
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA multi-faceted approach to probe organic phase composition in TODGA systems with 1-alcohol phase modifiers†
Allison A. Peroutka ,
Shane S. Galley and
Jenifer C. Shafer*
,
Shane S. Galley and
Jenifer C. Shafer*
Department of Chemistry, Colorado School of Mines, Golden, CO, USA. E-mail: jshafer@mines.edu
First published on 20th February 2023
Abstract
The effect of varying 1-alcohol alkyl chain length on extraction of lanthanides (Lns), H2O, and H+ was studied with tetraoctyl diglycolamide (TODGA) via solvent extraction coupled with FT-IR investigations. This multi-faceted approach provided understanding regarding the relationship between extracted Lns, H2O and H+, 1-alcohol volume fraction, and 1-alcohol alkyl chain length. Under acidic conditions there is competition with 1-alcohols and their ability to solubilize aggregates and incidentally induce third phase formation by increasing the extraction of H2O. At low 1-alcohol concentrations (5 vol%), the trend for 1-alcohol alkyl lengths in solubilizing the aggregates is 1-hexanol > 1-octanol > 1-decanol. Shorter alkyl chains suppress aggregation, ultimately resulting in lower H2O concentrations and less available TODGA to hydrogen bond with H+. Increasing the 1-alcohol concentration to 30 vol% results in the opposite trend, with longer alkyl chains suppressing aggregation. These results suggest this approach is effective at probing trends in the organic phase micro-structure, and indicates trends across the Ln period with various 1-alcohol alkyl chain lengths are a function of outer-sphere coordination.
Introduction
Lanthanides (Lns), which make up the 4f period, exhibit unique electronic properties that make them of interest for many applications – including digital technology, catalytic converters, phosphors, lasers, permanent magnets, and ceramics.1–3 With the vast array of applications comes the need for an abundance of Lns; however, the current worldwide shortage of Lns creates a supply-demand risk, where industrial product cannot meet the demand.1 Currently, trivalent Lns are recovered from ores through a hydrometallurgical process involving the dissolution of the ore in high concentrations of acid, where the Lns are recovered and separated via solvent extraction.1,4 Several different classes of extractants have been implemented for Ln separations, such as cationic, anionic, and solvating. Recent investigations have shown a promising solvating extractant, tetraoctyl diglycolamide (TODGA), for intra-Ln separations, which is shown in Fig. 1.5–9 At the industrial scale, solvating extractants are preferred because they can be used under high acid concentrations. This eliminates the need to dilute acid and reduces the volume of aqueous waste.1,7 TODGA is advantageous for hydrometallurgical processing because TODGA extracts Lns under highly acidic conditions and follows the CHON (Carbon, Hydrogen, Oxygen, Nitrogen) principle.7,8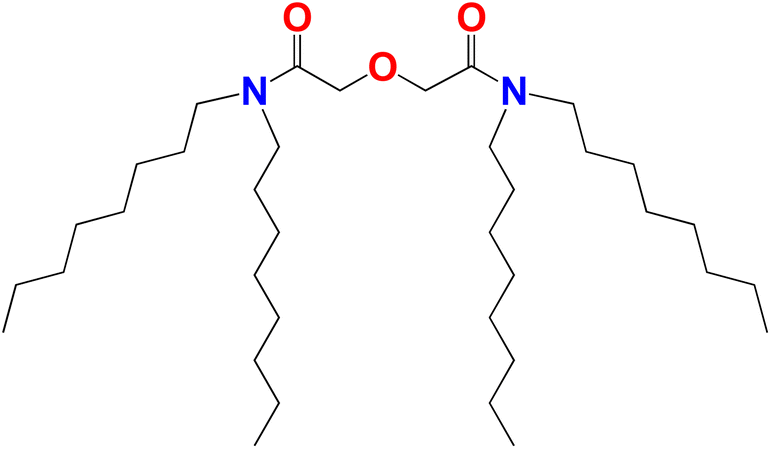 | ||
| Fig. 1 Molecular structure for tetraoctyl diglycolamide (TODGA). | ||
In the literature, the combination of solvent extraction, extended X-ray absorption fine structure (EXAFS), UV-vis, and FT-IR spectroscopy has led to an understanding of the Ln-TODGA structure in the organic phase.10–14 TODGA coordinates to Lns in a trischelate complex, [Ln(TODGA)3]3+, with outer-sphere nitrate anions (NO3−) positioned in the clefts of the octyl arms.13,15,16 These nitrates interact with co-extracted H2O molecules forming an extended hydrogen bonding network.10,11 Once this network extends beyond the clefts, NO3− and H2O molecules interact with (TODGA)·(HNO3)1–2 adducts – increasing the number of molecules associated with each Ln or aggregate. The size of these aggregates rapidly increases with higher HNO3 and Ln concentrations.10 In various solvent extraction systems, a threshold commonly referred to as the limiting organic concentration (LOC) describes the highest concentration of either Ln or HNO3 in the organic phase before the organic phase splits forming a third phase.17
Third phase formation (TPF) involves splitting the organic phase into light (diluent rich) and heavy (extractant rich) phases.18,19 Following TPF the extracted metal complexes are found in the heavy third phase. This results from extracting a large volume of H2O and HNO3 molecules, where the aggregates that form are no longer soluble in the organic diluent. It is important to prevent TPF as TPF is deleterious to the extraction process since industrial mixer-settlers are designed for biphasic extraction. Mitigation of TPF involves adding a phase modifier (PM), which is slightly more polar than the diluent, to the organic phase which solubilizes the aggregates.20 The aggregates are solubilized by PMs where the polar moiety of the PM interacts with the extracted H2O, HNO3, and Lns and displaces extractant molecules. As the concentration of PMs is large relative to the extractant concentration, adding PMs increases the complexity of the organic phase through changing both molecular and supramolecular interactions.21–23 Several investigations to date have used 1-alcohols as PMs14,20,24 with a particular emphasis on solvent extraction results paired with spectroscopy to probe the extracted Ln diglycolamide (DGA) complex. Systems containing 1-alcohol PMs have larger distribution ratios and the number of DGA molecules associated with the extracted Ln-DGA complex decreases in comparison to systems without PMs. As a result, the role of 1-alcohols in these systems has been attributed to solvation of the Ln-DGA complex through interactions with the outer-sphere hydrogen bonding network.3,25
In the current literature only select Lns are chosen for additional characterization methods. Techniques in addition to solvent extraction should be used across the entire 4f period aiming to highlight a relationship between organic phase bulk properties and the distribution ratios.26 This work performs a larger-scale investigation which combines the results obtained from solvent extraction, bulk organic phase characteristics, and spectroscopic techniques. In this article, the collection of distribution ratios, co-extraction of H2O, co-extraction of H+, and FT-IR spectra are compared to provide additional insight into these systems.
Experimental methods
Materials
Ln(NO3)3 were prepared from corresponding Ln oxides (99.99%, Treibacher Industries AG) for Ln = La, Ce, Pr, Nd, Sm, Eu, Gd, Dy, Ho, Er, Tm, Yb, Lu, by dissolving the Ln oxide in HNO3. The remaining materials were used as received, Tb(NO3)3 (99.9%, Sigma Aldrich), TODGA (97 wt%, Marshallton), 1-hexanol (98%, Sigma Aldrich), 1-octanol (98%, Sigma Aldrich), and 1-decanol (98%, Sigma Aldrich), nitric acid (70%, Avantor), isopropyl alcohol (99%, Pharmco), sodium hydroxide (98.5%, Fisher Scientific), potassium hydrogen phthalate (99.95%, Alfa Aesar), sodium chloride (98.5%, Fisher Scientific), lithium chloride (99%, Sigma Aldrich), ethylene glycol (99%, Sigma Aldrich), and tributylamine (99%, Thermo Scientific).Solvent extraction
Lns were extracted via solvent extraction, where equal volumes of organic and aqueous phases were used. Organic phases included 0.04 M TODGA (accounted for 97 wt%) with 1-alcohol PMs in n-dodecane, descriptions are listed in Table 1. As evident in Table 1 there are several gaps when comparing the concentration of 1-alcohol selected. 1-Octanol has been the most studied PM in literature, so a wide range of concentrations were selected (5, 10, 15, and 30 vol%) to further investigate the relationship between various 1-octanol concentrations. The concentrations of 10 and 15 vol% 1-alcohol were not selected for 1-hexanol, 1-decanol, and 1-dodecanol because the aim of this work was to determine the trend between varying 1-alcohol alkyl chain length at low and high concentrations. The low concentration of 5 vol% 1-alcohol was selected because 5 vol% is commonly used in literature to understand the fundamental chemistry in DGA systems. A high concentration of 30 vol% 1-alcohol was chosen as it is relevant to industrial applications. However, at 30 vol% 1-dodecanol a white precipitate formed at the interface during pre-equilibration. As 1-dodecanol has a melting point of 26 °C and the experiments were performed at room temperature (21 °C), this caused 1-dodecanol to form a precipitate. Since a precipitate formed, this prevented data collection for the 30 vol% 1-dodecanol system, and comparison between various 1-alcohol alkyl chain lengths with 1-dodecanol systems. The selected organic phases were pre-equilibrated with 1 M HNO3 for 5 min at 2000 rpm and centrifuged for 5 min at 2800 rpm. A 0.85 mL aliquot of the pre-equilibrated organic phase was contacted with 0.85 mL of 3 mM Ln(NO3)3 in 1 M HNO3 for 5 min at 2000 rpm and centrifuged for 5 min at 2800 rpm. Each Ln was individually contacted in triplicate for error analysis to evaluate trends in co-extracted solutes (Ln3+, H+, and H2O) and FT-IR data across the period. Following extraction, the organic phase was removed for analysis with Karl Fischer (KF), FT-IR, and potentiometric titration.| PM, vol% | 1-Hexanol | 1-Octanol | 1-Decanol | 1-Dodecanol |
|---|---|---|---|---|
| 5 | 0.34 | 0.32 | 0.26 | 0.22 |
| 10 | — | 0.64 | — | — |
| 15 | — | 0.95 | — | — |
| 30 | 2.39 | 1.91 | 1.57 | — |
ICP-OES
Inductively Coupled Plasma Optical Emission Spectroscopy (ICP-OES) was used to measure the concentration of Ln in the aqueous phase before and after solvent extraction. Samples analyzed by ICP-OES were dissolved in 2 wt% HNO3 prior to analysis on Avio 220 Max Concentric/Cyclonic. Initial samples were prepared for ICP-OES by adding a 0.2 mL aliquot of initial aqueous phase to 10 mL of 2 wt% HNO3. Then, a 1 ppm solution of each Ln was prepared in 2 wt% HNO3 to ensure sufficient signal on ICP-OES for each Ln measured. In post-extraction ICP-OES samples, an aliquot of 0.2 mL of aqueous phase post-extraction was added to 10 mL of 1 ppm of respective Ln. Data was collected for each Ln in both radial and axial mode for the two most sensitive wavelengths. Argon gas was used as the nebulizer gas and plasma gas with pressure maintained between 80–120 psig. Nitrogen gas was used as the shear gas at pressures between 40–120 psig. The instrument was then calibrated with 1 ppm and 10 ppm of individual Ln standards (Inorganic Ventures), in addition to a calibration blank (0 ppm Ln), all of which were in 2 wt% HNO3. Calibration curves of individual metals were generated using a linear regression model for each wavelength in each mode, resulting in a slope and y-intercept to convert the samples measured intensities into ppm. The ppm values are used to calculate distribution coefficient (DLn) as shown in eqn (1). Equilibrium DLn are the ratio of the Ln equilibrium concentration in the organic phase to the Ln equilibrium concentration in the aqueous phase. For ICP-OES measurements, where only the aqueous phase can be measured, it is assumed for mass balance purposes the concentration of Ln in the organic phase is equal to the difference in Ln concentration between the initial aqueous phase and the aqueous phase post-extraction. The error reported in the D values is propagated from instrumental and experimental errors, along with standard deviation from triplicate samples.
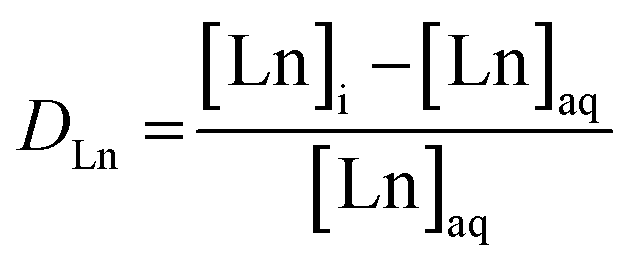 | (1) |
After the Ds were calculated, they were used in a subsequent calculation to determine the separation factor of select Ln as shown in eqn (2). The separation factor (SF) quantifies the ability for a specific system to separate two metals through a ratio of the D of metal 1 to the D of metal 2.
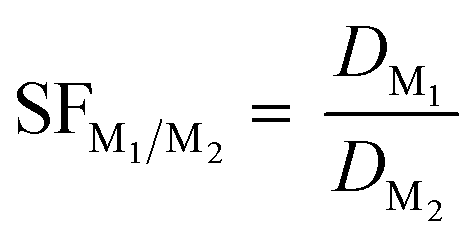 | (2) |
Potentiometric titrations
Mettler Toledo 888 Titrando was used with a Ross micro-electrode to calculate H+ concentration in the organic phase. The electrode fill solution was replaced with 1 M LiCl in ethylene glycol for efficient conductivity in n-dodecane. A solution containing 10 mM HNO3 in isopropyl alcohol was standardized with 0.608 M tributylamine as the titrant, from which a calibration curve was generated allowing the electrode's mV response to be converted into pcH. A 0.1 mL aliquot of each organic phase sample was diluted to 5 mL with isopropyl alcohol. The micro-electrode was then connected to a Thermo Scientific Orion Star A214 pH meter, to record the mV response of diluted organic phase after extraction, where the calibration curve was applied to calculate H+ concentration and account for dilution correction.Karl Fisher
Coulometric titrations were performed with a Mettler Toledo 852 Titrando Karl Fisher to measure the concentration of H2O in the organic phase before and after extraction. A 0.5 mL aliquot of organic phase was added to the KF cell using a 2 mL plastic syringe. The initial mass of the sample-loaded syringe was recorded, and the final mass of syringe following sample introduction to the KF cell. The water content was calculated according to eqn (3), where mH2O is the mass of H2O in μg in the sample, ms,i is the mass in g of the loaded syringe, and ms,f is the mass in g of the emptied syringe. The water content in ppm was then converted to molarity for comparison to H+ concentration.
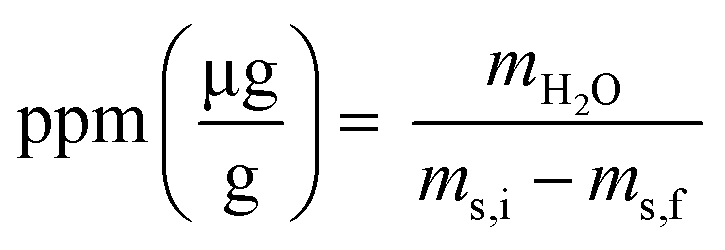 | (3) |
FT-IR
A Thermo Electron Nicolet 4700 FT-IR was used to analyze differences in coordination between TODGA, Ln, and HNO3 for the various studies conducted. The instruments spectral resolution was set to 6 (corresponding to 2 cm−1) and data was collected from 400 to 4000 cm−1. An aliquot of 5 μL of organic phase before and after extraction for each Ln, was analyzed, where the spectra was baseline corrected through the msbackadj MatLab function in the Bioinformatics Toolbox.27 The carbonyl band was further analyzed with OriginLab, through a more detailed analysis of peak fitting to deconvolute carbonyl bands and identify the corresponding wavenumber, area, height, and full width at half maximum (FWHM). The peak fitting results are provided as ESI.†Results and discussion
Solvent extraction
To understand Ln partitioning across several PM conditions, the Ds were measured across the Ln period and are reported in Fig. 2. In the absence of PMs, DLn trend is consistent with the results reported by Sasaki et al.,8 where the D increases from La–Dy with a plateau forming around Ho. With the addition of 1-octanol to the organic phase, the distribution trend is preserved but with an increase in D for each Ln measured. Increasing the 1-octanol concentration from 5 vol% to 30 vol% results in similar Ds across the Lns. At 30 vol% 1-alcohol, changing the length of the 1-alcohol alkyl chain results in a different trend across the Lns. For 1-hexanol (Fig. 2A), there is a decrease in D between Eu and Gd, whereas an increase in the D is observed across the remainder of the Lns. In the case of 1-decanol (Fig. 2C), there is a decrease in the D from Gd to Tb, with an increase in the D from Tb–Tm, with a plateau forming around Yb. The effect of 1-alcohols on DLn is most pronounced in the case of 30 vol% 1-decanol, where Ds for Tb–Lu are lower than the Ds for no modifier.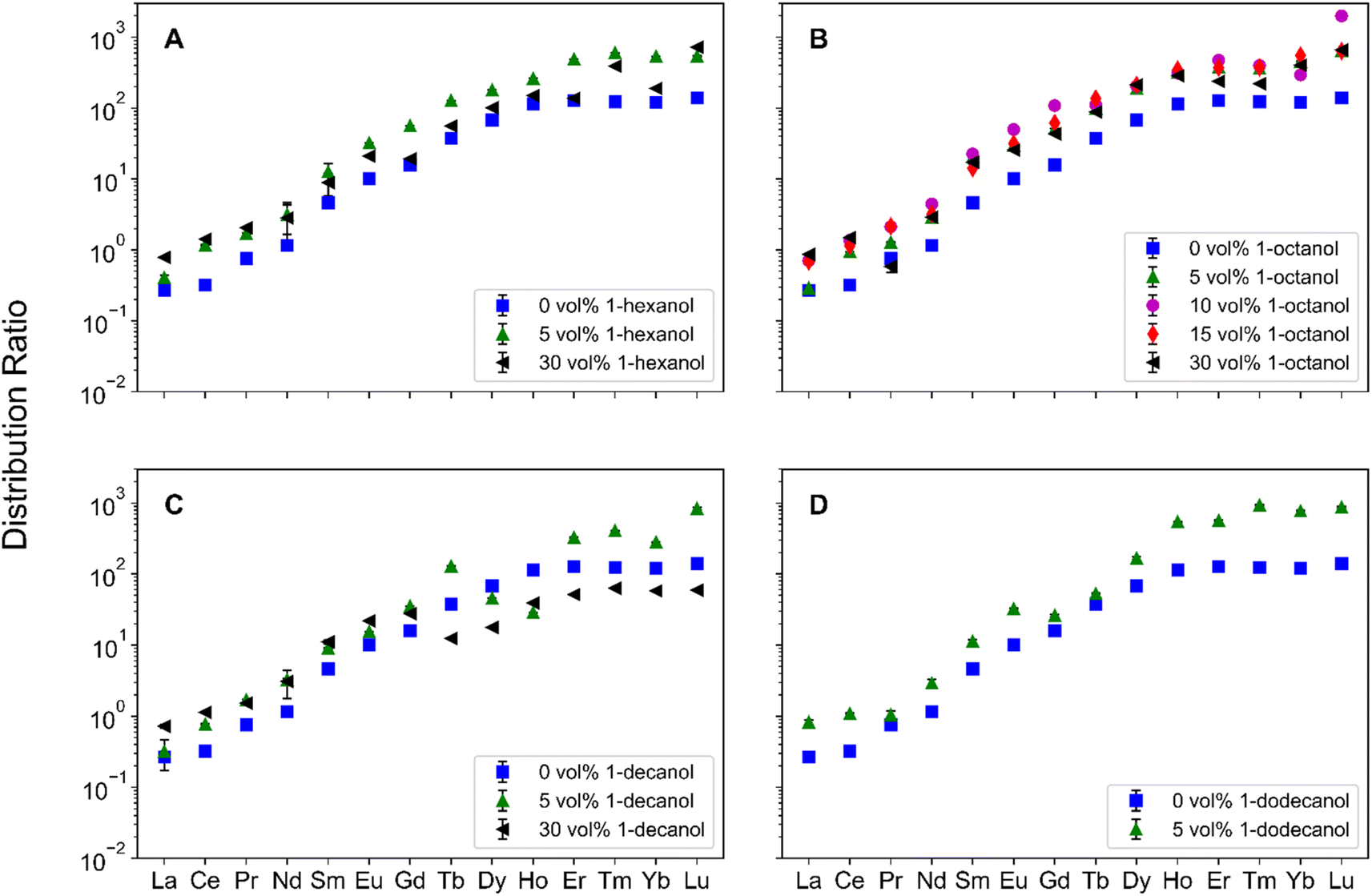 | ||
| Fig. 2 Ds across the Ln period for organic phases containing (A) 0 vol%, 5 vol%, or 30 vol% 1-hexanol, (B) 0 vol%, 5 vol%, 10 vol%, 15 vol%, or 30 vol% 1-octanol, (C) 0 vol%, 5 vol%, or 30 vol% 1-decanol, and (D) 0 vol% or 5 vol% 1-dodecanol. For each system tested, 0 vol% (blue, squares), 5 vol% (green triangles), 10 vol% (magenta circles), 15 vol% (red diamonds), and 30 vol% (black left-triangles). Error bars represent propagated uncertainty from preparing samples, instrument analysis, and sample standard deviation. | ||
Understanding which systems are better suited for practical applications requires the comparison of SF between systems. The two SFs of interest are Dy/Nd for applications to permanent magnets and Tb/Gd as Gd and Tb are the Lns where the D trend changes in 1-hexanol and 1-decanol systems. The Dy/Nd SFs are shown in Table 2, where a trend emerges between the various 1-alcohols, 1-octanol > 1-hexanol > 1-decanol. As the concentration of 1-octanol increases, the Dy/Nd SF also increases, whereas increasing the concentration of both 1-hexanol and 1-decanol decreases their respective Dy/Nd SFs.
| PM, vol% | SF | 1-hexanol | 1-octanol | 1-decanol |
|---|---|---|---|---|
| 0 | Dy/Nd | 58.5 | 58.5 | 58.5 |
| Tb/Gd | 2.38 | 2.38 | 2.38 | |
| 5 | Dy/Nd | 57.3 | 65.5 | 13.9 |
| Tb/Gd | 2.27 | 1.94 | 3.68 | |
| 30 | Dy/Nd | 35.9 | 74.1 | 5.75 |
| Tb/Gd | 2.92 | 2.03 | 0.44 |
The Tb/Gd SFs are also displayed in Table 2 and are significantly lower than the Dy/Nd SFs. This is a result of the difference in Ln Z, where Dy/Nd and Tb/Gd have differences of 6 and 1, respectively. For adjacent Lns, the corresponding SFs are typically lower with a value around 2. Increasing the concentration from 5 vol% to 30 vol% of 1-decanol decreases the Tb/Gd SF, however for both 1-hexanol and 1-octanol their Tb/Gd SFs increase. This results in two separate trends in Tb/Gd SFs for the different 1-alcohol concentrations. For 5 vol% 1-alcohol, the trend in SFs is, 1-decanol > 1-hexanol > 1-octanol, whereas the trend for 30 vol% 1-alcohol is, 1-hexanol > 1-octanol > 1-decanol. This suggests that changes in the trend are a factor of changes in 1-decanol behavior.
Additional characterizations of the organic phase were used to understand bulk phase properties, specifically extraction of H2O and H+. The extraction of H2O was measured via KF and reported in Fig. S2.† In each system, increasing the concentration of 1-alcohol increases H2O extraction, which is also seen following pre-equilibration. This suggests the increase in extraction of H2O molecules arises from the volume composition of 1-alcohol. An increase in H2O extraction is observed following Ln contact, with the largest change existing in the 30 vol% PM systems. It was found that at 30 vol%, adding Lns to the system increases H2O extraction from pre-equilibration contact by 0.05 M, except for 1-decanol which remains similar to pre-equilibration contact. This work suggests in 1-hexanol and 1-octanol systems Lns aid in H2O extraction. However, due to the large concentrations of extracted H2O molecules in comparison to the relatively small Ln concentrations, any trend that may exist across the Ln series is difficult to determine.
The different chain lengths of 1-alcohol have varying densities and molecular weights, so the number of 1-alcohols per H2O extracted was calculated to compare the systems to their corresponding PM concentrations. A decrease in 1-alcohol molecules per H2O molecule is associated with an increase in extracted H2O. These results are shown in Table 3. At 5 vol% 1-alcohol, the trend in number of 1-alcohol molecules per H2O molecule is, 1-decanol < 1-octanol < 1-hexanol. Increasing the concentration of 1-alcohol to 30 vol% has the opposite trend, 1-hexanol < 1-octanol < 1-decanol. It appears there is competition between how well 1-alcohol solubilizes the extracted complex versus how much 1-alcohol promotes TPF through extracting more H2O molecules.
| PM, vol% | 1-hexanol | 1-octanol | 1-decanol |
|---|---|---|---|
| 5 | 9.80 | 9.70 | 8.16 |
| 30 | 5.48 | 6.24 | 9.62 |
As 1-hexanol has a larger ratio of molecules per H2O molecule at low volume fractions, 1-hexanol suppresses aggregation better than 1-decanol. Increasing the concentration to 30 vol%, results in a lower 1-hexanol to H2O ratio from increasing the organic phase H2O concentration. This suggests at 5 vol% PM shorter alkyl chains suppress aggregation better, however at 30 vol% PM longer alkyl chains suppress aggregation better. This work supports current literature which determined the relationship between 1-alcohol alkyl chain lengths and aggregate sizes via dynamic light scattering measurements. Swami et al. showed that in tetra(2-ethylhexyl) diglycolamide (T2EHDGA) systems, the addition of 1 M 1-alcohols form smaller particles as the length of the 1-alcohol alkyl chain increases.24 Therefore, in DGA systems with 1-alcohol PMs, this work and previous literature provide insight to a relationship between 1-alcohol alkyl chain length, H2O extraction, and the ability to suppress aggregation. Additional understanding of the relationship between H2O extraction and aggregate size to elucidate the inverse trend for 5 vol% and 30 vol% PM is essential, requiring beamline techniques such as SAXS or SANS, providing a route for future investigations.
Further organic phase characterization utilized potentiometric titrations to determine the concentration of H+ in the organic phase. In general, there is a decrease in H+ extracted across the Ln period, as evident in Fig. S3.† Heavier Lns have an associated H+ concentration that approaches the H+ concentration after pre-equilibration. Additionally, there is not a consistent trend with H+ concentration across volume fractions for a specific 1-alcohol at constant TODGA and HNO3 concentrations. At 5 vol% 1-alcohol the trend is, 1-hexanol – 1-octanol > 1-decanol. The trend in H+ concentrations among the various alkyl lengths of 1-alcohol is opposite for the trend in H2O extraction. Therefore, with a lower concentration (5 vol%) of 1-alcohol a decrease in extraction of H2O corresponds to an increase in extraction of H+. Increasing the concentration to 30 vol% creates a different trend, 1-octanol > 1-decanol – 1-hexanol. At higher volume fractions of 1-alcohol, the extraction of H+ is related to extraction of Ln. More specifically, an increase in extraction of H+ increases extraction of Ln, particularly for the heavier Lns. This is reflected in the Dy/Nd separation factors, where 1-octanol has larger separation factors than both 1-hexanol and 1-decanol.
Relation between TODGA, H2O, and H+
To understand the relationship between molecules in the outer sphere involved in hydrogen bonding networks, the relationship between TODGA, H2O, and H+ was investigated. The ratio of unbound TODGA (TODGA not directly coordinating to Ln) to extracted H2O and H+ was calculated by eqn (4) and (5), respectively. These relationships provide information as to the number of TODGA molecules available to interact with either H2O or H+ molecules, to understand hydrogen bonding networks.
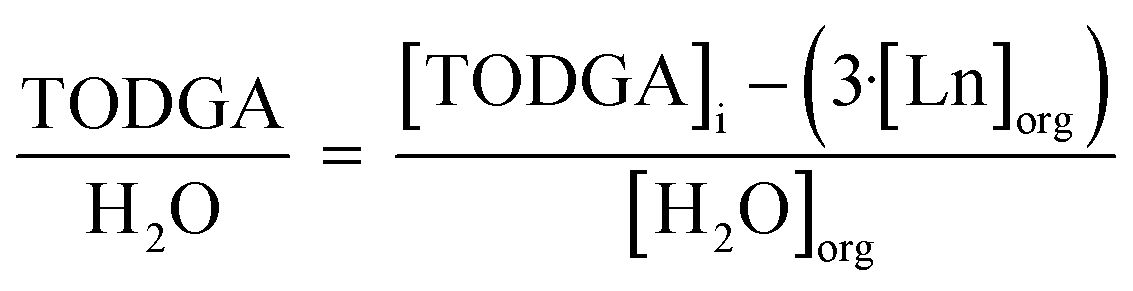 | (4) |
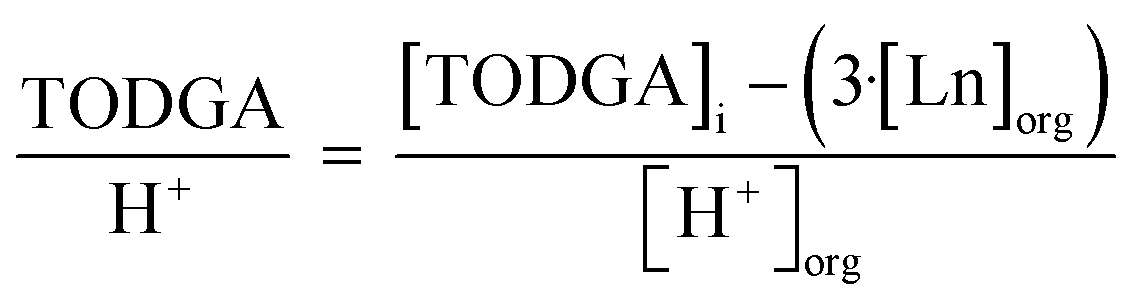 | (5) |
Similarly, the number of H2O molecules per H+ cation was calculated through eqn (6).
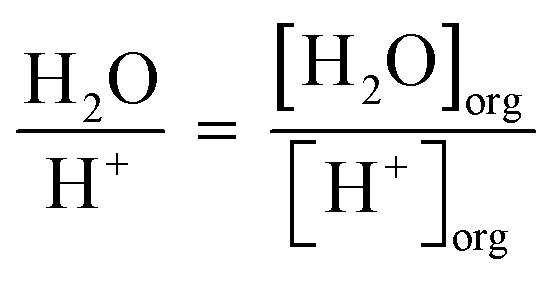 | (6) |
The number of extracted H2O molecules per acid is reported in Fig. S1.† There is an increase in the number of H2O molecules per H+ cation across the Ln period according to 1-hexanol > 1-octanol > 1-decanol. Larger volume fractions of 1-alcohol also have greater H2O per H+. These trends were then compared to those obtained by Baldwin et al., who reported that the trend in extracted H2O per NO3− closely mirrors the trend in D across the Ln period.15 It is important to note this was observed in a system with 0.001 M HNO3 and 0.5 M NaNO3. In comparison to the work by Baldwin et al., increasing the concentration of HNO3 results in different trends in extracted H2O per H+. These differences arise from the increase of H+ which has previously been reported to initiate hydrogen bonding networks crucial for extraction of Ln by TODGA.28,29
Understanding the relationship between unbound TODGA and extracted solutes gives insight into interactions in the hydrogen bonding network. The number of unbound TODGA molecules per extracted H2O molecule was calculated with eqn (4) and are displayed in Fig. 3. At each volume fraction of PM, the trend is 1-hexanol < 1-octanol < 1-decanol. Additionally, the number of unbound TODGA per H2O molecule decreases with increasing volume fraction of PM and decreases across the Ln period. As this parameter of unbound TODGA per solute reflects changes only in the outer-sphere, for a larger volume fraction of 1-alcohol, less TODGA molecules are involved in solubilizing the complex. In the current literature, it has been hypothesized the role of 1-alcohol PMs is solely to solubilize the aggregates.20,25 The results discussed from Fig. 3 support this hypothesis, where increasing the 1-alcohol concentration reduces the solubilization of H2O molecules by outer-sphere TODGA. This suggests an increase in 1-alcohol interactions with H2O molecules as the 1-alcohol concentration increases, resulting in better aggregate solubilization.
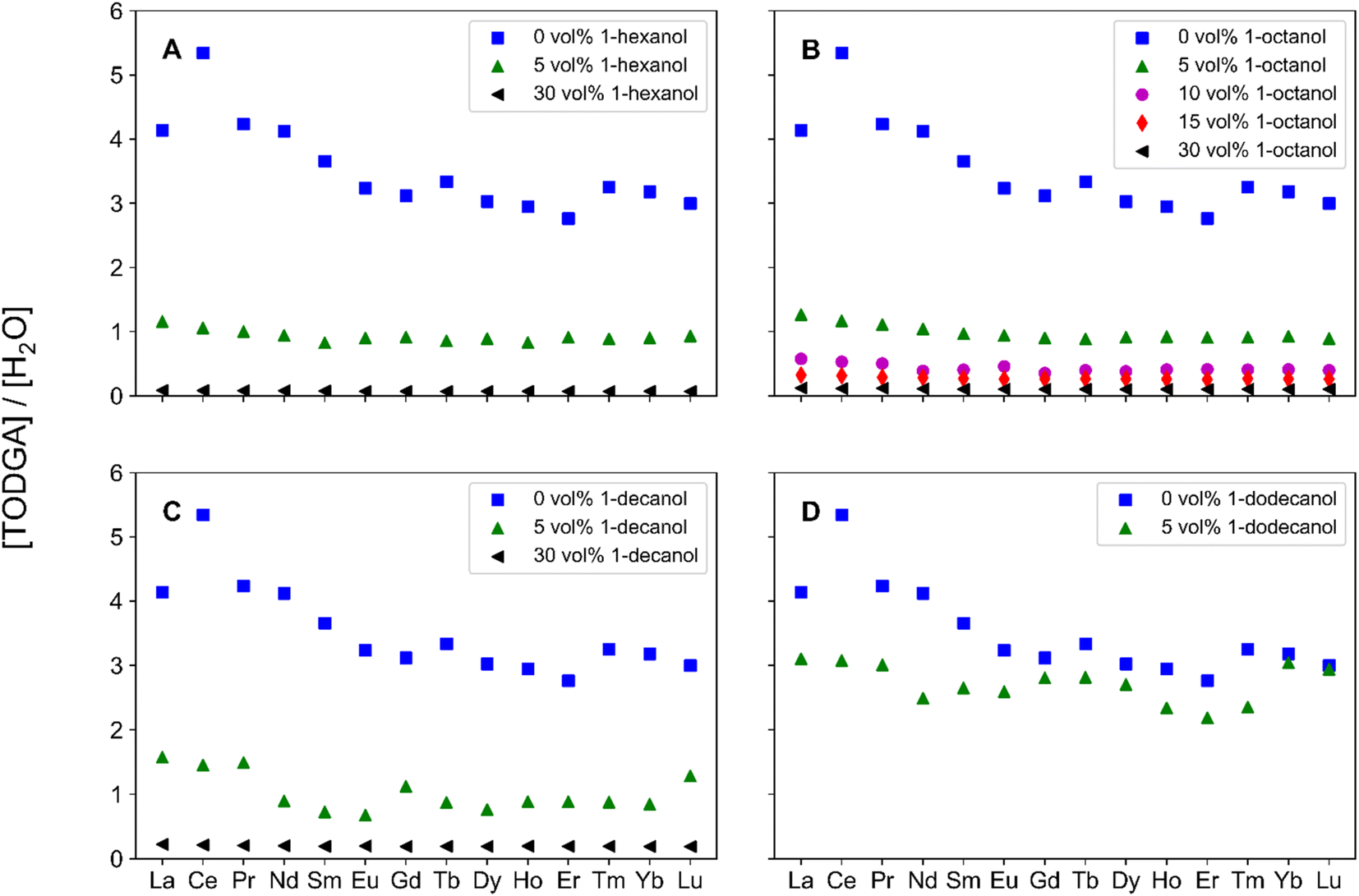 | ||
| Fig. 3 Calculated unbound TODGA per extracted H2O molecule, across the Ln period for organic phases containing (A) 0 vol%, 5 vol%, or 30 vol% 1-hexanol, (B) 0 vol%, 5 vol%, 10 vol%, 15 vol%, or 30 vol% 1-octanol, (C) 0 vol%, 5 vol%, or 30 vol% 1-decanol, and (D) 0 vol% or 5 vol% 1-dodecanol. For each system tested, 0 vol% (blue, squares), 5 vol% (green triangles), 10 vol% (magenta circles), 15 vol% (red diamonds), and 30 vol% (black left-triangles). | ||
The results for the number of unbound TODGA molecules per extracted H2O also show trends across the Ln period. In the early Lns, specifically for La and Ce it appears that there are different trends if organic phases contain 1-alcohols. These changes in H2O extraction across the Ln period for the no modifier system appear larger than the 1-alcohol systems because of the smaller quantities of H2O extracted, as shown in Fig. S2.† At each 1-alcohol concentration, with increasing Ln Z, a plateau forms, at a similar Ln Z to the plateau in the Ds. As Fig. 3 shows the ability for outer-sphere TODGA molecules to solubilize extracted H2O molecules, this gives insight into the D trends under acidic conditions. Specifically, the Lns with larger D are also the Lns with a decrease in solubilization of H2O molecules by TODGA.
Similarly, the number of unbound TODGA molecules per extracted H+ cation were determined, via eqn (5) and are reported in Fig. 4. Several trends emerge which are similar to those found for the number of 1-alcohol molecules per extracted H2O molecule. At 5 vol% PM the trend is, 1-hexanol < 1-octanol < 1-decanol, however increasing the concentration to 30 vol%, results in the following trend, 1-decanol < 1-octanol < 1-hexanol. The trends across 1-alcohol alkyl chain lengths are reversed at low and high-volume fractions. This follows that an increase in unbound TODGA per H+ involves more TODGA molecules that are available to interact with H+. In previous literature FT-IR and 1H-NMR investigations have determined that H+ are responsible for the formation of (DGA)(HNO3)1–2 adducts.11,29,30 Extracted HNO3 molecules likely interact with TODGA through either direct protonation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety or through hydrogen bonding interactions of the CO moiety to a hydronium nitrate ion-pair.29 From an understanding of the importance of HNO3 in the formation of (DGA)(HNO3)1–2 adducts, increasing the number of unbound TODGA molecules per extracted H+, results in more aggregation. It is important to note that while noticeable changes in the number of unbound TODGA molecules per H+ are observed with increasing the concentration of 1-hexanol and 1-decanol, there appears to be little change with varying the 1-octanol concentration. This suggests that 1-octanol can balance the competing interactions of solubilizing the aggregates and not incidentally promoting TPF through an increased extraction of H2O molecules.
O moiety or through hydrogen bonding interactions of the CO moiety to a hydronium nitrate ion-pair.29 From an understanding of the importance of HNO3 in the formation of (DGA)(HNO3)1–2 adducts, increasing the number of unbound TODGA molecules per extracted H+, results in more aggregation. It is important to note that while noticeable changes in the number of unbound TODGA molecules per H+ are observed with increasing the concentration of 1-hexanol and 1-decanol, there appears to be little change with varying the 1-octanol concentration. This suggests that 1-octanol can balance the competing interactions of solubilizing the aggregates and not incidentally promoting TPF through an increased extraction of H2O molecules.
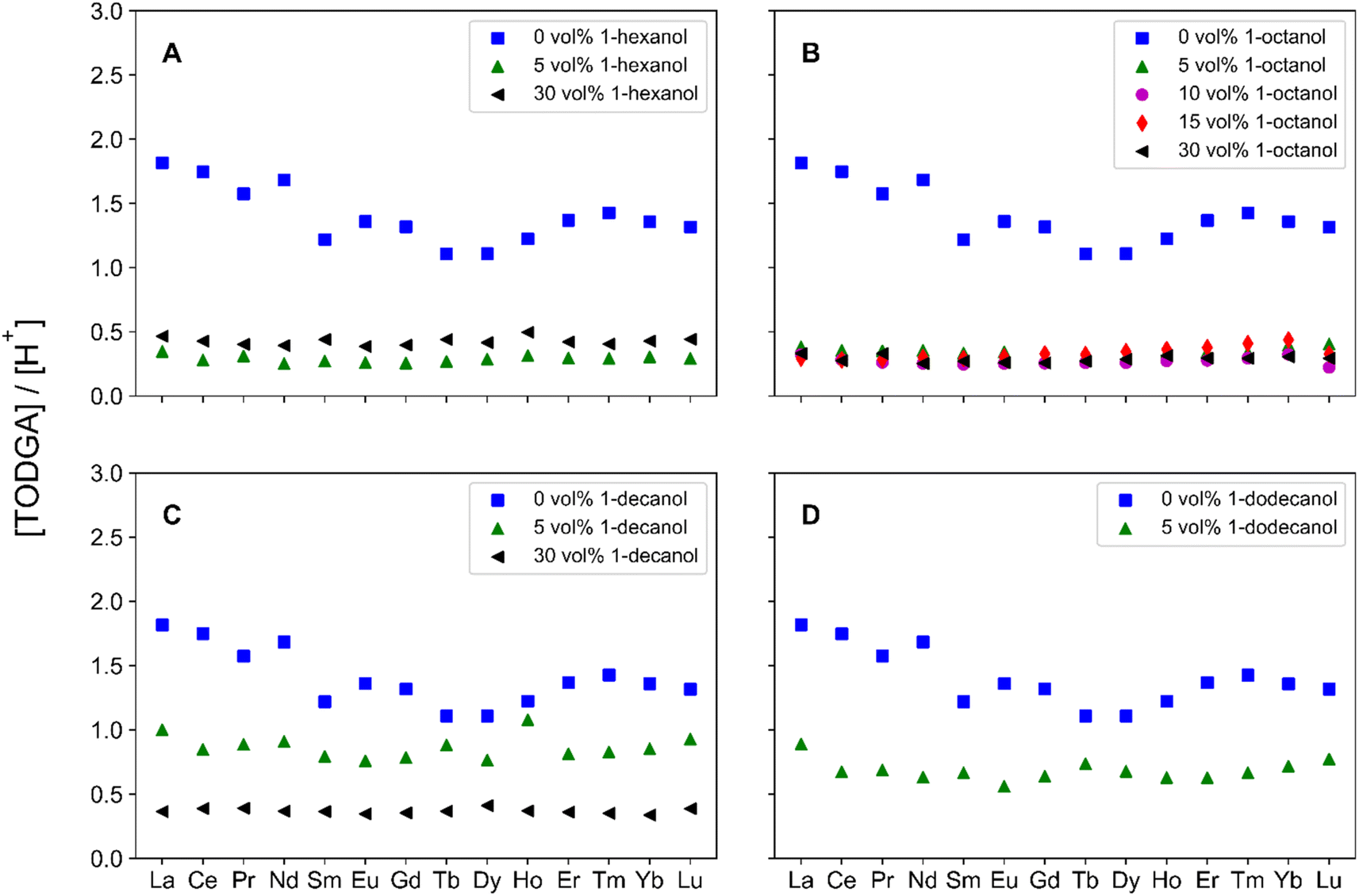 | ||
| Fig. 4 Calculated unbound TODGA per extracted H+ cation, across the Ln period for organic phases containing (A) 0 vol%, 5 vol%, or 30 vol% 1-hexanol, (B) 0 vol%, 5 vol%, 10 vol%, 15 vol%, or 30 vol% 1-octanol, (C) 0 vol%, 5 vol%, or 30 vol% 1-decanol, and (D) 0 vol% or 5 vol% 1-dodecanol. For each system tested, 0 vol% (blue, squares), 5 vol% (green triangles), 10 vol% (magenta circles), 15 vol% (red diamonds), and 30 vol% (black left-triangles). | ||
FT-IR analysis
To probe changes in molecular structure under the multitude of solution conditions studied FT-IR was used. A focus was placed on evaluating changes in the carbonyl, ether, and nitrate frequencies. Although the ether O is involved in coordination of TODGA to Ln, minor changes (1–2 cm−1) were observed after an in-depth evaluation of the FT-IR spectra, which could be the result of dative interactions. Under the conditions employed here, there were no changes in the asymmetric nitrate stretch across the Ln period, which was observed at 1301 cm−1. This suggests that nitrates are not “free” in solution but rather bound, likely participating in the hydrogen bonding network. Free nitrates are typically observed between 1320–1350 cm−1, whereas bound nitrates have a stretch between 1250–1310 cm−1.31 The frequency of nitrates in this system is consistent with the structure hypothesis that nitrates are involved in outer-sphere coordination. However, the role of nitrates in the outer-sphere is difficult to probe with FT-IR as the results cannot distinguish one interaction from another.32 For these reasons, only the carbonyl stretch was analyzed in detail. To develop a more in depth understanding of carbonyl peak features, the peaks were individually fit in OriginLab (Fig. S20–S179†) to determine the wavenumber, FWHM, and area. The frequency of the associated Ln-TODGA peak(s) are listed in Table S3,† for systems containing no modifier, 5 vol% modifier, and 30 vol% modifier.The CO stretch from the fresh organic phase with no modifiers appears at 1662 cm−1. This is consistent with previous TODGA and T2EHDGA literature values for the CO stretch.30,33,34 Upon the addition of 5 vol% 1-alcohol, 1-hexanol and 1-decanol have a shoulder that is red-shifted in comparison to the CO stretch of the no modifier system. A red shift (or decreasing frequency) is observed when the CO bond, in this case, weakens. In contrast a blue shift (or increasing frequency) is associated with the strengthening of CO bond or when dative bonds form.35,36 Increasing the 1-alcohol concentration to 30 vol%, results in a red-shifted shoulder for 1-octanol and 1-decanol systems. The red-shift of the CO stretch without contact with an aqueous phase suggests that 1-alcohols interact with the CO motif on TODGA. This likely occurs through hydrogen bonding interactions as 1-alcohols act as a hydrogen donor and the CO acts as a hydrogen acceptor.
After contact with 1 M HNO3, at 5 vol% the CO stretch of 1-hexanol remains unchanged, whereas both 1-octanol and 1-decanol experience red-shifts. At 30 vol% 1-alcohol, both 1-hexanol and 1-octanol have a red-shifted CO stretch, but 1-decanol is slightly blue-shifted. When a change is not observed following 1 M HNO3 contact, this suggests that the interactions in the organic phase are similar to the interactions prior to contact. The systems that experienced a red-shift are consistent with the literature which explains this slight red shift to be the result of hydrogen bond interactions of HNO3 molecules interacting with polar carbonyl group of TODGA, forming (DGA)(HNO3)1-2 adducts.30,37,38 Across the varying alkyl chain lengths of 1-alcohol, the trend in this red-shifted CO frequency for 5 vol% is 1-hexanol < 1-octanol < 1-decanol. Increasing the 1-alcohol concentration to 30 vol% results in the reverse trend, 1-decanol < 1-octanol < 1-hexanol. Between each of the 1-alcohol systems, the change in frequency is 1–2 cm−1, suggesting only subtle changes.
The effects across the Ln period on Ln-TODGA interactions are reflected the best in the modifier-free systems. In these, the initial peak at 1662 cm−1 decreases in intensity across the period, with a corresponding ingrowth of a peak at 1620 cm−1 that increases in intensity with increasing Ln Z, as shown in Fig. 5. The initial carbonyl stretch is associated with (DGA)(HNO3)1–2 adducts, and the red-shifted CO stretch is associated with the extracted Ln-DGA complex. As the Ln-DGA complex CO frequency is red-shifted, this indicates a weaker CO bond associated with the Ln-DGA complex compared to the stronger CO bond in the (DGA)(HNO3)1-2 adducts.33 This behavior is not isolated to the systems considered herein, but has been reported with DOHyA,33 D3DODGA,38 T2EHDGA,39 TODGA,33 TODGA (+DHOA),40 TODGA (+1-octanol),40 and TODGA (+DOHyA)33 systems. These studies involved increasing the concentration of Nd at constant extractant and HNO3 concentrations, where higher Nd concentrations resulted in larger Ds. This suggests for higher Ln Ds, there is a decrease in the carbonyl intensity corresponding to the (DGA)(HNO3)1–2 adducts and an increase in the carbonyl intensity corresponding to the Ln-DGA complex.
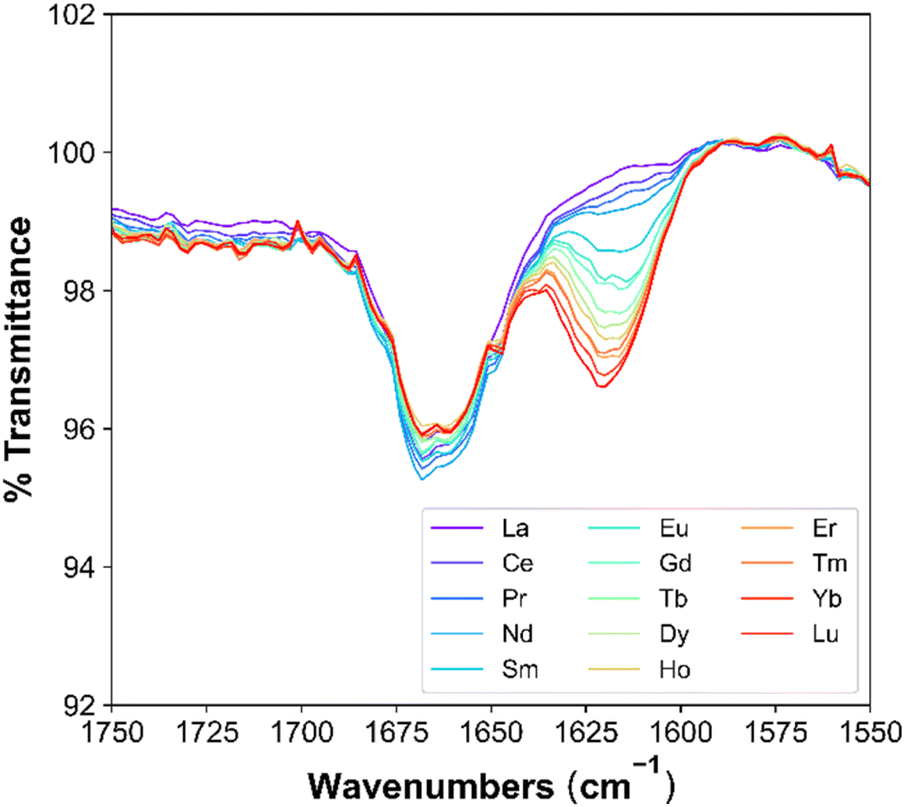 | ||
| Fig. 5 FT-IR spectra, highlighting the carbonyl stretch for a modifier-free system with 0.04 M TODGA in n-dodecane after contact with Lns. | ||
For the systems studied herein, this corresponds to trends across the Ln period. As Ln Z increases, there is a decrease in the number of (TODGA)(HNO3)1–2 adducts until Gd, where a plateau forms, as evident by the decrease in intensity of the initial carbonyl peak. A decrease in the number of (TODGA)(HNO3)1–2 adducts is accompanied by an increase in the number of Ln-TODGA complexes. The results obtained from FT-IR support Ds (Fig. 2), where traversing the Ln period results in higher extraction of Ln and therefore a larger intensity of the CO stretch that is attributed to Ln-TODGA interactions. Addition of PMs to the organic phase decreases the intensity of both carbonyl peaks, maintaining the same trend across the Ln period for the modifier-free system. The complete shift in both CO stretches occurs when the CO stretch associated with (TODGA)(HNO3)1-2 adducts is at a minimum and the CO stretch associated with the Ln-DGA complex is at a maximum, where neither are changing with the addition of Ln or HNO3. A complete shift of CO stretches, has been correlated with TPF, where polar–polar interactions of the extracted species dominate the solution.33 Across the Ln period, the LOC decreases,15 suggesting the behavior observed in FT-IR carbonyl intensities might be a product of organic phase micro-structure approaching that found in TPF.
In all systems studied the trend in carbonyl frequency for the red-shifted Ln–OC interaction, first decreases from La–Gd and then slightly increases from Tb–Lu. This suggests a stronger CO bond associated with Ln-TODGA complexes for the heavier Lns. Investigations with organophosphorus extractants reported that the strength of ligand interactions cannot be related to various M–O interactions.41,42 Instead, either Far-IR or Raman Spectroscopy are required to determine the frequency of M–O interactions. Without understanding the Ln–O bond strengths, it is difficult to make conclusions about the relationship of CO bond strength to the Ln–O bond strength.
The frequency of the Ln–OC stretch across the Ln period exhibits an interesting trend which cannot be explained by the reduced mass effect. If this were the case, increasing Z would result in a constant decrease in frequency.35 As that does not follow the observed behavior it can be concluded that these changes in frequency across the Ln period do not reflect the effect of reduced mass on the vibrations of Ln-TODGA complexes. The relationship of Ln–OC frequency with increasing Z resembles the trend in unbound TODGA per extracted H2O molecule (Fig. 3). These results provide insight to the frequency trend of the Ln–OC stretch across the Ln period might be a product of hydrogen bonding interactions.
Conclusions
The effects of alkyl chain length on 1-alcohols and its impact on Ds, concentration of H2O and H+ in the bulk organic phase of TODGA extraction systems, and probing coordination trends through FT-IR investigations, are reported. These results showed with 5 vol% 1-alcohol, there is a consistent increase in the D compared to systems with no PM. Increasing the PM concentration to 30 vol% leads to a different trend in Ds, except for 1-octanol. This trend differs by experiencing a decrease in the D at Gd, where the remainder of the Lns have Ds that are lower than expected.Comparison of Ds to extracted H2O and H+ provided a better understanding as to the importance of hydrogen bonding networks in TODGA systems. At low PM concentrations (5 vol%), the normalized organic phase concentration of H2O and number of unbound TODGA per extracted H+ suggests that 1-hexanol is better at suppressing aggregation through efficient solubilization of the Ln-TODGA complex. At higher concentrations of PM (30 vol%), the opposite trend in 1-alcohol alkyl chain lengths was established, which suggests 1-decanol forms the smallest particles. In support of this, the number of unbound TODGA per H+ resulted in less available TODGA to interact with H+ for longer 1-alcohol alkyl chains. The combination of these results suggests a competition of two effects, solubilization of the Ln-DGA aggregates and incidentally causing TPF.
The trend in Ds for a TODGA system under acidic conditions was found to resemble the trend in unbound TODGA per extracted H2O molecules. Similarly, to Ln Ds, a plateau forms around Tb in the number of unbound TODGA per H2O. This suggests that extraction of Ln may be related to the relationship between outer-sphere TODGA molecules and extracted H2O. Similarly, the ability for a system to suppress aggregation in the organic phase can be related to the number of unbound TODGA per extracted H+, where a decrease in the number of unbound TODGA per H+ is correlated with a suppression of aggregates.
The relationships that exist between Ln, TODGA, H2O, H+, and 1-alcohol are reflected in FT-IR results. Prior to contact with an aqueous phase, the red-shifted shoulder in CO stretch gives insight that the O–H moiety on 1-alcohol molecules interact with the CO moiety on TODGA through hydrogen bonding interactions. The trends in Ln–OC frequency across the Ln period reflected the trends in unbound TODGA per extracted H2O. This suggested that changes to the hydrogen bonding network are responsible for the trends established with increasing Ln Z. These results have showed the importance of the relationship between Ln, TODGA, H2O, H+, and 1-alcohol in establishing a hydrogen bonding network responsible for the unique trends in Ln extraction.
Overall, this study provided insight as to the importance of doing a multi-faceted approach to characterize the organic phase of these TODGA systems, through understanding the relationship between extraction of Ln, H+, H2O coupled with probing coordination. Based on the results provided herein, the 30 vol% 1-octanol system is of particular interest for Ln separations as is has the largest Dy/Nd separation factor. Application to industry (i.e., hydrometallurgical processes) requires additional studies in higher acid concentrations (3 M HNO3) to probe systems that are more relevant to practical applications. Coupling solvent extraction results with spectroscopic techniques to probe coordination will be instrumental in developing a relationship of organic phase structure and extraction of Lns.
Author contributions
AAP: methodology, investigation, data curation, formal analysis, visualization, writing – original draft, writing – review & editing. SSG: investigation, writing – review & editing. JCS: funding acquisition, project administration, conceptualization, methodology, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Mark R. Antonio for providing critical comments, perspectives, and insights. The authors were supported by the Department of Energy Basic Energy Science program (DE-SC0022217) for this work.Notes and references
- M. K. Jha, A. Kumari, R. Panda, J. Rajesh Kumar, K. Yoo and J. Y. Lee, Hydrometallurgy, 2016, 165, 2–26 CrossRef CAS.
- G. A. Picayo and M. P. Jensen, in Handbook on the Physics and Chemistry of Rare Earths, 2018, vol. 54, pp. 145–225 Search PubMed.
- I. Kajan, M. Florianova, C. Ekberg and A. v. Matyskin, RSC Adv., 2021, 11, 36707–36718 RSC.
- R. J. Ellis, D. M. Brigham, L. Delmau, A. S. Ivanov, N. J. Williams, M. N. Vo, B. Reinhart, B. A. Moyer and V. S. Bryantsev, Inorg. Chem., 2017, 56, 1152–1160 CrossRef CAS PubMed.
- E. A. Mowafy and H. F. Aly, Solvent Extr. Ion Exch., 2007, 25, 205–224 CrossRef CAS.
- E. A. Mowafy and D. Mohamed, Sep. Purif. Technol., 2014, 128, 18–24 CrossRef CAS.
- D. Xu, Z. Shah, Y. Cui, L. Jin, X. Peng, H. Zhang and G. Sun, Hydrometallurgy, 2018, 180, 132–138 CrossRef CAS.
- Y. Sasaki, Y. Sugo, K. Morita and K. L. Nash, Solvent Extr. Ion Exch., 2015, 33, 625–641 CrossRef CAS.
- D. Stamberga, M. R. Healy, V. S. Bryantsev, C. Albisser, Y. Karslyan, B. Reinhart, A. Paulenova, M. Foster, I. Popovs, K. Lyon, B. A. Moyer and S. Jansone-Popova, Inorg. Chem., 2020, 59, 17620–17630 CrossRef CAS PubMed.
- Y. Sasaki, T. Kimura and K. Oguma, J. Ion Exch., 2007, 18, 354–359 CrossRef CAS.
- D. Woodhead, F. McLachlan, R. Taylor, U. Müllich, A. Geist, A. Wilden and G. Modolo, Solvent Extr. Ion Exch., 2019, 37, 173–190 CrossRef CAS.
- E. R. Bertelsen, N. C. Kovach, B. J. Reinhart, B. G. Trewyn, M. R. Antonio and J. C. Shafer, CrystEngComm, 2020, 22, 6886–6899 RSC.
- M. R. Antonio, D. R. McAlister and E. P. Horwitz, Dalton Trans., 2015, 44, 515–521 RSC.
- Z. Chen, X. Yang, L. Song, X. Wang, Q. Xiao, H. Xu, Q. Feng and S. Ding, Inorg. Chim. Acta, 2020, 513, 1–11 CrossRef.
- A. G. Baldwin, A. S. Ivanov, N. J. Williams, R. J. Ellis, B. A. Moyer, V. S. Bryantsev and J. C. Shafer, ACS Cent. Sci., 2018, 4, 739–747 CrossRef CAS PubMed.
- D. M. Brigham, A. S. Ivanov, B. A. Moyer, L. H. Delmau, V. S. Bryantsev and R. J. Ellis, JACS Au, 2017, 139, 17350–17358 CrossRef CAS PubMed.
- M. B. Singh, S. R. Patil, A. A. Lohi and V. G. Gaikar, Sep. Sci. Technol., 2018, 53, 1361–1371 CrossRef CAS.
- T. Yaita, A. W. Herlinger, P. Thiyagarajan and M. P. Jensen, Solvent Extr. Ion Exch., 2004, 22, 553–571 CrossRef CAS.
- P. N. Pathak, S. A. Ansari, S. v. Godbole, A. R. Dhobale and V. K. Manchanda, SAA, 2009, 73, 348–352 CAS.
- L. Berthon, A. Paquet, G. Saint-Louis and P. Guilbaud, Solvent Extr. Ion Exch., 2021, 39, 204–232 CrossRef CAS.
- M. Spadina, J.-F. Dufreche, S. Pellet-Rostaing, S. Marcelja and T. Zemb, Langmuir, 2021, 37, 10637–10656 CrossRef CAS PubMed.
- P. N. Pathak, S. A. Ansari, S. Kumar, B. S. Tomar and V. K. Manchanda, J. Colloid Interface Sci., 2010, 342, 114–118 CrossRef CAS PubMed.
- K. R. Swami, K. A. Venkatesan, P. Sahu and S. M. Ali, J. Mol. Struct., 2020, 1221, 128795 CrossRef CAS.
- K. R. Swami and K. A. Venkatesan, J. Mol. Liq., 2019, 296, 111741 CrossRef CAS.
- D. Whittaker, A. Geist, G. Modolo, R. Taylor, M. Sarsfield and A. Wilden, Solvent Extr. Ion Exch., 2018, 36, 223–256 CrossRef CAS.
- A. E. Clark, P. Yang and J. C. Shafer, Experimental and Theoretical Approaches to Actinide Chemistry, From Fundamental Systems to Practical Applications, 2016, 237–282 Search PubMed.
- MATLAB and Bioinformatics Toolbox Release 2022b, The MathWorks, Inc., Natick, Massachusetts, United States Search PubMed.
- Y. Sasaki, P. Rapold, M. Arisaka, M. Hirata, T. Kimura, C. Hill and G. Cote, Solvent Extr. Ion Exch., 2007, 25, 187–204 CrossRef CAS.
- L. Lefrançois, J. J. Delpuech, M. Hébrant, J. Chrisment and C. Tondre, J. Phys. Chem. B, 2001, 105, 2551–2564 CrossRef.
- E. L. Campbell, V. E. Holfeltz, G. B. Hall, K. L. Nash, G. J. Lumetta and T. G. Levitskaia, Solvent Extr. Ion Exch., 2017, 35, 586–603 CrossRef CAS.
- D. J. Goebbert, E. Garand, T. Wende, R. Bergmann, G. Meijer, K. R. Asmis and D. M. Neumark, J. Phys. Chem. A, 2009, 113, 7584–7592 CrossRef CAS PubMed.
- N. Bessen, Q. Yan, N. Pu, J. Chen, C. Xu and J. Shafer, Inorg. Chem. Front., 2021, 8, 4177–4185 RSC.
- P. Narayanan, K. R. Swami, T. Prathibha and K. A. Venkatesan, J. Mol. Liq., 2020, 314, 113685 CrossRef CAS.
- E. Campbell, V. E. Holfeltz, G. B. Hall, K. L. Nash, G. J. Lumetta and T. G. Levitskaia, Solvent Extr. Ion Exch., 2018, 36, 331–346 CrossRef CAS.
- K. Nakamoto, Infrared and Raman spectra of inorganic and coordination compounds, Wiley, 1997 Search PubMed.
- N. Parvathy, K. R. Swami, T. Prathibha and K. A. Venkatesan, J. Mol. Liq., 2020, 317, 113940 CrossRef CAS.
- T. Prathibha, K. A. Venkatesan, B. Robert Selvan, M. P. Antony and P. R. Vasudeva Rao, Radiochim. Acta, 2012, 100, 907–913 CrossRef CAS.
- P. Narayanan, K. R. Swami, T. Prathibha and K. A. Venkatesan, ChemistrySelect, 2022, 7, e202202610 CrossRef CAS.
- K. R. Swami, A. S. Suneesh, R. Kumaresan, K. A. Venkatesan and M. P. Antony, ChemistrySelect, 2017, 2, 11177–11186 CrossRef CAS.
- T. Prathibha, K. A. Venkatesan and M. P. Antony, Colloids Surf., A, 2018, 538, 651–660 CrossRef CAS.
- M. Borkowski, J. R. Ferraro, R. Chiarizia and D. R. McAlister, Solvent Extr. Ion Exch., 2002, 20, 313–330 CrossRef CAS.
- A. W. Herlinger, J. R. Ferraro, J. A. Garcia and R. Chiarizia, Polyhedron, 1998, 17, 1471–1475 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra07786h |
| This journal is © The Royal Society of Chemistry 2023 |