
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLuminescent 1H-1,3-benzazaphospholes†
Sloane Evariste a,
Alexandra M. Harrisona,
Sunandan Sarkarb,
Arnold L. Rheingoldc,
Barry D. Dunietzd,
Joachim W. Heinickee,
Emalyn Delgado Rosarioa,
Sungwoon Yoonf,
Thomas S. Teetsf and
John D. Protasiewicz*a
a,
Alexandra M. Harrisona,
Sunandan Sarkarb,
Arnold L. Rheingoldc,
Barry D. Dunietzd,
Joachim W. Heinickee,
Emalyn Delgado Rosarioa,
Sungwoon Yoonf,
Thomas S. Teetsf and
John D. Protasiewicz*a
aDepartment of Chemistry, Case Western Reserve University, Cleveland, Ohio 44106, USA. E-mail: protasiewicz@case.edu
bDepartment of Chemistry, National Institute of Technology Tiruchirappalli, Tiruchirappalli, Tamil Nadu 620015, India
cDepartment of Chemistry and Biochemistry, University of California, La Jolla, San Diego, California 92093, USA
dDepartment of Chemistry and Biochemistry, Kent State University, Kent, Ohio 44242, USA
eInstitut für Biochemie, Anorganische Chemie, Ernst-Moritz-Arndt-Universität Greifswald, Felix-Hausdorff-Str. 4, D-17489, Greisfwald, Germany
fDepartment of Chemistry, University of Houston, 3585 Cullen Blvd. Room 112, Houston, TX 77204-5003, USA
First published on 22nd December 2022
Abstract
2-R-1H-1,3-Benzazaphospholes (R-BAPs) are an interesting class of σ2P heterocycles containing P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds. While closely related 2-R-1,3-benzoxaphospholes (R-BOPs) have been shown to be highly photoluminescent materials depending on specific R substituents, photoluminescence of R-BAPs has been previously limited to an example having a fused carbazole ring system. Here we detail the synthesis and structural characterization of a new R-BAP (3c, R = 2,2′-dithiophene), and compare its photoluminescence against two previously reported R-BAPs (3a, R, R′ = Me and 3b, R = 2-thiophene). The significant fluorescence displayed by the thiophene derivatives 3b (φ = 0.53) and 3c (φ = 0.12) stands in contrast to the weakly emissive methyl substituted analogue 3a (φ = 0.08). Comparative computational investigations of 3a–c offer insights into the interplay between structure–function relationships affecting excited state relaxation processes.
C bonds. While closely related 2-R-1,3-benzoxaphospholes (R-BOPs) have been shown to be highly photoluminescent materials depending on specific R substituents, photoluminescence of R-BAPs has been previously limited to an example having a fused carbazole ring system. Here we detail the synthesis and structural characterization of a new R-BAP (3c, R = 2,2′-dithiophene), and compare its photoluminescence against two previously reported R-BAPs (3a, R, R′ = Me and 3b, R = 2-thiophene). The significant fluorescence displayed by the thiophene derivatives 3b (φ = 0.53) and 3c (φ = 0.12) stands in contrast to the weakly emissive methyl substituted analogue 3a (φ = 0.08). Comparative computational investigations of 3a–c offer insights into the interplay between structure–function relationships affecting excited state relaxation processes.
Introduction
Organic π-conjugated materials continue to receive widespread attention; their optical and electronic properties make them potential candidates as semiconducting materials in optoelectronic devices.1–12 Many different strategies are being used to improve or modulate the electronic properties of π-conjugated oligomers or polymers, including incorporation of heterocyclopentadienes (such as thiophenes, pyrroles, and phospholes).13–17Phosphorus containing heterocycles can be subdivided by coordination number, with three coordinate and four coordinate geometries (σ3, σ4) being the most commonly studied systems.18–24 In particular, σ3,λ3-phospholes have drawn much attention, as their (i) trigonal pyramidal geometry and reduced aromaticity allow for unusual means to tune electronic delocalization into the π-system, and (ii) the lone pair on phosphorus offers additional opportunities to modulate HOMO and LUMO levels. Specifically, chemical modification of the P atom by oxidation, alkylation, or coordination by Lewis acids and metals has led to tunable new π-conjugated systems with diverse photophysical properties.25
In particular, σ4,λ5-phospholes can display impressive luminescence, and many different types of derivatives have been synthesized for diverse potential applications (Chart 1, A–D).26–35 For example, dibenzophospholes (A) have been used for OLEDs36 and dithienophosphole building blocks (B) have been used to generate white light emission.37 Fluorescent 2,2′-benzo[b]phosphole-benzo[b]heteroles (C)38 and super-photostable dyes based on naphthalene-fused phospholes (D)39 represent significant advances as well.
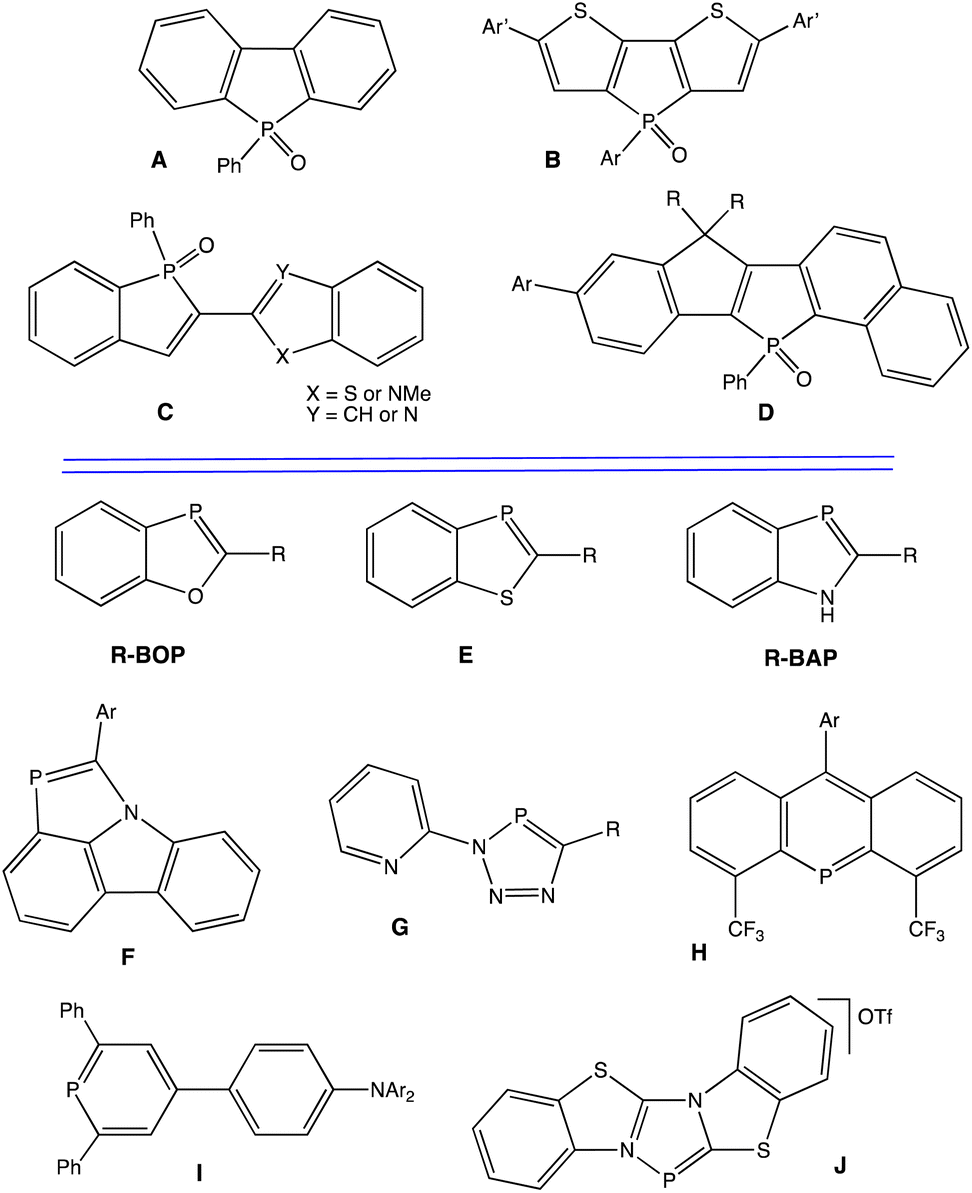 | ||
| Chart 1 Examples of luminescent phospholes. | ||
From this aspect, σ2,λ3-phospholes are much less studied. Such compounds feature PC pπ–pπ bonds directly analogous to conventional CC π bonds and offer further opportunities to explore π-conjugated materials. Our group has thus been interested in the development of luminescent phosphaalkenes featuring two-coordinate phosphorus atoms (σ2).40 In 2010,41 we reported investigations of highly luminescent 1,3-benzoxaphospholes (R-BOP, Chart 1), and have since documented other luminescent benzoxaphosphole based materials.42–46 The analogous sulfur-containing 1,3-benzothiaphospholes (E) are also luminescent.47 While numerous 1H-1,3-benzazaphospholes (R-BAP) have been reported, no significant photoluminescence studies have appeared.48–55 However, we have shown that a benzazaphosphole (F, Chart 1) adhered to carbazole (a common fluorophore) can display significant luminescence distinct from carbazole.56 The development of other luminescent materials featuring σ2,λ3-phosphorus is rapidly expanding, as recent examples G–J illustrate in Chart 1.57
These observations inspired re-examination of selected R-BAPs to probe their potential as luminescent materials. In this study previously reported 2-thiophene-1,3-benzazaphosphole58a is shown to be highly luminescent. Comparisons are also made to a previously reported 2-methyl substituted analogue,58b as well as to a new 2-dithiophene analogue. Modifications of the 2-substituent are shown to lead to significant changes in emission properties, and the underlying photophysical reasons for these changes have been analyzed by computational studies.
Results and discussion
1H-1,3-Benzazaphospholes 3a–c were identified for this study (Scheme 1). Compounds 3a and 3b were synthesized as previously reported.58 Following similar protocols, the new BAP derivative 3c was also prepared. The structure of 3c was readily confirmed by routine NMR spectroscopic analyses. For example, the 31P{1H} NMR resonance for 3c at δ 77.7 ppm is consistent with previously reported shifts for 1,3-benzazaphospholes, which typically range from δ 72.0 to 78.8 ppm.58 Absolute identification of 3c was made by an X-ray diffraction study.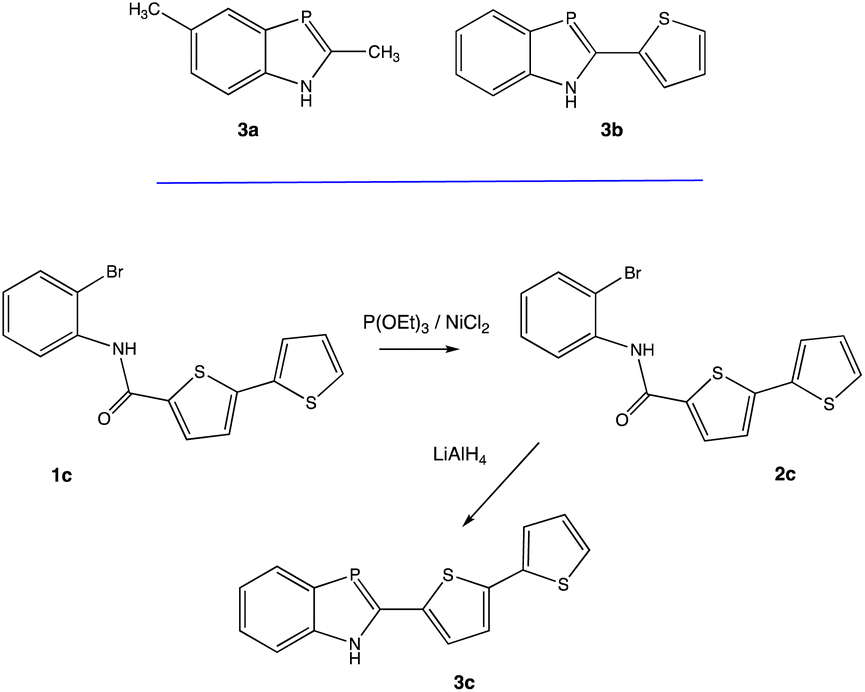 | ||
| Scheme 1 1H-1,3-benzazaphospholes 3a and 3b (top), preparation of 3c (bottom). | ||
Crystals of 3c suitable for X-ray diffraction studies were grown by diffusion of pentane into a concentrated CH2Cl2 solutions of 3c. The resulting solid-state structure is portrayed in Fig. 1, along with key bond lengths and angles (see ESI† for experimental details).
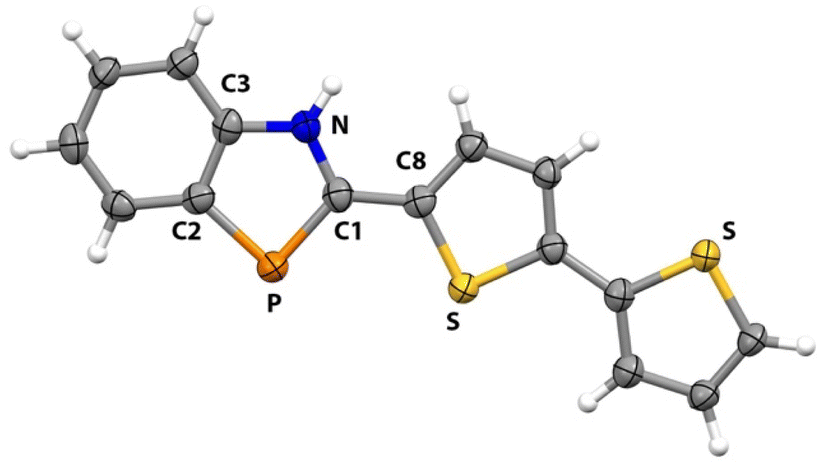 | ||
| Fig. 1 X-ray crystal structure of compound 3c. Selected bond lengths (Å) and bond angles (°) of 3c: PC1 1.745(5), P–C2 1.800(5), N–C1 1.353(7), N–C3 1.381(6), C2–C3 1.395(7), C1–C8 1.449(8), C1–P–C2 88.8(2), N–C1–P 113.1(4), C1–N–C3 114.8(4). | ||
The PC1 bond length in 3c (1.745(5) Å) is slightly longer than those distances found in related BAP derivatives (1.70–1.73 Å range).58,59 This value is between single and double bonds owing to delocalization across the NCP array. The C1–P–C2 angle in 3c (88.8(2)°) is consistent with such angles determined in related BAPs (88.2–90.1° range). Consistent with this hypothesis, all of the atoms in 3c lie in a nearly perfect plane (max deviation from mean plane is 0.16 Å for remote S atom).
The crystal packing diagram for compound 3c is represented in Fig. 2, and no π–π stacking interactions are noted in the crystalline state. Instead, a herringbone head-to-tail configuration along the a-axis (closest CH⋯centroid distance 3.0 Å) is observed. The centroid distance between two phosphole rings was found to be ca. 4.42 Å for 3c, which is larger than π–π interactions observed for some other BAP derivatives (ca. 3.45 Å).
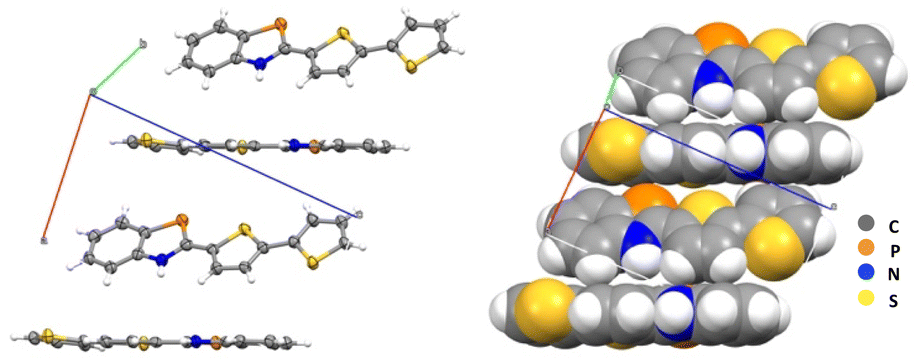 | ||
| Fig. 2 Packing diagrams for derivative 3c. | ||
Optical properties of BAP derivatives
The UV/Vis absorption and fluorescence spectra of the three compounds 3a–c were recorded in dry degassed CH2Cl2 (Table 1). All three BAP derivatives exhibit an absorption maximum in the UV-visible region (Fig. 3), attributed to π–π* transitions in the extended π-conjugated system. The nature of the substituent has a noticeable influence on the absorption maximum (λmax(3a) = 297 nm < λmax(3b) = 335 nm < λmax(3c) = 398 nm; Table 1), revealing enhanced π-conjugation that leads to the absorption wavelength being red-shifted. Thus, the extended system 3c possessing two thiophene rings absorbs at the longest wavelengths and across a wider UV-visible region compared to the others.| λmax (nm) | log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ε ε |
λF,max (nm) | ϕF | |
|---|---|---|---|---|
| a All measurements were performed in CH2Cl2.b Fluorescence quantum yield determined relative to 9,10-diphenylanthracene in cyclohexane.c Fluorescence quantum yield determined relative to anthracene in ethanol.d Fluorescence quantum yield determined relative to quinine sulfate in 0.5 M H2SO4 solution.e Data from ref. 60, n-hexane solvent. | ||||
| 3a | 297 | 3.81 | 407 | 0.08b |
| 3b | 335 | 4.14 | 470 | 0.53c |
| 3c | 398 | 4.30 | 508 | 0.12d |
| THOINe | 317 | — | 379 | 0.22 |
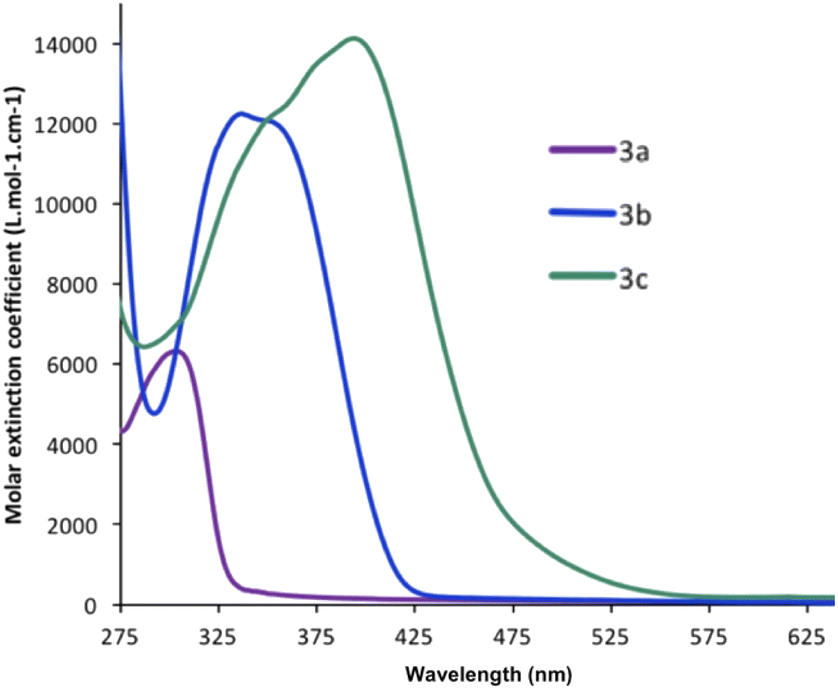 | ||
| Fig. 3 Absorption spectra recorded in CH2Cl2 for compounds 3a–c. | ||
On excitation at λmax, all the derivatives 3a–c emit in the visible region (Fig. 4). The same trend was observed with a red-shift in the emission spectra when the 2-R substituent is aromatic (λem(3b) = 470 nm), rather than being aliphatic (λem(3a) = 407 nm). The addition of a second thiophene to the first shifts the emission maximum further (λem(3c) = 508 nm), evidence that the π-conjugation is extended through the terminal thiophene ring. The reasons for the presence of a small shoulder for the emission for 3a are unclear, but it is unlikely to be due to aggregation. The fluorescence quantum yields ϕF in solution vary from 8% for 3a (2-R = CH3) to 53% for 3b (2-R = 2-thiophene). The quantum yield of 3c drops (ϕF = 12%) compared to the 3b derivative possessing only one thiophene unit. The emission maxima from 3a to 3b are also similar to luminescent benzoxaphospholes.41 The photophysics of the direct phosphorus-free (isolobal replacement of P for CH unit) indole analogue of 3b (THOIN, Chart 2) has been examined experimentally and theoretically in some detail.60
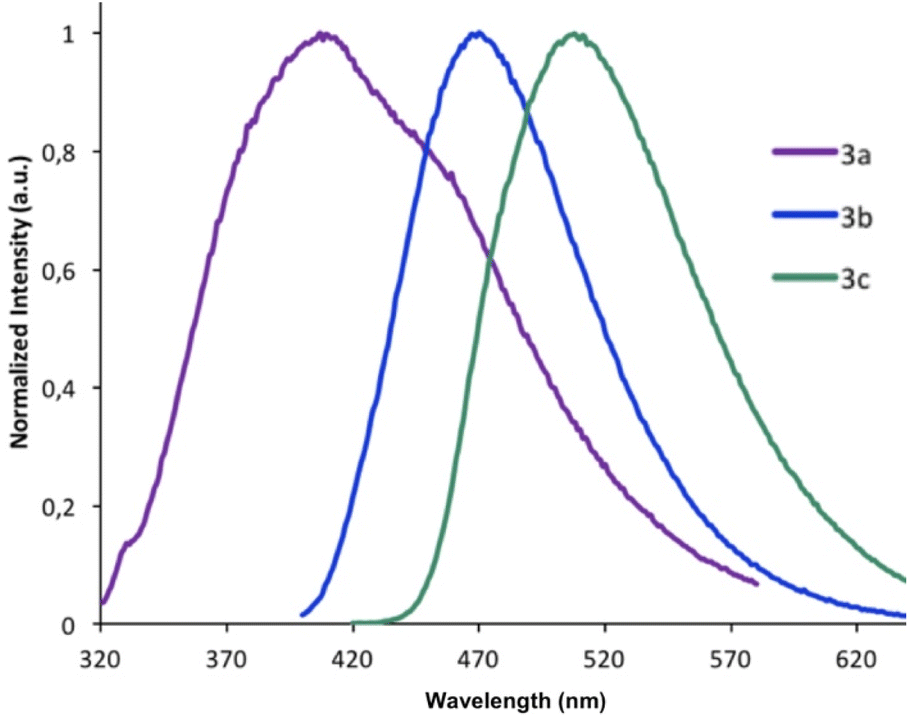 | ||
| Fig. 4 Emission spectra recorded in CH2Cl2 for compounds 3a–c. Each derivative was excited at its maximal absorbance λmax. | ||
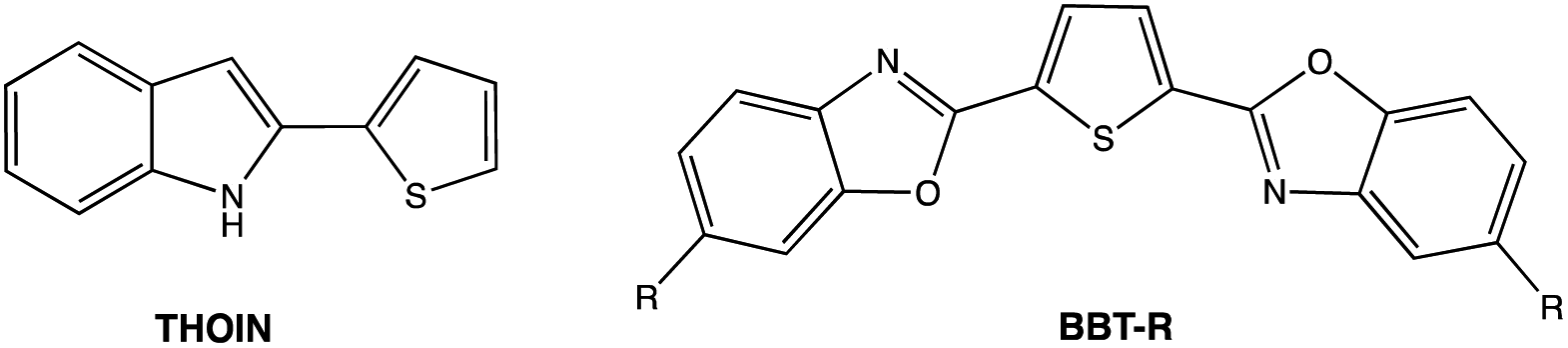 | ||
| Chart 2 Related luminescent molecules. | ||
The absorption maximum for compound 3b is red shifted compared to THOIN (Table 1) by 18 nm. The emission maxima, however, is shifted by 91 nm, yielding a significant Stokes shift of 135 nm (stokes shifts: THOIN: 5150 cm−1; 3b: 8574 cm−1; 3c: 5441 cm−1). Stokes shifts for Ar-BOPs range from 79 to 89 nm (5401–5852 cm−1). Thiophene substituted heterocycles such as BBT-R (Chart 2, R = NH2) have been developed showing even larger Stokes shifts, up to 186 nm (8752 cm−1).61,62 Large Stokes shifts have been associated with various features, such as intramolecular charge transfer (ICT), rotation of π-conjugated substituents, excited state intramolecular proton transfer (ESIPT) processes, and solvent effects.63 The larger Stokes shift observed for 3b might reflect significant changes in bond lengths upon excitation (vide infra).
Both absorption and emission spectra of compound 3b were recorded in several solvents (toluene, THF, dichloromethane, acetonitrile, methanol and trifluoroethanol; Fig. 5). Regardless of the solvent, there was no significant modification in its optical properties, suggesting that the absorption and emission processes for 3b do not involve significant charge transfer or special solvation effects. Indole derivative THOIN and Ar-BOPs also display solvent independent absorption and emission properties. Lifetime measurements conducted on 3b and 3c in CH2Cl2 revealed τ = 7.4 and 1.7 ns, respectively, which can be compared to fluorescence lifetimes determined for Ar-BOP (τ = 4.8–28.0)41 and THOIN (τ = 0.8).60
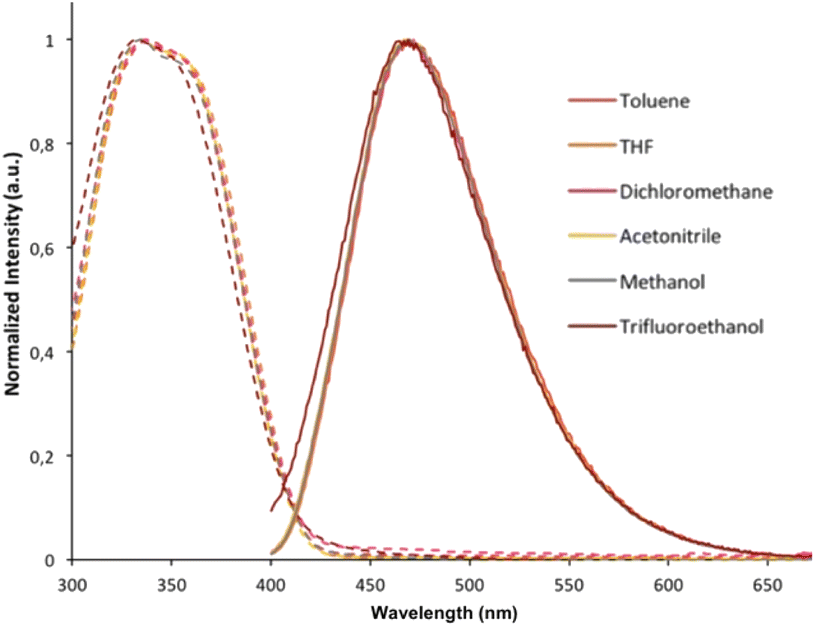 | ||
| Fig. 5 Absorption (dotted lines) and emission spectra (solid lines; λexc = 320 nm) for compound 3b recorded in different polarity solvents. | ||
Computational studies
Density functional theory (DFT) and time-dependent DFT (ωB97X-D/6-311++G(d,p) in polarizable continuum model (PCM)) calculations were employed to study the materials at the electronic structure level. The calculated ground state structural parameters (bond lengths and angles) for 3a and 3c correspond well with corresponding measured values from X-ray structure analyses. As shown in Table 2, the calculated bond lengths are within 0.15 Å of the measured data, and the calculated bond angles are within one degree of measured values.| Parameters | 3a (ref. 58b) | 3b | 3c |
|---|---|---|---|
| P–C1 | 1.728 (1.728) | 1.734 | 1.733 (1.745) |
| P–C2 | 1.799 (1.793) | 1.791 | 1.791 (1.800) |
| N–C1 | 1.355 (1.369) | 1.359 | 1.360 (1.353) |
| N–C3 | 1.379 (1.380) | 1.373 | 1.373 (1.381) |
| C2–C3 | 1.407 (1.413) | 1.408 | 1.408 (1.395) |
| C1–C8 | 1.495 (1.493) | 1.457 | 1.457 (1.449) |
| C1–P–C2 | 89.1 (89.5) | 88.8 | 88.7 (88.8) |
| N–C1–P | 113.5 (113.3) | 113.5 | 113.6 (113.1) |
| C1–N–C3 | 115.0 (114.6) | 114.7 | 114.6 (114.8) |
With the calculated geometries in excellent agreement with the crystallographic results, we proceeded to consider the excited states. Our approach follows our previous study on related organophosphorus systems64–67 and uses well benchmarked methods.64–66 The S0 → S1 excitations of 3a–c mainly correspond to the HOMO → LUMO transitions. The electronic densities associated with the HOMO and LUMO are delocalized throughout the π-systems for all compounds. In both 3b and 3c the π-conjugation is extended across the molecule including the thiophene unit. Hence, a decrease in the HOMO–LUMO energy gap is observed with the increase of the π-conjugation in the series (ΔEg(3a) = 8.32 eV > ΔEg(3b) = 7.35 eV > ΔEg(3c) = 7.08 eV; Fig. 6). As a result, the calculated lowest energy state (S1) is shifted towards longer wavelengths from 3a to 3c for both the absorption and emission processes (Table 3).
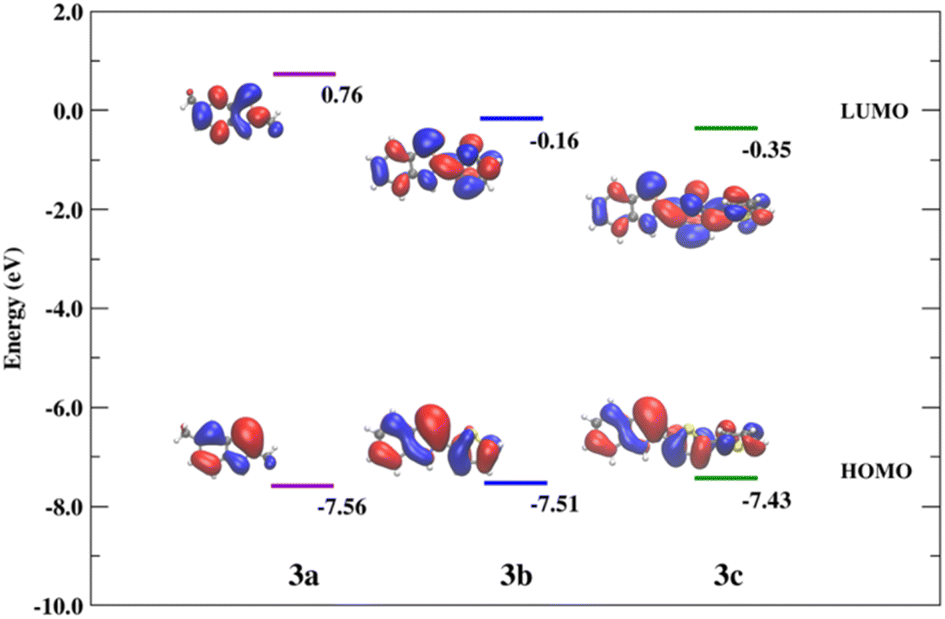 | ||
| Fig. 6 Spatial plots of the HOMO and LUMO of 3a–c. | ||
| λmax exp (nm) | λmax calc (nm) | F (H → L) | λem exp (nm) | λem calc (nm) | F (H ← L) | |
|---|---|---|---|---|---|---|
| 3a | 297 | 263 | 0.209 (0.94) | 407 | 302 | 0.198 (0.93) |
| 3b | 335 | 319 | 0.602 (0.96) | 470 | 404 | 0.597 (0.97) |
| 3c | 398 | 336 | 1.005 (0.95) | 508 | 464 | 1.118 (0.97) |
Upon excitation, longer PC bond lengths for 3b (Δ 0.112 Å) and 3c (Δ 0.074 Å) are predicted (see Fig. S5 in ESI† for mapping of other bond length changes). The greater change in PC bond length for 3b correlates with this material showing an enhanced Stokes shift. The CC bond bridging the BAP and directly connected thiophene shows corresponding bond length decreases (ca. 0.07 Å), which are consistent with the tendency of these systems to planarize in the excited state.
The alkyl substituted derivative displays significantly lower quantum yield for emission than found for the thiophene derivatives. Part of this can be attributed to the reduced oscillator strength f (0.21, Table 3) for the emission state of 3a, relative to those calculated for 3b (0.60) and 3c (1.00). If reduced oscillator strengths were the sole determining factor in controlling quantum yields, the quantum yield for 3c should be greater than that for 3b. However, 3b has greater than four times higher quantum yield than that associated with 3c. To explain this seeming contradiction, we need to explore also competing thermal non-radiative processes from the lowest lying excited states. Recently we analyzed computationally the relationships of structure and fluorescence yields in R-BOPs and R-BAPs.64 The intensity of fluorescence emission is dependent on the nature of the R substituent (alkyl < aryl) and the ability of the system to adopt a planar geometry in the excited state. Compared to R-BOPs, Ar-BAPs with potential N(R′)⋯H–Ar steric clashes can lead to deflection of the attached six- and five-membered rings from optimal geometry for π-conjugation and reduced photoluminescence. Steric clashes between two attached five-membered heterocyclic rings, however, are expected to be less severe. This expectation is supported by the observed planar X-ray structure of 3c. While this explanation alone may partly account for why compound 3b (Th-BAP) is luminescent and other BAPs have not been reported to be emissive, it does not explain why 3b is much more emissive than 3c.
Our working model involves comparing the calculated rates of fluorescence (kfl) and intersystem crossing (kisc) from the first excited state (S1). Effective emission must compete with non-radiative decay (knrd, thermal relaxation). Rigid ring systems can help minimize knrd relative to kfl. If, however, efficient conversion to nearby triplet states can occur, these longer lived states give more time for knrd to compete and reduce emission (Fig. 7). In most non-transition-metal compounds room temperature phosphorescence does not compete with non-radiative decay except in special situations or in the solid state.68 BBT-R (R = tBu), for example, has been shown to undergo competitive fluorescence quenching by intersystem crossover to nearby triplet state.61,62 This view is supported by the facts that (a) we have not observed lower wavelength (up to 800 nm) emissions attributable to phosphorescence in R-BOPs or R-BAPs, (b) emission spectra for both sets of compounds are similar, (c) lifetime measurements for 3b and 3c are consistent with fluorescence (neither of these compounds display visible luminescence under UV-light in the solid state), and (d) the predicted λem correlate with experimental data.
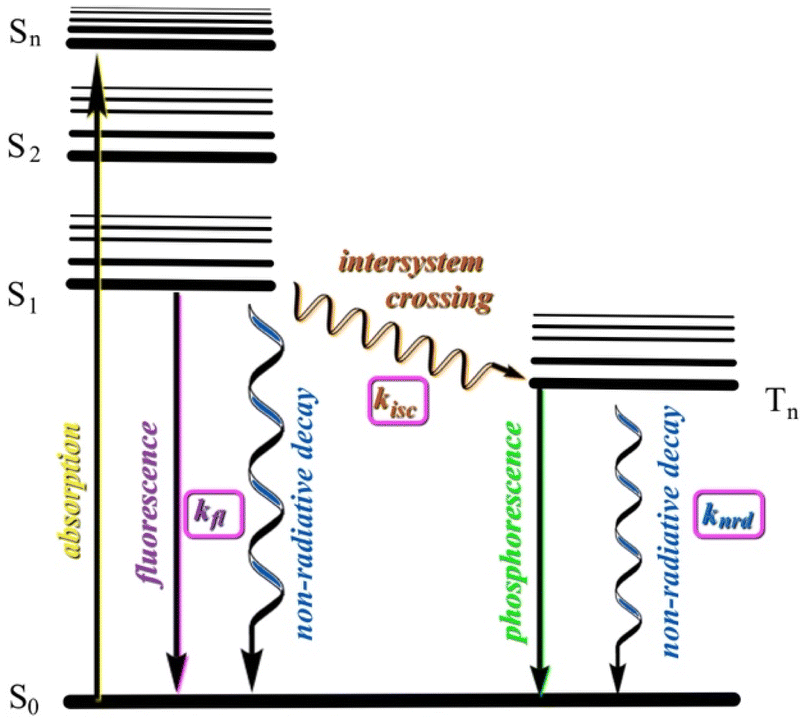 | ||
| Fig. 7 Jablonski diagram showing key events after absorption and internal conversion (not shown) to S1 excited state. Wavy lines represent non-radiative processes. | ||
Indeed, we found that higher fluorescence quantum yields are also associated with higher ratios of kfl:kisc. Radiative (fluorescence, blue bars) and non-radiative (intersystem crossing, orange bars) calculated rate constants of 3a–c are represented in Fig. 8. The fluorescence rates increase with the extension of the conjugation system, following the expected increasing trend with the oscillator strength of the emitting state (Table 3). The competing ISC non-radiative relaxation is the smallest for 3b. The ratio kfl:kisc is the largest for 3b, where it is less than two for 3c and close to one for 3a. This prediction is consistent with the experimental observation that 3b has the highest FL quantum yield.
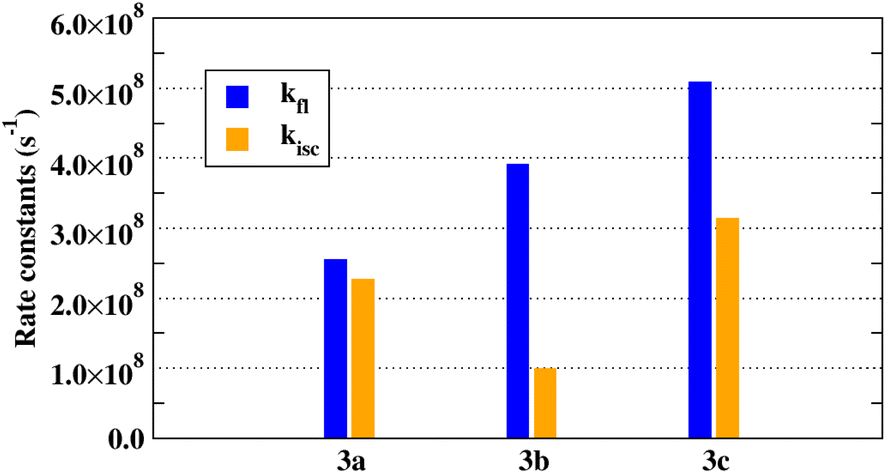 | ||
| Fig. 8 Radiative and non-radiative rate constants for 3a–c. | ||
To understand the trend of the ISC rates, the semiclassical Marcus theory of activated transition states was used as employed in our studies of related systems.64–66 The calculated parameters along with the rate constants are listed in Table 4. The processes are not in the far-inverted regime (comparing the energy bias [ΔG] to the reorganization energy [Er]), validating the semiclassical limit. In all cases the singlet-triplet spin–orbit coupling (SOC) is weak since all the states are of the same state character (π–π*). Nevertheless, the resulting intersystem crossing rate constants remain significant when compared to the fluorescence rate constants.
| SOC (cm−1) | ΔG (eV) | Er (eV) | Ea (eV) | kisc (s−1) | |
|---|---|---|---|---|---|
| 3a | 0.92 | −0.858 | 0.901 | 5.15 × 10−4 | 2.27 × 108 (S1–T3) |
| 3b | 0.55 | −0.113 | 0.222 | 1.33 × 10−2 | 0.99 × 108 (S1–T2) |
| 3c | 0.74 | −0.214 | 0.178 | 1.76 × 10−3 | 3.13 × 108 (S1–T2) |
The activation energy (Ea) for the singlet-triplet transition is low, while the energy for 3b is relatively the highest of the considered systems (Table 4). In case of 3b, the triplet excited state is ∼110 meV higher than the singlet excited state at lowest singlet excited state minima. These singlet-triplet splitting at lowest singlet excited state minima are smaller (around ± 30–40 meV) for the other two systems. This trend of singlet-triplet splitting energy leads to the higher ratio of reorganization (Er) to free energy (ΔG) for 3b and therefore to increase the activation energy, resulting with the lowest ISC rate.
Experimental
General materials and methods
All reactions were performed under an atmosphere of rigorously dry and oxygen-free nitrogen using either a MBraun Labmaster 130 glove box or standard Schlenk line techniques. Unless otherwise stated, all chemicals were purchased from commercial sources and used without further purification. Dry diethyl ether and hexanes were obtained from a MBraun SPS-800 solvent drying and purification system using filter material MB-KOL-A. The intermediate compounds N-(2-bromophenyl)-[2,2′-dithiophene]-5-carboxamide (1c) and {2-[((2,2′-dithiophene)-5-carbonyl)amino]phenyl} phosphonic acid diethyl ester (2c) were prepared according to literature procedures reported previously.58Dichloromethane-d2 was dried over basic alumina and degassed prior to use. NMR spectra were recorded on a Bruker Ascend Advanced III HD NMR spectrometer operating at 500.24 MHz and 202.5 MHz for 1H and 31P{1H} NMR, respectively. 31P{1H} NMR were referenced to 85% H3PO4. Dichloromethane was dried by distillation from calcium hydride prior to use for UV/Vis and fluorescence measurements. UV/Vis data were recorded by using a Cary 50 Bio UV/Vis spectrophotometer. Molar absorptivity determination was verified by linear least-squares fit of values obtained from at least three independent solutions at varying concentrations with absorbance in the range of 10−5 M. Emission spectra were recorded by using a Cary Eclipse spectrometer. The samples were excited at their absorption maximum. Emission quantum yields were determined by using as standards 9,10-diphenylanthracene in cyclohexane for compound 3a, anthracene in ethanol for 3b and quinine sulfate in 0.5 M H2SO4 solution for 3c, respectively (conc. 10−6 M). The excitation slit width for all measurements was kept at default settings (5 nm).
X-ray crystallography
Data were collected at 100K. The supplementary crystallographic data for 3c (CCDC 2126128). Tables S1–S5† contain selected data collection and refinement details-results. Fig. S3† offers a detailed packing diagram for 3c.Computational
The DFT calculations were implemented using Q-Chem as described in ESI.†Conclusions
1H-1,3-Benzazaphospholes (R-BAPs) are very well known materials with well-developed synthesis and applications, yet no photophysical studies have been reported despite close analogies to the related class of compounds 1,3-benzoxaphospholes (R-BOPs). In this work, we have demonstrated that selected R-BAPs can indeed display significant photoluminescence. The presence of one thiophene substituent (3b) or two (3c) leads to a better electron delocalization of the π-conjugated system and longer λmax values for absorption compared to the methyl-substituted compound (3a). X-ray diffraction studies show that the BAP ring and the connecting di-thiophene rings are highly coplanar in the solid state, also suggesting high degree of π-conjugation. The emission properties of 3c are also red-shifted towards the longest wavelengths compared to the others. TD-DFT calculations confirm the presence of π-conjugation throughout the BAP system. Computational efforts also revealed several factors that give rise to effective photoluminescence from R-BAPs. First, the presence of 5-membered thiophene units at the 2-position of the BAP group allows for easier adoption of an emissive planar configuration. Second, high oscillator strengths needed for emission are indicated. Third, the relative rates of fluorescence to intersystem crossing (a non-radiative decay pathway) are most favorable for compound 3b. Overall, this work provides a molecular understanding for means to develop BAP-based materials for optimal use in optoelectronic applications.Author contributions
JDP conceptualized the project. SE, AMH, and EDR carried out synthesis and spectral characterization of compounds. TST and SY measured excited state lifetimes. JH provided an initial sample of compound 3a, and intellectual guidance. BDD and SS performed all computational studies and provided interpretation of data. ALR conducted the crystallographic studies. All authors helped to write and to proofread manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
J. D. Protasiewicz thanks the National Science Foundation (CHE-1464855 and CHE-1955845) for funding. B. D. Dunietz acknowledges the Department of Energy (DOE), Basic Energy Sciences through Grant No. DE-SC0016501. We are also grateful to the college of Arts and Sciences, Kent State University for computing resources. T. S. Teets thanks the National Science Foundation (CHE-1846831) for support. We also thank Professor Zacharias Kinney (Oakland University) and reviewers for helpful comments.References
- J. R. Sheats and P. F. Barbara, Acc. Chem. Res., 1999, 32, 191–192 CrossRef CAS.
- J. L. Segura and N. J. Martín, Mater. Chem., 2000, 10, 2403–2435 RSC.
- H. Hoppe and N. S. Sariciftci, J. Mater. Res., 2004, 19, 1924–1945 CrossRef CAS.
- K. M. Coakley and M. D. McGehee, Chem. Mater., 2004, 16, 4533–4542 CrossRef CAS.
- A. P. Kulkarni, C. J. Tonzola, A. Babel and S. A. Jenekhe, Chem. Mater., 2004, 16, 4556–4573 CrossRef CAS.
- S. Günes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324–1338 CrossRef PubMed.
- S.-C. Lo and P. L. Burn, Chem. Rev., 2007, 107, 1097–1116 CrossRef CAS PubMed.
- G. Dennler, M. C. Scharber and C. J. Brabec, Adv. Mater., 2009, 21, 1323–1338 CrossRef CAS.
- (a) M. Helgesen, R. Søndergaard and F. C. Krebs, J. Mater. Chem., 2010, 20, 36 RSC; (b) A. Facchetti, Chem. Mater., 2011, 23, 733–758 CrossRef CAS.
- F. Riobe, R. Szűcs, P.-A. Bouit, D. Tondelier, B. Geffroy, F. Aparicio, J. Buendía, L. Sanchez, R. Reau, L. Nyulaszi and M. Hissler, Chem.–Eur. J., 2015, 21, 6547–6556 CrossRef CAS PubMed.
- M. P. Duffy, P.-A. Bouit, B. Geffroy, D. Tondelier and M. Hissler, Phosphorus Sulfur Relat. Elem., 2016, 190, 845–853 CrossRef.
- D. Joly, P.-A. Bouit and M. Hissler, J. Mater. Chem. C, 2016, 4, 3686–3698 RSC.
- B. M. Wong and J. G. Cordaro, J. Phys. Chem. C, 2011, 115, 18333–18341 CrossRef CAS PubMed.
- B. N. Norris, S. Zhang, C. M. Campbell, J. T. Auletta, P. Calvo-Marzal, G. R. Hutchison and T. Y. Meyer, Macromolecules, 2013, 46, 1384–1392 CrossRef CAS.
- H. Sun and J. Autschbach, J. Chem. Theory Comput., 2014, 10, 1035–1047 CrossRef CAS PubMed.
- T. Körzdörfer and J.-L. Brédas, Acc. Chem. Res., 2014, 47, 3284–3291 CrossRef PubMed.
- T. Fukushima, Mater. Chem. Front., 2018, 2, 1594 RSC.
- F. Mathey, Angew. Chem., Int. Ed. Engl., 2003, 42, 1578–1604 CrossRef CAS PubMed.
- T. Baumgartner and R. Réau, Chem. Rev., 2006, 106, 4681–4727 CrossRef CAS PubMed.
- M. Hissler, P. W. Dyer and R. Réau, Top. Curr. Chem., 2005, 250, 127–163 CrossRef CAS.
- D. Joly, D. Tondelier, V. Deborde, B. Geffroy, M. Hissler and R. Réau, New J. Chem., 2010, 34, 1603–1611 RSC.
- D. Joly, D. Tondelier, V. Deborde, W. Delaunay, A. Thomas, K. Bhanuprakash, B. Geffroy, M. Hissler and R. Réau, Adv. Funct. Mater., 2012, 22, 567–576 CrossRef CAS.
- J. Moon, M. Kim, J. S. Lim and J. Kim, Mol. Phys., 2018, 116, 1581–1588 CrossRef CAS.
- V. Iaroshenko and S. Mkrtchyan, Organophosphorus Chemistry: From Molecules to Applications, 2019, pp. 295–456 Search PubMed.
- M. P. Duffy, P.-A. Bouit and M. Hissler, in Main Group Strategies towards Functional Hybrid Materials, ed. T. Baumgartner and F. Jäkle, John Wiley & Sons Ltd., Hoboken, Chichester, 2018, ch. 12, pp. 295–327 Search PubMed.
- T. Baumgartner, Acc. Chem. Res., 2014, 47, 1613–1622 CrossRef CAS PubMed.
- M. A. Shameem and A. Orthaber, Chem.–Eur. J., 2016, 22, 10718–10735 CrossRef CAS PubMed.
- S. Yamaguchi, A. Fukazawa and M. Taki, J. Synth. Org. Chem., Jpn., 2017, 75, 1179–1187 CrossRef CAS.
- T. Higashino, K. Ishida, T. Sakurai, S. Seki, T. Konishi, K. Kamada, K. Kamada and H. Imahori, Chem.–Eur. J., 2019, 25, 6425–6438 CrossRef CAS PubMed.
- S. M. Parke, S. Tanaka, H. Yu, E. Hupf, M. J. Ferguson, Y. Zhou, K. Naka and E. Rivard, Macromolecules, 2019, 52, 7477–7488 CrossRef CAS.
- C. Wang, M. Taki, K. Kajiwara, J. Wang and S. Yamaguchi, ACS Mater. Lett., 2020, 2, 705–711 CrossRef CAS.
- Y. Xu, R. Xu, Z. Wang, Y. Zhou, Q. Shen, W. Ji, D. Dang, L. Meng and B. Z. Tang, Chem. Soc. Rev., 2021, 50, 667–690 RSC.
- S. Ikeda, A. Yoshimura, T. Shirahata, Y. Matano and Y. Misaki, Chem. Lett., 2021, 50, 1581–1585 CrossRef CAS.
- Y. Sugihara, N. Inai, M. Taki, T. Baumgartner, R. Kawakami, T. Saitou, T. Imamura, T. Yanai and S. Yamaguchi, Chem. Sci., 2021, 12, 6333–6341 RSC.
- K. Ishida, T. Higashino, Y. Wada, H. Kaji, A. Saeki and H. Imahori, ChemPlusChem, 2021, 86, 130–136 CrossRef CAS PubMed.
- H.-C. Su, O. Fadhel, C.-J. Yang, T.-Y. Cho, C. Fave, M. Hissler, C.-C. Wu and R. Réau, J. Am. Chem. Soc., 2006, 128, 983–995 CrossRef CAS PubMed.
- C. Romero-Nieto, S. Durben, I. M. Kormos and T. Baumgartner, Adv. Funct. Mater., 2009, 19, 3625–3631 CrossRef CAS.
- Y. Hayashi, Y. Matano, K. Suda, Y. Kimura, Y. Nakao and H. Imahori, Chem.–Eur. J., 2012, 18, 15972–15983 CrossRef CAS PubMed.
- C. Wang, M. Taki, Y. Sato, A. Fukazawa, T. Higashiyama and S. Yamaguchi, J. Am. Chem. Soc., 2017, 139, 10374–10381 CrossRef CAS PubMed.
- M. C. Simpson and J. D. Protasiewicz, Pure Appl. Chem., 2013, 85, 801–815 CrossRef CAS.
- M. P. Washington, V. B. Gudimetla, F. L. Laughlin, N. Deligonul, S. He, J. L. Payton, M. C. Simpson and J. D. Protasiewicz, J. Am. Chem. Soc., 2010, 132, 4566–4567 CrossRef CAS PubMed.
- F. L. Laughlin, A. L. Rheingold, N. Deligonul, B. J. Laughlin, R. C. Smith, L. J. Higham and J. D. Protasiewicz, Dalton Trans., 2012, 41, 12016–12022 RSC.
- F. L. Laughlin, N. Deligonul, A. L. Rheingold, J. A. Golen, B. J. Laughlin, R. C. Smith and J. D. Protasiewicz, Organometallics, 2013, 32, 7116–7121 CrossRef CAS.
- S. Wu, A. L. Rheingold and J. D. Protasiewicz, Chem. Commun., 2014, 50, 11036–11038 RSC.
- A. B. Grimm, K. Wang, A. L. Rheingold, C. E. Moore, D. Szieberth, L. Nyulaszi and J. D. Protasiewicz, Organometallics, 2021, 40, 3436–3444 CrossRef CAS.
- Z. T. Ekstrom, A. L. Rheingold and J. D. Protasiewicz, Phosphorus, Sulfur Silicon Relat. Elem., 2021, 197(5–6), 426–433 Search PubMed.
- (a) J. C. Worch, D. N. Chirdon, A. B. Maurer, Y. Qiu, S. J. Geib, S. Bernhard and K. J. T. Noonan, J. Org. Chem., 2013, 78, 7462–7469 CrossRef CAS PubMed; (b) Y. Qiu, J. C. Worch, D. N. Chirdon, A. Kaur, A. B. Maurer, S. Amsterdam, C. R. Collins, T. Pintauer, D. Yaron, S. Bernhard and K. J. T. Noonan, Chem.–Eur. J., 2014, 20, 7746–7751 CrossRef CAS PubMed.
- J. Heinicke, N. Gupta, S. Singh, A. Surana, O. Kuhl, R. K. Bansal, K. Karaghiosoff and M. Vogt, Z. Anorg. Allg. Chem., 2002, 628, 2869–2876 CrossRef CAS.
- B. Niaz, M. Ghalib, P. G. Jones and J. Heinicke, Dalton Trans., 2013, 42, 9523–9532 RSC.
- B. Niaz, F. Iftikhar, M. K. Kindermann, P. G. Jones and J. Heinicke, Eur. J. Inorg. Chem., 2013, 4220–4227 CrossRef CAS.
- N. Gupta, C. B. Jain, J. Heinicke, R. K. Bansal and P. G. Jones, Eur. J. Inorg. Chem., 1998, 1079–1086 CrossRef CAS.
- N. Gupta, C. B. Jain, J. Heinicke, N. Bharatiya, R. K. Bansal and P. G. Jones, Heteroat. Chem., 1998, 9, 333–339 CrossRef CAS.
- (a) G. Märkl and S. Pflaum, Tetrahedron Lett., 1987, 28, 1511–1514 CrossRef; (b) K. Issleib and R. Vollmer, Z. Anorg. Allg. Chem., 1981, 481, 22–32 CrossRef CAS.
- R. K. Bansal, N. Gupta, K. Karaghiosoff, A. Schmidpeter and C. Spindler, Chem. Ber., 1991, 124, 475–480 CrossRef CAS.
- (a) M. Ghalib, B. Niaz, P. G. Jones and J. W. Heinicke, Tetrahedron Lett., 2012, 53, 5012–5014 CrossRef CAS; (b) B. R. Aluri, M. K. Kindermann, P. G. Jones and J. Heinicke, Chem.–Eur. J., 2008, 14, 4328–4335 CrossRef CAS PubMed; (c) B. R. Aluri, P. G. Jones, I. Dix and J. W. Heinicke, Synthesis, 2014, 46, 1773–1778 CrossRef; (d) J. Heinicke, K. Steinhauser, N. Peulecke, A. Spannenberg, P. Mayer and K. Karaghiosoff, Organometallics, 2002, 21, 912–919 CrossRef CAS; (e) A. Surana, S. Singh, R. K. Bansal, N. Peulecke, A. Spannenberg and J. Heinicke, J. Organomet. Chem., 2002, 646, 113–124 CrossRef CAS; (f) M. Ghalib, P. G. Jones, S. Lysenko and J. W. Heinicke, Organometallics, 2014, 33, 804–816 CrossRef CAS; (g) B. R. Aluri, M. K. Kindermann, P. G. Jones, I. Dix and J. W. Heinicke, Inorg. Chem., 2008, 47, 6900–6912 CrossRef CAS PubMed; (h) B. R. Aluri, K. Shah, N. Gupta, O. S. Fomina, D. G. Yakhvarov, M. Ghalib, P. G. Jones, C. Schulzke and J. W. Heinicke, Eur. J. Inorg. Chem., 2014, 2014, 5958–5968 CrossRef CAS.
- S. Wu, A. L. Rheingold, J. A. Golen, A. B. Grimm and J. D. Protasiewicz, Eur. J. Inorg. Chem., 2016, 2016, 768–773 CrossRef CAS.
- (a) J. A. W. Sklorz, S. Hoof, N. Rades, N. De Rycke, L. Könczöl, D. Szieberth, M. Weber, J. Wiecko, L. Nyulaszi, M. Hissler and C. Müller, Chem.–Eur. J., 2015, 21, 11096–11109 CrossRef CAS PubMed; (b) S. Ito, K. Koshino and K. Mikami, Chem.–Asian J., 2018, 13, 830–837 CrossRef CAS PubMed; (c) R. Schoemaker, K. Schwedtmann, F. Hennersdorf, A. Bauzá, A. Frontera and J. J. Weigand, J. Org. Chem., 2020, 85, 14420–14434 CrossRef CAS PubMed; (d) N. Nagahora, S. Goto, T. Inatomi, H. Tokumaru, K. Matsubara, K. Shioji and K. Okuma, J. Org. Chem., 2018, 83, 6373–6381 CrossRef CAS PubMed.
- (a) B. R. Aluri, B. Niaz, M. K. Kindermann, P. G. Jones and J. Heinicke, Dalton Trans., 2011, 40, 211–224 RSC; (b) B. R. Aluri, S. Burck, D. Gudat, M. Niemeyer, O. Holloczki, L. Nyulaszi, P. G. Jones and J. Heinicke, Chem.–Eur. J., 2009, 15, 12263–12272 CrossRef CAS PubMed; (c) J. Heinicke, N. Gupta, A. Surana, N. Peulecke, B. Witt, K. Steinhauser, R. K. Bansal and P. G. Jones, Tetrahedron, 2001, 57, 9963–9972 CrossRef CAS.
- G. Becker, W. Massa, O. Mundt, R. E. Schmidt and C. Witthauer, Z. Anorg. Allg. Chem., 1986, 540, 336–344 CrossRef.
- V. Vetokhina, M. Kijak, T. M. Lipinska, R. P. Thummel, J. Sepiol, J. Waluk and J. Herbich, J. Phys. Chem. A, 2013, 117, 4898–4906 CrossRef CAS PubMed.
- M. A. Fourati, T. Maris, W. G. Skene, C. G. Bazuin and R. E. Prud’homme, J. Phys. Chem. B, 2011, 115, 12362–12369 CrossRef CAS PubMed.
- Z. Gao, Y. Hao, M. Zheng and Y. Chen, RSC Adv., 2017, 7, 7604–7609 RSC.
- (a) E. L. Mertz, V. A. Tikhomirov and L. I. Krishtalik, J. Phys. Chem. A, 1997, 101, 3433–3442 CrossRef CAS; (b) J. Yang, A. Dass, A.-M. M. Rawashdeh, C. Sotiriou-Leventis, M. J. Panzner, D. S. Tyson, J. D. Kinder and N. Leventis, Chem. Mater., 2004, 16, 3457–3468 CrossRef CAS; (c) X. Liu, Z. Xu and J. M. Cole, J. Phys. Chem. C, 2013, 117, 16584–16595 CrossRef CAS; (d) P. Horváth, P. Šebej, T. Šolomek and P. Klán, J. Org. Chem., 2015, 80, 1299–1311 CrossRef PubMed; (e) N. N. Mohd Yusof Chan, A. Idris, Z. H. Zainal Abidin, H. A. Tajuddin and Z. Abdullah, RSC Adv., 2021, 11, 13409–13445 RSC; (f) J. Zhao, S. Ji, Y. Chen, H. Guo and P. Yang, Phys. Chem. Chem. Phys., 2012, 14, 8803–8817 RSC.
- S. Sarkar, J. D. Protasiewicz and B. D. Dunietz, J. Phys. Chem. Lett., 2018, 9, 3567–3572 CrossRef CAS PubMed.
- S. Sarkar, H. P. Hendrickson, D. Lee, F. DeVine, J. Jung, E. Geva, J. Kim and B. D. J. Dunietz, J. Phys. Chem. C, 2017, 121, 3771–3777 CrossRef CAS.
- S. Sarkar, P. Durairaj, J. D. Protasiewicz and B. D. Dunietz, J. Photochem. Photobiol., 2021, 100089 CrossRef.
- (a) S. Bhandari, M. S. Cheung, E. Geva, L. Kronik and B. D. Dunietz, J. Chem. Theory Comput., 2018, 14, 6287–6294 CrossRef CAS PubMed; (b) S. Bhandari and B. D. Dunietz, J. Chem. Theory Comput., 2019, 15, 4305–4311 CrossRef CAS PubMed; (c) K. Begam, S. Bhandari, B. Maiti and B. D. Dunietz, J. Chem. Theory Comput., 2020, 16, 3287–3293 CrossRef CAS PubMed.
- Some reviews: (a) Kenry, C. Chen and B. Liu, Nat. Commun., 2019, 10, 2111 CrossRef CAS PubMed; (b) W. Zhao, Z. He, J. W. Y. Lam, Q. Peng, H. Ma, Z. Shuai, G. Bai, J. Hao and B. Z. Tang, Chem, 2016, 1, 592–602 CrossRef CAS; (c) S. Mukherjee and P. Thilagar, Chem. Commun., 2015, 51, 10988–11003 RSC; (d) Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng, X. Liu and W. Huang, Nat. Mater., 2015, 14, 685–690 CrossRef CAS PubMed; (e) L. Xiao and H. Fu, Chem.–Eur. J., 2019, 25, 714–723 CrossRef CAS PubMed; (f) A. D. Nidhankar, Goudappagouda, V. C. Wakchaure and S. S. Babu, Chem. Sci., 2021, 12, 4216–4236 RSC; (g) M. Shimizu and T. Sakurai, ChemPlusChem, 2021, 86, 446–459 CrossRef CAS PubMed; (h) W. Shao and J. Kim, Acc. Chem. Res., 2022, 55, 1573–1585 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra for compound 3c and X-ray analysis data (table and CIF files) for compound 3c. CCDC 2126128. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ra07226b |
| This journal is © The Royal Society of Chemistry 2023 |