
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrothiolation of alkynes with thiol–catechol derivatives catalysed by CuNPs/TiO2: exploring the reaction mechanism by DFT calculations†
Matías Capurso ,
Gabriel Radivoy,
Fabiana Nador and
Viviana Dorn*
,
Gabriel Radivoy,
Fabiana Nador and
Viviana Dorn*
Instituto de Química del Sur (INQUISUR-CONICET), Depto. de Química, Universidad Nacional del Sur, Av. Alem 1253, B8000CPB Bahía Blanca, Argentina. E-mail: vdorn@uns.edu.ar
First published on 10th March 2023
Abstract
Density functional theory (DFT) calculations were applied to describe the hydrothiolation reaction of activated alkynes with thiols bearing a catechol group. The thiol-yne click (TYC) process was efficiently catalysed by a CuNPs/TiO2 nanocatalyst giving the corresponding anti-Markovnikov vinyl sulphides with high Z-stereoselectivity. Based on the experimental results and DFT studies, a plausible reaction mechanism is proposed, which implies the activation of the carbon–carbon triple bond by coordination to the copper centre, followed by a stereoselective (external) nucleophilic attack to give preferentially the Z-vinyl sulphide isomer. Additionally, experimental and theoretical studies strongly correlate with the proposed synergistic role for the TiO2 support in the catalytic process.
Introduction
Thiol-yne click (TYC) reaction represents a simple and attractive approach to produce valuable synthetic intermediates such as vinyl sulphides, starting from readily available thiols and alkynes.1,2 This reaction is commonly promoted by the presence of free radicals,3 strong acids,4 bases5 or transition metals.6 Through these methodologies, branched and/or linear vinyl sulphides have been obtained in a Markovnikov or anti-Markovnikov fashion, respectively (Scheme 1).7,8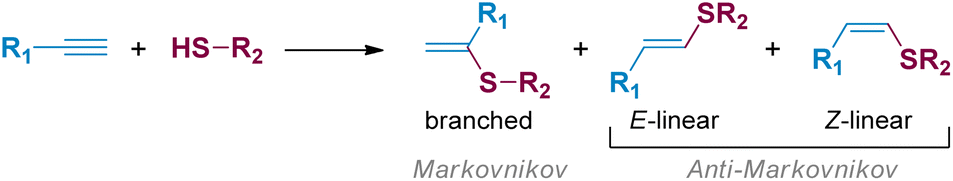 | ||
| Scheme 1 Schematic representation of alkyne hydrothiolation. | ||
Furthermore, due to the wide range of applications of these valuable derivatives in the field of materials science, an increasing interest has been focused on new synthetic methodologies for the synthesis of catechol-based molecules under smooth conditions, in a straightforward and atom-efficient way. Novel bioinspired functional materials based on catechol structures have been employed in the preparation of adhesives,9 capsules,10 coatings11 and in the formation of coordination polymer nanoparticles (CNPs),12 among others. Accordingly, in a recent paper published by our group,13 we described the TYC reaction between activated alkynes with adjacent electron-withdrawing groups, and thiols bearing catechols pre-synthesized through a versatile Michael addition methodology.14 The reaction was promoted by a heterogeneous and low-cost metal nanocatalyst composed of copper nanoparticles supported on TiO2 (CuNPs/TiO2), in 1,2-dichloroethane (1,2-DCE) as the solvent, under heating at 80 °C and in the absence of any added base. Through this novel methodology, thirteen new catechol-bearing vinyl sulphides were obtained, with conversions ranging from good to excellent. Moreover, the reaction was highly regio- and stereoselective towards the anti-Markovnikov Z-vinyl sulphide in most of the cases studied.
In terms of the possible mechanisms involved in the metal catalysed hydrothiolation of alkynes, two pathways have generally been proposed; a carbon–carbon triple bond activation followed by the nucleophilic attack of the thiol or an initial activation of the thiol followed by the insertion of the alkyne into the metal–sulphur bond. In this sense, two excellent review articles by Castarlenas, Oro et al.,2 and Beletskaya and Ananikov,15 highlighted the most important mechanistic aspects regarding the use of transition metal-based catalytic systems for the regio- and stereoselective control of this transformation.
It is known that copper catalysts are able to activate the acetylenic triple bond towards a nucleophilic attack by metal π-coordination. On this matter, Zhang et al. reported the CuI-catalysed stereoselective hydrothiolation of alkynes, in the presence of K2CO3 as base, under CO2 atmosphere and water as proton donor, working in DMSO as the solvent.16 The authors proposed the carboxylation of the terminal Csp atom promoted by copper-activation of the acetylenic group, and the formation of a cyclic alkene/carboxylate copper complex intermediate as the essential pathway for the observed stereoselectivity towards the Z-vinyl sulphides. Beletskaya and coworkers,17 reported the regio- and stereoselective formation of the corresponding Z-(β)-alkenyl sulphides starting from alkyl and aryl thiols and terminal alkynes in the presence of CuI and NEt3 as base. The authors suggested that CuI could activate the carbon–carbon triple bond by acting as a Lewis acid, also favouring the Z to E isomerisation of the vinyl sulphides, either in protic solvents or after prolonged heating. In another work, Kodomari et al.18 reported that the addition of thiols to methyl propiolate was notably accelerated by the presence of neutral alumina (Al2O3) to give the Z-isomer with high stereoselectivity. The authors suggested that alumina basic sites were responsible for the increase in thiol nucleophilicity, while the acid sites were proposed to be activating the carbon–carbon triple bond through coordination with the carboxymethyl group.
In line with these experimental observations and mechanistic proposals by other authors, in our recent work about the TYC reaction catalysed by CuNPs/TiO2,13 we observed that the nature of the support had a strong influence on the stereoselectivity of the reaction. We then speculated with a non-innocent role of the TiO2 support, that could be synergistically participating in the activation of the alkyne, the thiol or both. Additionally, we proposed that the TiO2 support could be acting as a fixer for the catechol moiety.19 In this regard, Terranova,20a and also Diebold and coworkers,20b computationally evaluated the interaction of one or both of the pyrocatechol hydroxyl groups with different rutile (TiO2) faces, by means of DFT methods. In agreement with the experimental data, they observed that in some cases the most stable mode of interaction was a monodentate one, while a bidentate coordination was favoured in other cases, suggesting that both coordination structures could easily interconvert.
From the above, it seems clear that the selectivity of the metal-catalysed hydrothiolation reaction could be influenced by many factors, not only by the nature of the metal catalyst but also by the structure of the starting alkyne, the use of additives, the reaction temperature and/or the solvent. In this scenario, performing theoretical studies regarding the possible intermediates and transition states involved in the reaction mechanism, could shed light on further rational designing of more selective catalytic systems.
In this regard, computational studies about the mechanism involved in metal-catalysed alkyne hydrothiolation are scarce, and only few papers can be found in the scientific literature. Zhang and Wang,21 have computationally studied the regiochemistry of the Au(I)/NHC-catalysed hydrothiolation of phenylacetylene with thiophenol. By using DFT methods, the authors proposed an alkyne insertion into the metal–sulphur bond as the rate-determining step, the Au(I)/NHC-catalyst showing a superior anti-Markovnikov selectivity when compared to Ag(I) and Cu(I) catalysts. More recently, Ananikov et al.22 reported the regioselective hydrothiolation of cyclopropyl acetylenes employing a Pd/NHC complex as precatalyst, in the presence of a base (NEt3), a radical trap (γ-terpinene) and toluene as the solvent, giving the corresponding Markovnikov vinyl sulphides. The authors also carried out an in-depth mechanistic study, by means of both experimental work and computational modelling, focused on the alkyne insertion into the palladium–sulphur bond aiming to disclose, the factors controlling the high regioselectivity observed. Regarding computational modelling studies on the alkyne hydrothiolation reaction promoted by heterogeneous metal-catalysts, Kalluraya and Turukarabettu informed about the hydrothiolation of two heteroaryl acetylenic ketones with 5-phenyl-1,3,4-oxadiazole-2-thiol catalysed by AuNPs/TiO2 in ethanol, yielding the corresponding Z-vinyl sulphides via an anti-addition process.23 By means of plane waves DFT methods, together with charges and OM interactions analyses, the authors proposed a reaction mechanism with a first step involving alkyne–gold coordination, a subsequent nucleophilic attack by the thiol at the β-carbon, and final protonation. It is worthy of note that no reaction coordinate was proposed, and both the thiol activation and the role of the metal support were not discussed.
In order to contribute to the rational design of more selective and active catalysts, it is crucial to have an in-depth understanding of the mechanistic aspects involved in each catalytic process. To this end, and inspired by our permanent interest in the use of computational methods for unveiling the main mechanistic aspects of organic transformations,24 specially those promoted by supported metal nanocatalysts,25 we present herein a computational theoretical study, based on DFT methods, to shed light on the high stereoselectivity observed in the hydrothiolation of activated alkynes with thiols bearing a catechol group catalysed by CuNPs/TiO2.
Computational methodology
The computational study was performed by using the ORCA software package.26 The density functional theory (DFT)27 energies were obtained with the PBE0 functional,28 applying the D3BJ Grimme's dispersion correction,29 which is known to be an appropriate methodology for similar mechanistic studies on alkyne hydrothiolation reactions22 as well as for the modelling of copper complexes,30 and the triple-ζ def2-TZVP basis set31 for all atoms. The energies in solution were obtained with the conductor-like polarizable continuum model (CPCM)32 using dichloromethane (DCM) as provided by the library of solvents of ORCA v. 4.2.1.Results and discussion
As we reported in our previous work,13 the TYC reaction between a series of thiols bearing a catechol group (1) and different activated alkynes with adjacent electron-withdrawing groups (2) catalysed by CuNPs/TiO2, led to the corresponding anti-Markovnikov vinyl sulphides (3) with high stereoselectivity towards the Z-isomer in most of the cases studied. The reaction conditions are summarised in Scheme 2, showing the reaction between 3-((4-mercaptobutyl)thio)benzene-1,2-diol (1) and propiolamide (2) as model starting compounds.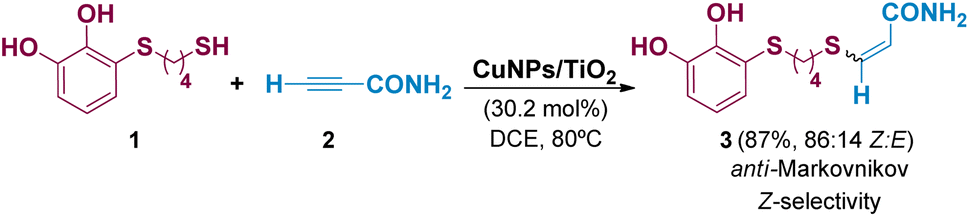 | ||
| Scheme 2 Reaction conditions for the synthesis of vinyl sulphides from activated alkynes and thiols bearing a catechol group. | ||
Although the exact mechanism involved for this reaction was difficult to ascertain, based on our previously reported results25 and those by other groups in the same area, and with the aid of some additional experiments, the formation of copper acetylides as reaction intermediates (deuterated-vinyl sulphide product was obtained when starting from 3-deuteriopropiolamide) and the participation of radical species (hydrothiolation was also efficient in the presence of TEMPO) could be disregarded.
It is worthy of note that the nature of the catalyst support proved to be essential for the hydrothiolation to take place efficiently, and consequently, it needs to be considered in any mechanistic proposal. As above mentioned, Lewis acidic (Ti4+) and/or Lewis basic (O2−) sites on the catalyst support could be acting as a fixer for the catechol moiety through coordination with the catechol hydroxyl groups (a and b in Fig. 1), while the basic sites could be also activating the thiol group (c in Fig. 1).
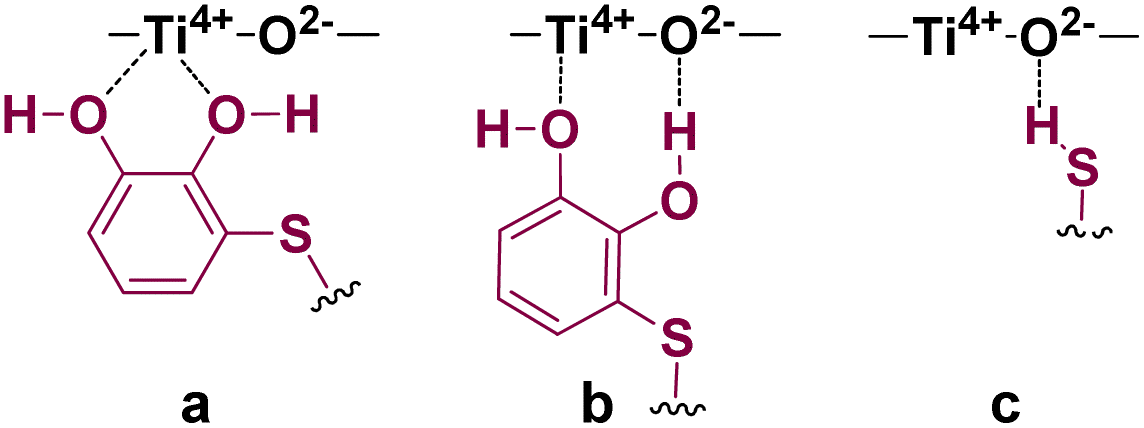 | ||
| Fig. 1 Possible interactions between thiol–catechol and TiO2 as catalyst support. | ||
In fact, other materials tested as catalyst supports such as cellulose, zeolite-Y, activated carbon and MCM-41 gave no conversion into the desired vinyl sulphide products, while lower conversion values were observed when ZnO, Al2O3, MgO were used (72%, 68% and 47% respectively, Table S1†). In line with the results previously reported by Kodomari,18 the use of Al2O3 as the catalyst support resulted in a remarkable selectivity towards the Z-isomer (95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, Table S1†).
:5, Table S1†).
In view of these observations, we proposed a plausible mechanistic pathway leading to the corresponding Z-vinyl sulphides, via a copper-catalysed anti-Markovnikov hydrothiolation process (Scheme 3), through an external nucleophilic attack from the thiol onto the carbon–carbon triple bond of the activated alkyne. Thus, we assume that both the alkyne and thiol are activated by copper, with the TiO2 support playing a non-innocent role in the catalytic process, probably through thiol S–H bond activation and by fixing the catechol moiety on the catalyst surface. As shown in Scheme 3, the protonated TiO2 would be responsible for the proton transfer step to give the final product and subsequent release of the catalyst to restart the catalytic cycle. In Scheme 3 the proposed structures for the reactive complex (RC), intermediates (IN), transition states (TS) and product complex (PC) are shown.
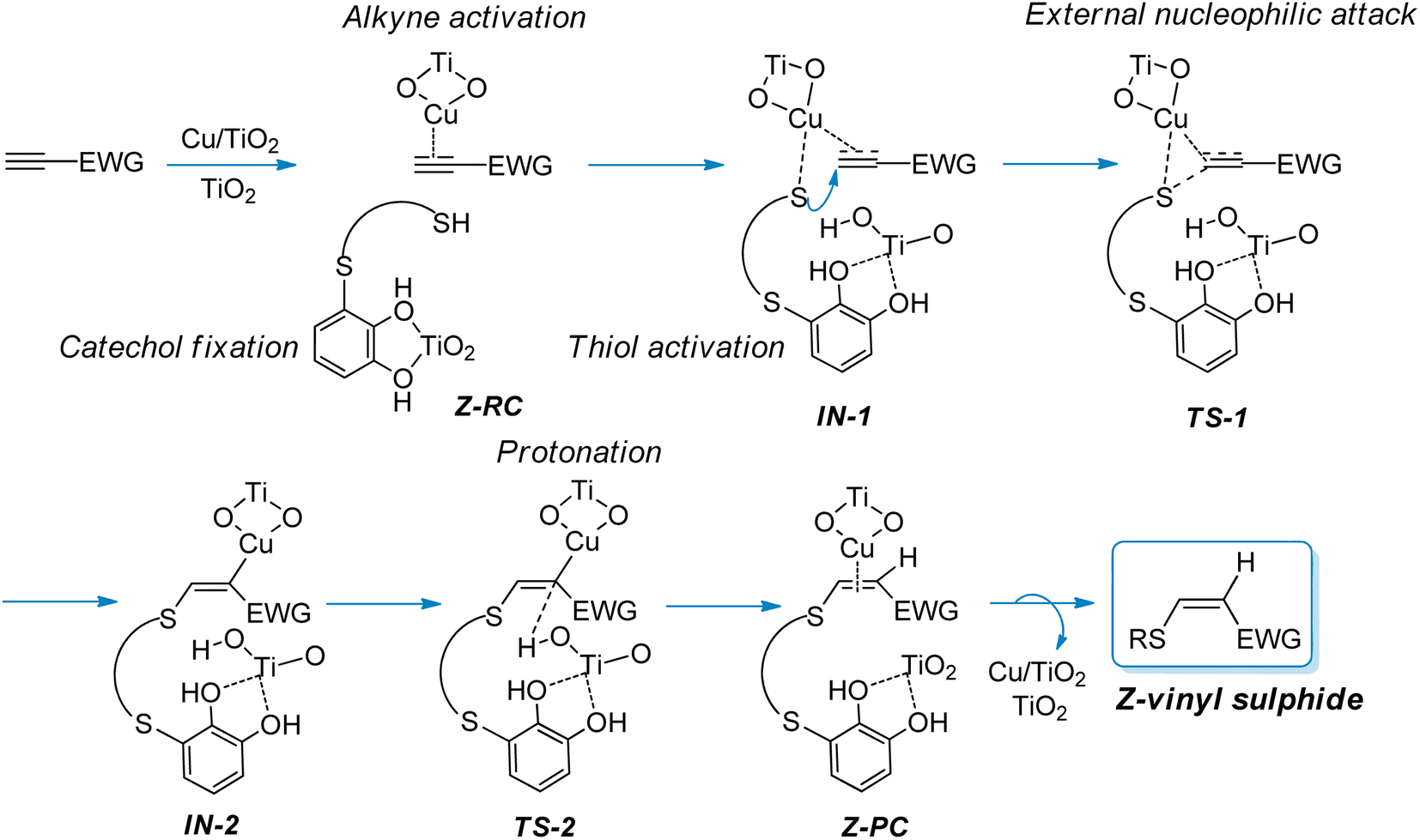 | ||
| Scheme 3 Proposed mechanistic pathway for the synthesis of Z-vinyl sulphides from activated alkynes and thiols bearing a catechol group catalysed by CuNPs/TiO2. | ||
With the aim of better understanding the experimental results and also to find additional information on the reaction mechanism, we performed a computational study by using 3-((4-mercaptobutyl)thio)benzene-1,2-diol and propiolamide as model starting materials. Since the reaction could be considered to start with the CuNPs/TiO2 nanocatalyst activating the alkyne and/or the thiol, it was necessary to establish the way in which the support surface could be attached to copper so as to stabilize the metal nanoparticles. Taking into account our previous results on computational calculations for a similar copper nanocatalyst,25 and assuming that copper would be attached to the support through both TiO2 oxygen atoms, we modelled different plausible structures for our CuNPs/TiO2 nanocatalyst. As can be seen from Fig. 2, this led us to propose four stable structures, by including different TiO2 monomeric (I) and dimeric species (II–IV), being the planar monomeric structure I less exothermic than the dimeric ones.
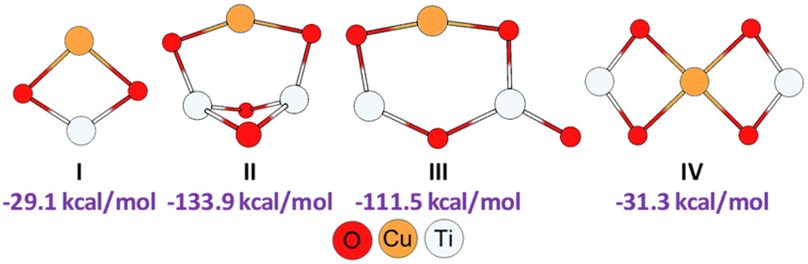 | ||
| Fig. 2 Geometries and formation energies for the different Cu/(TiO2)n (n = 1, 2) modelled structures (PBE0-D3BJ/Def2-TZVP/CPCM = DCM). | ||
Regarding the dimeric frameworks, planar structures III and IV were energetically less favourable than non-planar structure II, which resulted to be thermochemically more favoured, showing an exothermicity of −133.9 kcal mol−1.
As shown in Scheme 3, an external nucleophilic attack by a sulphur nucleophile onto the alkynyl copper complex, from the opposite side of the copper catalyst, would be the key step for the selective Z-vinyl sulphide formation. In order to facilitate the modelling of the stationary structures and to locate the transition states in a simpler way, we initially considered the formation of a reactive-like structure that we called reactive-complex (RC). In the Z-RC structure, the alkyne is being activated by copper, and the thiol–catechol is being fixed to the TiO2 support but is not yet coordinated with copper at this stage. For this purpose, we considered the formation of a π-complex between propiolamide and the structures I and II of the modelled nanocatalysts (Fig. 2), and we found that complex I-A was energetically more stable than structure II-A (−41.8 and −25.5 kcal mol−1 respectively, Fig. 3). Regarding the thiol–catechol, as discussed above, it would be fixed to the TiO2 support through coordination with the catechol hydroxyl groups, rendering the structure named S-T in Fig. 3, which was found to occur with an exothermicity of −24.8 kcal mol−1.
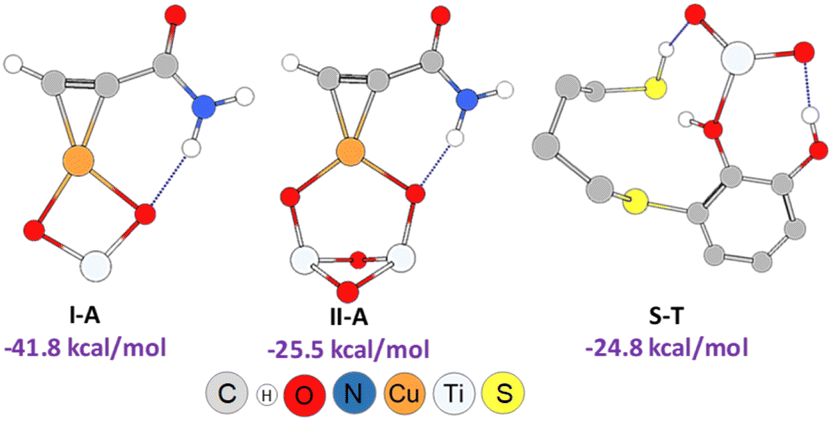 | ||
| Fig. 3 Geometries and formation energies for the I-A, II-A and S-T structures (PBE0-D3BJ/Def2-TZVP/CPCM = DCM). | ||
The Z-RC intermediate was then optimised (−110.3 kcal mol−1) showing strong interactions between thiol–catechol and the TiO2 support, as set out in Fig. 1b. Specifically, one of the O atoms of the catechol hydroxyl groups would be coordinating with a Ti atom in the support (2.17 Å), while the other hydroxyl group would be interacting through a hydrogen bond with one of the TiO2 oxygen atoms (1.45 Å). Stabilizing electrostatic interactions were also observed in all intermediates and transition states structures, both between the HNH2 atom of propiolamide and one of the TiO2 oxygen atoms (Z-RC, 1.86 Å), and between a Ti atom and the OC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O atom of the propiolamide (2.09 Å). By scanning the Sthiol–Cacetylenic reactive coordinate, starting from Z-RC, we found that thiol deprotonation (thiol activation) by Lewis basic sites of the support, would take place exothermically (−4.7 kcal mol−1) without any activation barrier, to give the intermediate IN-1 (Fig. 4), being this process also assisted by the formation of S–Cu bond (IN-1, 2.19 Å). All the mentioned interatomic distances are detailed in blue in Fig. 4.
O atom of the propiolamide (2.09 Å). By scanning the Sthiol–Cacetylenic reactive coordinate, starting from Z-RC, we found that thiol deprotonation (thiol activation) by Lewis basic sites of the support, would take place exothermically (−4.7 kcal mol−1) without any activation barrier, to give the intermediate IN-1 (Fig. 4), being this process also assisted by the formation of S–Cu bond (IN-1, 2.19 Å). All the mentioned interatomic distances are detailed in blue in Fig. 4.
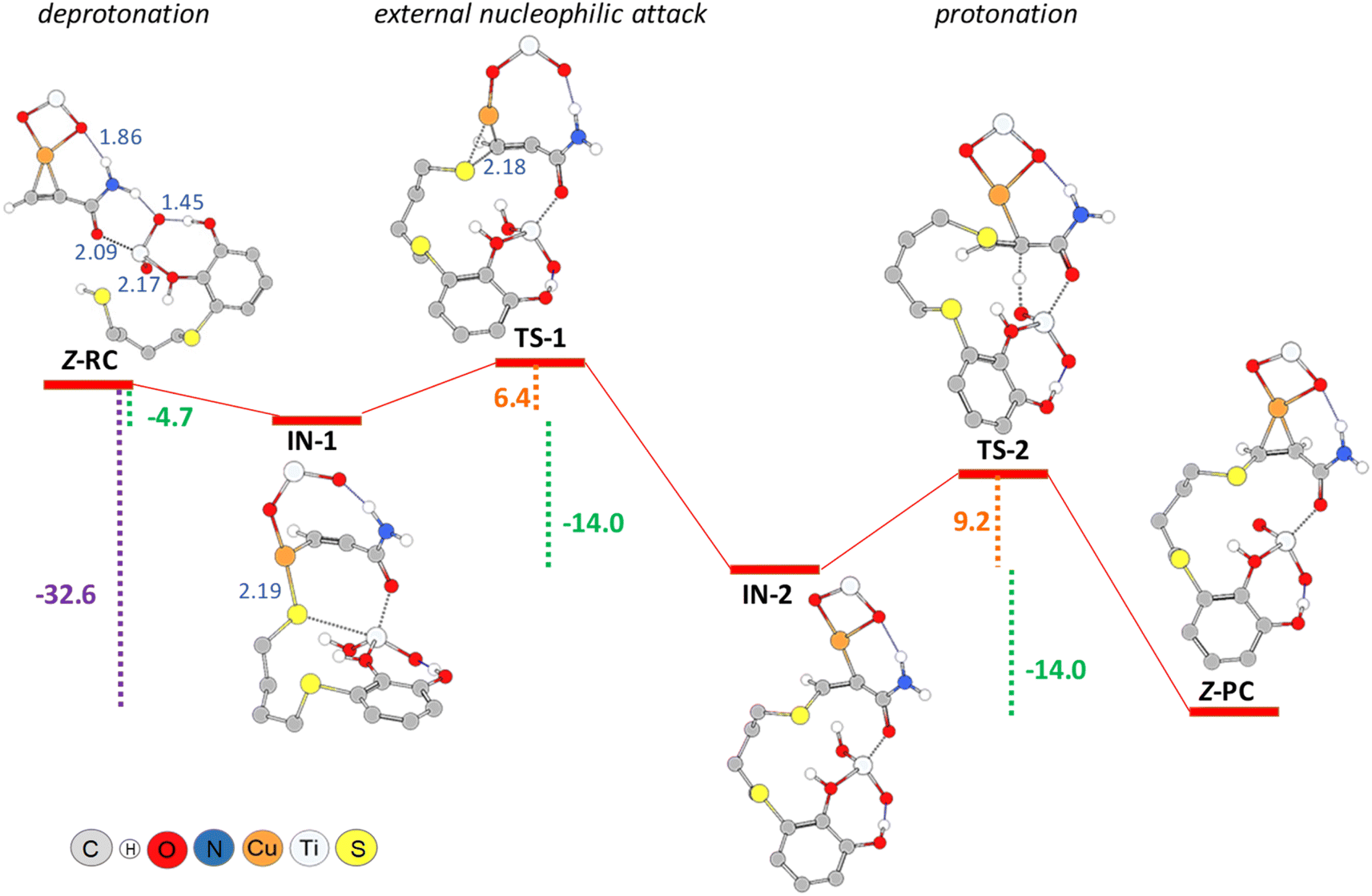 | ||
| Fig. 4 PBE0-D3BJ/Def2-TZVP/CPCM = DCM potential energy profile (kcal mol−1) for thiol deprotonation, nucleophilic addition and protonation steps in the formation of Z-vinyl sulphide, showing interatomic distances (Å, detailed in blue). | ||
In a subsequent step, through a nucleophilic addition, the S–C bond would be formed exothermically (−14.0 kcal mol−1) with very low activation energy (6.4 kcal mol−1), leading to the intermediate IN-2 through a cyclic three-membered (S–Cu–C) transition state (TS-1). For the sake of comparison, the same reaction step was modelled by using another widely known computational methodology for this kind of chemical reactive systems, such as the B3LYP functional33 and 6-311+G* basis set, and applying the same solvation model (CPCM = DCM). Thus, we found a relatively similar value for the activation energy (8.9 kcal mol−1) but the process showed to be much less exothermic (−7.1 kcal mol−1).
Turning again to the PBE0 functional method, we observed that the proton transfer step from TiO2–H species to give the Z-PC intermediate was kinetically less favourable, and proceeded with an activation energy of 9.2 kcal mol−1, thus being equally exothermic as the previous step (−14.0 kcal mol−1). It is important to highlight that the full process, from Z-RC to Z-PC, was exothermic in 32.6 kcal mol−1. Finally, both the active copper nanocatalyst and the support were regenerated, leading to the Z-vinyl sulphide product which showed a strong interaction between the O atom of the amide and one of the hydroxyl H atoms of catechol (Z-VS, 1.68 Å, Fig. 5).
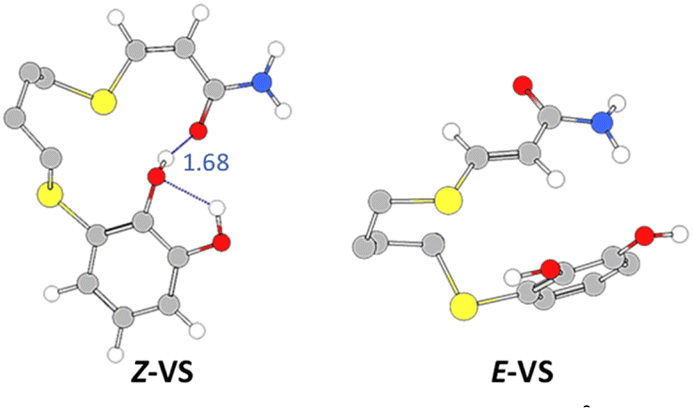 | ||
| Fig. 5 Geometries and interatomic distances (Å, detailed in blue) for the vinyl sulphide isomers (PBE0-D3BJ/Def2-TZVP/CPCM = DCM). | ||
On the other hand, taking into account that in the hydrothiolation of propiolamide the formation of minor amounts of the E-vinyl sulphide was experimentally observed during the hydrothiolation of propiolamide, we proposed that the formation of the E-isomer could proceed via the mechanism shown in Scheme 4. Since we considered that E-vinyl sulphide formation could proceed through an internal nucleophilic attack (from the same side of the copper catalyst) by a copper-coordinated sulphur nucleophile onto an alkynyl–copper complex, we employed the modelled structure II (Fig. 2) for the Cu/TiO2 nanocatalyst. In this case, as can be seen from E-RC structure, both the alkyne and thiol would be coordinated to copper, and the catechol hydroxyl groups would be fixed to the TiO2 support.
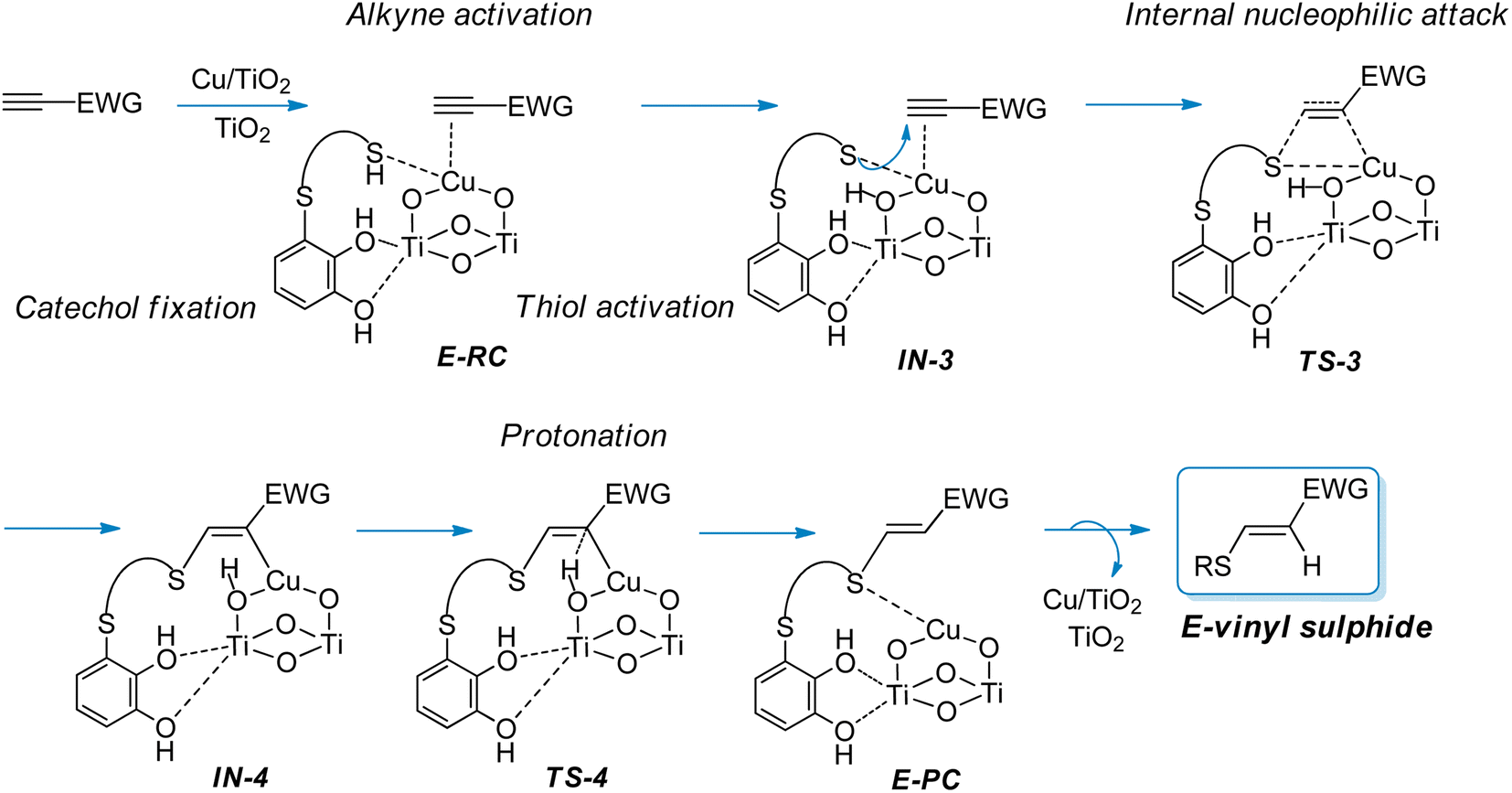 | ||
| Scheme 4 Proposed mechanistic pathway for the synthesis of E-vinyl sulphides from activated alkynes and thiols bearing a catechol group catalysed by CuNPs/TiO2. | ||
The DFT calculations showed that E-RC intermediate formation was much less exothermic (−57.1 kcal mol−1) than Z-RC, with both O atoms of the catechol hydroxyl groups coordinated to a Ti atom of the support (2.25 and 2.29 Å), and the SH atom coordinated with copper (2.35 Å), as represented in Fig. 1a and 6, respectively. As shown in Fig. 6, a stabilizing electrostatic interaction (E-RC, IN-3, TS-3, IN-4), between the HN atom of propiolamide and one of the O atoms of the TiO2 support (E-RC, 1.95 Å) was found.
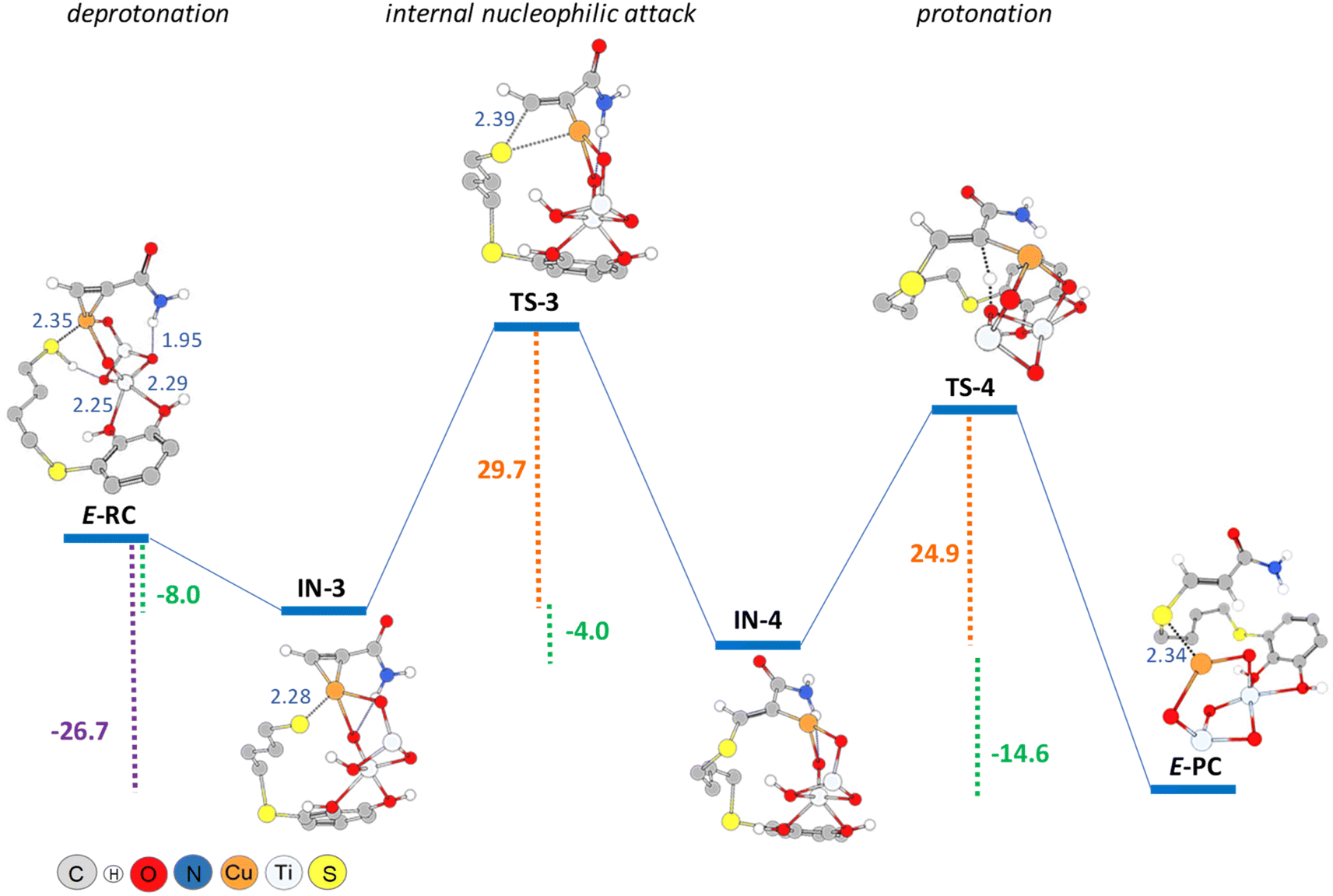 | ||
| Fig. 6 PBE0-D3BJ/Def2-TZVP/CPCM = DCM potential energy profile (kcal mol−1) for thiol deprotonation, nucleophilic addition and protonation steps in the formation of E-vinyl sulphide, showing interatomic distances (Å, detailed in blue). | ||
As in the case of the Z-isomer, here again thiol deprotonation was shown to be exothermic (−8.0 kcal mol−1) giving intermediate IN-3 without any activation barrier, and through a pathway calculated to be 3.3 kcal mol−1 more exothermic than the deprotonation of the Z-isomer.
When modelling the nucleophilic addition step, we observed that formation of the S–C bond leading to the intermediate IN-4, occurred with an activation energy of 29.7 kcal mol−1, that is more than 20 kcal mol−1 higher than that of the formation of IN-2 (Z-isomer). As shown in Fig. 6, intermediate IN-4 would be formed through a four-membered cyclic (C–S–Cu–C) transition state (TS-3), being also exothermic (−4.0 kcal mol−1) but significantly less than that of the formation of IN-2 (−14.0 kcal mol−1, Z-isomer). We consider that this could be probably due to the lack of coordination between titanium and the O atom of the amide group both in the corresponding intermediates and the transition states for the formation of the E-isomer. Another factor that could be energetically favouring the nucleophilic attack conducting to the Z-isomer, could be the distance between sulphur and carbon atoms in the corresponding transition states structures, which is 2.18 Å in TS-1 (Z-isomer) and 2.39 Å in TS-3 (E-isomer).
Here again, we also modelled this reaction step by employing B3LYP/6-311+G* methodology. Thus, we found a very similar value for the activation energy to that observed when modelled with PBE0 (29.2 kcal mol−1) the process showing to be less exothermic (−1.8 kcal mol−1).
As shown in Fig. 6, the last step involving the protonation from TiO2 support to give the E-PC intermediate, was observed to be kinetically similar (activation energy of 24.9 kcal mol−1) but much more exothermic (−14.6 kcal mol−1) than the previous step. The observed activation barrier was notably higher than that of the same proton transfer step found for the formation of Z-isomer (24.9 vs. 9.2 kcal mol−1). In the E-PC modelled structure, copper from the nanocatalyst would be coordinated to sulphur atom at an interatomic distance of 2.34 Å, leading to the E-vinyl sulphide product that has no stabilizing interactions (Fig. 5).
It is important to note that the full process, from E-RC to E-PC, was exothermic in 26.7 kcal mol−1, i.e. 6 kcal mol−1 less exothermic than that of the formation of the Z-isomer.
On the other hand, as previously reported by some of us,13 when we carried out the hydrothiolation reaction between the thiol–catechol 1 and dimethyl acetylenedicarboxylate, catalysed by CuNPs/TiO2 under the same reaction conditions, a near 1:1 ratio (52:48) of the Z- and E-vinyl sulphide isomers were obtained. In order to explain this lack of selectivity, we computationally modelled the proposed mechanisms for the nucleophilic addition step leading to the formation of the corresponding Z- and E-vinyl sulphides (see ESI Fig. S1 and S2† for the complete mechanisms).
In agreement with the experimental results, as can be seen from Fig. 7, the activation barriers for the nucleophilic attack resulted energetically comparable, 26.9 kcal mol−1 (Z-isomer) and 23.6 kcal mol−1 (E-isomer). In addition, transition state structures also show a similar sulphur–carbon distance, being 2.44 Å (TS-1′) and 2.52 Å (TS-3′), respectively. Although the mentioned reaction step is thermodynamically favoured for the formation of the E-isomer (−14.7 kcal mol−1 vs. −0.2 kcal mol−1), the subsequent protonation step is much more energetically demanding for this isomer (39.2 kcal mol−1, see Fig. S2,† formation of E-CP′ structure) than the protonation step for the formation of Z-CP′ structure (21.5 kcal mol−1, see Fig. S1†).
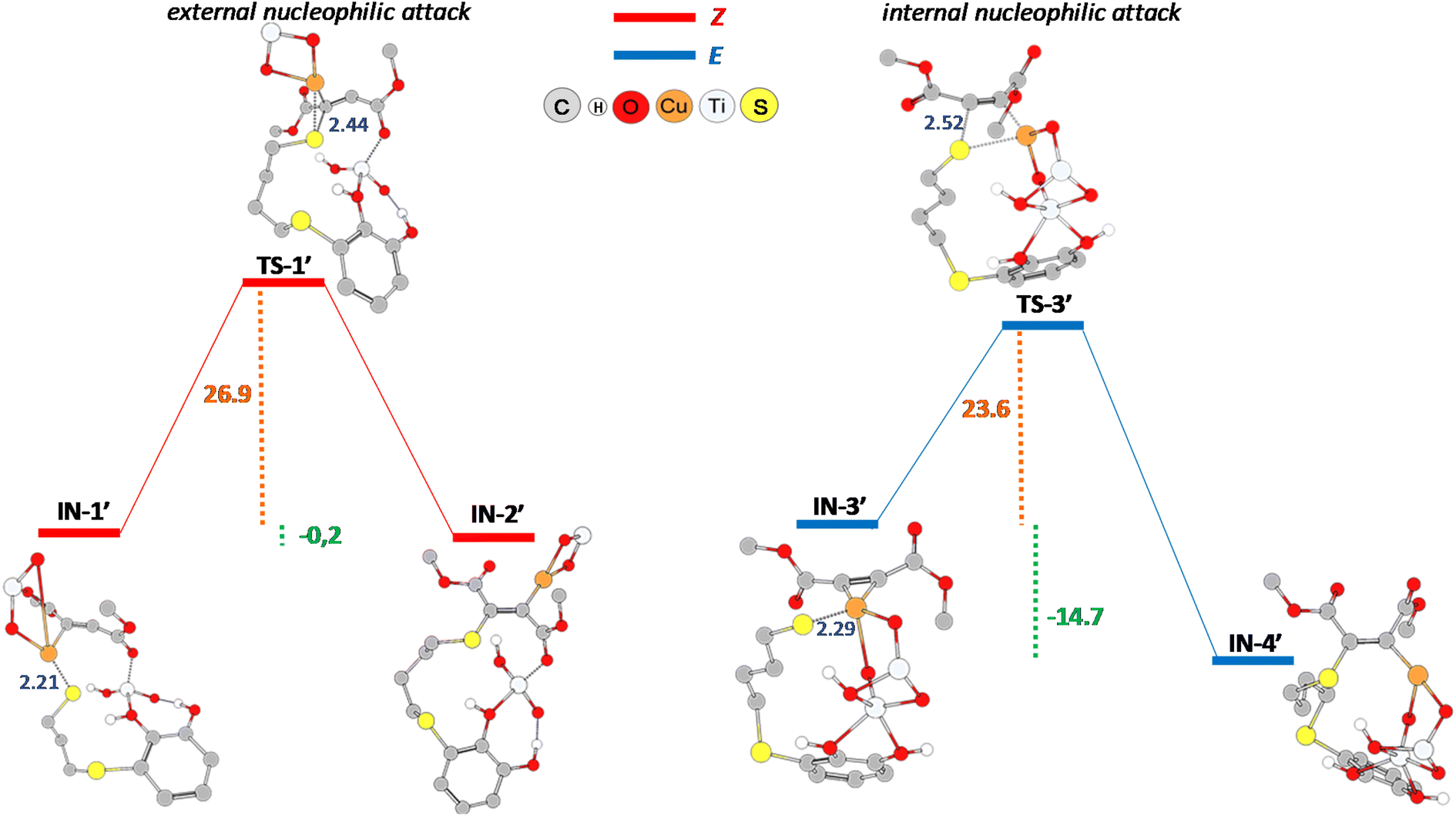 | ||
| Fig. 7 PBE0-D3BJ/Def2-TZVP/CPCM = DCM potential energy profile (kcal mol−1) for the nucleophilic addition step in the formation of Z- (red line) and E-vinyl sulphide (blue line) derived from thiol–catechol 1 and dimethyl acetylenedicarboxylate. Interatomic distances (Å) are detailed in blue. | ||
Finally, by comparing the nucleophilic addition steps leading to the formation of the corresponding Z-vinyl sulphides in the hydrothiolation of propiolamide (IN-1 to IN-2, Fig. 4) and dimethyl acetylenedicarboxylate (IN-1′ to IN-2′, Fig. 7), we found that the activation barrier for the former was notably lower (6.4 kcal mol−1 for TS-1 vs. 26.9 kcal mol−1 for TS-1′, see Fig. 4 and 7). This observation is in agreement with the shorter distance between sulphur and carbon atoms in TS-1 transition state structure (2.18 Å for TS-1 and 2.44 Å for TS-1′). Moreover, even when this reaction step was exothermic for both starting alkynes, the formation of IN-1 intermediate derived from propiolamide was exothermic by −14 kcal mol−1 (Fig. 4), while the formation of IN-1′ derived from dimethyl acetylenedicarboxylate was exothermic by only −0.2 kcal mol−1 (Fig. 7). We think that this could be related, to a some extent, to the lack of extra stabilizing electrostatic interactions (HNH2–OTiO2) as those observed for all the modelled structures in the formation of the Z-vinyl sulphide derived from the hydrothiolation of propiolamide.
Conclusions
In sum, based on experimental results and DFT studies, we presented a detailed reaction mechanism for the CuNPs/TiO2-catalysed hydrothiolation of activated alkynes with thiols bearing a catechol group, leading to the corresponding vinyl sulphides with high Z-stereoselectivity. We have suggested a plausible reaction mechanism that implies the activation of the carbon–carbon triple bond by coordination to a copper centre, followed by either an external or internal nucleophilic attack by the thiol–catechol, to give the corresponding Z or E isomer via a three membered cyclic S–Cu–C transition state or a four membered cyclic C–S–Cu–C transition state, respectively. It is important to mention that the computationally predicted selectivity for the reaction resulted higher than that observed experimentally, with both following the same trend. Besides, the proposed mechanism revealed the crucial synergistic role of the TiO2 support by: (i) fixing and activating the thiol–catechol, (ii) acting as a proton transfer agent in the deprotonation–protonation steps, and (iii) assisting in the control of stereoselectivity through a strong Ti–OCO coordination for the preferential formation of the Z-isomer.
Additionally, we proved that both functional PBE0 and B3LYP provided very similar and consistent energy profiles for the modelled reaction.
The mechanistic understanding of heterogeneous catalytic processes represents a big challenge and a need for both academic and industrial areas. As far as we know, this is the first computational mechanistic study about the alkyne hydrothiolation reaction by using thiol–catechols as functionalised nucleophiles. We consider that the comprehensive mechanistic knowledge obtained from our work could be of relevance as a contribution for further rational design of new and efficient copper-based catalytic systems for the selective construction of S–Csp2 bonds. The significance of the metal-catalysed hydrothiolation reaction in different fields, such as stereoselective organic synthesis, pharmaceuticals and functional materials, makes it essential to continue gaining better understanding of the mechanistic pathways involved, thus contributing to the rational development of improved catalytic systems.
Author contributions
VD carried out the conceptualization, investigation, methodology and writing. FN participated in the investigation, methodology and writing. MC participated in the methodology. GR carried out the funding acquisition, the project administration, investigation and writing. All authors read and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was generously supported by the Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET, PIP N°1665), Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT, PICT-2018-2471) and Universidad Nacional del Sur (UNS, PGI 24/Q106) from Argentina. MC thanks the ANPCyT for a doctoral fellowship.Notes and references
- K. Choudhuri, M. Pramanik, A. Mandal and P. Mal, Asian J. Org. Chem., 2018, 7, 1849–1855 CrossRef CAS.
- R. Castarlenas, A. Di Giuseppe, J. J. Pérez-Torrente and L. A. Oro, Angew. Chem., Int. Ed., 2013, 52, 211–222 CrossRef CAS PubMed.
- (a) M. Lo Conte, S. Pacifico, A. Chambery, A. Marra and A. Dondoni, J. Org. Chem., 2010, 75, 4644–4647 CrossRef CAS PubMed; (b) L. Benati, L. Capella, P. C. Montevecchi and P. Spagnolo, J. Chem. Soc., Perkin Trans. 1, 1995, 1035–1038 RSC.
- (a) S. Kanagasabapathy, A. Sudalai and B. C. Benicewicz, Tetrahedron Lett., 2001, 42, 3791–3794 CrossRef CAS; (b) C. G. Screttas and M. Micha-Screttas, J. Org. Chem., 1979, 44, 713–719 CrossRef CAS.
- (a) A. Kondoh, K. Takami, H. Yorimitsu and K. Oshima, J. Org. Chem., 2005, 70, 6468–6473 CrossRef CAS PubMed; (b) Z. L. Wang, R. Y. Tang, P. S. Luo, C. L. Deng, P. Zhong and J. H. Li, Tetrahedron, 2008, 64, 10670–10675 CrossRef CAS; (c) N. Zhao, C. Lin, L. Wen and Z. Li, Tetrahedron, 2019, 75, 3432–3440 CrossRef CAS.
- (a) S. N. Riduan, J. Y. Ying and Y. Zhang, Org. Lett., 2012, 14, 1780–1783 CrossRef CAS PubMed; (b) H. Ma, X. Ren, X. Zhou, C. Ma, Y. He and G. Huang, Tetrahedron Lett., 2015, 56, 6022–6029 CrossRef CAS.
- Y. Yang and R. M. Rioux, Chem. Commun., 2011, 47, 6557–6559 RSC.
- A. Dondoni and A. Marra, Eur. J. Org. Chem., 2014, 3955–3969 CrossRef CAS.
- (a) Z. Li, S. Zhao, Z. Wang, S. Zhang and J. Li, J. Hazard. Mater., 2020, 396, 122722 CrossRef CAS PubMed; (b) K. Wei, B. Senturk, M. T. Matter, X. Wu, I. K. Herrmann, M. Rottmar and C. Toncelli, ACS Appl. Mater. Interfaces, 2019, 11, 47707–47719 CrossRef CAS PubMed.
- (a) F. Nador, E. Guisasola, A. Baeza, M. A. Moreno-Villaécija, M. Vallet-Regí and D. Ruiz-Molina, Chem.–Eur. J., 2016, 23, 2753–2758 CrossRef PubMed; (b) J. Cui, Y. Yan, G. K. Such, K. Liang, C. J. Ochs, A. Postma and F. Caruso, Biomacromolecules, 2012, 13, 2225–2228 CrossRef CAS PubMed.
- (a) M. A. Moreno-Villaécija, J. Sedó-Vegara, E. Guisasola, A. Baeza, M. V. Regí, F. Nador and D. Ruiz-Molina, ACS Appl. Mater. Interfaces, 2018, 10, 7661–7669 CrossRef PubMed; (b) K. G. Malollari, P. Delparastan, C. Sobek, S. J. Vachhani, T. D. Fink, R. H. Zha and P. B. Messersmith, ACS Appl. Mater. Interfaces, 2019, 11, 43599–43607 CrossRef CAS PubMed.
- (a) F. Nador, F. Novio and D. Ruiz-Molina, Chem. Commun., 2014, 50, 14570–14572 RSC; (b) F. Nador, K. Wnuk, J. García-Pardo, J. Lorenzo, R. Solorzano, D. Ruiz-Molina and F. Novio, ChemNanoMat, 2018, 4, 183–193 CrossRef CAS; (c) J. García-Pardo, F. Novio, F. Nador, I. Cavaliere, S. Suárez-García, S. Lope-Piedrafita, A. P. Candiota, J. Romero-Gimenez, B. Rodríguez-Galván, J. Bové, M. Vila, J. Lorenzo and D. Ruiz-Molina, ACS Nano, 2021, 15, 8592–8609 CrossRef PubMed.
- F. Nador, J. Mancebo-Aracil, D. Zanotto, D. Ruiz-Molina and G. Radivoy, RSC Adv., 2021, 11, 2074–2082 RSC.
- (a) J. Mancebo-Aracil, C. Casagualda, M. A. Moreno-Villaécija, F. Nador, J. García-Pardo, A. Franconetti-García, F. Busqué, R. Alibés, M. J. Esplandiu, D. Ruiz-Molina and J. Sedó-Vegara, Chem.–Eur. J., 2019, 25, 12367–12379 CrossRef CAS PubMed; (b) J. Mancebo-Aracil, J. Sedó-Vegara and D. Ruiz-Molina, ICN2-CSIC, WO2019025498, 2019.
- I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2022, 122, 16110–16293 CrossRef CAS PubMed.
- S. N. Riduan, J. Y. Ying and Y. Zhang, Org. Lett., 2012, 14, 1780–1783 CrossRef CAS PubMed.
- I. G. Trostyanskaya and I. P. Beletskaya, Synlett, 2012, 23, 535–540 CrossRef CAS.
- M. Kodomari, G. Saitoh and S. Yoshitomi, Bull. Chem. Soc. Jpn., 1991, 64, 3485–3487 CrossRef.
- J. Saiz-Poseu, J. Mancebo-Aracil, F. Nador, F. Busqué and D. Ruiz-Molina, Angew. Chem., Int. Ed., 2019, 58, 696–714 CrossRef CAS PubMed and references therein..
- (a) U. Terranova and D. R. Bowler, J. Phys. Chem., 2010, 114, 6491–6495 CAS; (b) S. C. Li, J. G. Wang, P. Jacobson, X. Q. Gong, A. Selloni and U. Diebold, J. Am. Chem. Soc., 2009, 131, 980–984 CrossRef CAS PubMed.
- X. Zhang and K. Wang, RSC Adv., 2015, 5, 34439–34446 RSC.
- L. T. Sahharova, E. G. Gordeev, D. B. Eremin and V. P. Ananikov, ACS Catal., 2020, 10, 9872–9888 CrossRef CAS.
- T. Vishwanath and K. Balakrishna, J. Phys. Org. Chem., 2020, 33, e4040 Search PubMed.
- (a) V. B. Dorn, M. A. Badajoz, M. T. Lockhart, A. B. Chopa and A. B. Pierini, J. Organomet. Chem., 2008, 693, 2458–2462 CrossRef CAS; (b) M. E. Budén, V. B. Dorn, M. Gamba, A. B. Pierini and R. A. Rossi, J. Org. Chem., 2010, 75, 2206–2218 CrossRef PubMed; (c) F. Nador, Y. Moglie, A. Ciolino, A. Pierini, V. Dorn, M. Yus, F. Alonso and G. Radivoy, Tetrahedron Lett., 2012, 53, 3156–3160 CrossRef CAS; (d) V. B. Dorn, G. F. Silbestri, M. T. Lockhart, A. B. Chopa and A. B. Pierini, New J. Chem., 2013, 37, 1150–1156 RSC; (e) M. J. Lo Fiego, V. B. Dorn, M. A. Badajoz, M. T. Lockhart and A. B. Chopa, RSC Adv., 2014, 4, 49079–49085 RSC.
- L. Fortunato, Y. Moglie, V. Dorn and G. Radivoy, RSC Adv., 2017, 7, 18707–18713 RSC.
- (a) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS; (b) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2017, 8, e1327 Search PubMed.
- W. Kohn and I. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6169 CrossRef CAS.
- (a) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed; (b) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- B. Dereli, M. A. Ortuño and C. J. Cramer, ChemPhysChem, 2018, 19, 959–966 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem., 2005, 7, 3297–3305 CAS.
- V. Barone and M. J. Cossi, Phys. Chem., 1998, 102, 1995–2001 CAS.
- (a) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed; (b) A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed; (c) E. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Computational procedure, xyz coordinates and total energies in atomic units for all of the calculated structures. See DOI: https://doi.org/10.1039/d3ra00169e |
| This journal is © The Royal Society of Chemistry 2023 |