
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrgano-photocatalytic C–H bond oxidation: an operationally simple and scalable method to prepare ketones with ambient air†
Ky Nguyen*,
Van Nguyen,
Hieu Tran and
Phong Pham *
*
Faculty of Chemistry, University of Science, Vietnam National University, Hanoi, Vietnam. E-mail: phvpham@hus.edu.vn; nvknguyen.hus@gmail.com
First published on 6th March 2023
Abstract
Oxidative C–H functionalization with O2 is a sustainable strategy to convert feedstock-like chemicals into valuable products. Nevertheless, eco-friendly O2-utilizing chemical processes, which are scalable yet operationally simple, are challenging to develop. Here, we report our efforts, via organo-photocatalysis, in devising such protocols for catalytic C–H bond oxidation of alcohols and alkylbenzenes to ketones using ambient air as the oxidant. The protocols employed tetrabutylammonium anthraquinone-2-sulfonate as the organic photocatalyst which is readily available from a scalable ion exchange of inexpensive salts and is easy to separate from neutral organic products. Cobalt(II) acetylacetonate was found to be greatly instrumental to oxidation of alcohols and therefore was included as an additive in evaluating the alcohol scope. The protocols employed a nontoxic solvent, could accommodate a variety of functional groups, and were readily scaled to 500 mmol scale in a simple batch setting using round-bottom flasks and ambient air. A preliminary mechanistic study of C–H bond oxidation of alcohols supported the validity of one possible mechanistic pathway, nested in a more complex network of potential pathways, in which the anthraquinone form – the oxidized form – of the photocatalyst activates alcohols and the anthrahydroquinone form – the relevant reduced form of the photocatalyst – activates O2. A detailed mechanism, which reflected such a pathway and was consistent with previously accepted mechanisms, was proposed to account for formation of ketones from aerobic C–H bond oxidation of both alcohols and alkylbenzenes.
Introduction
Ketone is a chemical function group frequently found in bioactive molecules,1 and ketone-containing compounds are routinely employed as building blocks in syntheses of complex molecules.2 Among different means of preparation,3 C–H bond oxidation, converting alcohols and alkylbenzenes to corresponding ketones, offers a direct route towards this class of compounds. Despite successes of traditional stoichiometric oxidants,4,5 molecular oxygen (O2), because of its abundance, low cost, and environmentally benign nature, is preferred as a waste-free oxidant for C–H bond oxidation of alkylbenzenes and alcohols.6 However, due to the inertness of O2 towards closed-shell organic molecules and limited solubility of O2 in organic solvents under ambient pressure,7 a unified approach, capable of delivering scalable aerobic C–H bond oxidation in an operationally simple fashion such as batch setting with ambient air, is highly valuable but challenging to develop. Many thermal methods for C–H bond oxidation of alcohols and alkylbenzenes require pure O2 atmosphere or high pressure of inert-gas-diluted O2,8 elevated temperature,9 toxic/corrosive solvents,10 expensive transition metal catalysts,11 and prolonged reaction time12 which are undesirable from cost and safety considerations especially when large-scale applications are concerned (Fig. 1A).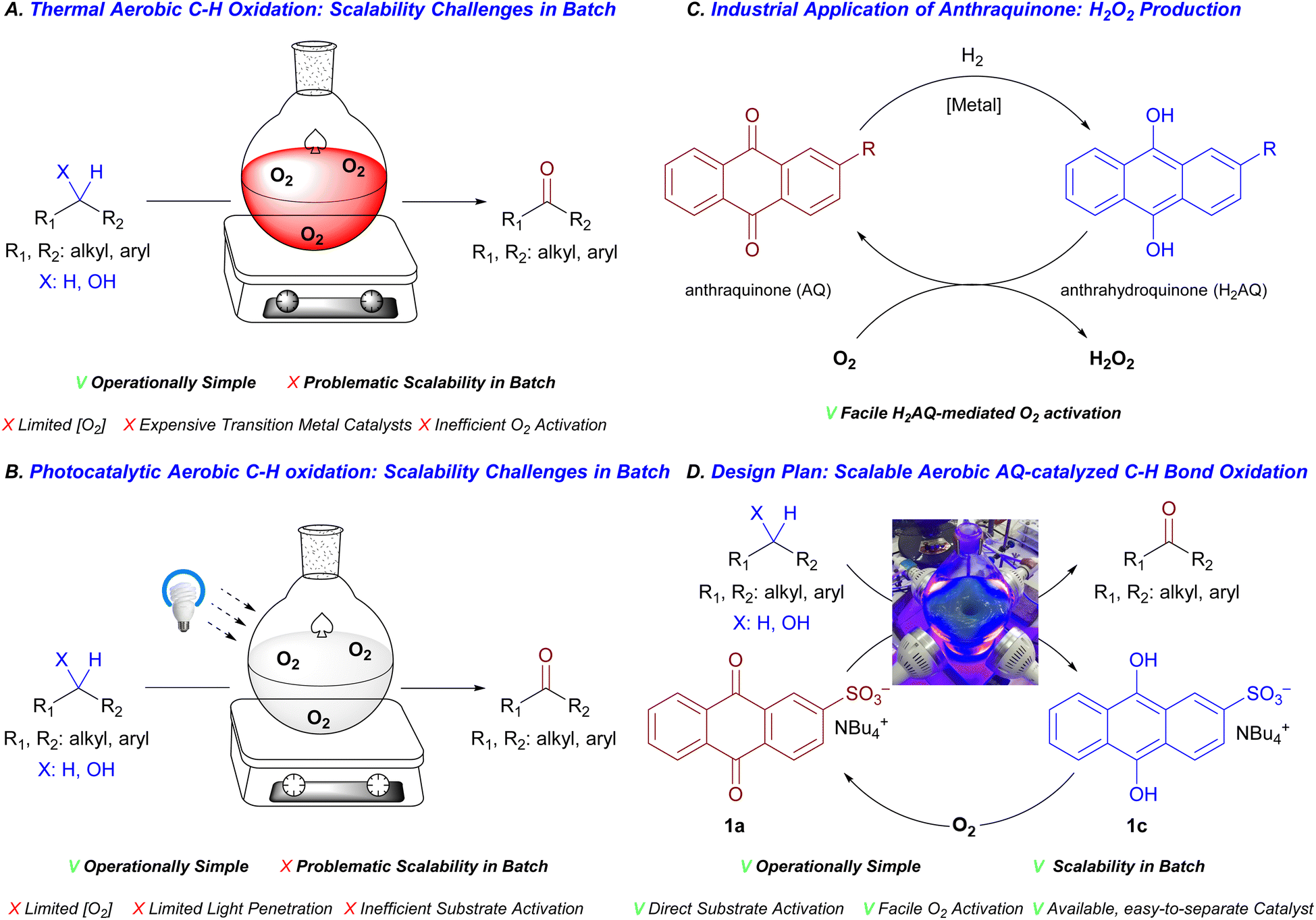 | ||
| Fig. 1 Challenges in developing scalable yet operationally simple protocols for catalytic C–H bond oxidation with ambient air. | ||
Over the last two decades, various photocatalytic modes of activation have emerged to activate otherwise unreactive molecules through intermediacy of C-centered organic radicals.13 By rendering organic molecules open-shell, energetic barriers was significantly lowered for O2 incorporation step into organic substrates.14 Many photocatalytic procedures, capable of activating C–H bonds in unactivated aliphatic alcohols and less electron-rich alkylbenzene derivatives, relied on indirect hydrogen atom transfer catalysis (HAT)15 due to inertness of the aforementioned substrates toward photo-induced electron transfer. In this activation mode, “secondary” reactive species, which activate C–H bonds of organic substrates such as singlet O2 or heteroatom-centered radicals, formed from O2 and radical precursors upon quenching a photocatalyst in its excited state.16 These redox cascades, despite being elegant and capable of effectively harnessing alcohols and alkylbenzenes into many chemical transformations, might be kinetically compromised from a low concentration of “secondary” reactive species, rendering substrate activation step inefficient. This, along with inherent challenges associated with an aerobic photochemical process such as light penetration17 and low dissolved oxygen concentration,7 might hamper the scalability of developed aerobic photocatalytic procedures (Fig. 1B). Indeed, scalable yet operationally simple procedures for aerobic C–H bond photo-oxidation of alcohols and alkylbenzenes are rarely reported.
We anticipated that an efficient catalytic approach for C–H bond photo-oxidation would require a facile direct hydrogen atom transfer (HAT) between the excited state of the photocatalyst and organic substrates. Accordingly, we expected that a strong thermodynamic driving force for HAT step would be necessary for efficient catalysis. In addition, an effective oxidative recycling of the photocatalyst by O2 is crucial to the overall success of the catalytic protocol. Anthraquinone (AQ) derivatives have been employed as mediators for industrial hydrogen peroxide production, which takes advantages of a spontaneous aerobic oxidation of anthrahydroquinone (H2AQ) – one reduced form of anthraquinone (Fig. 1C).18 Therefore, we envisioned that thanks to the facile redox reaction of H2AQ with O2, AQ derivatives, which are commercially and synthetically available with diverse substituents and substitution patterns,19 represent a rich pool of potential catalysts for oxidative transformations with O2. Furthermore, the triplet state of photoexcited AQ* possesses high triplet energy of 62.9 kcal mol−1 and therefore should have enough thermodynamic driving force for a hydrogen atom transfer with alcohols and alkylbenzenes.20,21 Therefore, in this study, we sought to develop scalable yet operationally simple protocols for aerobic C–H bond oxidation of alcohols and alkylbenzenes based on photocatalytic reactivity of anthraquinone derivatives (with an oversimplified design plan outlined tentatively in Fig. 1D).
Results and discussion
We initiated our study by subjecting different anthraquinone derivatives to catalytic oxidation of 4-tert-butylcyclohexanol (commercially available as mixture of cis and trans diastereomers) at 2 mmol scale using ambient air as the oxidant and acetone as the solvent (Table 1). To our delight, while catalytic oxidations mediated by AQ derivatives with strong electron-donating substituents provided diminished yields of ketones (Table 1, entries 1–3, 0–11% yield), those employed AQ derivatives with weak electron-donating or electron-withdrawing substituents afforded higher than 30% yield of ketone and with almost 100% selectivity (Table 1, entries 4–8, 34–54% yield). This was promising, given only 5 mol% of catalysts were used and samples were merely irradiated for 50 min. This result prompted us to try another cheap commercially available anthraquinone derivative, sodium anthraquinone-2-sulfonate monohydrate (2-AQ-SO3Na·H2O) which possesses a strong electron-withdrawing group, for catalytic oxidation. Nevertheless, due to low solubility of this salt in acetone, only 15% yield of ketone was achieved (Table 1, entry 9). By adding 5 mol% tetrabutylammonium chloride, NBu4Cl, as a phase transfer catalyst, the catalytic oxidation afforded 57% of ketone (Table 1, entry 10) which is in accordance with catalytic trend of other AQ derivatives (Table 1, entries 4–8). Independent ion exchange between 2-AQ-SO3Na·H2O and NBu4Cl (see ESI†) provided a previously undocumented compound, tetrabutylammonium anthraquinone-2-sulfonate (1a) (Fig. 1D), which was as effective at catalysing alcohol oxidation (Table 1, entry 11, 53% yield) as an equimolar mixture of 2-AQ-SO3Na·H2O and NBu4Cl (Table 1, entry 10, 57% yield). Among these three best catalytic systems 2-Me-AQ, an equimolar mixture of 2-AQ-SO3Na·H2O and NBu4Cl, and 1a (Table 1, entries 5, 10 and 11), we considered 1a to be most convenient for preparative synthesis for two reasons. First, 1a is easy to make in large scale, bench-stable, highly soluble, and non-hygroscopic so it should be easier to handle than an equimolar mixture of insoluble 2-AQ-SO3Na·H2O and a highly hygroscopic NBu4Cl salt in both small scale synthesis and upscale synthesis using flow reactors. Second, 1a is ionic and should be easy to separate from the neutral organic substrates and products by simple filtration through a silica gel plug, while neutral anthraquinone derivatives are notorious for low solubility in conventional organic solvents and might present a purification challenge when used in higher loadings.
|
|||
|---|---|---|---|
| Entry | AQ derivatives | Conversionb | Yieldb |
| a 2 mmol scale in 10 mL acetone, ambient air, with one 25 W 400–410 nm Lamp.b 1H NMR yield with 1,3,5-trimethoxybenzene as internal standard.c Prestir 30 min.d With 5 mol% NBu4Cl, prestir 30 min.e With 1 mol% Co(acac)2.f With 5 mol% NBu4Cl and 1 mol% Co(acac)2, prestir 30 min. | |||
| 1 | 2-NH2-AQ | 2% | 0% |
| 2 | 2-OH-AQ | 2% | 2% |
| 3 | 2-OMe-AQ | 12% | 11% |
| 4 | 2-t-Bu-AQ | 42% | 38% |
| 5 | 2-Me-AQ | 59% | 54% |
| 6 | AQ | 35% | 34% |
| 7 | 2-Cl-AQ | 35% | 34% |
| 8 | 2-AQ-COOH | 38% | 37% |
| 9 | 2-AQ-SO3Na·H2Oc | 18% | 15% |
| 10 | 2-AQ-SO3Na·H2Od | 59% | 57% |
| 11 | 1a | 54% | 53% |
| 12 | 1ae | 100% | 94% |
| 13 | 2-AQ-SO3Na·H2Of | 100% | 90% |

For the aforementioned reasons, 1a was employed for further optimization. We were pleased to find that an inclusion of merely 1 mol% of Co(acac)2 in catalytic oxidation with 1a gave full conversion of 4-tert-butylcyclohexanol and 94% of ketone (Table 1, entry 12). A similar effect of Co(acac)2 was also observed when an equimolar mixture of 2-AQ-SO3Na·H2O and NBu4Cl was employed to afford a comparable ketone yield of 90% (Table 1, entry 13).
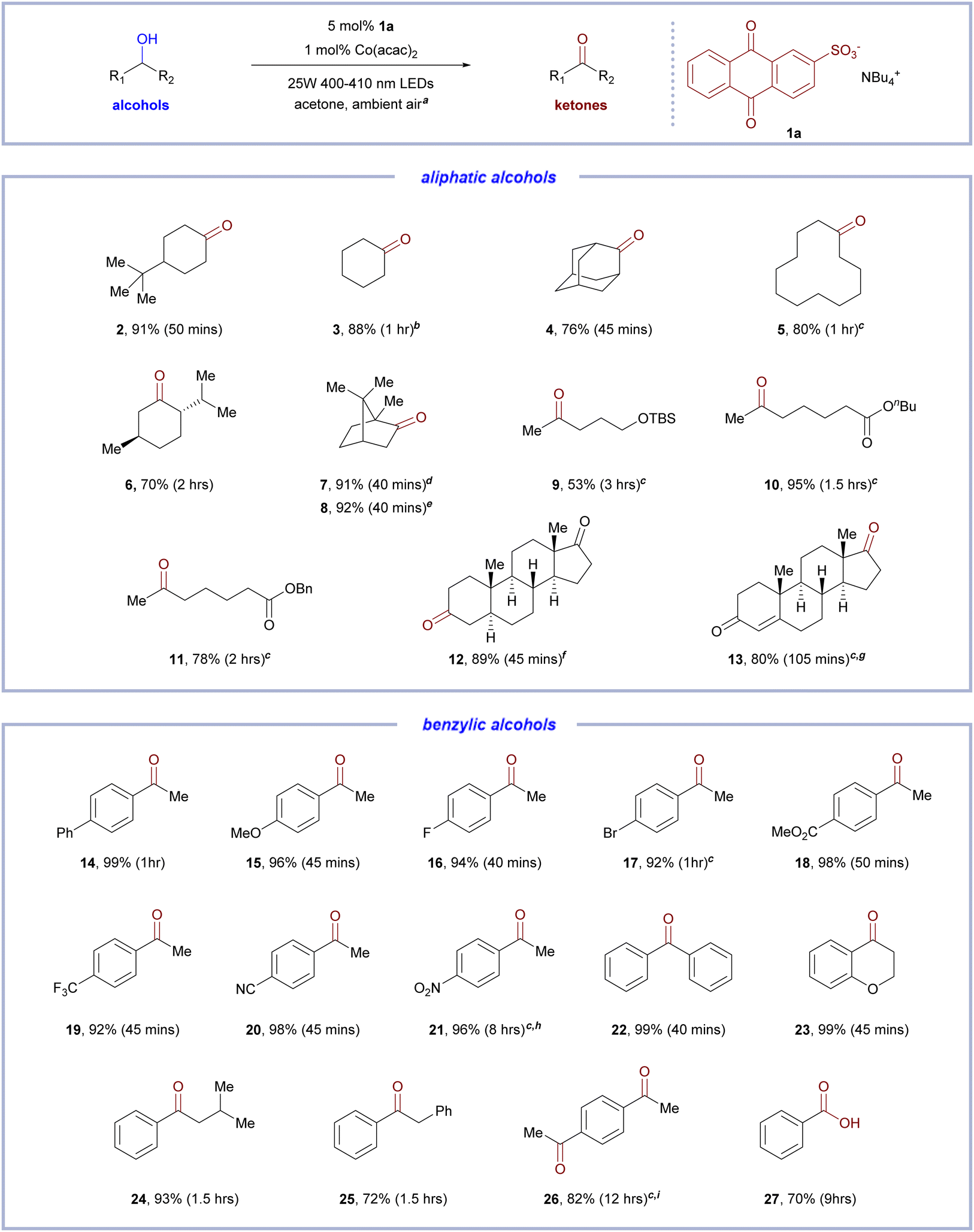
With the optimized condition in hands, we sought to explore the substrate scope of the developed C–H bond oxidation protocol for alcohol using 5 mol% of 1a and 1 mol% of Co(acac)2. Secondary unactivated aliphatic alcohols showed excellent reactivity under the optimized condition to afford ketones in good to excellent yields (Table 2). Catalytic oxidations were fast with virtually full conversion in 1–2 mmol scales within less than 2 h in all cases, even in challenging cases of sterically hindered alcohols (Table 2, entries 6–8, 13, 70–92% yield) which previously required pure O2 atmosphere and either prolonged irradiation or elevated temperature.8c,16a,22 A range of functional groups were well tolerated, including large-ring cyclic secondary alcohol (Table 2, entry 5, 80% yield), tertiary C–H bonds (Table 2, entries 2, 4, 6–8, 12 and 13, 76–92% yield), TBS-protected ether (Table 2, entry 9, 53% yield), ester (Table 2, entries 10 and 11, 78–95% yield), electronically deactivated benzylic C–H bond (Table 2, entry 11, 78% yield), ketone (Table 2, entry 12, 89% yield), and enone (Table 2, entry 13, 80% yield). After assessing the scope of aliphatic alcohols, a variety of secondary benzylic alcohols were subjected to the developed alcohol oxidation protocol. Secondary benzylic alcohols, bearing both electron-donating and electron-withdrawing substituents, was readily oxidized in excellent yields (>90%) in 1–2 mmol scale (Table 2, entries 14–20 and 22–23, 92–99% yield) with time to completion of less than 1 h. Nitro-group-bearing benzylic alcohol (Table 2, entry 21), which is electronically deactivated by a strong electron-withdrawing group and has a relatively colored ketone product, while proved to be challenging to other aerobic photo-oxidation,16a was amenable to our developed procedure to yield 96% of ketone. Secondary benzylic alcohol substrates with aromatic ether (Table 2, entry 23, 99% yield) and tertiary C–H bond (Table 2, entry 24, 94% yield) were well tolerated and afforded excellent yields of ketones. Benzylic alcohol substrates containing benzylic C–H bonds were more challenging with a diminished 72% yield of ketone (Table 2, entry 25), and the analogous oxidation of 1-(4-ethylphenyl)ethan-1-ol (Table 2, entry 26) provided 82% of 1,4-diacetylbenzene, where alcohol oxidation was concurrent with oxygenation of benzylic C–H bonds. As a representative example of primary benzylic alcohol (Table 2, entry 27), catalytic oxidation of benzyl alcohol resulted in a good 70% yield of benzoic acid in 2 mmol scale.
| a 2 mmol scale in 10 mL acetone with one 25 W 400–410 nm LEDs lamp.b In MeCN, isolated as 2,4-dinitrophenylhydrazone due to product volatility.c 1 mmol scale in 10 mL acetone.d From rac-borneol.e from rac-isoborneol.f 2 mmol scale in 15 mL acetone.g With 3 mol% Co(acac)2.h With two 25 W 400–410 nm LEDs Lamps.i From 1-(4-ethylphenyl)ethan-1-ol. All yield values are yields of the isolated compounds. |
|---|
 |
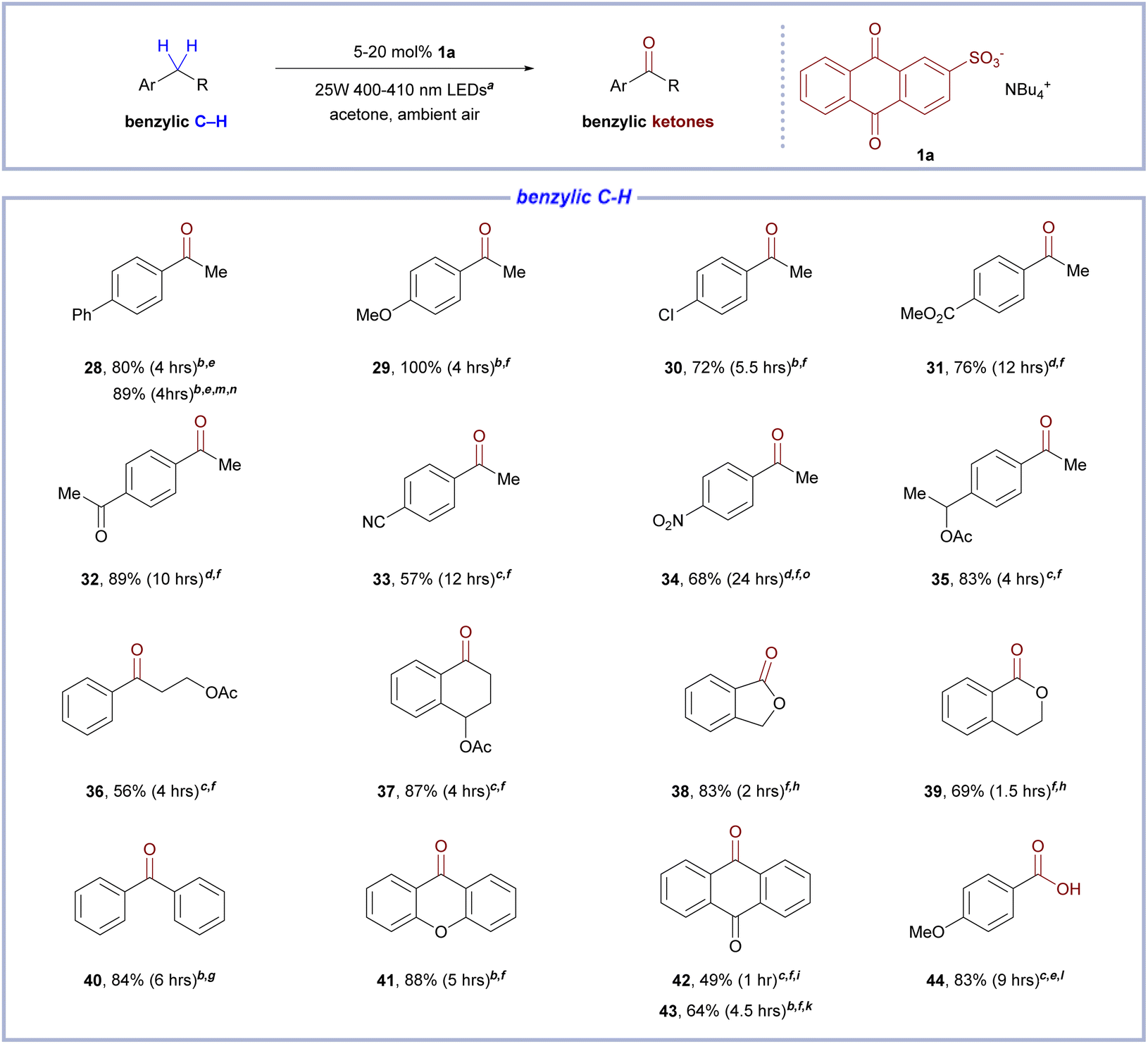
From the results of catalytic oxidation of 1-(4-ethylphenyl)ethan-1-ol (Table 2, entry 26), we noticed the reactivity of benzylic C–H bonds toward oxidation under the standard condition. Further control experiments suggested that while Co(acac)2 was still instrumental to photochemical aerobic benzylic oxidation, its effect in benzylic oxidation is less dramatic than that in alcohol oxidation (Table 3, entry 28). Therefore, we decided to explore the scope of alkylbenzene oxidation using a sole organocatalytic approach with 1a as the catalyst and no additives (Table 3). To our delight, ethylbenzene derivatives bearing a variety of functional groups were amenable to catalytic aerobic oxidation with 1a (Table 3, entries 28–34, 57–100% yield). Noticeably, ethylbenzene derivatives with electron-withdrawing groups, which were normally recalcitrant to or absent in scope of other aerobic benzylic photooxidation protocols,23 could be converted to ketones in average to good yields (Table 3, entries 30–34, 57–89% yield). Benzylic substrates with acetyl-protected benzylic alcohols (Table 3, entries 35 and 37, 83–87% yield) underwent aerobic oxidation smoothly to afford good yields of ketones, while oxidation of a substrate with acetyl-protected alcohol (Table 3, entry 36, 56% yield), which is positioned in close proximity of benzylic C–H bonds, was less effective. Using 2-methoxyanthraquinone (1b) as catalyst, oxygenation can be affected in substrates with benzylic C–H bonds that is at alpha position to aliphatic ether (Table 3, entries 38–39, 69–83% yield). Highly activated benzylic C–H substrates with extended conjugate systems in the ketone products were also amenable to catalytic oxidation with 1a (Table 3, entries 40–43, 49–88% yield). As a representative example of primary alkylbenzenes (Table 3, entry 44), catalytic oxidation of 4-methylanisole resulted in 83% yield of 4-methoxybenzoic acid in 2 mmol scale.
| a With one 25 W 400–410 nm LEDs lamp.b With 5 mol% 1a.c With 10 mol% 1a.d With 20 mol% 1a.e 2 mmol scale in 10 mL acetone.f 1 mmol scale in 10 mL acetone.g 2 mmol scale in 20 mL acetone.h With 5 mol% 2-OMe-AQ in place of 1a.i From anthrone.k From 9,10-dihydroanthracence.l From 4-methylanisole.m With 1 mol% Co(acac)2.n HPLC yield with 1,4-dimethoxybenzene as internal standard.o With two 25 W 400–410 nm LEDs lamps. All yield values are yields of the isolated compounds, unless noticed. |
|---|
 |
Mechanistic investigation
After evaluating the substrate scope, we sought to study the mechanism by which 1a catalyzes aerobic C–H bond oxidation of organic substrates. With alcohols as chosen substrates, we conducted a set of control experiments to evaluate the role of reaction variables (Table 4). A significant difference in yields between experiments under air (Table 4, entries 1 and 4, 53–94% yield) and experiments under Ar (Table 4, entries 6, 8 and 9, 5–7% yield) hinted at a critical role of O2 as the terminal oxidant. A control experiment without 1a (Table 4, entry 3, 0% yield) confirmed the necessity of 1a in the optimized procedure, and no-light experiments (Table 4, entries 2, 5, 7 and 10, 0% yield) confirmed an exclusive photochemical, not thermal, oxidation of alcohols under conditions of the developed protocol. Non-zero yields in control experiments under Ar (Table 4, entries 8 and 9, 5–7% yield) suggested that 1a in the presence of a light source can oxidize alcohols even in the absence of O2. Therefore, we investigated closely the direct photochemical reaction of 1a and alcohols.
|
|||
|---|---|---|---|
| Entry | Deviations | Conversionb | Yieldb |
| a 2 mmol scale in 10 mL acetone, ambient air, with one 25 W 400–410 nm LEDs lamp.b 1H NMR yield with 1,3,5-trimethoxybenzene as internal standard. | |||
| 1 | None | 100% | 94% |
| 2 | No light | 0% | 0% |
| 3 | No 1a | 0% | 0% |
| 4 | No Co(acac)2 | 54% | 53% |
| 5 | No light, no Co(acac)2 | 0% | 0% |
| 6 | Under Ar | 5% | 5% |
| 7 | Under Ar, no light | 1% | 0% |
| 8 | Under Ar, no Co(acac)2, 5 mol% 1a | 5% | 5% |
| 9 | Under Ar, no Co(acac)2, 10 mol% 1a | 8% | 7% |
| 10 | Under Ar, no Co(acac)2, no light, 10% 1a | 0% | 0% |

A stoichiometric photo-assisted reaction between 1a and cyclooctanol under inert atmosphere afforded tetrabutylammonium anthrahydroquinone-2-sulfonate (1c) as a yellow precipitate (Fig. 2), of which the structural assignment was consistent with 1H and 13C NMR data of the isolated solid (see ESI†). This corresponds to a formal reduction of AQ core by one alcohol molecule. It is worth mentioning that the Hollman group proposed a putative mechanism for aerobic oxidation with sodium anthraquinone-2-sulfonate in which the catalyst cycles between AQ form and 9,10-dihydroxy-9,10-dihydroanthracence form which corresponds to a formal reduction of AQ core by two alcohol molecules.21f We therefore studied the photochemical reaction between 1a and alcohols under a more catalytically relevant condition in order to address the following mechanistic questions:
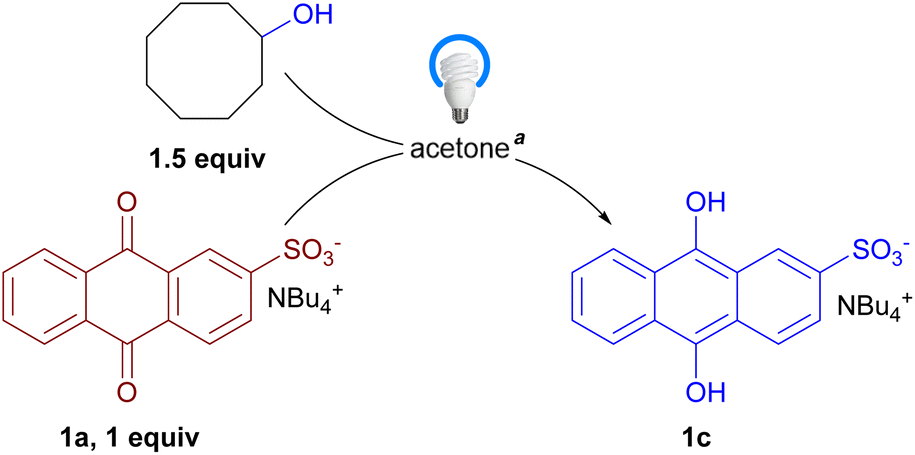 | ||
| Fig. 2 Photochemical stoichiometric reaction of 1a and cyclooctanol. a 0.4 mmol 1a and 0.6 mmol cyclooctanol in 6 mL acetone, under argon atmosphere, irradiated by one 25 W 400–410 nm LEDs lamp for 16 h. | ||
(1) Are 1a and alcohols cleanly converted to 1c and ketones, respectively, under a catalytically relevant condition?
(2) If 1c is formed, can 1c be cleanly oxidized to 1a by ambient air?
We anticipated that a qualitative test that could detect characteristic features of 1c and a quantitative test that correlated the amount of 1c and ketone formed over a range of reaction conversion would unambiguously address question 1. Specifically, a high fidelity of 1a-to-1c and alcohol-to-ketone conversion under catalytically relevant condition would predict a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of 1c and ketone formed over the course of the photo-oxidation under inert atmosphere.
:1 ratio of 1c and ketone formed over the course of the photo-oxidation under inert atmosphere.
Indeed, upon irradiating a 0.2 M acetone-d6 solution of cyclooctanol and 10 mol% 1a (Fig. 3A) under Ar and taking 1H NMR of the resultant mixture at indicated time points (Fig. 3B–D), a set of new signals in the aromatic region appeared that were consistent with the structural assignment of 1c.24 Also, relative intensity between peaks in this set of signals were essentially constant over the course of reaction (Fig. 3B–D) which confirmed that this set of signals indeed belonged to a single molecule and did not appear by chance. This set of signals co-evolved with a characteristic signal of cyclooctanone at δ = 2.34 ppm across reaction conversion and a 2:1 ratio of intensity between signals at δ = 2.34 ppm of cyclooctanone and δ = 7.40 ppm of 1c were maintained over the same set of experiments (Fig. 3B–D). This corresponded to 1:1 ratio of 1c and cyclooctanone formed across different reaction conversions. This observation supported a clean conversion of 1a to 1c that is concomitant with alcohol oxidation to ketones upon irradiation. Therefore, at least under argon atmosphere, photoirradiation of a mixture of 1a and alcohols resulted in a clean putative transfer of 2 hydrogen atoms from alcohols to AQ core to form ketone products and H2AQ core. A quick comparison of NMR data of the final reaction mixture upon exposure to air (Fig. 3E) and that of the initial mixture (Fig. 3A) confirmed a clean oxidation of 1c to 1a by ambient air.25 Overall, these observations supported one possible mechanistic pathway for alcohol oxidation in which 1a functions as an alcohol-activating photocatalyst and 1c, the reduced form of 1a, acts as a mediator for O2 activation (Fig. 4A and B). Based on these, we proposed a detailed mechanistic pathway that reflect both potential validity of the aforementioned mechanistic pathways and explanatory function of previously proposed mechanisms for aerobic C–H bond oxidation of both alcohols and alkylbenzenes with anthraquinone derivatives (Fig. 4).26
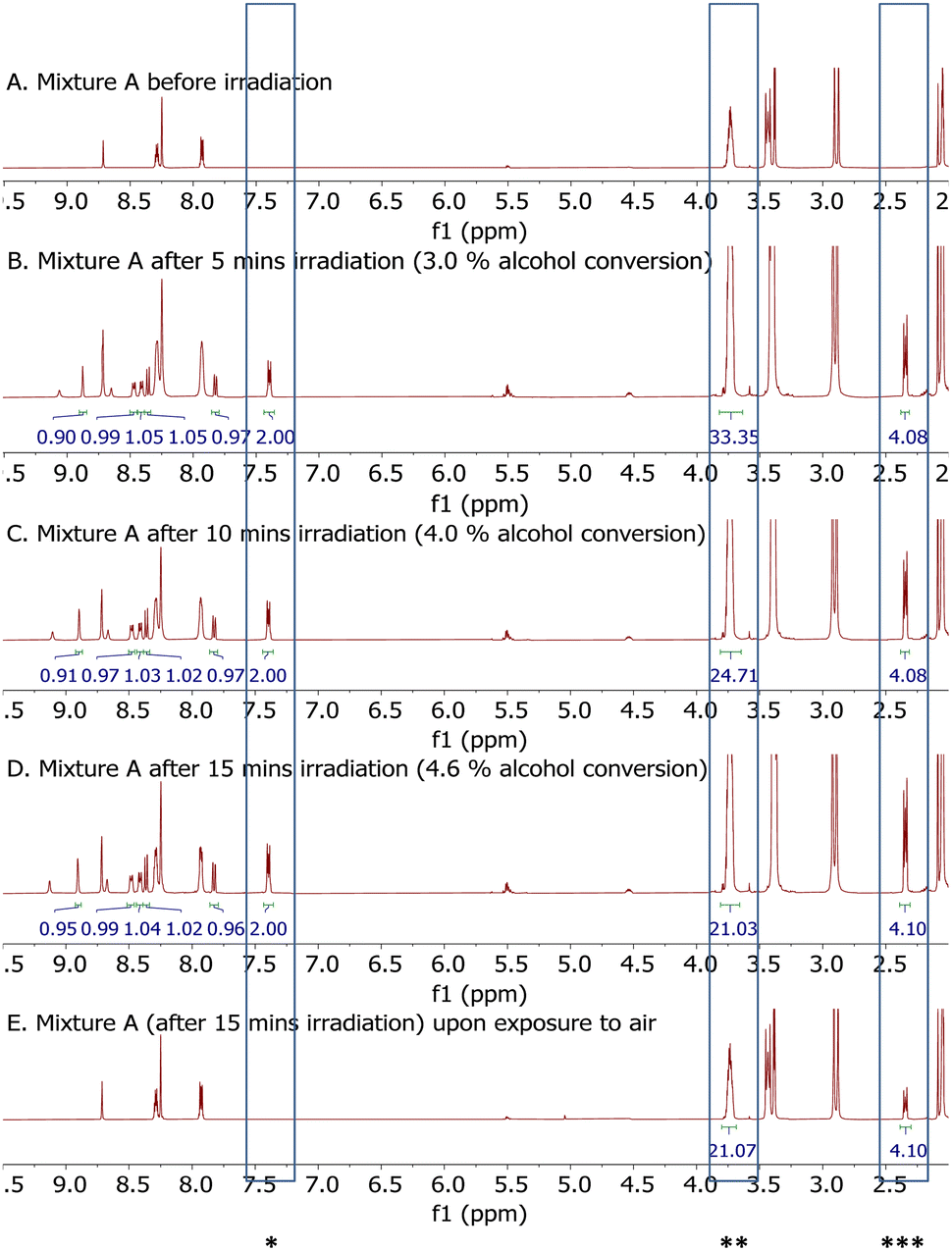 | ||
| Fig. 3 NMR study of a photochemical reaction between 1a and cyclooctanol. Condition: 0.016 mmol 1a (10 mol%) and 0.16 mmol cyclooctanol in 0.8 mL acetone-d6 (mixture A) in a NMR tube under argon atmosphere, irradiated by one 25 W 400–410 nm LEDs lamp (see ESI†). *1c; **cyclooctanol (CH(OH)); ***cyclooctanone (CH2–CO). | ||
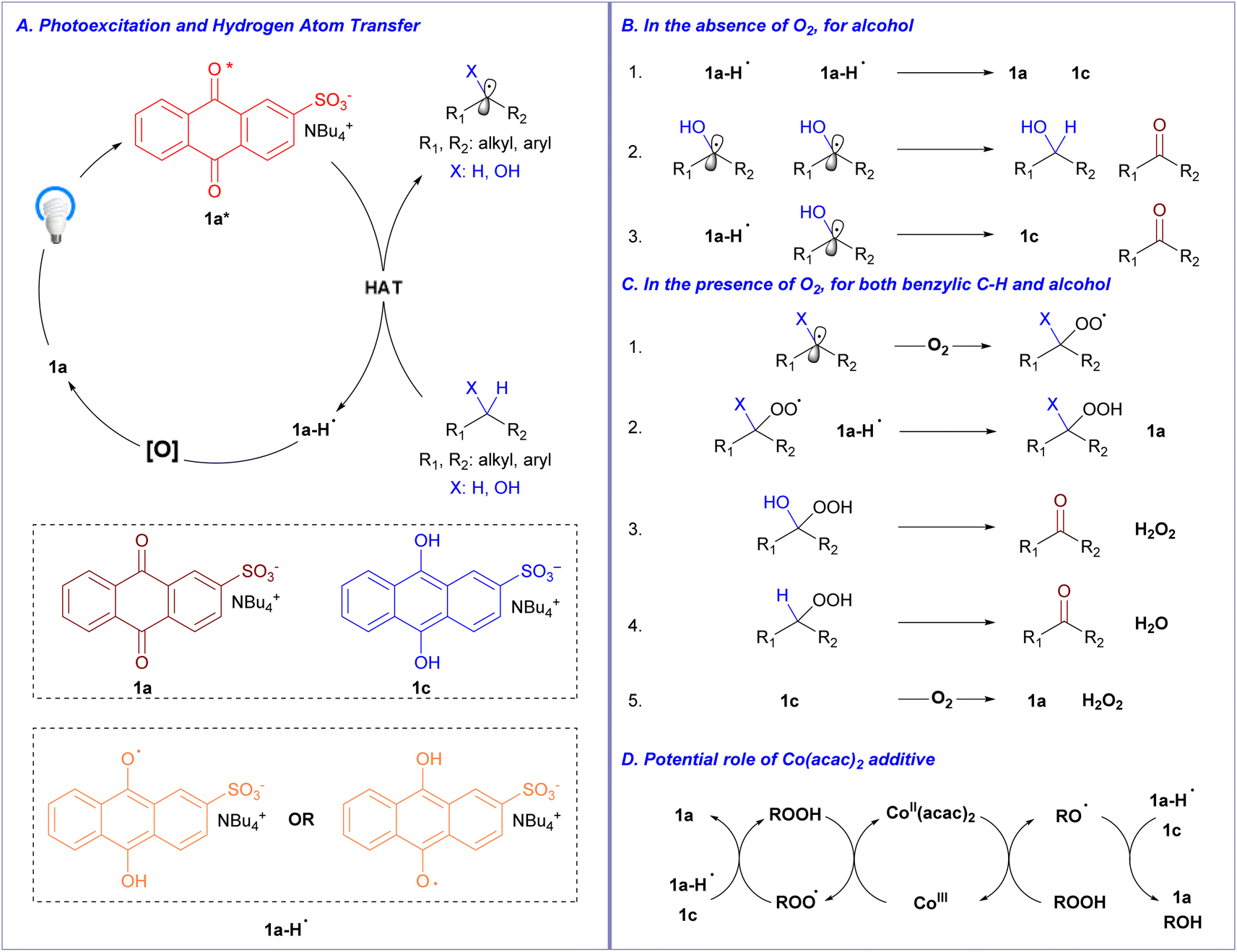 | ||
| Fig. 4 Proposed mechanism of aerobic C–H bond oxidation of alcohols and alkylbenzenes. | ||
Initially, 1a absorbs light and is excited, with intersystem crossing, to a triplet excited state 1a*. The triplet excited state 1a* might participate in a thermodynamically favorable hydrogen atom transfer with organic substrates (alcohols and alkylbenzenes) to yield the semi-anthrahydroquinone (1a-H˙) and a C-centered organic radical (Fig. 4A).20,27 Based on our study of photochemical reaction of 1a and alcohol under inert atmosphere (Fig. 3) which establish a clean 1:1 ratio of 1c and ketone formed upon irradiation, we proposed elementary steps in Fig. 4B to be operative in high fidelity, at least in the absence of O2 for oxidation of alcohols. The disproportionations of 1a-H˙ (Fig. 4B1) and ketyl radical (Fig. 4B2) yield 1c and ketone, respectively, and alternatively a second hydrogen atom transfer between 1a-H˙ and ketyl radical (Fig. 4B3) can give rise to 1c and ketone. In the presence of O2, we hypothesize a more complex set of elementary steps to be in operation (Fig. 4C),26 in addition to potentially operative elementary steps outlined in Fig. 4B. The C-centered organic radical, formed from HAT between 1a* and organic substrate (Fig. 4A), could trap O2 to yield organic peroxy radical (Fig. 4C1), which subsequently oxidizes the semi-anthrahydroquinone (1a-H˙) through hydrogen atom transfer to yield organic peroxide and give back 1a (Fig. 4C2). The decomposition of the resulting organic peroxide to ketone product might depend on the identity of the substituent X (Fig. 4C3 and C4), with expulsion of hydrogen peroxide if X is hydroxy group21a and with simple dehydration if X is hydrogen as in the case of benzylic C–H oxidation.21c Lastly, 1c, if formed when elementary steps in Fig. 4B are kinetically competitive, could be oxidized by O2 back to 1a and hydrogen peroxide (Fig. 4C5).
Regarding the role of Co(acac)2, based on reported works on interaction of Co(acac)2 with peroxides,28 it is proposed that Co(acac)2 assisted the decomposition of organic peroxides and/or hydrogen peroxide, which should be generated in higher amount in alcohol oxidation than in benzylic C–H oxidation, to produce more oxidizing equivalents that help recycle the catalyst in its oxidized form and accelerate the overall process (Fig. 4D). This might explain why the benefit of Co(acac)2 inclusion was much more pronounced in the case of alcohol oxidation where 1c might be formed through elementary steps of Fig. 4B and an assistance of Co(acac)2, which utilizes in situ formed peroxides to recycle 1a, might provide rate enhancement of the overall process.
Upscale oxidation
In order to evaluate the plausibility of the developed protocols for large-scale applications, we performed three experiments of gram-scale oxidations, all of which were conducted in simple batch setting using round-bottom flasks and ambient air as the oxidant (Fig. 5). Additionally, although the production of 1a is scalable and 1a is non-hygroscopic and easy to handle, we chose to further simplify the reaction setup, at least in batch setting, by employing a 1:1 combination of sodium anthraquinone-2-sulfonate monohydrate and tetrabutylammonium chloride in place of 1a. A 100 mmol-scale oxidation of 4-tert-butylcyclohexanol (commercially available as a mixture of cis and trans diastereomers) afforded 95% yield of 4-tert-butylcyclohexanone in less than 4 h (Fig. 5) which presented no drop in yield compared to a small scale synthesis (Table 2, entry 2, 91% yield). As an example of secondary benzylic alcohol, 1-phenylethanol could be effectively oxidized to acetophenone (Figure 5, 98% yield), at 500 mmol scale with time to completion of 11 h. It is worth mentioning that for this upscale experiment, we employed a lower catalyst/additive loading (2 mol% 2-AQ-SO3Na·H2O, 2 mol% NBu4Cl, and 0.4 mol% Co(acac)2) and the reaction was conducted at a more concentrated mixture (500 mmol alcohol in 1 L acetone), compared to our standard condition for benzylic alcohol oxidation (Table 2, entries 14–27). Finally, xanthene could be converted to xanthone at 10 mmol scale with excellent 91% yield (Fig. 5) within 2 h, which is comparable to reaction at 1 mmol scale (Table 3, entry 41, 88% yield). Since our developed procedures could accommodate upscale oxidation for three representative examples of aliphatic/benzylic alcohols and benzylic C–H bonds in simple batch setting with ambient air, we expect few hurdles exists in adopting the protocols in large-scale production, especially when a more sophisticated reaction setup such as flow chemistry is employed.
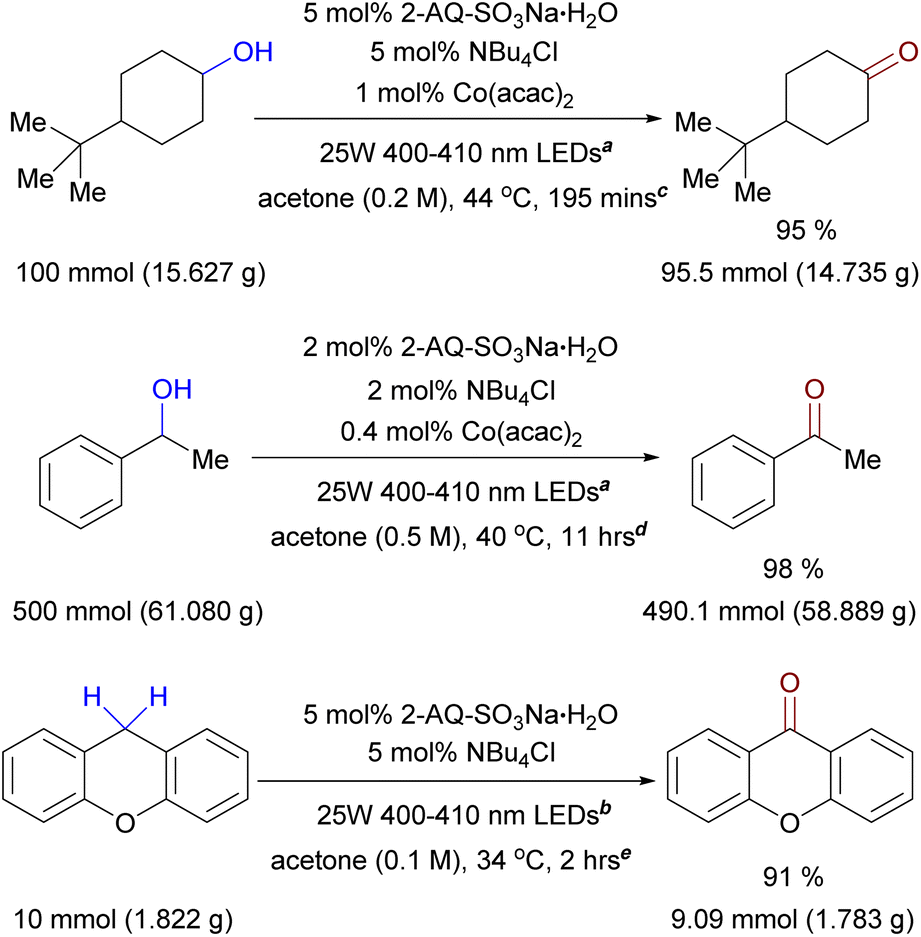 | ||
| Fig. 5 Representative upscale C–H bond oxidation of aliphatic alcohol, benzylic alcohol, and benzylic C–H. a With four 25 W 400–410 nm LEDs lamps; b with two 25 W 400–410 nm LEDs lamps; c in a 1 L round-bottom flask, open to air; d in a 2 L round-bottom flask, open to air; e in a 500 mL round-bottom flask, open to air; all yields values are yields of the isolated compounds. | ||
Conclusion
We described herein organo-photocatalytic protocols for aerobic C–H bond oxidation of alcohols and alkylbenzenes to corresponding ketones with ambient air as the source of oxidant. The protocols employed an easy-to-separate organic photocatalyst and were readily scalable even in simple batch setting with round-bottom flasks and ambient air. A preliminary investigation was conducted to study the role of the organic photocatalyst – tetrabutylammonium anthraquinone-2-sulfonate – in aerobic C–H bond oxidation. From this study, we found experimental evidences that support plausibility of one mechanistic pathway for alcohol oxidation in which the aromatic core of the catalyst cycles back and forth between anthraquinone form and anthrahydroquinone form, in addition to previously proposed mechanisms. The anthraquinone form acts as the substrate-activating photocatalyst and the anthrahydroquinone form activates O2. Combined together with previously reported mechanisms, a detailed mechanism was proposed to rationalize the formation of ketones from activation of both alcohol and benzylic C–H bond. We anticipate that the devised protocols will provide compelling alternatives to traditional methods for C–H bond oxidation of alcohols and alkylbenzenes. Furthermore, we expect anthraquinone derivatives will serve as a fruitful catalyst platform for other C–H functionalization procedures.Author contributions
K. V. N. conceived of the project, developed the idea, designed and performed experiments, and wrote the manuscript. V. T. N. and H. M. T. assisted with substrates preparation. P. V. P. acquired funding, supervised the project, and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is funded by Vietnam National University, Hanoi (VNU) under project number QG.21.06. The authors thank colleagues at the Faculty of Chemistry, University of Science, Vietnam National University, Hanoi; Prof. Binh Chu at Hanoi University of Science and Technology; Mr Dong Le at Military Technical Academy; Dr Trang Nguyen and Ms Hoa Phung at National Institute of Medicinal Materials for their kind supports.References
- P. Ertl, E. Altmann and J. F. McKenna, J. Med. Chem., 2020, 63, 8408–8418 CrossRef CAS PubMed.
- D. J. Foley and H. Waldmann, Chem. Soc. Rev., 2022, 51, 4094–4120 RSC.
- J. Cossy, Science of Synthesis: Houben-Weyl Methods of Molecular Transformations, Thieme Chemistry, 2004, vol. 26: Ketones Search PubMed.
- For stoichiometric oxidants for alcohol oxidation, see: (a) E. J. Corey and G. Schimidt, Tetrahedron Lett., 1979, 20, 399–402 CrossRef; (b) E. J. Corey and J. W. Suggs, Tetrahedron Lett., 1975, 16, 2647–2650 CrossRef; (c) D. B. Dess and J. C. Martin, J. Org. Chem., 1983, 48, 4155–4156 CrossRef CAS; (d) M. Uyanik and K. Ishihara, Chem. Commun., 2009, 16, 2086–2099 RSC; (e) A. J. Mancuso, S. Huang and D. Swern, J. Org. Chem., 1978, 43, 2480–2482 CrossRef CAS; (f) G. Tojo, Oxidation of Alcohols to Aldehydes and Ketones: A Guide to Current Common Practice, Springer Science, New York, 2006 Search PubMed.
- For stoichiometric oxidants for oxidation of alkylbenzenes, see: (a) A. Shaabani, A. Bazgir, F. Teimouri and D. G. Lee, Tetrahedron Lett., 2002, 43, 5165–5167 CrossRef CAS; (b) Y. Sarrafi, M. Tajbakhsh, R. Hosseinzadeh, M. Sadatshahabi and K. Alimohammadi, Synth. Commun., 2012, 42, 678–685 CrossRef CAS; (c) R. Hosseinzadeh, M. Tajbakhsh and H. Vahedi, Synlett, 2005, 18, 2769–2770 CrossRef; (d) K. K. Laali, M. Herbert, B. Cushnyr, A. Bhatt and D. Terrano, J. Chem. Soc., Perkin Trans. 1, 2001, 6, 578–583 RSC; (e) H. Firouzabadi, P. Salehi, A. R. Sardarian and M. Seddighi, Synth. Commun., 1991, 21, 1121–1127 CrossRef CAS; (f) K. C. Nicolaou, T. Montagnon, P. S. Baran and Y. L. Zhong, J. Am. Chem. Soc., 2002, 124, 2245–2258 CrossRef CAS PubMed; (g) T. Dohi, N. Takenaga, A. Goto, H. Fujioka and Y. Kita, J. Org. Chem., 2008, 73, 7365–7368 CrossRef CAS PubMed; (h) S. Kamijo, Y. Amaoka and M. Inoue, Synthesis, 2010, 14, 2475–2489 Search PubMed; (i) D. P. Lubov, O. Y. Lyakin, D. G. Samsonenko, T. V. Rybalova, E. P. Talsi and K. P. Bryliakov, Dalton Trans., 2020, 49, 11150–11156 RSC; (j) J. Zhang, Z. Wang, Y. Wang, C. Wan, X. Zheng and Z. Wang, Green Chem., 2009, 11, 1973–1978 RSC; (k) A. Alanthadka, E. S. Devi, S. Nagarajan, V. Sridharan, A. Suvitha and C. U. Maheswari, Eur. J. Org. Chem., 2016, 28, 4872–4880 CrossRef; (l) J. A. Marko, A. Durgham, S. L. Bretz and W. Liu, Chem. Commun., 2019, 55, 937–940 RSC; (m) T. Nagano and S. Kobayashi, Chem. Lett., 2008, 37, 1042–1043 CrossRef CAS; (n) A. J. Catino, J. M. Nichols, H. Choi, S. Gottipamula and M. P. Doyle, Org. Lett., 2005, 7, 5167–5170 CrossRef CAS PubMed; (o) J. J. Dong, D. Unjaroen, F. Mecozzi, E. C. Harvey, P. Saisaha, D. Pijper, J. W. de Boer, P. Alsters, B. L. Feringa and W. R. Browne, ChemSusChem, 2013, 6, 1774–1778 CrossRef CAS PubMed; (p) H. R. Mardani and H. Golchoubian, J. Mol. Catal. A: Chem., 2006, 259, 197–200 CrossRef CAS; (q) X.-F. Wu, Tetrahedron Lett., 2012, 53, 6123–6126 CrossRef CAS; (r) Y. Hu, L. Zhou and W. Lu, Synthesis, 2017, 49, 4007–4016 CrossRef CAS; (s) L. Yin, J. Wu, J. Xiao and S. Cao, Tetrahedron Lett., 2012, 53, 4418–4421 CrossRef CAS; (t) L. R. Ojha, S. Kudugunti, P. P. Maddukuri, A. Kommareddy, M. R. Gunna, P. Dokuparthi, H. B. Gottam, K. K. Botha, D. R. Parapati and T. K. Vinod, Synlett, 2009, 1, 117–121 Search PubMed; (u) P. Liu, Y. Liu, E. L. Wong, S. Xiang and C.-M. Che, Chem. Sci., 2011, 2, 2187–2195 RSC.
- M. G. Clerici and O. A. Khodeeva, Liquid Phase Oxidation via Heterogeneous Catalysis: Organic Synthesis and Industrial Applications, John Wiley & Sons, Inc., New Jersey, 2013 Search PubMed.
- (a) I. B. Golovanov and S. M. Zhenodarova, Russ. J. Gen. Chem., 2005, 75, 1795–1797 CrossRef CAS; (b) C. Franco and J. Olmsted III, Talanta, 1990, 37, 905–909 CrossRef CAS PubMed; (c) R. Battino, T. R. Rettich and T. Tominaga, J. Phys. Chem. Ref. Data, 1983, 12, 163–178 CrossRef CAS; (d) T. Sato, Y. Hamada, M. Sumikawa, S. Araki and H. Yamamoto, Ind. Eng. Chem. Res., 2014, 53, 19331–19337 CrossRef CAS.
- (a) T. Iwahama, Y. Yoshino, T. Keitoku, S. Sakaguchi and Y. Ishii, J. Org. Chem., 2000, 65, 6502–6507 CrossRef CAS PubMed; (b) I. E. Markó, A. Gautier, R. Dumeunier, K. Doda, F. Philippart, S. M. Brown and C. J. Urch, Angew. Chem., Int. Ed., 2004, 43, 1588–1591 CrossRef PubMed; (c) M. B. Lauber and S. S. Stahl, ACS Catal., 2013, 3, 2612–2616 CrossRef CAS; (d) T. Katsina, L. Clavier, J.-F. Giffard, N. M. P. da Silva, J. Fournier, R. Tamion, C. Copin, S. Arseniyadis and A. Jean, Org. Process Res. Dev., 2020, 24, 856–860 CrossRef CAS; (e) N. Jiang and A. J. Ragauskas, Org. Lett., 2005, 7, 3689–3692 CrossRef CAS PubMed; (f) X. Ye, M. D. Johnson, T. Diao, M. H. Yates and S. S. Stahl, Green Chem., 2012, 12, 1180–1186 RSC; (g) A. Dijksman, A. Marino-González, A. M. Payeras, I. W. C. E. Arends and R. A. Sheldon, J. Am. Chem. Soc., 2001, 123, 6826–6833 CrossRef CAS PubMed; (h) I. W. C. E. Arends, G.-J. ten Brink and R. A. Sheldon, J. Mol. Catal. A: Chem., 2006, 251, 246–254 CrossRef CAS; (i) S. Lee, S. A. Kim and H.-Y. Jang, ACS Omega, 2019, 4, 17934–17938 CrossRef CAS PubMed; (j) Z. Zhang, Y. Gao, Y. Liu, J. Li, H. Xie, H. Li and W. Wang, Org. Lett., 2015, 17, 5492–5495 CrossRef CAS PubMed; (k) R. D. Patil, B. Fuchs, N. Taha and Y. Sasson, ChemistrySelect, 2016, 1, 3791–3796 CrossRef CAS.
- (a) V. B. Sharma, S. L. Jain and B. Sain, Tetrahedron Lett., 2003, 44, 383–386 CrossRef CAS; (b) S. Velusamy and T. Punniyamurthy, Org. Lett., 2004, 6, 217–219 CrossRef CAS PubMed; (c) M. Lee and S. Chang, Tetrahedron Lett., 2000, 41, 7507–7510 CrossRef CAS; (d) V. R. Choudhary, A. Dhar, P. Jana, R. Jha and B. S. Uphade, Green Chem., 2005, 7, 768–770 RSC; (e) G. Urgoitia, A. Maiztegi, R. SanMartin, M. T. Herrero and E. Domínguez, RSC Adv., 2015, 5, 103210–103217 RSC; (f) G. Urgoitia, A. Maiztegi, R. SanMartin, M. T. Herrero and E. Domínguez, Green Chem., 2011, 13, 2161–2166 RSC.
- (a) S. Mozina and J. Iskra, J. Org. Chem., 2019, 84, 14579–14586 CrossRef CAS PubMed; (b) M. Shibuya, Y. Osada, Y. Sasano, M. Tomizawa and Y. Iwabuchi, J. Am. Chem. Soc., 2011, 133, 6497–6500 CrossRef CAS PubMed; (c) Y. Sasano, H. Sato, S. Tadokoro, M. Kozawa and Y. Iwabuchi, Org. Process Res. Dev., 2019, 4, 571–577 CrossRef; (d) E. Gaster, S. Kozuch and D. Pappo, Angew. Chem., Int. Ed., 2017, 56, 5912–5915 CrossRef CAS PubMed; (e) Y. Yoshino, Y. Hayashi, T. Iwahama, S. Sakaguchi and Y. Ishii, J. Org. Chem., 1997, 62, 6810–6813 CrossRef CAS.
- (a) B. Guan, D. Xing, G. Cai, X. Wan, N. Yu, Z. Fang, L. Yang and Z. Zhi, J. Am. Chem. Soc., 2005, 127, 18004–18005 CrossRef CAS PubMed; (b) T. Iwasawa, M. Tokunaga, Y. Obora and Y. Tsuji, J. Am. Chem. Soc., 2004, 126, 6554–6555 CrossRef CAS PubMed; (c) D. R. Jensen, M. J. Schultz, J. A. Mueller and M. S. Sigman, Angew. Chem., Int. Ed., 2003, 42, 3810–3813 CrossRef CAS PubMed; (d) M. Shibuya, S. Nagasawa, Y. Osada and Y. Iwabuchi, J. Org. Chem., 2014, 79, 10256–10268 CrossRef CAS PubMed; (e) A. Abad, C. Almela, A. Corma and H. García, Tetrahedron, 2006, 62, 6666–6672 CrossRef CAS; (f) Y. Kadoh, K. Oisaki and M. Kanai, Chem. Pharm. Bull., 2016, 64, 737–753 CrossRef CAS PubMed.
- (a) X. Liu, Q. Xia, Y. Zhang, W. Chen and C. Chen, J. Org. Chem., 2013, 78, 8531–8536 CrossRef CAS PubMed; (b) A. Dhakshinamoorthy, M. Alvaro and H. Garcia, ACS Catal., 2011, 1, 48–53 CrossRef CAS; (c) H. Yu, J. Ren, Y. Xie, X. Su, A. Wang, L. Yan, F. Jiang and Y. Wei, Green Chem., 2022, 24, 6511–6516 RSC; (d) Q. Zhu, S. Yin, M. Zhou, J. Wang, C. Chen, P. Hu, X. Jiang, Z. Zhang, Y. Li and W. Ueda, ChemCatChem, 2021, 13, 1763–1771 CrossRef CAS; (e) N. Nagarjun and A. Dhakshinamoorthy, ChemistrySelect, 2018, 3, 12155–12162 CrossRef CAS.
- (a) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed; (b) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed; (c) L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2022, 122, 1875–1924 CrossRef CAS PubMed; (d) L. Capaldo and D. Ravelli, Eur. J. Org. Chem., 2017, 15, 2056–2071 CrossRef PubMed; (e) A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, D. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS PubMed; (f) P. R. D. Murray, J. H. Cox, N. D. Chiappini, C. B. Roos, E. A. McLoughlin, B. G. Hejna, S. T. Nguyen, H. H. Ripberger, J. M. Ganley, E. Tsui, N. Y. Shin, B. Koronkiewicz, G. Qiu and R. R. Knowles, Chem. Rev., 2022, 122, 2017–2291 CrossRef CAS PubMed; (g) F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC; (h) F. Juliá, T. Constantin and D. Leonori, Chem. Rev., 2022, 122, 2292–2352 CrossRef PubMed; (i) F. Juliá, ChemCatChem, 2022, 14, e202200916 CrossRef; (j) J. Liu, L. Lu, D. Wood and S. Lin, ACS Cent. Sci., 2020, 6, 1317–1340 CrossRef CAS PubMed.
- (a) Y. Zhang, W. Schilling and S. Das, ChemSusChem, 2019, 12, 2898–2910 CrossRef CAS PubMed; (b) X. Zhang, K. P. Rakesh, L. Ravindar and H.-L. Qin, Green Chem., 2018, 20, 4790–4833 RSC.
- For review of HAT catalysis, see: (a) L. Capaldo and D. Ravelli, Eur. J. Org. Chem., 2017, 15, 2056–2071 CrossRef PubMed; (b) L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2022, 122, 1875–1924 CrossRef CAS PubMed; (c) H. Cao, X. Tang, H. Tang, Y. Yuan and J. Wu, Chem Catal., 2021, 1, 523–598 CrossRef CAS.
- (a) N. F. Nikitas, D. I. Tzaras, I. Triandafillidi and C. G. Kokotos, Green Chem., 2020, 22, 471–477 RSC; (b) J. L. Jeffrey, J. A. Terrett and D. W. C. MacMillan, Science, 2015, 349, 1532–1536 CrossRef CAS PubMed; (c) J. Twilton, M. Christensen, D. A. DiRocco, R. T. Ruck, I. W. Davies and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2018, 57, 1–6 CrossRef PubMed; (d) K. Sakai, K. Oisaki and M. Kanai, Adv. Synth. Catal., 2020, 362, 337–343 CrossRef CAS; (e) K. Merkens, N. Sanosa, I. Funes-Ardoiz and A. Gosmez-Suárez, ACS Catal., 2022, 12, 13186–13192 CrossRef CAS; (f) V. Dimakos, H. Y. Su, G. E. Garrett and M. S. Taylor, J. Am. Chem. Soc., 2019, 141, 5149–5153 CrossRef CAS PubMed; (g) M. Zidan, A. O. Morris, T. McCallum and L. Barriault, Eur. J. Org. Chem., 2020, 10, 1453–1458 CrossRef; (h) S. Rohe, A. O. Morris, T. McCallum and L. Barriault, Angew. Chem., Int. Ed, 2018, 57, 15664–15669 CrossRef CAS PubMed; (i) H. Fuse, H. Mitsunuma and M. Kanai, J. Am. Chem. Soc., 2020, 142, 4493–4499 CrossRef CAS PubMed; (j) J. Jin and D. W. C. MacMillan, Nature, 2015, 525, 87–90 CrossRef CAS PubMed; (k) L.-M. Zhao, Q.-Y. Meng, X.-B. Fan, C. Ye, X.-B. Li, B. Chen, V. Ramamurthy, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed, 2017, 56, 3020–3024 CrossRef CAS PubMed; (l) W. Zhang, K. L. Carpenter and S. Lin, Angew. Chem., Int. Ed., 2020, 59, 409–417 CrossRef CAS PubMed; (m) T. Hering, T. Slanina, A. Hancock, U. Wille and B. Konig, Chem. Commun., 2015, 51, 6568–6571 RSC; (n) M. Xiang, Z.-K. Xin, B. Chen, C.-H. Tung and L.-Z. Wu, Org. Lett., 2017, 19, 3009–3012 CrossRef CAS PubMed; (o) G. Pandey, R. Laha and P. K. Mondal, Chem. Commun., 2019, 55, 9689–9692 RSC; (p) D.-M. Yan, Q.-Q. Zhao, L. Rao, J.-R. Chen and W.-J. Xiao, Chem.–Eur. J., 2018, 24, 16895–16901 CrossRef CAS PubMed; (q) G. Pandey and R. Laha, Angew. Chem., Int. Ed., 2015, 54, 14875–14879 CrossRef CAS PubMed; (r) P. T. G. Rabet, G. Fumagalli, S. Boyd and M. F. Greaney, Org. Lett., 2016, 18, 1646–1649 CrossRef CAS PubMed; (s) T. Kawasasi, N. Ishida and M. Murakami, J. Am. Chem. Soc., 2020, 142, 3366–3370 CrossRef PubMed; (t) X. Cheng, H. Lu and Z. Lu, Nat. Commun., 2019, 10, 3549–3555 CrossRef PubMed; (u) H.-P. Deng, Q. Zhou and J. Wu, Angew. Chem., Int. Ed., 2018, 130, 12843–12847 CrossRef; (v) X. Liu, L. Lin, X. Ye, C.-H. Tan and Z. Jiang, Asian J. Org. Chem., 2017, 6, 422–425 CrossRef CAS; (w) A. Vasilopoulos, S. W. Krska and S. S. Stahl, Science, 2021, 372, 398–403 CrossRef CAS PubMed; (x) S. Guo, D. I. AbuSalim and S. P. Cook, J. Am. Chem. Soc., 2018, 140, 12378–12382 CrossRef CAS PubMed.
- (a) O. Shvydkiv, S. Gallagher, K. Nolan and M. Oelgemoller, Org. Lett., 2010, 12, 5170–5173 CrossRef CAS PubMed; (b) D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noel, Chem. Rev., 2016, 116, 10276–10341 CrossRef PubMed.
- (a) G. Goor, J. Glenneberg and S. Jacobi, Ullmann's Encycl. Ind. Chem., 2007, 18, 393–427 Search PubMed; (b) T. Nishimi, T. Kamchi, K. Kato, T. Kato and K. Yoshizawa, Eur. J. Org. Chem., 2011, 2, 4113–4120 CrossRef; (c) H.-G. Korth and P. Mulder, J. Org. Chem., 2020, 85, 2560–2574 CrossRef CAS PubMed.
- (a) H.-S. Bien, J. Stawitz and K. Wunderlich, Ullmann's Encycl. Ind. Chem., 2000, 3, 513–578 Search PubMed; (b) J. Cervantes-González, D. A. Vosburg, S. E. Mora-Rodriguez, M. A. Vázquez, L. G. Zepeda, C. V. Gómez and S. Lagunas-Rivera, ChemCatChem, 2020, 12, 3811–3827 CrossRef.
- F. Wilkinson, J. Phys. Chem., 1962, 66, 2569–2574 CrossRef CAS . Also, according to ref. 18c, the BDE of O–H bond in 10-OH-9-anthroxyl, the product from anthraquinone core upon a hydrogen atom transfer step, is: 42.9 kcal mol−1. This means the triplet state of anthraquinone can have a thermoneutral hydrogen atom transfer with a H-atom donor with BDE of 105.8 kcal mol−1..
- (a) Y. Shimada, K. Hattori, N. Tada, T. Miura and A. Itoh, Synthesis, 2013, 45, 2684–2688 CrossRef CAS; (b) Y. Matsusaki, T. Yamaguchi, N. Tada, T. Miura and A. Itoh, Synlett, 2012, 23, 2059–2062 CrossRef CAS; (c) N. Tada, K. Hattori, T. Nobuta, T. Miura and A. Itoh, Green Chem., 2011, 13, 1669–1671 RSC; (d) N. Tada, Y. Ikebata, T. Nobuta, S.-I. Hirashima, T. Miura and A. Itoh, Photochem. Photobiol. Sci., 2012, 11, 616–619 CrossRef CAS PubMed; (e) L. Cui, N. Tada, H. Okubo, T. Miura and A. Itoh, Green Chem., 2011, 13, 2347–2350 RSC; (f) W. Zhang, J. Gacs, I. W. C. E. Arends and F. Hollmann, ChemCatChem, 2017, 9, 3821–3826 CrossRef CAS PubMed; (g) B. Yuan, D. Mahor, Q. Fei, R. Wever, M. Alcalde, W. Zhang and F. Hollmann, ACS Catal., 2020, 10, 8277–8284 CrossRef CAS PubMed; (h) L. Zhao, W. Cai, G. Ji, J. Wei, Z. Du, C. He and C. Duan, Inorg. Chem., 2022, 61, 9493–9503 CrossRef CAS PubMed.
- J. E. Steves and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 15742–15745 CrossRef CAS PubMed.
- (a) B. Muhldorf and R. Wolf, Chem. Commun., 2015, 51, 8425–8428 RSC; (b) J. Zelenka, E. Svobodová, J. Tarábek, I. Hoskovcová, V. Boguschoá, S. Bailly, M. Sikorski, J. Roithová and R. Cibulka, Org. Lett., 2019, 21, 114–119 CrossRef CAS PubMed; (c) L.-Y. Jiang, J.-J. Ming, L.-Y. Wang, Y.-Y. Jiang, L.-H. Ren, Z.-C. Wang and W.-C. Cheng, Green Chem., 2020, 22, 1156–1163 RSC; (d) X. Liu, L. Lin, X. Ye, C.-H. Tan and Z. Jiang, Asian J. Org. Chem., 2017, 6, 422–425 CrossRef CAS.
- It is proposed that H-to-D exchange in acetone-d6 diminished phenolic signals of 1c in NMR study of Fig. 3 which seemed to appear as singlets at δ = 8.67 ppm and at δ = 9.13 ppm (Fig. 3B–D).
- See ESI† for study of oxidative recycling of 1a from 1c in DMSO.
- For previously proposed mechanisms, see ref. 21a and 21c.
- For references about interaction of triplet anthraquinone and alcohols and benzylic C–H, see: (a) C. F. Wells, Trans. Faraday Soc., 1961, 57, 1703–1718 RSC; (b) C. F. Wells, Trans. Faraday Soc., 1961, 57, 1719–1731 RSC; (c) M.-J. Zhou, L. Zhang, G. Liu, C. Xu and Z. Huang, J. Am. Chem. Soc., 2021, 143, 16470–16485 CrossRef CAS PubMed.
- (a) E. Spier, U. Neuenschwander and I. Hermans, Angew. Chem., Int. Ed., 2013, 52, 1581–1585 CrossRef CAS PubMed; (b) T. Mukaiyama, S. Isayama, S. Inoki, K. Kato, T. Yamada and T. Takai, Chem. Lett., 1989, 18, 449–452 CrossRef; (c) K. Kato, T. Yamada, T. Takai, S. Inoki and S. Isayama, Bull. Chem. Soc. Jpn., 1990, 63, 179–186 CrossRef CAS; (d) M. S. Alam, B. S. M. Rao and E. Janata, Radiat. Phys. Chem., 2003, 67, 723–728 CrossRef CAS; (e) T. Matsue, M. Fujihira and T. Osa, J. Electrochem. Soc., 1981, 128, 2565–2569 CrossRef CAS . Additionally, regarding the great works of Prof. Mukaiyama (ref. 28b) and Prof. Yamada (ref. 28c) where Co(acac)2 was also believed to activate O2 to eventually form Co(acac)2-OOH, we believe that this specific process might be less relevant for our alcohol activation because our chemistry proceeded well under lower temperature than the condition (75 °C) reported in the aforementioned two references. Moreover, as demonstrated in our manuscript, 1c (and the semi-anthrahydroquinone radical) can activate O2, this might be the dominant O2 activation pathway; and in entry 3 of Table 4, a 0% yield and 0% conversion showed that under our condition, a Co(acac)2-initiated-oxidation with O2 of alcohol might not contribute to the success of the alcohol oxidation protocol..
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra00332a |
| This journal is © The Royal Society of Chemistry 2023 |