs and SV were implemented on CHI 660E electrochemical workstation. Pt and Ag/AgCl were utilized as the counter electrode and reference electrode. For the preparation of working electrode, 5 mg CN, SVCs or SV was mixed firstly with 0.2 mL Nafion in 1.8 mL ethanol by sonication and then dropped on 1 cm2 of FTO glass. TPC and EIS of the catalysts were detected in 0.2 M Na2SO4 electrolyte solution.2.4 Photocatalytic degradation reaction
100 mg of CN, SVCs or SV was dispersed in 200 mL of 12.0 mg L−1 LVX, 5.0 mg L−1 MB or 5.0 mg L−1 RhB, respectively. The solution was continuously stirred in the dark for 30 min to reach adsorption desorption equilibrium of the contaminants on the catalysts. The photocatalytic degradation of contaminants was evaluated under the irradiation of 40 W white LED. The average intensity of the visible light was about 20 mW cm−2 (CEL-NP2000-2, CEAULIGHT, Beijing). 5 mL solution was fetched out at intervals and the catalyst was removed through 0.22 μm PTFE filter membrane. The contaminant concentration in degradation solution was detected by UV-vis spectrophotometer at 294 nm (LVX), 664 nm (MB) and 552 nm (RhB). The temperature was fixed at 25 °C in the photocatalysis process. The degradation experiments were repeated three times. The apparent quantum yield of LVX degradation was estimated according to the literature.39 The stability of 10%SVC was appraised by 4 cycles of photocatalytic degradation experiments. 10%SVC was centrifuged and desorbed in deionized water for several times after each cycle experiment to eliminate absorbed contaminant. Then, 10%SVC was separated and freeze-dried for the usage in next cycle experiment. The reproducibility of 10%SVC was evaluated by repeating the above process twice. NaNO3, EDTA, isopropyl alcohol (IPA) and p-benzoquinone (pBQ) were selected as the scavengers to trap e−, h+, ˙OH and ˙O2−, respectively.3. Results and discussion
The XRD patterns of CN, SVCs and SV were shown in Fig. 1. Two characteristic diffraction peaks at 13.0° and 27.6° were clearly observed in the XRD patterns of CN and SVCs. It respectively corresponded to (100) and (002) crystal planes of CN, suggesting the existence of CN in the as-prepared SVCs. The peaks at 13.0° originated from the in-plane repeating structural units and the peaks at 27.6° were attributed to the inter-planar structure stacking of aromatic systems.40 The broad feature of the peaks indicated low crystallinity of the samples. All diffraction peaks of SV were in accord well with tetragonal phase Sc2VO5 (PDF#28-1014). The main peaks at 21.6°, 26.4°, 31.6°, 36.5°, 41.0°, 44.4°, 46.9°, 48.6°, 52.6°, 52.9°, 62.6° and 63.2° could be clearly observed, which was respectively correspond to (112), (201), (203), (220), (223), (224), (320), (322), (400) and (217) crystal plane of tetragonal phase. The diffraction peaks of SV could not be detected in 2%SVC and 5%SVC. It was mainly due to the relative lower dosage of SV in SVCs, which could not initiate the chemical skeleton and structure changes of CN. It was similar to the previous result.41 The diffraction peak at 31.6° of SV was observed in 10%SVC and 20%SVC. The weak and broad diffraction peaks at 31.6° of 10%SVC and 20%SVC demonstrated the low crystallinity of SV in SVCs. Hence, the XRD result confirmed the successful preparation of SVCs with certain of amorphous phase.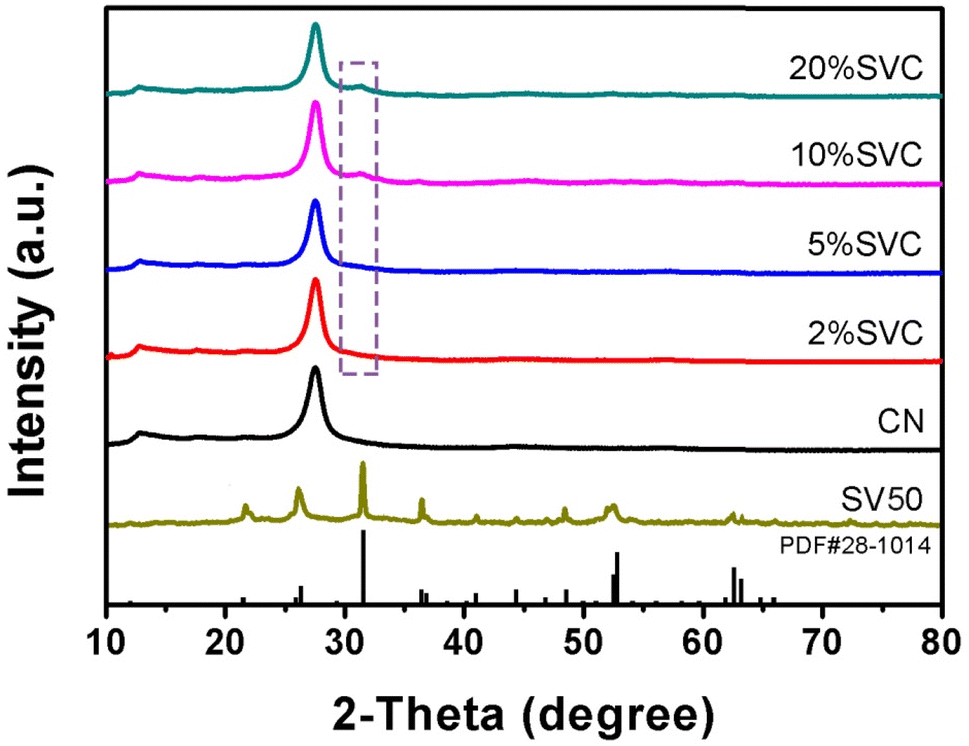 | ||
| Fig. 1 XRD patterns of CN, 2%SVC, 5%SVC, 10%SVC, 20%SVC and SV. | ||
As reported in the literature, the amorphous phase in the catalyst can significantly influence its crystallinity and further affect its surface OVs.33 Actually, exorbitant crystallinity resulted in lower concentration of surface OVs, which was not conducive to the separation of photogenerated charge. Conversely, excessive low crystallinity caused more bulk OVs, which was also detrimental to the improvement of photocatalytic performance. In general, the calcination temperature range between 350 to 500 °C is advantageous to achieve the optimal surface OVs concentration. Thus, the appropriate crystallinity of SVCs could be conveniently generated by optimizing the calcination temperature. And then the optimal surface OVs concentration was achieved by regulating the crystallinity of the catalyst. To corroborate the influence of calcination temperature on crystallinity and OVs concentration of the catalyst, 10%SVC was synthesized at 400, 450, 500 and 550 °C respectively. The crystallinity was investigated by XRD patterns and the surface OVs were detected by XPS O1s spectra. As the XRD results depicted in Fig. S1,† the diffraction peak at 31.6° appeared stronger and sharper with the increase of the calcination temperature, indicating the crystallinity of 10%SVC was gradually ameliorated.
The morphologies of bulk CN, CN nanosheets and SVCs were determined by SEM. As displayed in Fig. 2a, the bulk CN exhibited blocky aggregate structure, in which lamellar structure of CN nanosheets could not be observed. The characteristic layered structure of CN was clearly revealed in Fig. 2b, indicating that CN nanosheets could effectively be generated by the exfoliation effect in the ultrasonic treatment and the thermal etching in the reheating synthetic procedure. As shown in Fig. 2c and d, 10%SVC had the structural feature of CN nanosheets. The SEM result clearly revealed that SV nanoparticles were located on CN nanosheets (Fig. 2c). 2%SVC, 5%SVC and 20%SVC also presented the same layered stacking structure (Fig. S2†). The thin-layer structure of SVCs could increase the specific surface area and reaction active sites, which was instrumental in accelerating photocatalytic degradation reaction. The EDS spectra and elemental mapping techniques were employed to appraise the chemical composition and the element distribution of SVCs. 10%SVC was selected as the target heterojunction. C, N, O, Sc and V were identified by EDS in selected region (Fig. S3†). The corresponding weight ratio of C, N, O, Sc and V in 10%SVC was listed in Fig. S3.† 9.54% of SV mass percentage in 10%SVC was calculated from the EDS result. It was consistent with the dosage of Sc(NO3)3·6H2O and NH4VO3 in the precursor. The distribution of C, N, O, Sc and V in 10%SVC was assessed by the elemental mappings shown in Fig. 2e–i. The uniformity of the element mapping images suggested that Sc, V and O were distributed evenly in 10%SVC. This result demonstrated that SV was loaded homogeneously on CN nanosheets.
 | ||
| Fig. 2 SEM images of (a) bulk CN, (b) CN nanosheets and (c), (d) 10%SVC, and the element mapping of (e) C, (f) N, (g) O, (h) Sc and (i) V. Scale bar = 2 μm. | ||
The morphology and structure of CN, SVCs and SV were further characterized by TEM and HRTEM. The results were displayed in Fig. 3 and S4.† The ultrathin layered structures of CN nanosheets with wrinkles in 10%SVC were distinctly observed in Fig. 3a and b. SV exhibited irregular granular morphology with a certain degree of aggregation. The particle size of SV was about 25 to 70 nm estimated from the TEM images. SV nanoparticles were loaded on CN nanosheets. TEM images evidently clarified that the intimate contact between CN nanosheets and SV nanoparticles, meaning the successful formation of SVCs heterostructure. The lattice fringes of SV in 10%SVC were definitely displayed in HRTEM (Fig. 3c). The d-spacing of 0.28 nm was indexed to the (203) lattice planes of tetragonal SV, which was in accordance with the XRD result discussed in Fig. 1. The similar layered structure of CN and irregular granular morphology of SV in 2%SVC, 5%SVC and 20%SVC could also be observed in Fig. S4.† These TEM results sufficiently confirmed the formation of SVC heterojunction.
 | ||
| Fig. 3 (a), (b) TEM and (c) HRTEM images of 10%SVC. | ||
FTIR and Raman spectra were conducted to investigate the chemical structure of as-prepared catalysts. Fig. 4a manifested the FTIR spectra of CN nanosheets and SVCs. Four typical adsorption bands were observed in all spectra. The adsorption peaks at 806 and 886 cm−1 were assigned to breathing vibration and N–H deformation vibration mode of CN s-triazine, respectively.42 A characteristic adsorption band in the range of 1200–1700 cm−1 was ascribed to the stretching vibrations of C–N and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N of aromatic CN heterocycles.43 A weak adsorption band at 2174 cm−1 exhibited in 5%SVC, 10%SVC and 20%SVC, which was originated from the terminal cyano groups. Similar adsorption band generally could be found in many CN-based composites.44 A broad adsorption band at 2900–3500 cm−1 was attributed to N–H stretching vibration of residual unpolymerized amino groups and O–H stretching related to physically adsorbed water molecule.44 All the peak positions of 2%SVC, 5%SVC, 10%SVC and 20%SVC in the FTIR spectra did not shift compared with CN. It was consistent with the XRD results, suggesting that the formation of SVCs heterojunction did not vary its chemical skeleton and structure of CN nanosheets.
N of aromatic CN heterocycles.43 A weak adsorption band at 2174 cm−1 exhibited in 5%SVC, 10%SVC and 20%SVC, which was originated from the terminal cyano groups. Similar adsorption band generally could be found in many CN-based composites.44 A broad adsorption band at 2900–3500 cm−1 was attributed to N–H stretching vibration of residual unpolymerized amino groups and O–H stretching related to physically adsorbed water molecule.44 All the peak positions of 2%SVC, 5%SVC, 10%SVC and 20%SVC in the FTIR spectra did not shift compared with CN. It was consistent with the XRD results, suggesting that the formation of SVCs heterojunction did not vary its chemical skeleton and structure of CN nanosheets.
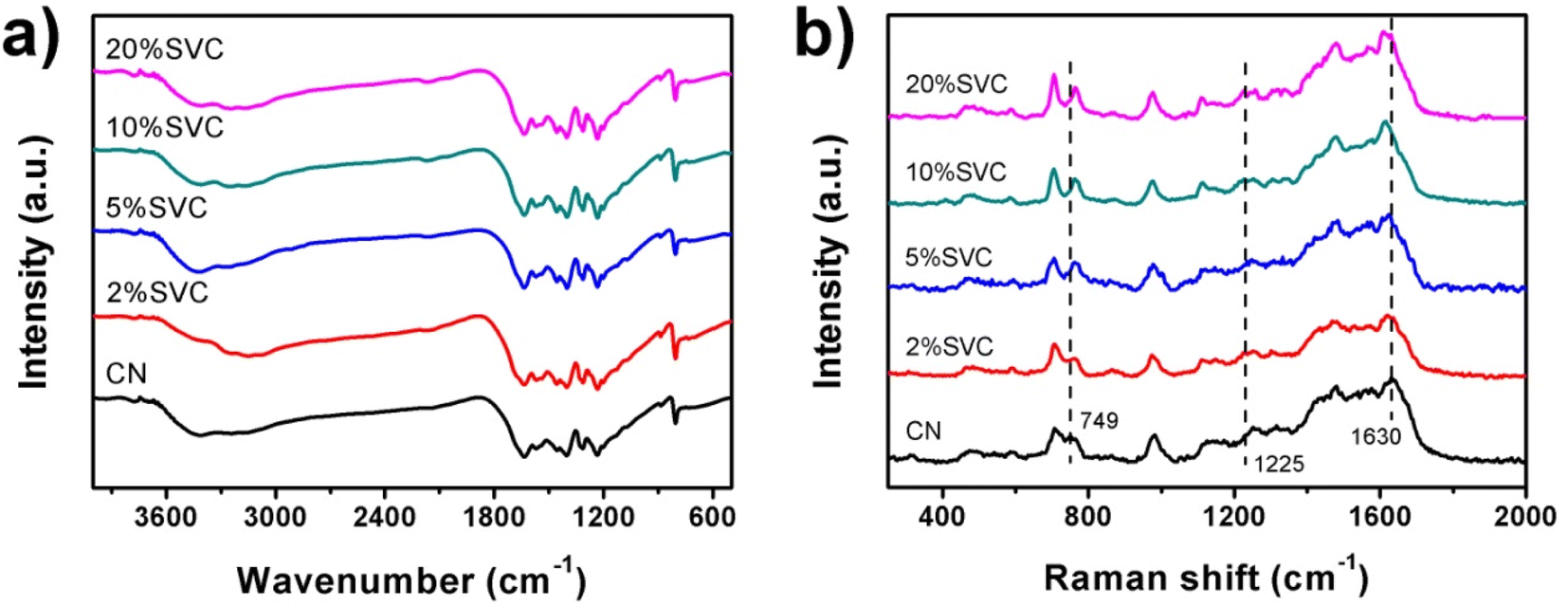 | ||
| Fig. 4 (a) FTIR and (b) Raman spectra of CN, 2%SVC, 5%SVC, 10%SVC and 20%SVC. | ||
The Raman spectra of CN nanosheets and SVCs were depicted in Fig. 4b. The characteristic peaks of CN were presented at 486, 588, 704, 749, 976, 1108, 1250, 1315, 1480, 1565 and 1630 cm−1. It was consistent with the Raman results reported in the literature.43,45,46 In detail, the peak at 486 cm−1 was originated from the heptazine ring breathing mode,46 and the 588 cm−1 band caused by the ring bending vibration.43 The peaks centered at 704, 749 and 976 cm−1 were respectively assigned to in plane bending vibration,45 out-of-plane bending vibration of graphitic domain and the breathing mode of triazine ring.47 Peaks around 1250 and 1630 cm−1 were corresponded to the CN stretching vibration mode of triazine ring.43 The 1315, 1480 and 1565 cm−1 bands were distributed to semi-circle stretching,43 the bending vibration of –NH2 group and the graphitic G band.48 As shown in Fig. 4b, the Raman peaks of CN at 749 and 1630 cm−1 were shifted to 762 and 1614 cm−1 after the SVC heterojunction was formed. Meanwhile, the intensity of 1225 cm−1 peak was gradually strengthened with the increase of SV content in SVCs. These results clarified that the intimate contact between CN nanosheets and SV nanoparticles. It was agreed with the TEM and HRTEM results and signified the successful formation of SVCs heterostructure, which was advantageous to the spatial separation of the photogenerated electrons and holes during the photocatalytic degradation reaction.49
The surface chemical composition and chemical state of CN, 10%SVC and SV were analyzed by XPS spectra. As illustrated in Fig. 5a, the XPS survey spectrum of 10%SVC revealed the existence of C, N, O, Sc and V, further demonstrating the successful formation of SVC heterojunction. In Fig. 5b, four peaks at the binding energy of 284.8, 285.8, 288.3 and 288.5 eV were observed in the high resolution C1s XPS spectrum of pristine CN. These peaks were respectively designated to the graphitic C–C bond of polluted carbon, C–OH, CN and OC–O groups.50 The C1s peaks shifted to 284.9, 286.2, 288.2 and 288.7 eV after the formation of SVC heterojunction. The N1s peaks of pristine CN located at 398.8, 399.3, 400.9 and 404.7 eV (Fig. 5c), corresponding to the pyridinic nitrogen (CN–C), pyrrolic nitrogen (N–(C)3), surface amino nitrogen (C–NH2) and the charge effect in the heterocycles.51 In contrast with pristine CN, the N1s peaks of 10%SVC were located at 398.7, 399.4, 400.8 and 404.8 eV. The variations of the binding energy implied the change of electron cloud density of C and N atoms.41 It might be attributed to the strong interaction between SV and CN. These XPS results supplied the convincing evidence to corroborate the formation of SVC heterojunctions. The Sc2p XPS spectra of SV and 10%SVC supported this verdict. As displayed in Fig. S5,† pure SV had the peaks at 398.4 and 403.1 eV relating to Sc2p3/2 and Sc2p1/2, which could originate from the Sc–O bond.52,53 These two deconvoluted peaks shifted to 398.0 and 402.6 eV for 10%SVC, respectively. The chemical shift towards lower binding energy was observed after CN was incorporated with SV. The increase of the electron cloud density of Sc might be the reason for this reduction of the binding energy.54 This result further indicated the strong interaction between SV nanoparticles and CN nanosheets.
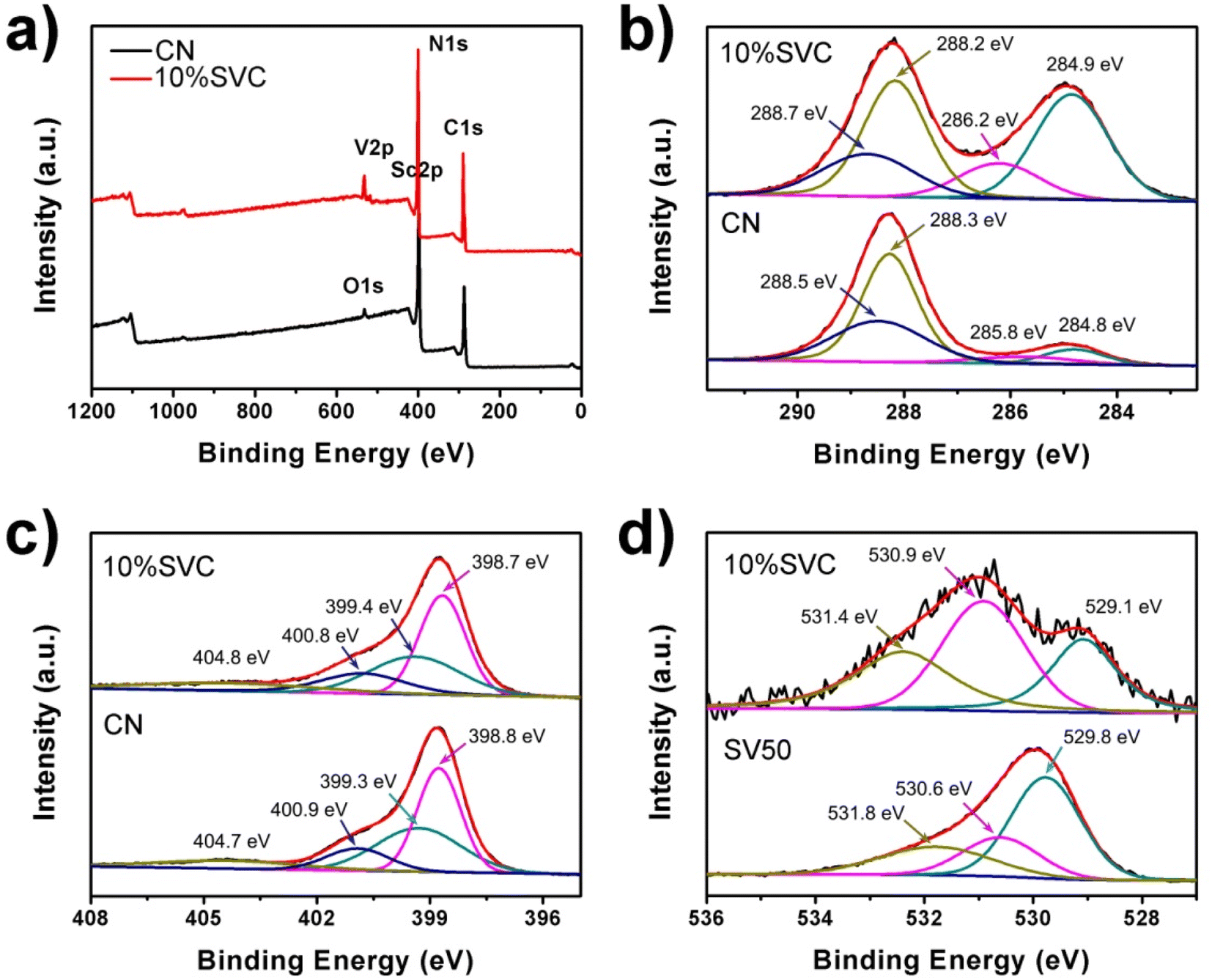 | ||
| Fig. 5 (a) XPS survey spectra, high resolution XPS spectra of (b) C1s, (c) N1s and (d) O1s of CN, 10%SVC and SV50. | ||
The O1s spectra of 10%SVC and SV were depicted in Fig. 5d. For SV, three deconvoluted peaks at 529.8, 530.6 and 531.8 eV were obtained, respectively attributing to the lattice oxygen (OL), surface oxygen vacancies (OV) and surface adsorption oxygen (OA). The peak area percentage of OL, OV and the ratio of OV/OL in SVCs and SV samples were estimated and listed in the Table S1.† The result distinctly revealed that the surface OVs concentration in 10%SVC was higher than other SVCs and pure SV. The peak intensity of 10%SVC at 529.8 eV related to the lattice oxygen was lower than that of SV. It was presumably resulted from the partial coverage of SV by CN nanosheets, which also led to the decrease of the XRD diffraction peak intensity of SV in 2%SVC and 5%SVC.41 Furthermore, the O1s binding energies of SV40 and SV50 were lower than that of values reported previously, which might be associated with its lower crystallinity. In order to investigate the effect of calcination temperature on the surface OVs, the precursors were calcinated at 400, 500 and 600 °C for 4 h to synthesize SV. The corresponding O1s spectra of SV40, SV50 and SV60 were comparatively exhibited in Fig. S6.† The deconvoluted peaks of SV60 located at the binding energy of 529.8, 531.5 and 532.8 eV, which was very consistent with the literature.55 It indicated that SV60 had high crystallinity. The peak area percentage of OL, OV and OV/OL values were presented in Table S1.† The result manifested that the OVs concentration in SV was in negative correlation with the calcination temperature. It was reasonably inferred that the OVs concentration in SV was bound up with the content of amorphous phase.33 The calcination temperature was the crucial and convenient parameter to regulate the amorphous phase and the OVs concentration.
A series of 10%SVC samples were prepared at 400, 450, 500 and 550 °C for 2 h to further confirm the influence of calcination temperature on the OVs concentration. The surface OVs concentration was calculated based on XPS O1s spectra and the results were listed in Table S2.† As displayed in Table S2,† the surface OVs concentration of 10%SVC was increased with the rise of calcination temperature. It reached the maximum when the calcination temperature was 500 °C. Conversely, the surface OVs concentration then decreased while the calcination temperature exceeded 500 °C. These results signified that the optimal surface OVs concentration was achieved at 500 °C, which are adequately clarified that the calcination temperature can effectively control the crystallinity and the surface OVs of the catalyst.33 EPR spectra can provide the direct evidence to confirm the presence of OVs in CN, SV and 10%SVC.56 As shown in Fig. 6a, the typical EPR signals of OVs at g = 2.003 were observed. This signal could be attributed to the electrons trapped by the OVs. The weak EPR signal of pristine SV indicated that SV sample had low OVs concentration. Meanwhile, 10%SVC exhibited strong signal at g = 2.003, suggesting that the OVs were successfully introduced into 10%SVC.
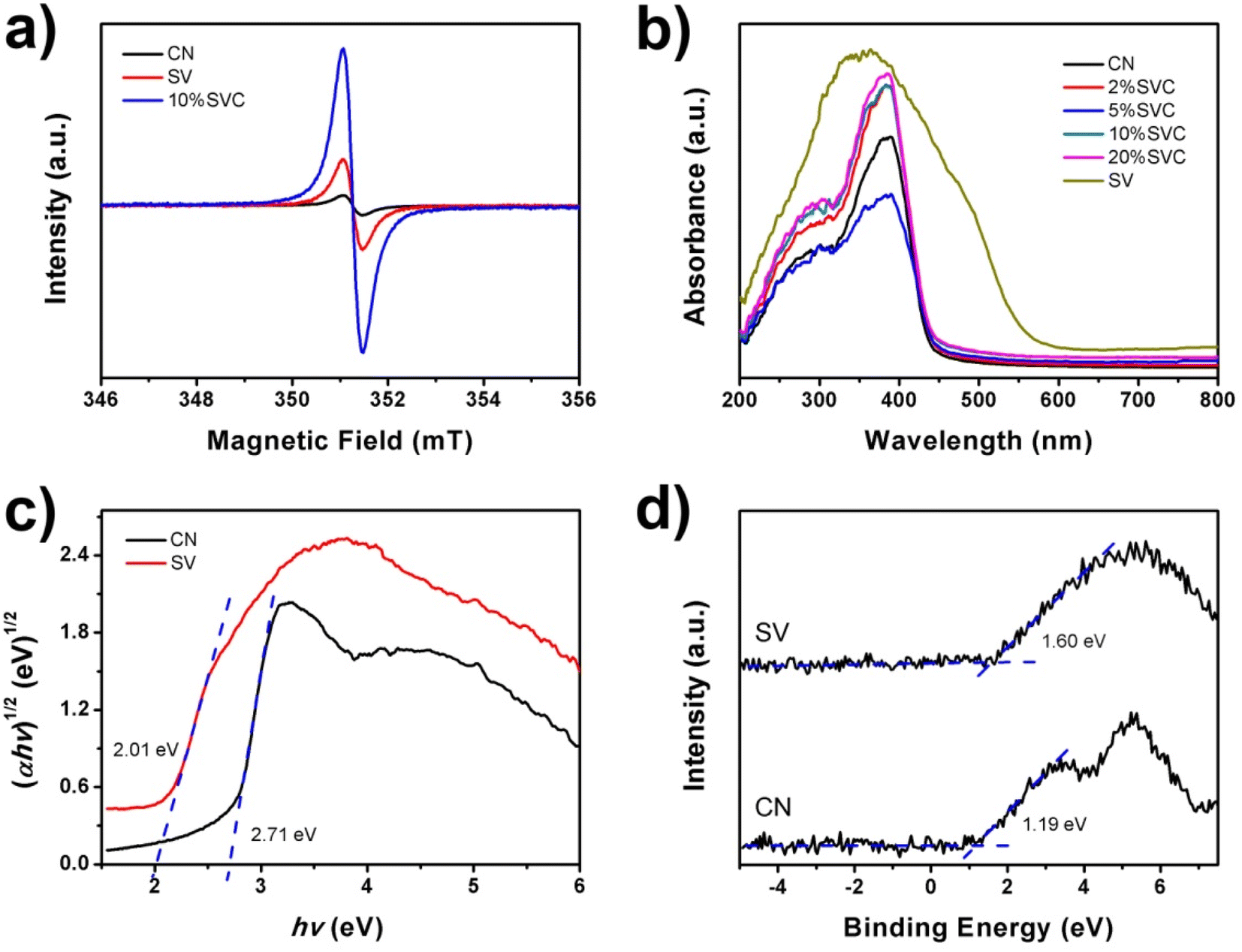 | ||
| Fig. 6 (a) EPR spectra of CN, SV and 10%SVC, (b) UV-vis diffuse reflectance spectra of CN, SV and SVCs, (c) XPS VB spectra and (d) band gap energies of CN and SV. | ||
UV-vis DRS of CN, SV and SVCs were measured to assess its optical properties. Fig. 6b revealed that the absorption edge of CN was about 457 nm. It was equivalent to 2.71 eV of band gap energy (Eg), which implied that CN nanosheets had certain visible light absorption capacity. This result was similar to the Eg of pure CN in the literature.14 SVCs samples exhibited almost identical absorption edge, indicating that the incorporation of SV and CN did not change the band gap energy. SV possessed higher absorbance in the UV and visible light region than that of CN and SVCs samples. The absorption edge of SV was around 617 nm, corresponding to 2.01 eV of Eg. Conversely, the visible light absorption of SVCs has not been effectively improved. In addition, the small shoulder was found in the visible light region in DRS spectra of SVCs, which might derive from the abundant OVs in the catalysts.56 The Eg of pristine SV and CN were determined by the Kubelka–Munk function. As shown in Fig. 6c, the Eg of SV and CN were respectively 2.01 and 2.71 eV, which were consist with the DRS results. The VB positions of SV and CN were +1.60 and +1.19 eV according to the XPS VB spectra depicted in Fig. 6d. The corresponding CB positions of SV and CN could be consequently estimated based on the formula: Eg = EVB − ECB. It was −0.41 and −1.52 eV, respectively.
The visible light driven photocatalytic degradation activities of LVX, MB and RhB over as-prepared CN, SVCs and SV samples were investigated by using a 40 W White LED as the light source. The photodegradation kinetic curves of LVX on CN, SVCs and SV were illustrated in Fig. 7a. 10%SVC exhibited best photocatalytic performance among these samples. 89.1% of LVX was eliminated within 80 min over 10%SVC. The apparent quantum yield for LVX degradation was 2.01%, which was 4.3 and 17.8 times of pristine CN and SV, respectively. Compared to CN and SV, all SVC heterojunctions presented enhanced photocatalytic activities, indicating that the formation of the SVC heterostructures and the optimization of the surface OVs concentration had accelerated the separation of the photogenerated charge carriers and finally improved its photocatalysis performance. MB and RhB were also selected as the target pollutants to further verify the excellent visible light driven photocatalytic activities of SVC heterojunctions. The photodegradation kinetic curves of MB and RhB on CN, SVCs and SV were displayed in Fig. S7a and S8a.† 10%SVC also manifested highest degradation efficiency. 98.8% of MB and 99.0% of RhB were removed, which was 2.9 and 8.1 times of pure SV, respectively. SVC heterojunctions all emerged higher photocatalytic activities than that of CN and SV. Furthermore, the photocatalytic degradation efficiency of MB, RhB and LVX on 10%SVC was compared with various recent reported results. As depicted in Table S4,† its degradation activity was higher than that of literature values, signifying that 10%SVC is probably a valuable catalyst of practical application in the environmental sewage treatment.
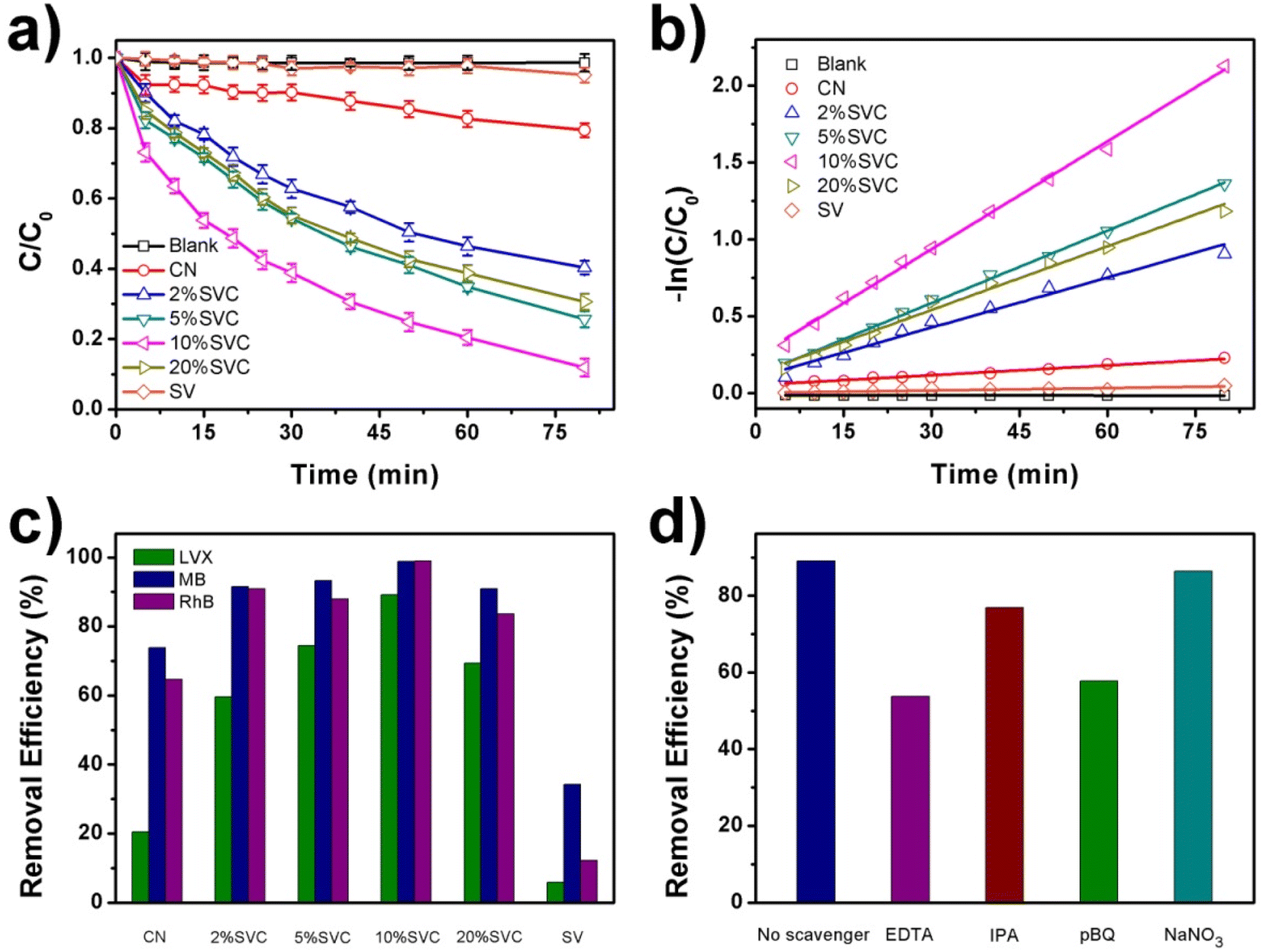 | ||
| Fig. 7 (a) Photocatalytic degradation of LVX over CN, SVCs and SV under visible light irradiation, (b) corresponding first-order kinetics fitting plots, (c) removal efficiency of LVX, MB and RhB over 10%SVC and (d) the effect of radical scavengers on the photocatalytic degradation of LVX over 10%SVC [experimental conditions: V = 200 mL; LVX = 12.0 mg L−1; catalyst dosage = 0.5 g L−1]. | ||
The photodegradation data of LVX over the as-prepared catalysts followed the pseudo first order kinetic equation (Fig. 7b). The order of the apparent rate constants (kapp) was as follows: 0.00213 min−1 (CN) ˂ 0.00313 min−1 (SV) ˂ 0.0108 min−1 (2%SVC) ˂ 0.0138 min−1 (20%SVC) ˂ 0.0156 min−1 (5%SVC) ˂ 0.0234 min−1 (10%SVC). The kapp of 10%SVC was 11.0 and 7.5 times of pristine CN and SV, respectively. The photodegradation reaction data of MB and RhB over CN, SVCs and SV were also fitted by the pseudo first order kinetic equation (Fig. S7b and S8b†) and the kapp was listed in Table S3.† It was observed that the kapp of MB degradation on 10%SVC was 2.5 and 8.3 times of pristine CN and SV. Meanwhile, the kapp of RhB degradation on 10%SVC was 3.4 and 40.0 times of pristine CN and SV. In addition, the enhanced photocatalytic activities of SVC heterojunctions could be confirmed in Fig. 7c. The removal efficiencies of LVX, MB and RhB over CN, SVCs and SV lucidly revealed that SVCs possessed much higher photocatalytic performance than that of pristine CN and SV under the same degradation conditions.
Furthermore, we synthesized a series of 10%SVC samples at different calcination temperature and characterized the surface OVs by O1s XPS spectra. The results demonstrated that the surface OVs of 10%SVC were significantly affected by the calcination temperature. Herein, LVX photocatalytic degradation experiments over this series of 10%SVC samples were conducted to analyze the effect of surface OVs on the photocatalytic performance. The results were exhibited in Fig. S9.† It was observed that the photodegradation performance of 10%SVC increased firstly and then decreased with the calcination temperature, the optimum removal efficiency was acquired when the calcination temperature reached 500 °C. Comparing with the surface OVs concentration of 10%SVC samples from O1s XPS spectra results (Table S2†), in which 10%SVC50 possessed the optimal surface OVs concentration. The LVX photocatalytic degradation performance was positive correlation with the surface OVs concentration in 10%SVC. Therefore, combining the photocatalytic experiments, O1s XPS and EPR spectra results, we could reasonably infer that the formation of the SVC heterostructures and the optimization of the surface OVs concentration were two major factors to accelerate the separation of the photogenerated charge carriers and finally to improve its photocatalysis performance. The photodegradation of LVX on 10%SVC was implemented for four cycles to assess the structural and photocatalytic stability of the catalyst. The recycling experiments and the FTIR, XRD results of 10%SVC before and after the photodegradation reaction were displayed in Fig. S10.† The negligible decrease of photodegradation efficiency signified the superior photocatalytic stability of 10%SVC. Meanwhile, the variations were hardly observed in the XRD and FTIR results, indicating 10%SVC possessed excellent structural stability.
To identify the reactive species involved in the photocatalytic degradation of LVX on 10%SVC, NaNO3, EDTA, IPA, and pBQ were selected as the scavengers to trap e−, h+, ˙OH and ˙O2−, respectively. The radical scavenger experiments were implemented under the same photodegradation conditions. As the results exhibited in Fig. 7d, the photodegradation efficiency of LVX on 10%SVC were remarkably reduced by the addition of EDTA and pBQ, indicating that h+ and ˙O2− were the main reactive species participated in the LVX photodegradation process. Furthermore, the addition of IPA also slightly inhibited the photodegradation of LVX, suggesting ˙OH played the minor role in the degradation process. The presence of NaNO3 hardly affected the degradation, which proved that e− did not directly involved in the photodegradation reaction of LVX.
ESR spectra can provide a direct evidence to authenticate the participation of the ˙O2− and ˙OH in the photodegradation reaction of LVX. In this technique, DMPO was utilized as the capture agent for the measurement of ˙O2− and ˙OH with the presence of CN, 10%SVC and SV. As shown in Fig. 8a, the DMPO-˙O2− signal of 10%SVC was clearly stronger than that of pristine SV and CN, revealing that more ˙O2− radicals were generated with the existence of 10%SVC. The DMPO-˙O2− signal of SV was extremely weak, indicating the poor yield of ˙O2−. Similar results were observed in the ESR spectra of DMPO-˙OH with the presence of CN, 10%SVC and SV (Fig. 8b). 10%SVC had stronger DMPO-˙O2− and DMPO-˙OH signals, demonstrating the formation of the SVC heterostructures and the optimization of the surface OVs concentration was advantageous to accelerate the production of the ˙O2− and ˙OH radicals, even if the photocatalytic activity of pristine CN and SV was finite. It might result from the higher spatial separation efficiency of the photogenerated charge in SVCs. It also was proved by the photodegradation of LVX, MB and RhB. Actually, in these photodegradation experiments, CN exhibited better photocatalytic activity than SV, which was very in accordance with the ESR results.
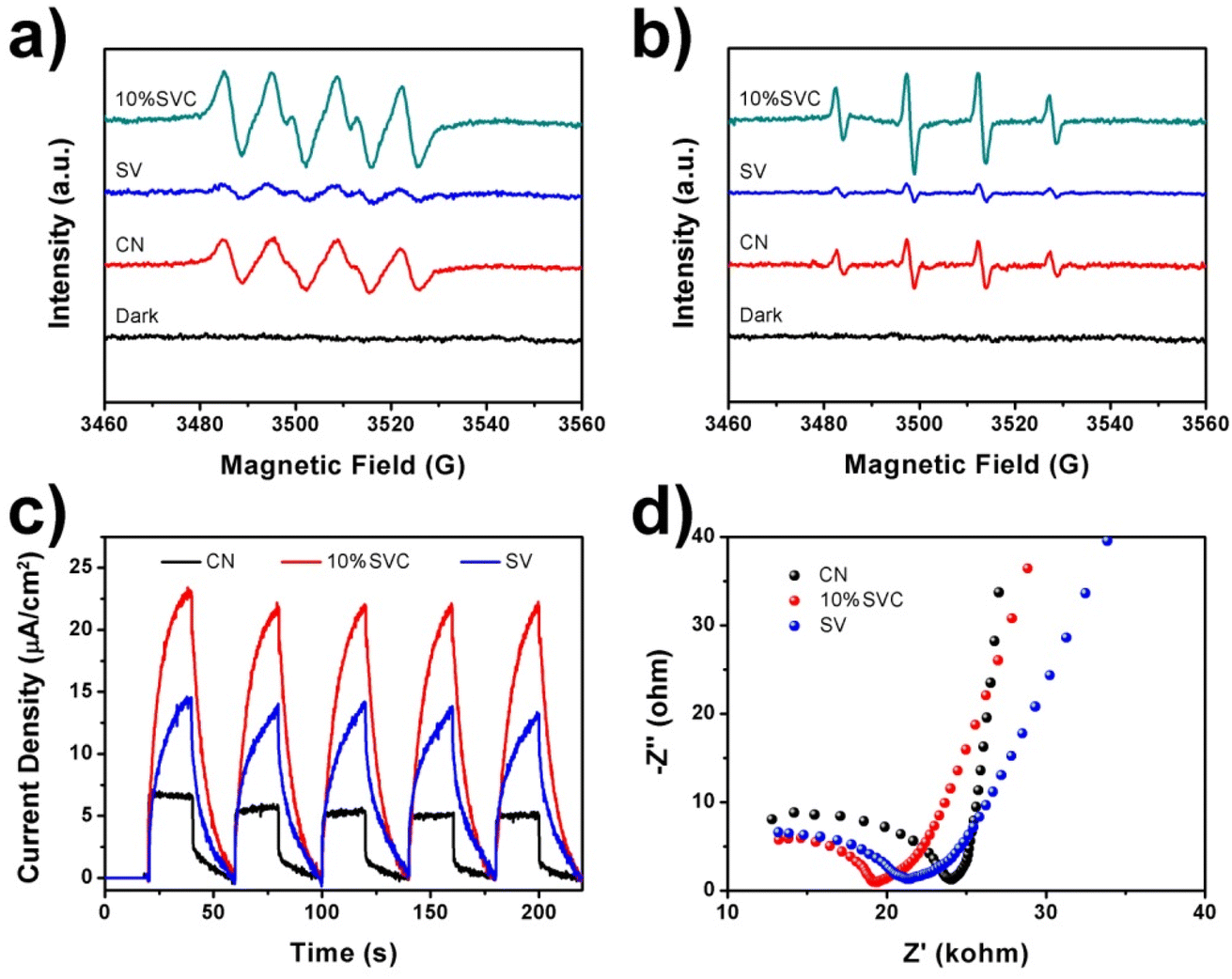 | ||
| Fig. 8 ESR spectra of (a) DMPO-˙O2− in methanol and (b) DMPO-˙OH in water with the presence of CN, 10%SVC and SV, (c) TPC spectra and (d) EIS Nyquist plots of CN, 10%SVC and SV. | ||
To further appraise the spatial separation efficiency of the photogenerated charge carriers, TPC spectra, EIS Nyquist plots and photoluminescence spectra (PL) of CN, 10%SVC and SV were measured. TPC spectra of these samples were illustrated in Fig. 8c. The photocurrent response curves of CN, 10%SVC and SV was stable and repeatable within 5 cycles of the on–off switching period, suggesting the high photo-stability of as-prepared catalysts. 10%SVC manifested higher photocurrent density than that of pristine CN and SV. The higher photocurrent implied the faster migration and the slower recombination of the photogenerated charge carriers, which finally resulted in better photocatalytic performance.57 Therefore, the enhanced photodegradation activity of LVX, MB and RhB on 10%SVC can attribute to the higher spatial separation efficiency of the electron–hole pairs in 10%SVC. The formation of the SVC heterostructures and the optimization of the surface OVs concentration might be two major factors to accelerate the separation of the charge carriers. EIS Nyquist plots of CN, 10%SVC and SV were depicted in Fig. 8d. It clearly showed that 10%SVC had much smaller arc radius than pristine CN, meaning that 10%SVC possessed lower charge transfer resistance58 and consequently contributed to reduce recombination rate of photogenerated carriers. Furthermore, the PL spectra of CN, 10%SVC and SV were displayed in Fig. S11.† The PL intensity decreased as compared to CN when the 10%SVC heterojunction formed. It was another evidence to indicate that the photogenerated carrier separation efficiency of 10%SVC was higher than that of CN.59 It could therefore effectively enhance photocatalytic degradation activity of LVX.
According to the results analyzed above, the schematic diagram of energy band structures of CN, SV and the possible mechanism of the photodegradation of LVX on 10%SVC were proposed and schemed in Fig. 9. The EVB of SV and CN was +1.60 and +1.19 eV, corresponded to the ECB of −0.41 and −1.52 eV, respectively. Therefore, the SVCs samples were type II heterojunction. The photogenerated electrons and holes were firstly generated in SVCs under the stimulation of the visible light (eqn (1) and (2)). In consideration of SVCs were belonged to type II heterojunction, the photogenerated electrons on the CB of CN migrated onto the CB of SV, and the photogenerated holes on the VB of SV transferred to the VB of CN. The VB potential of SV was negative than the redox potentials of ˙OH/OH⁻ (1.99 eV vs. NHE) and ˙OH/H2O (2.27 eV vs. NHE),60 revealing that H2O and OH⁻ could not be oxidized directly to form ˙OH by the holes on the VB of CN. Comparatively, the CB potential of SV was negative enough than the redox potential of O2/˙O2− (−0.33 eV vs. NHE). Therefore, the photogenerated electrons could reduce O2 to generate ˙O2− (eqn (3)). The ˙O2− could subsequently react with H+ to produce H2O2 (+0.68 eV vs. NHE) (eqn (4)). And H2O2 was then reduced by the photogenerated electrons to get ˙OH61 (eqn (5)). Finally, ˙OH, ˙O2− and h+ triggered the photodegradation of LVX (eqn (6)).
| CN + hν → e− + h+ | (1) |
| SV + hν → e− + h+ | (2) |
| O2 + e− (SV) → ˙O2− | (3) |
| ˙O2− + 2H+ + e− (SV) → H2O2 | (4) |
| H2O2 + e− (SV) → ˙OH + OH− | (5) |
| ˙OH/˙O2−/h+ (SVCs) + LVX → degraded products | (6) |
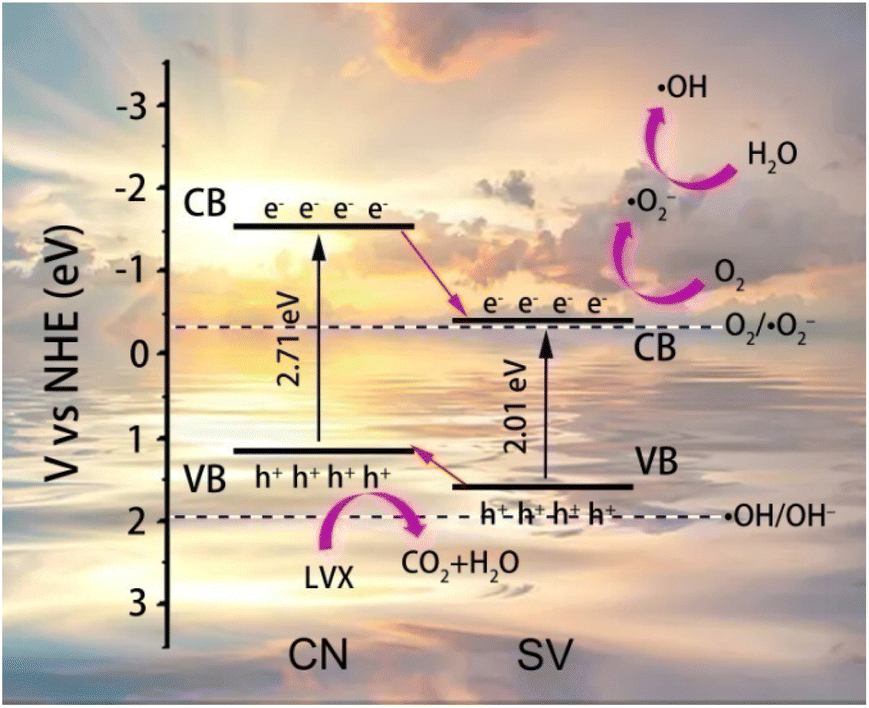 | ||
| Fig. 9 The possible mechanism of the photodegradation of LVX on 10%SVC. | ||
4. Conclusion
In summary, the Sc2VO5−δ/g-C3N4 heterojunctions with abundant OVs were successfully manufactured. The surface OVs of the catalyst can be effectively regulated by adjusting the calcination temperature. XRD, TEM, HRTEM, SEM, FTIR, Raman and XPS results clarified the successful formation of SVCs and its ultrathin structure. DRS and XPS VB spectra revealed it was type II heterojunction. EPR confirmed the existence of OVs and XPS O1s spectra determined the OVs concentration. PL, TPC and EIS assessed the promotion of the separation efficiency and the inhibition of the recombination of the photogenerated charge carriers in SVCs. The influence of OVs and SV content on the photocatalytic degradation efficiency of LVX, MB and RhB was investigated. Due to the introduction of surface OVs and the formation of heterojunction, the as-prepared SVCs manifested highly enhanced photocatalytic degradation efficiency. The as-prepared 10%SVC exhibited best photocatalytic performance. 89.1% of LVX, 98.8% of MB and 99.0% of RhB were effectively eliminated and the photodegradation data followed the pseudo first order kinetic equation. The kapp of LVX degradation on 10%SVC was 11.0 and 7.5 times of pristine CN and SV. The h+, ˙OH and ˙O2− were the major reactive species in the photodegradation process. This study supplied a new paradigm for further exploring the synergistic effect of surface OVs and heterostructure on the photocatalytic activity.Author contributions
Jian Feng: conceptualization, methodology, supervision, writing – review & editing, resources, funding acquisition. Liyao Zu: investigation. Hongrong Yang: investigation. Yuanyuan Zheng: investigation. Ziying Chen: investigation. Wei Song: investigation. Ran Zhao: investigation. Li Wang: investigation, data curation, visualization. Xia Ran: investigation, visualization. Bo Xiao: project administration.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This research was funded by the National Natural Science Foundation of China (Grant No. 21865006), the Key Project of Basic Research Program of Guizhou Province, China (No. ZK[2021]022), the Science and Technology Project of Guizhou Province, China (No. [2018]5779-18).References
- H. Anwer, A. Mahmood, J. Lee, K. Kim, J. Park and A. C. K. Yip, Photocatalysts for degradation of dyes in industrial effluents: opportunities and challenges, Nano Res., 2019, 12, 955–972 CrossRef CAS
.
- G. Wang, Q. Chang, X. Han and M. Zhang, Removal of Cr(VI) from aqueous solution by flocculant with the capacity of reduction and chelation, J. Hazard. Mater., 2013, 248–249, 115–121 CrossRef CAS PubMed
- M. K. Uddin, A review on the adsorption of heavy metals by clay minerals, with special focus on the past decade, Chem. Eng. J., 2017, 308, 438–462 CrossRef CAS
- M. Li, Y. Liu, G. Zeng, N. Liu and S. Liu, Graphene and graphene-based nanocomposites used for antibiotics removal in water treatment: a review, Chemosphere, 2019, 226, 360–380 CrossRef CAS PubMed
- F. Yi, J. Ma, C. Lin, L. Wang, H. Zhang, Y. Qian and K. Zhang, Insights into the enhanced adsorption/photocatalysis mechanism of a Bi4O5Br2/g-C3N4 nanosheet, J. Alloys Compd., 2020, 821, 153557 CrossRef CAS
- Y. Li, Z. Lai, Z. Huang, H. Wang, C. Zhao, G. Ruan and F. Du, Fabrication of BiOBr/MoS2/graphene oxide composites for efficient adsorption and photocatalytic removal of tetracycline antibiotics, Appl. Surf. Sci., 2021, 550, 149342 CrossRef CAS
- S. K. Kassahun, Z. Kiflie, H. Kim and A. F. Baye, Process optimization and kinetics analysis for photocatalytic degradation of emerging contaminant using N-doped TiO2-SiO2 nanoparticle: Artificial Neural Network and Surface Response Methodology approach, Environ. Technol. Innovat., 2021, 23, 101761 CrossRef CAS
- F. Yi, J. Liu, G. Liang, X. Xiao and H. Wang, Insight into the enhanced degradation mechanism of g-C3N4/g-C3N5 heterostructures through photocatalytic molecular oxygen activation in Van der Waals junction and excitation, J. Alloys Compd., 2022, 905, 164064 CrossRef CAS
- M. S. Anantha, S. Olivera, C. Hu, B. K. Jayanna, N. Reddy, K. Venkatesh, H. B. Muralidhara and R. Naidu, Comparison of the photocatalytic, adsorption and electrochemical methods for the removal of cationic dyes from aqueous solutions, Environ. Technol. Innovat., 2020, 17, 100612 CrossRef
- S. Zhang, P. Gu, R. Ma, C. Luo, T. Wen, G. Zhao, W. Cheng and X. Wang, Recent developments in fabrication and structure regulation of visible-lightdriven g-C3N4-based photocatalysts towards water purification: a critical review, Catal. Today, 2019, 335, 65–77 CrossRef CAS
- F. Yi, H. Gan, H. Jin, W. Zhao, K. Zhang, H. Jin, H. Zhang, Y. Qian and J. Ma, Sulfur- and chlorine-co-doped g-C3N4 nanosheets with enhanced active species generation for boosting visible-light photodegradation activity, Sep. Purif. Technol., 2020, 233, 115997 CrossRef CAS
- S. Wang, J. Zhang, B. Li, H. Sun and S. Wang, Engineered Graphitic Carbon Nitride-Based Photocatalysts for Visible-Light-Driven Water Splitting: A Review, Energy Fuel., 2021, 35, 6504–6526 CrossRef CAS
- K. Xu, X. Yang, L. Ruan, S. Qi, J. Liu, K. Liu, S. Pan, G. Feng, Z. Dai, X. Yang, R. Li and J. Feng, Superior Adsorption and Photocatalytic Degradation Capability of Mesoporous LaFeO3/g-C3N4 for Removal of Oxytetracycline, Catalysts, 2020, 10, 301 CrossRef CAS
- J. Fu, J. Yu, C. Jiang and B. Cheng, g-C3N4-Based Heterostructured Photocatalysts, Adv. Energy Mater., 2017, 7, 1701503 Search PubMed
- J. Zhu, P. Xiao, H. Li and S. A. C. Carabineiro, Graphitic Carbon Nitride: Synthesis, Properties, and Applications in
Catalysis, ACS Appl. Mater. Interfaces, 2014, 6, 16449–16465 CrossRef CAS PubMed
- V. Sydorchuk, S. Khalameida, N. Shcherban, V. Hreb, V. Mykhaylyk, Y. Zhydachevskyy and L. Vasylechko, Phase and structural behavior and photocatalytic properties of new mixed bismuth-praseodymium vanadates, J. Solid State Chem., 2021, 296, 122002 CrossRef CAS
- P. Mazierski, J. Sowik, M. Miodyńska, G. Trykowski, A. Mikołajczyk, T. Klimczuk, W. Lisowski, J. Nadolna and A. Zaleska-Medynska, Shape-controllable synthesis of GdVO4 photocatalysts and their tunable properties in photocatalytic hydrogen generation, Dalton Trans., 2019, 48, 1662–1671 RSC
- I. A. Mkhalid, R. M. Mohamed, M. Alhaddad, A. Basaleh, L. A. Al-Hajji and A. A. Ismail, Construction of mesoporous lanthanum orthovanadate/carbon nitride heterojunction photocatalyst for the mineralization of trichloroethylene, Ceram. Int., 2022, 48, 14899–14912 CrossRef CAS
- J. Ding, X. Liu, M. Wang, Q. Liu, T. Sun, G. Jiang and Y. Tang, Controlled synthesis of CeVO4 hierarchical hollow microspheres with tunable hollowness and their efficient photocatalytic activity, CrystEngComm, 2018, 20, 4499–4505 RSC
- M. Vosoughifar, Dysprosium vanadate nanoparticles: morphology-controlled preparation, characterization and investigation of photocatalytic properties, J. Mater. Sci.: Mater. Electron., 2017, 28, 13976–13982 CrossRef CAS
- C. Reitz, B. Smarsly and T. Brezesinski, General Synthesis of Ordered Mesoporous Rare-Earth Orthovanadate Thin Films and Their Use as Photocatalysts and Phosphors for Lighting Applications, ACS Appl. Nano Mater., 2019, 2, 1063–1071 CrossRef CAS
- A. Alborzi and M. Nasiri, Novel size-controlled fabrication of pure HoVO4 nanoparticles via a new approach, J. Mater. Sci.: Mater. Electron., 2017, 28, 13829–13835 CrossRef CAS
- M. Vosoughifar, Synthesis, characterization, and investigation magnetic and photocatalytic property of neodymium vanadate nanoparticles, J. Mater. Sci.: Mater. Electron., 2016, 27, 7384–7388 CrossRef CAS
- B. Sriram, J. N. Baby, Y.-F. Hsu, S.-F. Wang and M. George, Surfactant-Assisted Synthesis of Praseodymium Orthovanadate Nanofiber-Supported NiFe-Layered Double Hydroxide Bifunctional Catalyst: The Electrochemical Detection and Degradation of Diphenylamine, Inorg. Chem., 2022, 61, 5824–5835 CrossRef CAS PubMed
- T.-A. Quach, J. Becerra, D.-T. Nguyen, M. Sakar, M.-H. Vu, F. Dion, H. Abou-Rachid and T.-O. Do, Direct Z-scheme mediated SmVO4/UiO-66-NH2 heterojunction nanocomposite for the degradation of antibiotic tetracycline hydrochloride molecules under sunlight, Chemosphere, 2022, 303, 134861 CrossRef CAS PubMed
- R. K. Selvan, A. Gedanken, P. Anilkumar, G. Manikandan and C. Karunakaran, Synthesis and Characterization of Rare Earth Orthovanadate (RVO4; La, Ce, Nd, Sm, Eu & Gd) Nanorods/Nanocrystals/Nanospindles by a Facile Sonochemical Method and Their Catalytic Properties, J. Clust. Sci., 2009, 20, 291–305 CrossRef
- J. Guan, J. Li, Z. Ye, D. Wu, C. Liu, H. Wang, C. Ma, P. Huo and Y. Yan, La2O3 media enhanced electrons transfer for improved CeVO4@halloysite nanotubes photocatalytic activity for removing tetracycline, J. Taiwan Inst. Chem. Eng., 2019, 96, 281–298 CrossRef CAS
- Y. Shen, Y. Huang, S. Zheng, X. Guo, Z.-X. Chen, L. Peng and W. Ding, Nanocrystals of CeVO4 Doped by Metallic Heteroions, Inorg. Chem., 2011, 50, 6189–6194 CrossRef CAS PubMed
- P. A. Deshpande and G. Madras, Photocatalytic degradation of phenol by base metal-substituted orthovanadates, Chem. Eng. J., 2010, 161, 136–145 CrossRef CAS
- W. Zhai, J. He, S. Hu, Y. Liang, F. Chen, Y. Wang, G. He and Q. He, Enhanced photocatalytic degradation of tetracycline over magnetic La0.7Sr0.3MnO3/g-C3N4 p–n heterojunction arising from the synergistic effects of oxygen vacancy defects and high-potential photogenerated electrons, J. Alloys Compd., 2022, 918, 165699 CrossRef CAS
- W. Liu, X. Li, X. Chu, S. Zuo, B. Gao, C. Yao, Z. Li and Y. Chen, Boosting photocatalytic reduction of nitrate to ammonia enabled by perovskite/biochar nanocomposites with oxygen defects and O-containing functional groups, Chemosphere, 2022, 294, 133763 CrossRef CAS PubMed
- H. Mai, D. Chen, Y. Tachibana, H. Suzuki, R. Abe and R. A. Caruso, Developing sustainable, high-performance perovskites in photocatalysis: design strategies and applications, Chem. Soc. Rev., 2021, 50, 13692–13729 RSC
- P. Wang, X. Ma, X. Hao, B. Tang, A. Abudula and G. Guan, Oxygen vacancy defect engineering to promote catalytic activity toward the oxidation of VOCs: a critical review, Catal. Rev., 2022 DOI:10.1080/01614940.2022.2078555
- J. Low, J. Yu, M. Jaroniec, S. Wageh and A. A. Al-Ghamdi, Heterojunction Photocatalysts, Adv. Mater., 2017, 29, 1601694 CrossRef PubMed
- P. K. Kaur, R. Badru, P. P. Singh and S. Kaushal, Photodegradation of organic pollutants using heterojunctions: a review, J. Environ. Chem. Eng., 2020, 8, 103666 CrossRef
- Y.-A. Chen, Y.-T. Wang, H. S. Moon, K. Yong and Y.-J. Hsu, Yolk–shell nanostructures: synthesis, photocatalysis and interfacial charge dynamics, RSC Adv., 2021, 11, 12288–12305 RSC
- C.-W. Tsao, M.-J. Fang and Y.-J. Hsu, Modulation of interfacial charge dynamics of semiconductor heterostructures for advanced photocatalytic applications, Coord. Chem. Rev., 2021, 438, 213876 CrossRef CAS
- X. Zou, C. Yuan, Y. Dong, H. Ge, J. Ke and Y. Cui, Lanthanum orthovanadate/bismuth oxybromide heterojunction for enhanced photocatalytic air purification and mechanism exploration, Chem. Eng. J., 2020, 379, 122380 CrossRef CAS
- Y.-H. Chiu, T.-F. M. Chang, C.-Y. Chen, M. Sone and Y.-J. Hsu, Mechanistic Insights into Photodegradation of Organic Dyes Using Heterostructure Photocatalysts, Catalysts, 2019, 9, 430 CrossRef CAS
- Z. Lin and X. Wang, Nanostructure Engineering and Doping of Conjugated Carbon Nitride Semiconductors for Hydrogen Photosynthesis, Angew. Chem., Int. Ed., 2013, 52, 1735–1738 CrossRef CAS PubMed
- L. Wang, X. Ran, B. Xiao, L. Lei, J. Zhu, X. Xi, G. Feng, R. Li and J. Feng, Visible light assisted Fenton degradation of oxytetracycline over perovskite ErFeO3/porous g-C3N4 nanosheets p–n heterojunction, J. Environ. Chem. Eng., 2022, 10, 108330 CrossRef CAS
- B. Rani, A. K. Nayak and N. K. Sahu, Degradation of mixed cationic dye pollutant by metal free melem derivatives and graphitic carbon nitride, Chemosphere, 2022, 298, 134249 CrossRef CAS PubMed
- Y. Yuan, L. Zhang, J. Xing, M. I. B. Utama, X. Lu, K. Du, Y. Li, X. Hu, S. Wang, A. Genç, R. Dunin-Borkowski, J. Arbiold and Q. Xiong, High-yield synthesis and optical properties of g-C3N4, Nanoscale, 2015, 7, 12343–12350 RSC
- Z. Chu, J. Li, Y.-P. Lan, C. Chen, J. Yang, D. Ning, X. Xia and X. Mao, KCl–LiCl molten salt synthesis of LaOCl/CeO2-g-C3N4 with excellent photocatalytic-adsorbed removal performance for organic dye pollutant, Ceram. Int., 2022, 48, 15439–15450 CrossRef CAS
- A. Kumar, S. Singh and M. Khanuja, A comparative photocatalytic study of pure and acid-etched template free graphitic C3N4 on different dyes: an investigation on the influence of surface modifications, Mater. Chem. Phys., 2020, 243, 122402 CrossRef CAS
- P. Kumar, E. Vahidzadeh, U. K. Thakur, P. Kar, K. M. Alam, A. Goswami, N. Mahdi, K. Cui, G. M. Bernard, V. K. Michaelis and K. Shankar, C3N5: A Low Bandgap Semiconductor Containing an Azo-Linked Carbon Nitride Framework for Photocatalytic, Photovoltaic and Adsorbent Applications, J. Am. Chem. Soc., 2019, 141, 5415–5436 CrossRef CAS PubMed
- Y. Shiraishi, S. Kanazawa, Y. Sugano, D. Tsukamoto, H. Sakamoto, S. Ichikawa and T. Hirai, Highly Selective Production of Hydrogen Peroxide on Graphitic Carbon Nitride (g-C3N4) Photocatalyst Activated by Visible Light, ACS Catal., 2014, 4, 774–780 CrossRef CAS
- A. Ibrahim, U. B. Memon, S. P. Duttagupta, I. Mahesh, R. K. S. Raman, A. Sarkar, G. Pendharkar and S. S. V. Tatiparti, Nano-structured palladium impregnate graphitic carbon nitride composite for efficient hydrogen gas sensing, Int. J. Hydrogen Energy, 2020, 45, 10623–10636 CrossRef CAS
- X. Jiang, Q. Xing, X. Luo, F. Li, J. Zou, S. Liu, X. Li and X. Wang, Simultaneous photoreduction of uranium (VI) and photooxidation of arsenic (III) in aqueous solution over g-C3N4/TiO2 heterostructured catalysts under simulated sunlight irradiation, Appl. Catal. B Environ., 2018, 228, 29–38 CrossRef CAS
- T. Lv, M. Wu, M. Guo, Q. Liu and L. Jia, Self-assembly photocatalytic reduction synthesis of graphene-encapusulated LaNiO3 nanoreactor with high efficiency and stability for photocatalytic water splitting to hydrogen, Chem. Eng. J., 2019, 356, 580–591 CrossRef CAS
- S. Li, X. Yang, Z. Cui, Y. Xu, Z. Niu, P. Li, D. Pan and W. Wu, Efficient photoreduction strategy for uranium immobilization based on graphite carbon nitride/perovskite oxide heterojunction nanocomposites, Appl. Catal. B Environ., 2021, 298, 120625 CrossRef CAS
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Sc, Ti, V, Cu and Zn, Appl. Surf. Sci., 2010, 257, 887–898 CrossRef CAS
- M. Sakar, S. Balakumar, P. Saravanan and S. Bharathkumar, Particulates vs. fibers: dimension featured magnetic and visible light driven photocatalytic properties of Sc modified multiferroic bismuth ferrite nanostructures, Nanoscale, 2016, 8, 1147–1160 RSC
- X. Wang, X. Chen, A. Thomas, X. Fu and M. Antonietti, Metal-containing carbon nitride compounds: a new functional organic-metal hybrid material, Adv. Mater., 2009, 21, 1609–1612 CrossRef CAS
- T. Chen, Z. Zhu, Z. Wang, H. Zhang, Y. Qiu, D. Yin and G. Zhao, 3D hollow sphere-like Cu-incorporated LaAlO3 perovskites for peroxymonosulfate activation: Coaction of electron transfer and oxygen defect, Chem. Eng. J., 2020, 385, 123935 CrossRef CAS
- Y. Chen, W. Yang, S. Gao, C. Sun and Q. Li, Synthesis of Bi2MoO6 Nanosheets with Rich Oxygen Vacancies by Postsynthesis Etching Treatment for Enhanced Photocatalytic Performance, ACS Appl. Nano Mater., 2018, 1, 3565–3578 CrossRef CAS
- Y. Yao, J. Fan, M. Shen, W. Li, B. Du, X. Li and J. Dai, One-step synthesis of hexylresorcinol calix[4]arene-capped ZnO–Ag nanocomposites for enhanced degradation of organic pollutants, J. Colloid Interface Sci., 2019, 546, 70–82 CrossRef CAS PubMed
- Y. Zhang, A. Sun, M. Xiong, D. K. Macharia, J. Liu, Z. Chen, M. Li and L. Zhang, TiO2/BiOI p–n junction-decorated carbon fibers as weavable photocatalyst with UV-vis photoresponsive for efficiently degrading various pollutants, Chem. Eng. J., 2021, 415, 129019 CrossRef CAS
- J. Feng, X. Ran, L. Wang, B. Xiao, L. Lei, J. Zhu, Z. Liu, X. Xi, G. Feng, Z. Dai and R. Li, The synergistic effect of adsorption-photocatalysis for removal of organic pollutants on mesoporous Cu2V2O7/Cu3V2O8/g-C3N4 heterojunction, Int. J. Mol. Sci., 2022, 23, 14264 CrossRef CAS PubMed
- B. Liu, W. Guo, Q. Si, W. Jia, S. Zheng, H. Wang, Q. Zhao, H. Luo, J. Jiang and N. Ren, Atomically dispersed cobalt on carbon nitride for peroxymonosulfate activation: Switchable catalysis enabled by light irradiation, Chem. Eng. J., 2022, 446, 137277 CrossRef CAS
- Y. Q. He, F. Zhang, B. Ma, N. Xu, L. B. Junior, B. Yao, Q. Yang, D. Liu and Z. Ma, Remarkably enhanced visible-light photocatalytic hydrogen evolution and antibiotic degradation over g-C3N4 nanosheets decorated by using nickel phosphide and gold nanoparticles as cocatalysts, Appl. Surf. Sci., 2020, 517, 146187 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra07484b |
| This journal is © The Royal Society of Chemistry 2023 |