
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of polynorbornadienes by ring-opening metathesis polymerization and their saturated derivatives bearing various ester groups and carboxyl groups†
Xiaoxue Lin a,
Jianjun Shi*ab and
Satomi Niwayama*b
a,
Jianjun Shi*ab and
Satomi Niwayama*b
aCollege of Chemistry and Chemical Engineering, Hainan Normal University, Haikou, Hainan 571158, P. R. China
bDivision of Sustainable and Environmental Engineering, Graduate School of Engineering, Muroran Institute of Technology, 27-1, Mizumoto-cho, Muroran, Hokkaido 050-8585, Japan. E-mail: niwayama@mmm.muroran-it.ac.jp
First published on 24th January 2023
Abstract
Various symmetric and non-symmetric polynorbornadienes having a variety of ester groups and carboxyl groups were synthesized by ring-opening metathesis polymerization (ROMP) with Grubbs' third generation catalyst (G3 or [Ru]-III catalyst) in a controlled living manner from half-esters prepared by the selective monohydrolysis of symmetric diesters that we previously reported. The half-esters thus obtained can be directly submitted to ROMP with the G3 catalyst, leading to mostly the trans structure and narrow polydispersity indexes. The subsequent hydrogenation yielded saturated polymers, improving the thermostabilities according to the T5d results. Our selective monohydrolysis reactions combined with ROMP initiated by the G3 catalyst have proven to be an efficient tool for the production of a variety of homopolymers with well-controlled structures in a living manner.
1 Introduction
Polyolefins are among the most widely utilized materials in a variety of research fields. Ring-opening metathesis polymerization (ROMP) is a metal carbene-catalyzed polymerization reaction starting from strained cyclic alkenes, which produce a wide range of polyolefins,1 and has been attracting a great deal of attention because of its ability to enable living polymerization and hence a wide range of applicability in polymer materials.2 In particular, Chauvin, Grubbs, and Schrock et al.3 developed ROMP with [Ru]-based catalysts, taking advantage of their general insensitivities to air or moisture and their high tolerance for various functional groups. The driving force for the polymerization is explained to be the strain of the [Ru] catalysts in addition to the strain of alkenes.4 Among these catalysts, 2nd generation (G2 or [Ru]-II)5 and 3rd generation (G3 or [Ru]-III)3f,6 are widely utilized because of their stability (Scheme 1). Functionalized polymers bearing polar functional groups7 have also been synthesized by the highly reactive G3 catalyst, and have been applied to various materials.8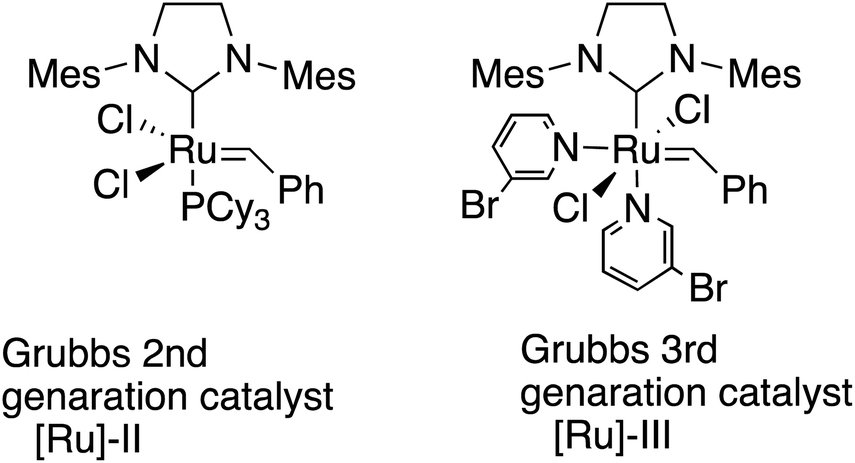 | ||
| Scheme 1 Grubbs 2nd and 3rd generation catalysts. | ||
We previously reported the synthesis of polymer libraries by ROMP with the Grubbs 2nd generation catalyst using the half-esters obtained by the selective monohydrolysis of symmetric diesters we developed earlier.9 This selective monohydrolysis reaction enables hydrolysis of one of the two identical ester groups in various symmetric diesters, producing half-esters in high yields (Scheme 2A).10 The symmetric diesters having a norbornadiene skeleton were efficiently monohydrolyzed for the production of the corresponding half-esters, and their various derivatives, bis(alkoxycarbonyl)norbornadienes, were polymerized with the Grubbs 2nd generation catalyst in a well-controlled manner. We therefore demonstrated that this selective monohydrolysis is a powerful tool for the construction of libraries of polymers with a variety of functional groups including amphiphilic polymers having carboxyl groups (Scheme 2B).
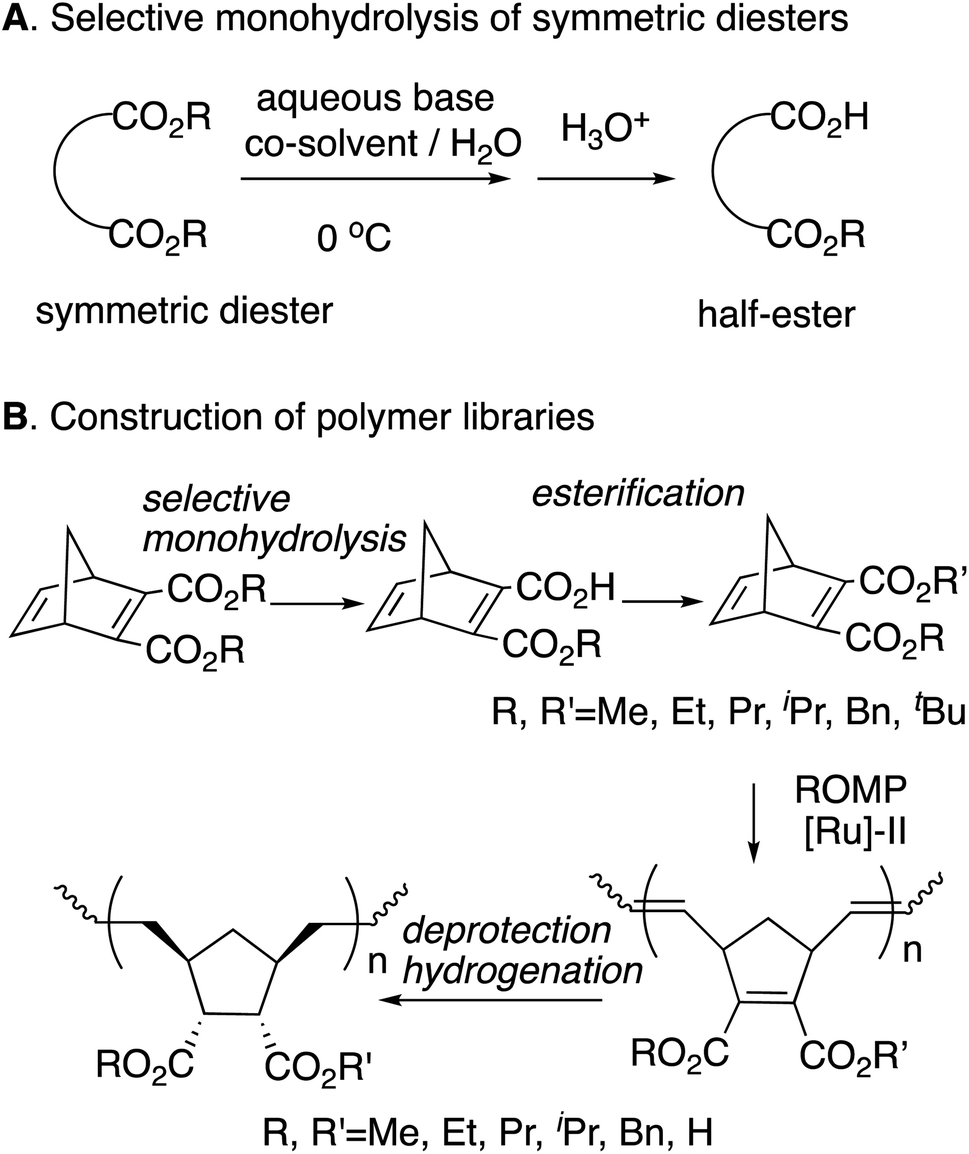 | ||
| Scheme 2 Selective monohydrolysis and the application to polymer synthesis. | ||
However, these polymers exhibited rather broad polydispersity indexes ranging from around 1.2 to 1.9. The molecular weights of the obtained polymers also appeared rather uncontrolled in some cases because of the slower reaction rates in the initiation step and faster propagation rates.11,12 The [Ru]-II catalyst also appears to be less tolerant of a polar functional group like COOH. Since the Grubbs 3rd generation catalyst is known to enhance the reactivities in the initiation steps of the polymerization by the 3-bromopyridine ligand, resulting in improved living polymerization, here we report a more precise and controlled living ROMP method for the construction of libraries of polymers with the Grubbs 3rd generation catalyst from symmetric and non-symmetric diesters bearing the norbornadiene skeleton that were obtained by our selective monohydrolysis reactions. We also found that the amphiphilic homopolymers can be synthesized directly from the half-esters obtained by the selective monohydrolysis reaction because of the higher tolerance of the catalyst.
2 Results and discussion
2.1 Polymerization, characterization, and thermal properties of norbornadienes bearing various ester groups
We first studied the ROMP of six symmetric diesters 1–6 to examine the influence of the [M]/[I] ratios with the [Ru]-III catalyst at −15 °C. The results of the isolated yields, number average molecular weight (Mn), the trans contents around the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds in the main chain of polymers, the thermogravimetric analysis (TGA) results, and the differential scanning calorimetry (DSC) of polymers 1a–6a are shown in Table 1.
C bonds in the main chain of polymers, the thermogravimetric analysis (TGA) results, and the differential scanning calorimetry (DSC) of polymers 1a–6a are shown in Table 1.
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Diester | R | [M]/[I] (mole ratio) | Yielda (%) | Mcalcdb (kDa) | Mn (GPC)c (kDa) | PDIsd | Trans contente (%) | Tg (°C) | T5d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) f (°C) f (°C) |
| a Isolated yield after precipitation.b Calculated theoretical molecular weight obtained by a formula: Wmonomer × [M]/[I] × yield%, where Wmonomer is the molecular weight of monomer.c Determined by GPC with polystyrene standards.d Values of Mw/Mn determined by GPC.e Determined by 1H NMR or 13C NMR.f Temperature at 5% weight loss.g Solid polymer did not form after reprecipitation. | ||||||||||
| 1 | 1 | Me | 333 | —g | —g | —g | —g | —g | —g | |
| 2 | 1 | Me | 200 | 82 | 3.4 | 7.0 | 1.05 | >99 | 114 | 229 |
| 3 | 1 | Me | 100 | 72 | 1.5 | 1.3 | 1.09 | >99 | 77 | 205 |
| 4 | 2 | Et | 200 | 76 | 3.6 | 6.7 | 1.08 | >99 | 105 | 231 |
| 5 | 3 | nPr | 200 | 71 | 3.7 | 5.7 | 1.06 | >99 | 92 | 232 |
| 6 | 4 | iPr | 200 | 72 | 3.8 | 5.2 | 1.04 | 95 | 99 | 184 |
| 7 | 5 | nBu | 200 | 67 | 3.9 | 3.7 | 1.05 | >99 | 84 | 217 |
| 8 | 6 | tBu | 200 | 64 | 3.7 | 2.1 | 1.10 | 90 | 117 | 125 |
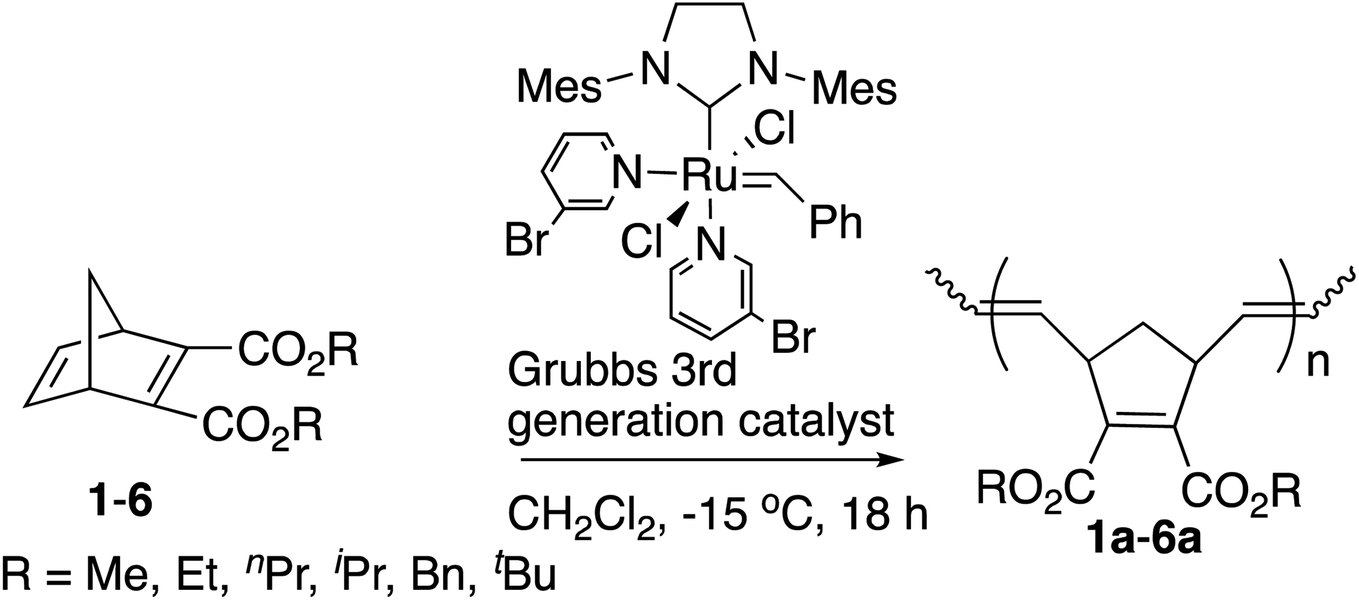
Solid polymer 1a was not obtained under the common [M]/[I] ratio of 33313 after the quench and evaporation of the solvent (entry 1, Table 1). Extending the reaction time from 18 to 48 hours and raising the polymerization temperature from −15 °C to room temperature did not help the polymerization either. However, when the amount of [Ru]-III catalyst was increased ([M]/[I] ratio = 200), the yield improved to 82% and a rather narrow polydispersity index of 1.05 was observed at −15 °C after 18 hours (entry 2, Table 1), indicating that corresponding polymer 1a was produced in a controlled living manner as has been reported by others.3f,14 Therefore, this molar ratio (200) was adopted for the polymerization of the remaining monomers in Table 1.
The yields tended to decrease with the increased size of the alkyl groups. This tendency is consistent with the previous studies by us9 and others.13,15 When the reaction time took more than 18 hours, the Mn values and the PDIs increased with the reaction time (data not shown). Polymer 2a was obtained with a high yield (76%) and narrow PDI (1.08) at −15 °C under the same reaction conditions as 1a (Table 1, entry 4), which is better than the reported ROMP of 1 and 2 with the Ru-based catalysts16 and the Mo-based catalysts17 (1.2–1.8 of PDI values). The comparison also demonstrates that the polymerization was initiated by the [Ru]-III catalyst rapidly even at −15 °C, suppressing the chain-transfer reactions despite the increased steric hindrance and possible intramolecular/intermolecular coordination of ester groups. Moreover, more sterically hindered monomers 3–6 showed high reactivities of ROMP in a living manner with narrow PDIs of 1.04–1.10 (Table 1, entries 5–8). The observed Mn values of 2a–6a showed better matches with the calculated theoretical values than polymer 1a, indicating that the controlled Mn was within a reasonable scope under the [M]/[I] ratio of 200. The fact that the observed Mn values show somewhat broader ranges than the theoretical values was perhaps derived from the imbalanced rates between the initiation and the propagation.
Polymers 1a–6a from symmetric diesters with the norbornadiene backbone often favor the “trans” configuration in ROMP initiated by Ru-based catalysts, and NMR spectroscopic studies have also been reported for this stereochemistry.16a,18 Generally, it is perceived that the “trans” isomers are more thermally stable, but the trans content of polymers 1a–6a decreased slightly with the increase in the size of side groups, perhaps due to the increased steric hindrance of the ester functional groups in the process of forming metallacylobutane intermediates.13,15,16
Polymers 1a and 2a obtained here revealed the presence of one sharp doublet at near 5.40 ppm, suggesting the all-trans structures. Polymers 4a–6a, with an increased size in the alkyl substituent, slightly lacked solid microstructures, reducing the trans ratios for the junctions between two adjacent cyclopentene units. The trans content (%) was determined from the 1H NMR or 13C NMR spectra as reported in the literature.16
The glass transition temperature (Tg) and the 5% decomposition temperature (T5d) exhibited a broad range, 77–117 °C and 125–232 °C, respectively. Various Tg values are necessary for practical applications for the synthesis of libraries of polynorbornadienes. These polymers from symmetric diesters displayed good thermal stabilities, and the crystalline melting point was not detected. In general, the Tg decreased with the extended size of alkyl pendant chains and increased with the branching of alkyl groups. The Tg values of polymers 4a (R = iPr, 99 °C) and 6a (R = tBu, 117 °C) bearing the branched alkyl pendant were higher than the corresponding polymers with a linear structure, polymer 3a (R = nPr, 92 °C) and 5a (R = nBu, 84 °C). This effect can be explained by the plasticizing effect of the hydrocarbon chain and the softer side groups of the flexible longer alkyl pendant.13,15 The T5d of polymer 6a was found to have the lowest temperature, 125 °C, resulting from the facile decomposition of tert-Bu substituted polymers due to the release of the isobutene molecule.
Next various non-symmetric diesters, 7–11, were prepared from the half-esters obtained by the highly efficient selective monohydrolysis of symmetric diesters 1–6, and were polymerized by the [Ru]-III catalyst under the same conditions as above. The results are summarized in Table 2. The tert-Bu group was introduced into the COOH of the half-esters with the use of magnesium chloride in high yields.19 As these diesters were racemic mixtures, the tert-butyl and alkyl ester groups were interchangeable in these structures.
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Diester | R | Yielda (%) | Mcalcd (kDa) | Mn (GPC) (kDa) | Mw/Mnb | Trans contentc (%) | Tg (°C) | T5dd (°C) |
| a Isolated yield after precipitation.b Values of Mw/Mn determined by GPC with polystyrene standards.c Determined by 1H NMR.d Temperature at 5% weight loss. | |||||||||
| 1 | 7 | Me | 68 | 3.6 | 7.11 | 1.08 | 53 | 121 | 231 |
| 2 | 8 | Et | 62 | 3.9 | 5.96 | 1.07 | 51 | 114 | 235 |
| 3 | 9 | nPr | 58 | 4.0 | 5.07 | 1.05 | 47 | 107 | 236 |
| 4 | 10 | iPr | 57 | 4.1 | 4.89 | 1.09 | 45 | 109 | 182 |
| 5 | 11 | nBu | 49 | 4.3 | 2.73 | 1.09 | 45 | 96 | 162 |
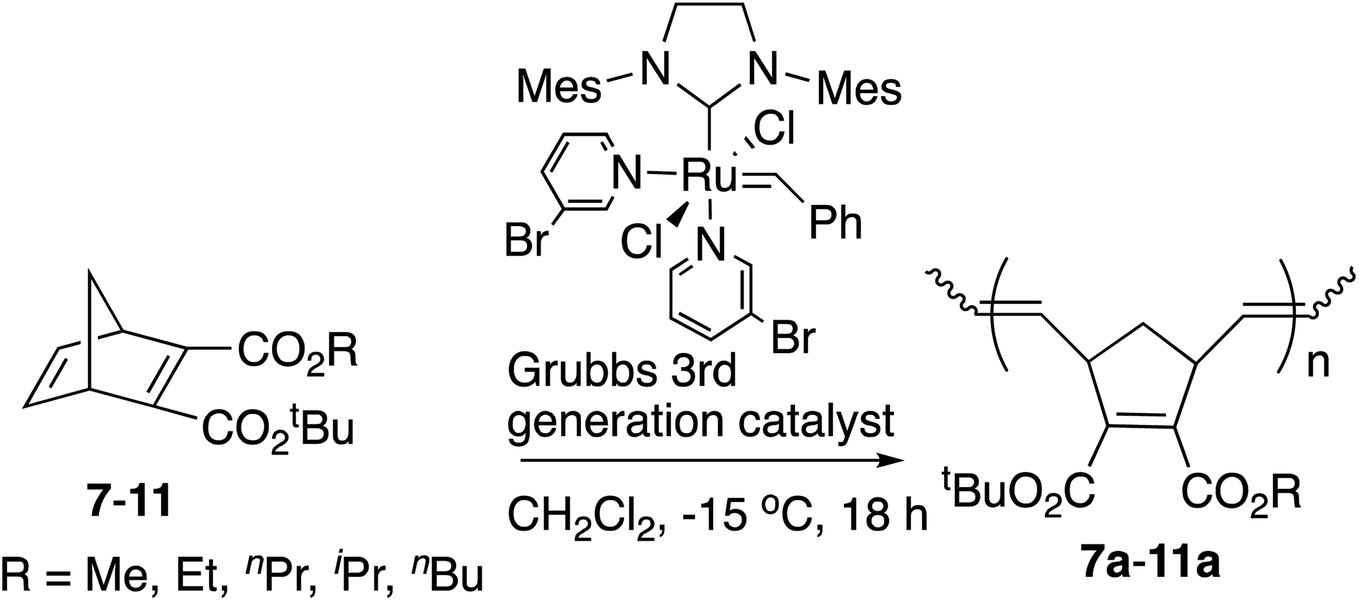
Decreased yields of polymers 7a–11a with an increase in the size of alkyl groups in ester substituents were also observed. The yields of these polymers from non-symmetric monomers were lower than those of polymers from symmetric monomers due to the steric hindrance of tert-Bu ester groups. The Mn observed by GPC for polymer 7a, which is similar to polymer 1a, was somewhat higher than theoretical values. The correlations between the calculated theoretical Mn (ranging from 3.6–4.3 kDa for polymers 8a–11a) and the observed Mn values from GPC (ranging from 2.73–5.96 kDa for polymers 8a–11a) were better than for the symmetric polymers, 1a–6a. Despite the less-than-ideal yields, the narrow PDIs (1.05–1.09) of the resulting polymers implies living polymerization. The structure of the non-symmetric polymer unit was determined by the 1H NMR and 13C NMR spectra. The trans/cis ratios were near 1:1 according to the integral ratios of olefinic protons in the 1H NMR spectra, without showing the diastereoselectivities. One probable reason is that the possibilities for the coordination of the active center of metal carbene at the position of exo-face or endo-face in the ring-opening step are equal. The other probable reason is the steric hindrance or the increased stability of the cis configuration. Again an increase in Tg values was observed with branched alkyl groups, and a decrease in Tg values was observed with longer alkyl chains.
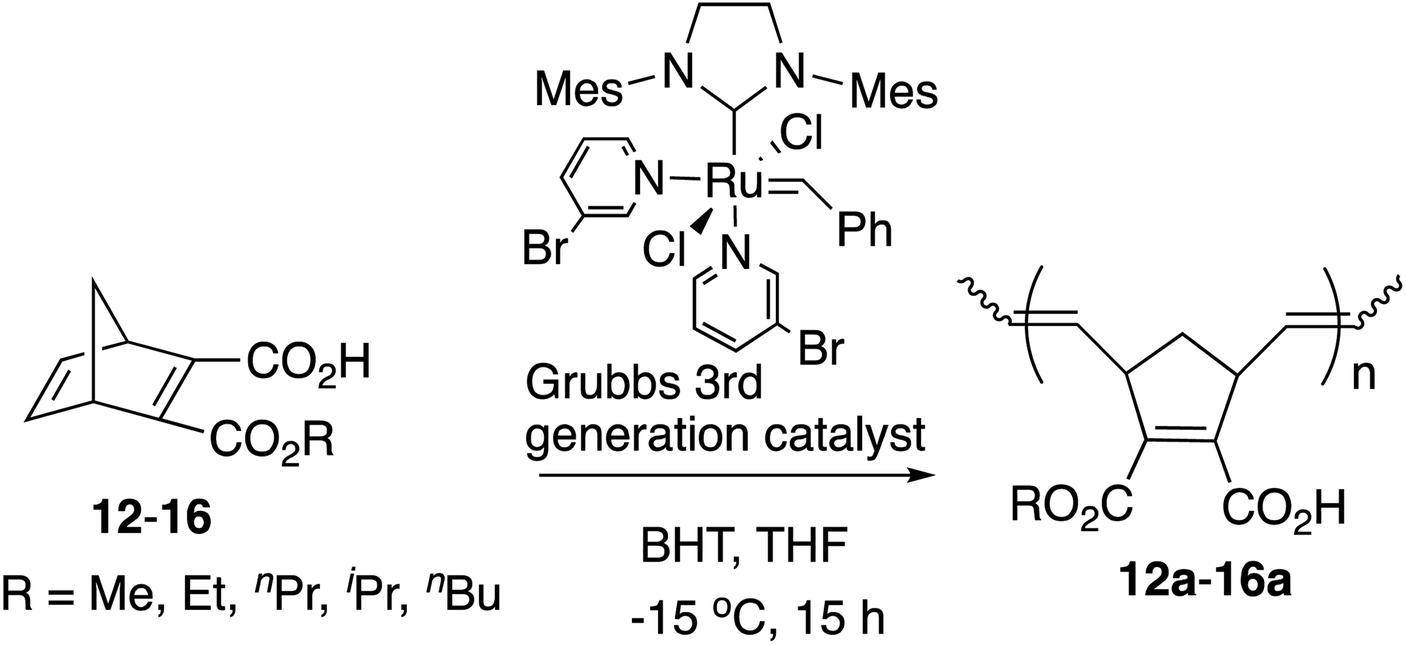
2.2 Synthesis of amphiphilic polymers with half-esters by Grubbs' 3rd generation catalyst
In our previous studies,9 the direct polymerization of half-ester monomers by the [Ru]-II catalyst led to the production of only a trace amount of polymers even after more than 3 days due to the catalyst sensitivity with the carboxyl group. Therefore, the –COOH was protected as the tert-Bu ester before ROMP and was deprotected to the –COOH group successfully with trifluoroacetic acid (TFA) afterwards as shown in Scheme 3.10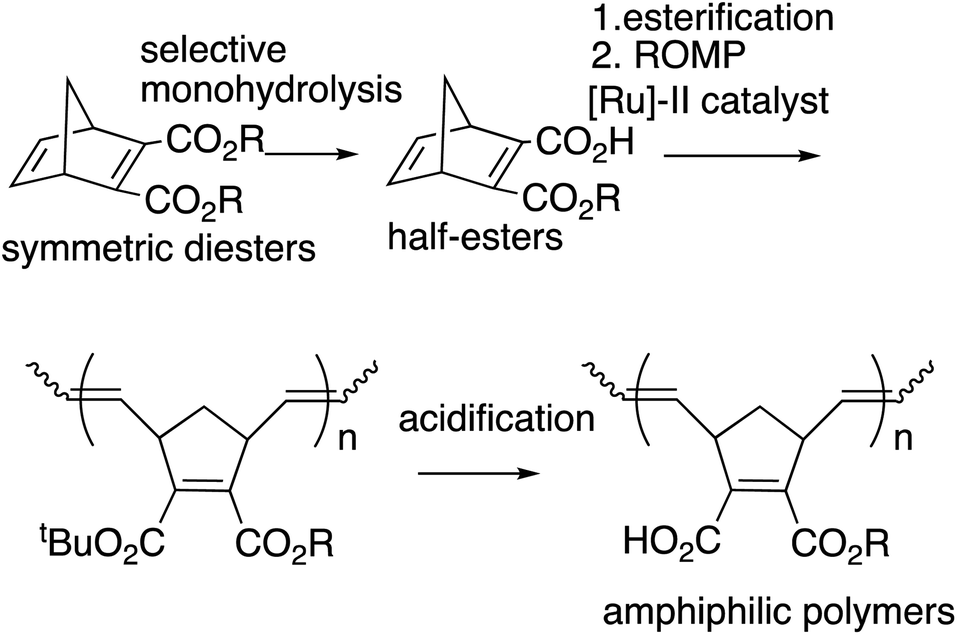 | ||
| Scheme 3 Synthetic route of amphiphilic polymers with the [Ru]-II catalyst. | ||
However, half-esters 12–16 were able to be polymerized by the [Ru]-III catalyst directly because of the catalyst tolerance for carboxylic groups as shown in Table 3. The polymerization was conducted in anhydrous THF because of the low solubility of the obtained polymers in CH2Cl2.
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Half-ester | R | [M]/[I] (mole ratio) | Yielda (%) | Mcalcd (kDa) | Mn (GPC) (kDa) | Mw/Mnb | Trans contentc (%) | Tg (°C) | T5dd (°C) |
| a Isolated yield after precipitation.b Values of Mw/Mn determined by GPC with polystyrene standards.c Determined by 1H NMR.d Temperature at 5% weight loss.e Solid polymer did not form after reprecipitation. | ||||||||||
| 1 | 12 | Me | 100 | 58.3 | 1.13 | 1.93 | 1.13 | >99 | 73 | 175 |
| 2 | 12 | Me | 50 | 77.2 | 0.75 | 2.64 | 1.24 | >99 | 76 | 184 |
| 3 | 13 | Et | 100 | 32.6 | 0.68 | 1.85 | 1.16 | >99 | 72 | 154 |
| 4 | 13 | Et | 50 | 70.9 | 0.74 | 2.57 | 1.27 | >99 | 74 | 201 |
| 5 | 14 | nPr | 100 | 18.1 | 0.40 | 0.92 | 1.20 | 99 | 67 | 179 |
| 6 | 14 | nPr | 50 | 61.4 | 0.68 | 1.91 | 1.29 | >99 | 69 | 208 |
| 7 | 15 | iPr | 100 | 14.5 | 0.32 | 0.66 | 1.18 | >99 | 69 | 175 |
| 8 | 15 | iPr | 50 | 62.5 | 0.69 | 2.21 | 1.23 | 98 | 70 | 183 |
| 9 | 16 | nBu | 100 | —e | —e | —e | —e | —e | —e | —e |
| 10 | 16 | nBu | 50 | 37.4 | 0.42 | 0.75 | 1.22 | 98 | 68 | 177 |
The [M]/[I] ratio of 200, which was effective in the ROMP of 1–11, yielded only a low (∼27%) yield in the ROMP of 12 (data not shown). However, an increased catalyst ratio of 50 led to the production of the corresponding amphiphilic homopolymers. The yields obviously decreased with the increase in the size of the alkyl groups. It appears that addition of a radical scavenger, 2,6-di-tert-butyl-4-methylphenol (BHT), helped improve the polymerization as has been reported for polymerization of other carboxyl group-containing monomers.7a Although the PDIs of these amphiphilic polymers showed a marked increase compared to the polymers from the diester monomers above, these values remained within a favorable range (1.13–1.29). The observed Mn values from GPC (0.75–2.64 kDa) were comparable to the theoretical Mn values (0.42–0.75 kDa). The higher molecular weights also indicate good polymerization control and a “living” manner. The olefinic protons in the monomers at around 6.40 ppm disappeared, and new olefinic protons were found at 5.63–5.37 ppm, indicating the trans and cis structures around the main chain in the polymers. Other significant differences in the polymers above from the non-symmetric monomers include their high selectivities towards the trans structures, probably due to the mutual interaction of carboxylic groups in the polymer chains. Judging from the results of Tg and T5d, their thermal stabilities decreased slightly compared to polymers 1a–11a above with the same ester groups, perhaps due to the decreased Mn values.
2.3 Synthesis of saturated polynorbornadienes by hydrogenation
Next we performed hydrogenation of the polymers above in order to obtain saturated polymers, which is expected to improve thermal properties as well as to provide useful information for structural elucidation.16b The hydrogenations were performed by the rapid in situ addition of diimide (NHNH) with the use of p-toluenesulfonyl hydrazide (TSH) and 2,6-di-tert-butyl-4-methylphenol in xylene at 135 °C. The results are summarized in Table 4.
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Polymer | R | R′ | Yielda (%) | Mn(GPC) (kDa) | Mw/Mnb | Tg (°C) | T5dc (°C) |
| a Isolated yield after precipitation.b Values of Mw/Mn determined by GPC with polystyrene standards.c Temperature at 5% weight loss. | ||||||||
| 1 | 1a | Me | Me | 99.4 | 6.65 | 1.04 | 72 | 233 |
| 2 | 2a | Et | Et | 97.8 | 7.13 | 1.07 | 66 | 230 |
| 3 | 3a | nPr | nP | 96.6 | 5.72 | 1.07 | 58 | 242 |
| 4 | 4a | iPr | iP | 96.3 | 5.34 | 1.09 | 69 | 195 |
| 5 | 5a | nBu | nBu | 95.5 | 3.96 | 1.08 | 50 | 221 |
| 6 | 6a | tBu | tBu | 94.4 | 2.11 | 1.07 | 74 | 129 |
| 7 | 7a | Me | tBu | 90.4 | 6.77 | 1.07 | 89 | 243 |
| 8 | 8a | Et | tBu | 86.1 | 6.25 | 1.08 | 75 | 237 |
| 9 | 9a | nPr | tBu | 83.4 | 4.94 | 1.08 | 62 | 239 |
| 10 | 10a | iPr | tBu | 79.6 | 5.12 | 1.09 | 65 | 190 |
| 11 | 11a | nBu | tBu | 72.7 | 2.93 | 1.10 | 48 | 174 |
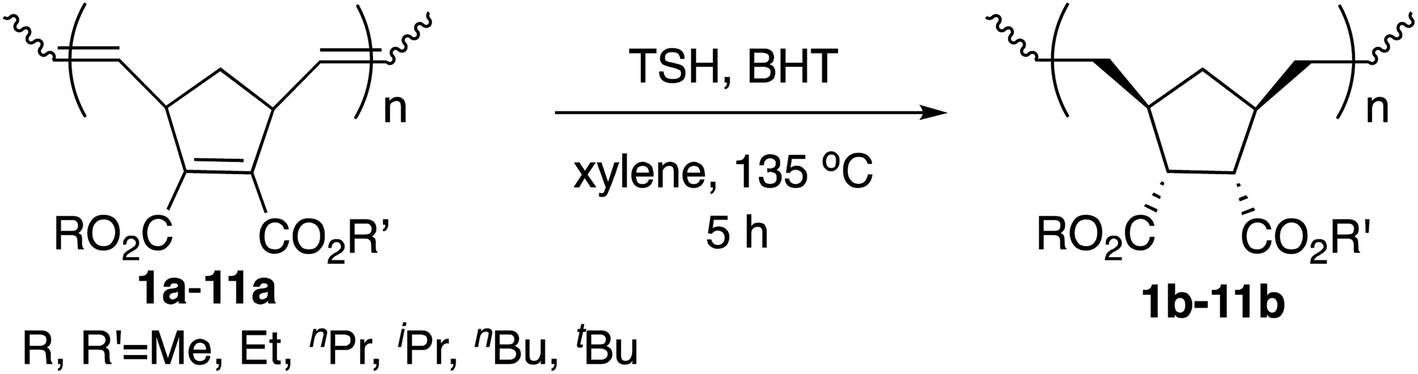
The yields were high, and the PDIs remained similarly narrow compared to those of the unsaturated polymers, ranging from 1.04 to 1.10. The isolated yields also similarly decreased with the increase in the size of the ester groups. The proton signals of the CC bond in the olefinic region disappeared, and both the CC bonds in the main chains and the cyclopentene ring were completely hydrogenated without side reactions according to 1H NMR and 13C NMR analysis as in our previous studies with the [Ru]-II initiated ROMP.9 Due to the concerted pericyclic mechanism, the addition mode of hydrogen atoms is known to be specifically “syn,” and the addition occurred from the exo face of the cyclopentene ring to afford thermodynamically stable and less hindered products.16b,20 There was no splitting of signal peaks as investigated in 13C NMR spectroscopy after hydrogenation, which also suggests the high tacticity of the polymers. The carbon atoms of C5,6 in cyclopentane associated with ester groups showed sharp singlets, which indicates that the stereoselective cis-addition of two hydrogen atoms occurred on the endocyclic CC bond and without introducing new tacticity splitting. Although the Tg values decreased after hydrogenation because of the increased mobilities of the polymer chains, the thermostabilities were indeed improved, based on the T5d values.
Hydrogenation was also performed on the amphiphilic polymers 12a–16a under the same reaction conditions, and the results are shown in Table 5. The hydrogenation was complete on both the polymer main chain and cyclopentane rings judging from the 1H NMR analysis.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Polymer | R | Yielda (%) | Mn(GPC) (kDa) | Mw/Mnb |
Tg (°C) | T5dc (°C) |
| a Isolated yield after precipitation.b Values of Mw/Mn determined by GPC with polystyrene standards.c Temperature at 5% weight loss. | |||||||
| 1 | 12a | Me | 93.1 | 2.67 | 1.26 | 73 | 185 |
| 2 | 13a | Et | 89.2 | 2.58 | 1.28 | 62 | 211 |
| 3 | 14a | nPr | 74.8 | 2.06 | 1.28 | 54 | 213 |
| 4 | 15a | iPr | 76.5 | 2.35 | 1.29 | 55 | 186 |
| 5 | 16a | nBu | 61.3 | 0.98 | 1.27 | 48 | 182 |
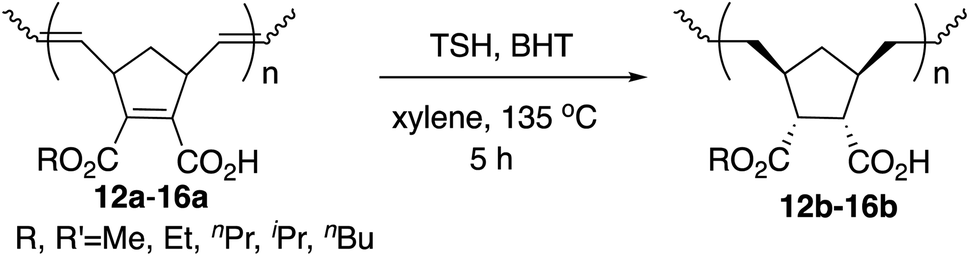
Again, high yields and narrow PDIs were observed. Compared with the results in Table 3, the thermal properties were indeed improved for all the cases judging from the values of T5d, although Tg values decreased as above. Although it is generally known that undesirable side reactions often occur with polymers, these hydrogenation reactions took place smoothly and completely, which is remarkable as in our previous studies.9
3 Conclusions
We have constructed libraries of polymers by ROMP initiated by the [Ru]-III catalyst from half-esters prepared by the selective monohydrolysis of various symmetric diesters. The polymers from symmetric diesters were obtained with mainly the trans olefinic microstructure in high yields. The Tg values decreased with the increased size of the ester groups, and the Tg values increased with the branched alkyl groups. The polymers from non-symmetric monomers did not exhibit selectivities toward the trans structures. In addition, various amphiphilic homopolymers bearing both ester groups and carboxyl groups were directly prepared from the half-esters by the [Ru]-III catalyzed ROMP. These amphiphilic homopolymers also exhibited high trans ratios for the olefinic stereochemistry and thermal properties similar to other polymers. Their thermostabilities were improved by the hydrogenation judging from the T5d results.Amphiphilic polymers generally constitute a wide range of advanced materials applied to tissue engineering, drug delivery, surface modifications, etc., and many amphiphilic block copolymers consisting of hydrophobic and hydrophilic monomers have been reported by tandem ROMP or ATRP, often combined with protection and deprotection of polar functional groups. Although these copolymers can have well-defined structures, the composition distribution is not necessarily uniform, leading to low thermal stabilities. Therefore, the direct synthesis of amphiphilic homopolymers from amphiphilic monomers such as half-esters makes a significant contribution to materials development. Our selective monohydrolysis reactions coupled with ROMP initiated by the [Ru]-III catalyst serve as a versatile tool for the production of a variety of amphiphilic homopolymers with well-controlled structures in a living manner.
4 Experiment
4.1 General
All the reactions were carried out in a nitrogen atmosphere. The glassware was dried in an oven (180 °C) and heated under reduced pressure before use.Nuclear magnetic resonance (1H NMR and 13C NMR) spectra were characterized on a Bruker AV 600 mHz NMR spectrometer with chloroform-d as a solvent; chemical shifts (in ppm) were reported using trimethylsilane as the internal standard. The gel permeation chromatography (GPC) was performed at 30 °C in CHCl3 (0.85 mL min−1) with the use of a JASCO PU-2080 system equipped with a set of Shodex K-804 and Shodex K-805 columns. The number average molecular weights (Mn), weight average molecular weights (Mw), and polydispersity indexes (PDI) of the obtained polymers were calculated on the basis of a polystyrene calibration. For polymers 12a–16a and 12b–16b, the GPC was performed on the methylated samples with trimethylsilyl diazomethane at 40 °C in THF (1.0 mL min−1) with the use of a Waters 150C and a Waters 410 refractive index detector. TGA analyses were carried out on a Shimadzu TGA-50 instrument in a nitrogen atmosphere (flow rate of 50 mL min−1) for the determination of 5% weight decomposition temperatures (T5d) at which 5% weight loss was observed (heating rate of 10 °C min−1). DSC analyses were carried out with a Shimadzu DSC-60 instrument in a nitrogen atmosphere (flow rate of 50 mL min−1) with liquid N2 as a refrigerant for the determination of the glass transition temperature (Tg) (heating rate of 10 °C min−1).
4.2 Materials
The third generation Grubbs' catalyst was purchased from Sigma Aldrich and used without further purification. Functionalized norbornenes 1–16 were synthesized according to our procedures reported in the literature.9,104.3 Synthesis of polymers
CH), 3.93 (br, 2H, CHCH), 3.73 (s, 6H, OCH3), 2.53 and 1.45 (m, 2H, CHCH2CH); 13C NMR (125 MHz, CDCl3) δ = 38.89 (–CH2–), 44.38 (CC–CH), 52.21 (CH3), 131.60 (CC), 142.42 (CC–CO), 165.40 (CO).CH), 4.18 (q, 4H, OCH2–CH3), 3.94 (br, 2H, CHCH), 2.53 and 1.45 (m, 2H, CHCH2CH), 1.26 (t, 6H, OCH2CH3); 13C NMR (125 MHz, CDCl3) δ = 14.20 (CH3), 38.94 (–CH2–), 44.46 (CC–CH), 61.09 (OCH2), 131.60 (CC), 142.17 (CC–CO), 164.95 (CO).CH), 4.10 (m, 4H, OCH2–CH2), 3.93 (d, 2H, CHCH), 2.48 and 1.46 (m, 2H, CHCH2CH), 1.64 (m, 4H, –CH2CH2CH3), 0.93 (t, 6H, CH3); 13C NMR (125 MHz, CDCl3) δ = 10.56 (CH3), 21.92 (–CH2–CH3), 38.93 (–CH2–), 44.48 (CC–CH), 66.73 (OCH2), 131.61 (CC), 142.09 (CC–CO), 165.00 (CO).CH), 5.06 (m, 2H, OCH(CH3)2), 3.92 (d, 2H, CHCH), 2.51 and 1.45 (m, 2H, CHCH2CH), 1.24 (d, 12H, OCH(CH3)2); 13C NMR (125 MHz, CDCl3) δ = 21.95 (CH3), 39.21 (–CH2–), 44.55 (CC–CH), 68.80 (OCH(CH3)2), 131.61 (CC), 142.05 (CC–CO), 164.48 (CO).CH), 4.11 (m, 4H, OCH2–CH2), 3.95 (d, 2H, CHCH), 2.49 and 1.43 (m, 2H, CHCH2CH), 1.60 (m, 4H, –OCH2CH2CH2), 1.35 (m, 4H, –OCH2CH2CH2), 0.91 (t, 6H, CH3); 13C NMR (125 MHz, CDCl3) δ = 13.82 (CH3), 19.21 (–CH2–CH3), 30.57 (–CH2–CH2–CH3), 38.86 (–CH2–), 44.48 (CC–CH), 64.99 (OCH2), 131.57 (CC), 142.02 (CC–CO), 164.96 (CO).CH), 3.84 (d, 2H, CHCH), 2.50 and 1.48 (m, 2H, CHCH2CH), 1.45 (s, 18H, OC(CH3)3); 13C NMR (125 MHz, CDCl3) δ = 28.34 (C(CH3)3), 39.75 (–CH2–), 44.65 (CC–CH), 81.79 (C(CH3)2), 131.71 (CC), 142.25 (CC–CO), 164.35 (CO).CH), 3.90 (s, 3H, O–CH3), 3.73 (d, 2H, CHCH), 2.49 and 1.58 (m, 2H, CHCH2CH), 1.45 (s, 9H, –C(CH3)3); 13C NMR (125 MHz, CDCl3) δ = 28.12 (–CH3), 39.20 (–CH2–), 44.28, 44.53 (CC–CH), 51.89 (O–CH3), 81.91 (–C(CH3)3), 131.36, 131.79 (C–CC–C), 141.23, 143.34 (CC–CO), 163.93, 165.64 (CO).CH), 4.20 (m, 2H, O–CH2–) 3.89 (m, 1H, CHCH), 3.56 (d, 1H, CHCH), 2.49 and 1.56 (m, 2H, CHCH2CH), 1.44 (s, 9H, –C(CH3)3), 1.28 (3H, t, –CH3); 13C NMR (125 MHz, CDCl3) δ = 14.40 (–CH3), 28.29 (–C(CH3)3), 39.33 (–CH2–), 44.44, 44.64 (CC–CH), 61.07 (O–CH2–), 81.96 (–C(CH3)3), 131.57, 131.78 (C–CC–C), 141.92, 142.42 (CC–CO), 163.97, 165.50 (CO).CH), 4.11 (m, 2H, O–CH2) 3.90 (m, 2H, CHCH), 2.50 (s, 1H, CHCH2CH), 1.65 (m, 3H, CHCH2CH and –CH2CH3), 1.44 (s, 9H, –C(CH3)3), 0.91 (t, 3H, –CH3); 13C NMR (125 MHz, CDCl3) δ = 10.51 (–CH3), 21.91 (–CH2CH3), 28.11 (–C(CH3)3), 39.19 (C–CH–CH2–CH–C), 44.21, 44.47 (CC–CH), 66.60 (–CH2–O2C), 81.82 (–C(CH3)3), 131.20, 131.63 (C–CC–C), 142.03, 142.30 (CC–CO), 163.81, 165.38 (CO).CH), 5.04 (m, 1H, CH(CH3)2) 3.87 (s, 2H, CHCH), 2.48, 1.70 (m, 2H, CHCH2CH), 1.42 (s, 9H, –C(CH3)3), 1.22 (d, 6H, CH(CH3)2); 13C NMR (125 MHz, CDCl3) δ = 21.94 (CH(CH3)2), 28.21 (–C(CH3)3), 39.42 (C–CH–CH2–CH–C), 44.51, 44.58 (CC–CH), 68.61 (CH(CH3)2), 81.85 (–C(CH3)3), 131.41, 131.81 (C–CC–C), 141.74, 142.43 (CC–CO), 164.00, 164.72 (CO).CH), 4.11 (m, 2H, O–CH2) 3.89 (m, 2H, CHCH), 2.49 (s, 1H, CHCH2CH), 2.16 (m, 2H, –CH2CH2CH2), 1.68 (m, 3H, CHCH2CH and –CH2CH3), 1.43 (s, 9H, –C(CH3)3), 0.92 (–CH3); 13C NMR (125 MHz, CDCl3) δ = 10.42 (–CH3), 17.10 (–CH2CH3), 21.84 (–CH2CH3), 28.11 (–C(CH3)3), 39.32 (C–CH–CH2–CH–C), 44.41, 44.48 (CC–CH), 68.51 (–CH2–O2C), 81.75 (–C(CH3)3), 131.31, 131.71 (C–CC–C), 141.64, 142.33 (CC–CO), 163.90, 164.62 (CO).CH), 3.97 (s, 3H, O–CH3), 3.61 (d, 2H, CHCH), 2.55 and 1.35 (m, 2H, CHCH2CH); 13C NMR (125 MHz, (CD3)2CO) δ = 39.77 (–CH2–), 44.83, 52.20 (CC–CH), 55.25 (O–CH3), 132.01, 132.47 (C–CC–C), 143.26, 143.34 (CC–CO), 165.82, 166.19 (CO).CH), 4.12 (s, 2H, O–CH2–) 4.00 (s, 1H, CHCH), 3.60 (d, 1H, CHCH), 2.60 and 1.39 (m, 2H, CHCH2CH), 1.16 (t, 3H, –CH3); 13C NMR (125 MHz, (CD3)2CO) δ = 14.32 (–CH3), 39.98 (–CH2–), 45.18, 55.37 (CC–CH), 61.48 (O–CH2–), 132.40, 132.75 (C–CC–C), 142.86, 143.92 (CC–CO), 165.80, 165.95 (CO).CH), 4.09 (m, 2H, O–CH2), 3.63 (m, 2H, CHCH), 2.66, 1.62 (m, 2H, CHCH2CH), 1.66 (m, 2H, –CH2CH3), 0.93 (m, 3H, –CH3); 13C NMR (125 MHz, (CD3)2CO) δ = 10.69 (–CH3), 22.33 (–CH2CH3), 39.84 (C–CH–CH2–CH–C), 45.05, 66.95 (CC–CH), 67.87 (–CH2–O2C), 132.31, 132.42 (C–CC–C), 142.95, 143.63 (CC–CO), 165.64, 165.83 (CO).CH), 5.03 (m, 1H, CH(CH3)2), 3.62 (m, 2H, CHCH), 2.66, 1.46 (m, 2H, CHCH2CH), 1.23 (m, 6H, CH(CH3)2); 13C NMR (125 MHz, (CD3)2CO) δ = 21.94 (CH(CH3)2), 40.07 (C–CH–CH2–CH–C), 45.28, 55.39 (CC–CH), 69.20 (CH(CH3)2), 132.33, 132.58 (C–CC–C), 142.54, 144.55 (CC–CO), 165.65, 165.93 (CO).CH), 4.19 (m, 2H, O–CH2), 3.67 (m, 2H, CHCH), 2.67, 1.27 (m, 2H, CHCH2CH), 1.89 (m, 2H, –CH2CH2CH3), 1.65 (m, 2H, –CH2CH3), 0.95 (m, 3H, –CH3); 13C NMR (125 MHz, (CD3)2CO) δ = 9.78 (–CH3), 21.09 (–CH2CH3), 28.87 (–CH2CH2CH3), 29.49 (C–CH–CH2–CH–C), 45.17, 67.24 (CC–CH), 68.36 (–CH2–O2C), 134.33, 134.72 (C–CC–C), 143.46, 144.73 (CC–CO), 165.44, 165.33 (CO).O).O).O).O).O).O).O).O).O).O).O).O).O).O).O).O).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (Grant No. 22168017), the Hainan Provincial Natural Science Foundation of China (No. 521RC1058), the Program of Hainan Association for Science and Technology Plans to Youth R & D Innovation (No. QCXM201925), the Natural Science Foundation of Hainan Province (222CXTD513, 2019RC187), The Iwatani Naoji Foundation Grant, The Ogasawara Toshiaki Memorial Foundation Grant, and Grants-in-Aid for Scientific Research (22K05106).References
- (a) T. A. Davidson, A. Wagener and D. Priddy, Polymerization of dicyclopentadiene: A tale of two mechanisms, Macromolecules, 1996, 29(2), 786–788 CrossRef CAS; (b) M. R. Buchmeiser, Homogeneous metathesis polymerization by well-defined group VI and group VIII transition-metal alkylidenes: Fundamentals and applications in the preparation of advanced materials, Chem. Rev., 2000, 100(4), 1565–1604 CrossRef CAS PubMed; (c) D. S. Belov, L. Mathivathanan, M. J. Beazley, W. B. Martin and K. V. Bukhryakov, Stereospecific ring-opening metathesis polymerization of norbornene catalyzed by iron complexes, Angew. Chem., Int. Ed., 2021, 60(6), 2934–2938 CrossRef CAS PubMed.
- (a) Q. C. Chen, Surface-initiated ring-opening metathesis polymerization (SI-ROMP): History, general features, and applications in surface engineering with polymer brushes, Int. J. Polym. Sci., 2021, 6677049 CAS; (b) S. K. Podiyanachari, M. Barłóg, M. Comí, S. Attar, S. Al-Meer, M. Al-Hashimi and H. S. Bazzi, Living ring-opening metathesis polymerization of norbornenes bay-functionalized perylene diimides, J. Polym. Sci., 2021, 59(24), 3150–3160 CrossRef CAS; (c) B. R. Huang, M. F. Wei, E. Vargo, Y. W. Qian, T. Xu and F. D. Toste, Backbone-photodegradable polymers by incorporating acylsilane monomers via ring-opening metathesis polymerization, J. Am. Chem. Soc., 2021, 143(43), 17920–17925 CrossRef CAS PubMed; (d) J. D. Feist, D. C. Lee and X. Yan, A versatile approach for the synthesis of degradable polymers via controlled ring-opening metathesis copolymerization, Nat. Chem., 2022, 14, 53–58 CrossRef CAS PubMed.
- (a) Y. Chauvin, Olefin metathesis: The early days (Nobel Lecture), Angew. Chem., Int. Ed., 2006, 45(23), 3740–3747 CrossRef PubMed; (b) R. R. Schrock, Multiple metal-carbon bonds for catalytic metathesis reactions (Nobel Lecture), Angew. Chem., Int. Ed., 2006, 45, 3748–3759 CrossRef CAS PubMed; (c) E. L. Dias, S. T. Nguyen and R. H. Grubbs, Well-defined ruthenium olefin metathesis catalysts: Mechanism and activity, J. Am. Chem. Soc., 1997, 119(17), 3887–3897 CrossRef CAS; (d) S. T. Nguyen, L. K. Johnson, R. H. Grubbs and J. W. Ziller, Ring-opening metathesis polymerization (ROMP) of norbornene by a Group VIII carbene complex in protic media, J. Am. Chem. Soc., 1992, 114(10), 3974–3975 CrossRef CAS; (e) G. C. Fu, S. B. T. Nguyen and R. H. Grubbs, Catalytic ring-closing metathesis of functionalized dienes by a ruthenium carbene complex, J. Am. Chem. Soc., 1993, 115(21), 9856–9857 CrossRef CAS; (f) T. L. Choi and R. H. Grubbs, Controlled living ring-opening-metathesis polymerization by a fast-initiating ruthenium catalyst, Angew. Chem., Int. Ed., 2003, 42, 1743–1746 CrossRef CAS PubMed.
- (a) R. H. Grubbs, Olefin-metathesis catalysts for the preparation of molecules and materials (Nobel Lecture), Angew. Chem., Int. Ed., 2006, 45, 3760–3765 CrossRef CAS PubMed; (b) K. J. Ivin, A. M. Kenwright and E. Khosravi, The Ru(=CHPh)Cl2(PCy3)2-initiated ring-opening metathesis polymerization of 7-tert-butoxybicyclo[2.2.1]hepta-2,5-diene: regeneration of initiator and the implied formation of macrocycles, Chem. Commun., 1999, 1209–1210 RSC; (c) P. Schwab, R. H. Grubbs and J. W. Ziller, Synthesis and applications of RuCl2(=CHR′)(PR3)2: The influence of the alkylidene moiety on metathesis activity, J. Am. Chem. Soc., 1996, 118, 100–110 CrossRef CAS; (d) Y. Schrodi and R. L. Pederson, Evolution and applications of second-generation ruthenium olefin metathesis catalysts, Aldrichimica Acta, 2007, 40(2), 45–52 CAS; (e) P. Schwab, M. B. France, J. W. Ziller and R. H. Grubbs, A series of well-defined metathesis catalysts-synthesis of [RuCl2(=CHR')(PR3)2] and its reactions, Angew. Chem., Int. Ed. Engl., 1995, 34, 2039–2041 CrossRef CAS; (f) B. R. Maughon and R. H. Grubbs, Ruthenium alkylidene initiated living ring-opening metathesis polymerization (ROMP) of 3-substituted cyclobutenes, Macromolecules, 1997, 30, 3459–3469 CrossRef CAS.
- (a) D. R. Anderson, T. Ung, G. Mkrtumyan, G. Bertrand, R. H. Grubbs and Y. Schrodi, Kinetic selectivity of olefin metathesis catalysts bearing cyclic (alkyl)(amino)carbenes, Organometallics, 2008, 27(4), 563–566 CrossRef CAS PubMed; (b) R. M. Thomas, B. K. Keitz, T. M. Champagne and R. H. Grubbs, Highly selective ruthenium metathesis catalysts for ethenolysis, J. Am. Chem. Soc., 2011, 133(19), 7490–7496 CrossRef CAS PubMed; (c) T. P. Montgomery, T. S. Ahmed and R. H. Grubbs, Stereoretentive olefin metathesis: An avenue to kinetic selectivity, Angew. Chem., Int. Ed. Engl., 2017, 56(37), 11024–11036 CrossRef CAS PubMed.
- O. M. Ogba, N. C. Warner, D. J. O'Leary and R. H. Grubbs, Recent advances in ruthenium-based olefin metathesis, Chem. Soc. Rev., 2018, 47, 4510–4544 RSC.
- (a) K. Lienkamp, C. F. Kins, S. F. Alfred, A. E. Madkour and G. N. Tew, Water-soluble polymers from acid-functionalized norbornenes, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1266–1273 CrossRef CAS; (b) J. K. Pontrello, M. J. Allen, E. S. Underbakke and L. L. Kiessling, Solid-phase synthesis of polymers using the ring-opening metathesis polymerization, J. Am. Chem. Soc., 2005, 127, 14536–14537 CrossRef CAS PubMed; (c) S. A. Isarov and J. K. Pokorski, Protein ROMP: Aqueous graft-from ring-opening metathesis polymerization, ACS Macro Lett., 2015, 4(9), 969–973 CrossRef CAS PubMed; (d) A. E. Madkour, A. H. R. Koch, K. Lienkamp and G. N. Tew, End-functionalized ROMP polymers for biomedical applications, Macromolecules, 2010, 43, 4557–4561 CrossRef CAS PubMed.
- (a) J. Chen, H. F. Li, H. C. Zhang, X. J. Liao, H. J. Han, L. D. Zhang, R. Y. Sun and M. R. Xie, Blocking-cyclization technique for precise synthesis of cyclic polymers with regulated topology, Nat. Commun., 2018, 9, 5310 CrossRef CAS PubMed; (b) A. A. Nagarkar and A. F. M. Kilbinger, Catalytic living ring-opening metathesis polymerization, Nat. Chem., 2015, 718–723 CrossRef CAS PubMed; (c) A. L. Liberman-Martin, C. K. Chu and R. H. Grubbs, Application of bottlebrush block copolymers as photonic crystals, Macromol. Rapid Commun., 2017, 38, 1700058 CrossRef PubMed.
- J. Shi, Y. Hayashishita, T. Takata, Y. Nishihara and S. Niwayama, Syntheses of polynorbornadienes by ring-opening metathesis polymerizations of symmetric and nonsymmetric 2,3-bis(alkoxycarbonyl) norbornadienes and their conversion to half-ester derivatives, Org. Biomol. Chem., 2020, 18, 6634–6642 RSC.
- (a) S. Niwayama, Highly efficient selective monohydrolysis of symmetric diesters, J. Org. Chem., 2000, 65, 5834–5836 CrossRef CAS PubMed; (b) S. Niwayama, H. Cho, M. Zabet-Moghaddam and B. R. Whittlesey, Remote exo/endo selectivity in selective monohydrolysis of dialkyl bicyclo[2.2.1]heptane-2,3-dicarboxylate derivatives, J. Org. Chem., 2010, 75, 3775–3780 CrossRef CAS PubMed; (c) S. Niwayama, H. Cho and C. Lin, Highly efficient selective monohydrolysis of dialkyl malonates and their derivatives, Tetrahedron Lett., 2008, 49, 4434–4436 CrossRef CAS; (d) J. Shi, T. Zhao and S. Niwayama, Practical selective monohydrolysis of bulky symmetric diesters: Comparing with sonochemistry, Tetrahedron, 2018, 74(47), 6815–6820 CrossRef CAS; (e) T. Barsukova, T. Sato, H. Takumi and S. Niwayama, Efficient and practical synthesis of monoalkyl oxalates under green conditions, RSC Adv., 2022, 12, 25669–25674 RSC.
- M. S. Sanford, M. Ulman and R. H. Grubbs, New insights into the mechanism of ruthenium-catalyzed olefin metathesis reactions, J. Am. Chem. Soc., 2001, 123(4), 749–750 CrossRef CAS PubMed.
- M. S. Sanford, J. A. Love and R. H. Grubbs, Mechanism and activity of ruthenium olefin metathesis catalysts, J. Am. Chem. Soc., 2001, 123, 6543–6554 CrossRef CAS PubMed.
- Y. Nishihara, S. Izawa, Y. Inoue, Y. Nakayama, T. Shiono and K. Takagi, Synthesis, characterization, and thermal properties of ring-opening metathesis polynorbornenes and their hydrogenated derivatives bearing various ester and cyano groups, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3314–3325 CrossRef CAS.
- Y. Nishihara, Y. Doi, S. Izawa, H.-Y. Li, Y. Inoue, M. Kojima, J.-T. Chen and K. Takagi, Enantioseparation of doubly functionalized polar norbornenes by HPLC and their ruthenium-catalyzed ring-opening methathesis polymerization. Rapid Communication, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 485–491 CrossRef CAS.
- (a) Y. Nishihara, Y. Inoue, Y. Nakayama, T. Shiono and K. Takagi, Comparative reactivity of exo- and endo-isomers in the Ru-initiated ring-opening metathesis polymerization of doubly functionalized norbornenes with both cyano and ester groups, Macromolecules, 2006, 39, 7458–7460 CrossRef CAS; (b) Y. Nishihara, Y. Inoue, A. T. Saito, Y. Nakayama, T. Shiono and K. Takagi, Living ring-opening metathesis polymerization of exo-norbornenes bearing both cyano and ester functionalities by a well-defined ruthenium catalyst, Polym. J., 2007, 39, 318–329 CrossRef CAS.
- (a) L. Delaude, A. Demonceau and A. F. Noels, Highly stereoselective Ruthenium-catalyzed ring-opening metathesis polymerization of 2,3-difunctionalized norbornadienes and their 7-oxaanalogues, Macromolecules, 1999, 32(7), 2091–2103 CrossRef CAS; (b) L. Delaude, A. Demonceau and A. F. Noels, Probing the stereoselectivity of the Ruthenium-catalyzed ring-opening metathesis polymerization of norbornene and norbornadiene diesters, Macromolecules, 2003, 36, 1446–1456 CrossRef CAS.
- (a) E. Khosravi, W. J. Feast, A. A. Al-Hajaji and T. Leejarkpai, ROMP of n-alkyl norbornene dicarboxyimides: from classical to well-defined initiators, an overview, J. Mol. Catal. A: Chem., 2000, 160, 1–11 CrossRef CAS; (b) E. Khosravi and A. A. Al-Hajaji, Ring opening metathesis polymerisation of n-alkyl norbornene dicarboxyimides using well-defined initiators, Polymer, 1998, 39(23), 5619–5625 CrossRef CAS.
- A. D. Benedicto, B. M. Novak and R. H. Grubbs, Microstructural studies of poly(7-oxabicyclo[2.2.1]hept-2-ene) derivatives prepared from selected ruthenium catalysts, Macromolecules, 1992, 25, 5893–5900 CrossRef CAS.
- G. Bartoli, M. Bosco, A. Carlone, R. Dalpozzo, E. Marcantoni, P. Melchiorre and L. Sambri, Reaction of dicarbonates with carboxylic acids catalyzed by weak Lewis acids: General method for the synthesis of anhydrides and esters, Synthesis, 2007, 22, 3489–3496 CrossRef.
- V. Amir-Ebrahimi, D. A. K. Corry, J. G. Hamilton and J. J. Rooney, Determination of the tacticities of ring-opened metathesis polymers of symmetrical 5,6-disubstituted derivatives of norbornene and norbornadiene from the 13C NMR spectra of their hydrogenated derivatives, J. Mol. Catal. A: Chem., 1998, 133, 115–122 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The NMR spectra for all the polymers. See DOI: https://doi.org/10.1039/d2ra07779e |
| This journal is © The Royal Society of Chemistry 2023 |