
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmidinoquinoxaline N-oxides: synthesis and activity against anaerobic bacteria†
Nadia Gruber a,
Liliana Fernández-Canigia*b,
Natalia B. Kilimcilera,
Pierluigi Stipac,
Juan A. Biscegliaa,
María B. Garcíaa,
Daniel H. Gonzalez Magliod,
Mariela L. Pazd and
Liliana R. Orelli*a
a,
Liliana Fernández-Canigia*b,
Natalia B. Kilimcilera,
Pierluigi Stipac,
Juan A. Biscegliaa,
María B. Garcíaa,
Daniel H. Gonzalez Magliod,
Mariela L. Pazd and
Liliana R. Orelli*a
aUniversidad de Buenos Aires, CONICET, Química Orgánica II, Departamento de Ciencias Químicas, Facultad de Farmacia y Bioquímica, Junín 956, (1113) Buenos Aires, Argentina. E-mail: lorelli@ffyb.uba.ar
bLaboratorio de Microbiología, Hospital Alemán, Av. Pueyrredón 1640, (1118) Buenos Aires, Argentina
cSIMAU Departament – Chemistry Division, Università Politecnica delle Marche, Via Brecce Bianche 12, Ancona (I-60131), Italy
dUniversidad de Buenos Aires, Instituto de Estudios de la Inmunidad Humoral (IDEHU), Cátedra de Inmunología, Facultad de Farmacia y Bioquímica, Junín 956, (1113) Buenos Aires, Argentina
First published on 13th September 2023
Abstract
We present herein an in-depth study on the activity of amidinoquinoxaline N-oxides 1 against Gram-positive and Gram-negative anaerobic bacteria. Based on 5-phenyl-2,3-dihydropyrimidoquinoxaline N-oxide 1a, the selected structural variations included in our study comprise the substituents α− to the N-oxide function, the benzofused ring, substitution and quaternization of the amidine moiety, and the amidine ring size. Compounds 1 showed good to excellent antianaerobic activity, evaluated as the corresponding CIM50 and CIM90 values, and an antimicrobial spectrum similar to metronidazole. Six out of 13 compounds 1 had CIM90 values significantly lower than the reference drug. Among them, imidazoline derivatives 1i–l were the most active structures. Such compounds were synthesized by base-promoted ring closure of the corresponding amidines. The N-oxides under study showed no significant cytotoxicity against RAW 264.7 cells, with high selectivity indexes. Their calculated ADME properties indicate that the compounds are potentially good oral drug candidates. The antianaerobic activity correlated satisfactorily with the electron affinity of the compounds, suggesting that they may undergo bioreductive activation before exerting their antibacterial activity.
1 Introduction
Anaerobic bacteria are the main component of the bacterial microbiota of normal human skin and mucous membranes and can behave as opportunistic pathogens under an appropriate environment. Infections have commonly an endogenous origin and, comparatively, a small number of diseases are associated with exogenous anaerobic pathogens. These bacteria can lead to a broad spectrum of infections, which in some cases are life-threatening: septicemia, endocarditis, brain, lung and liver abscesses and aspiration pneumonias, among others.1,2Among the antimicrobials that can be chosen according to the infection localization and the anaerobic species, metronidazole continues to be one of the drugs of choice3 showing generally low resistance levels among pathogenic anaerobes.4 However, resistant strains have been reported over the past decades5–9 and decreased in vitro susceptibility has been observed in recent years.10,11 In addition, even if metronidazole resistance was first reported limited to Bacteroides spp, it now includes Gram-positive cocci and other bacilli.12 Several mechanisms of metronidazole resistance in anaerobic bacteria have been proposed,13–16 including specific genes (nim), that encode an alternative reductase that can convert 4- or 5-nitroimidazoles to a non-bactericidal derivative by reduction of the nitro group to an amino function.17,18 These genes have been isolated in Gram-positive and Gram-negative anaerobic bacteria.19–22 Thus, as resistance to metronidazole emerges, the development of new specific anti-anaerobe agents becomes necessary.
Among anaerobic bacteria, Clostridioides difficile, an anaerobe responsible for intestinal infections associated with life-threatening severe diarrhea, abdominal pain and fever, is currently a topic of concern, given that virulent strains are causing nosocomial outbreaks in North America, Canada and Europe.23 It is also an important agent of diarrheal illness in outpatients. Infections with C. difficile have been classified as an urgent public health threat because of the number of infections and deaths directly attributable to it.24 The first line treatments for this pathogen are metronidazole or oral vancomycin. However, a recent review reports 22.4% and 14.2% treatment failure and 27.1% and 24.0% recurrence after treatment with metronidazole and vancomycin, respectively.25 Metronidazole, vancomycin, and fidaxomicin drug resistance in C. difficile is fortunately not widespread at this time,26 but given the increasing prevalence of C. difficile infections over the past decade, the requirement for new antimicrobials effective against C. difficile is an important preventive measure.27
Several research groups have sought for alternatives to treat anaerobic infections, including traditional antimicrobials and other biological therapeutics. In recent years, metronidazole derivatives were reported, which in many cases involve modification in the hydroxyethyl chain such as the replacement of the hydroxyl group by an N-piperazino carbamoyl,28 a triazole29 or a triazolylthio group.30 Other modifications of this chain include the preparation of ester and ether derivatives using terpenes.31 Nitroheterocycles including secnidazole derivatives and their copper(II) complexes,32 substituted nitroimidazoles,31,33 nitazoxanide analogs34–36 and nitrofuranylsemicarbazones37 were also tested on anaerobes. Since most of the developed compounds are modifications of existing antimicrobials, they are only short term solutions that cannot usually overcome multiple resistance mechanisms.38
Amidinoquinoxaline N-oxides represent a heterocyclic core of interest due to their pharmacological properties. Some suitably substituted derivatives possess antineoplastic activity,39–41 in particular against hypoxic tumors, while others behave as antiamoebic agents.42 The activity of some related compounds against a small number of anaerobic bacterial strains is also described in the literature.43–46 In addition, recent results of our group show that these nitrones behave as antioxidants47 due to their ability to act as spin traps48,49 as well as to undergo single electron transfer reactions. The pharmacological interest of this heterocyclic core, the versatility of the N-oxide function and the results of our recent research, prompted us to study the activity of amidinoquinoxaline N-oxides 1 (Fig. 1) against anaerobic bacteria as an alternative to metronidazole. Since the molecular size and functional groups present in our compounds are different from those of metronidazole, it could be expected that they would circumvent the action of reductases encoded by nim genes. As a substantial difference, the functional group to be reduced in metronidazole is the nitro function, while in the nitrones the bioreducible functionality is the N-oxide.
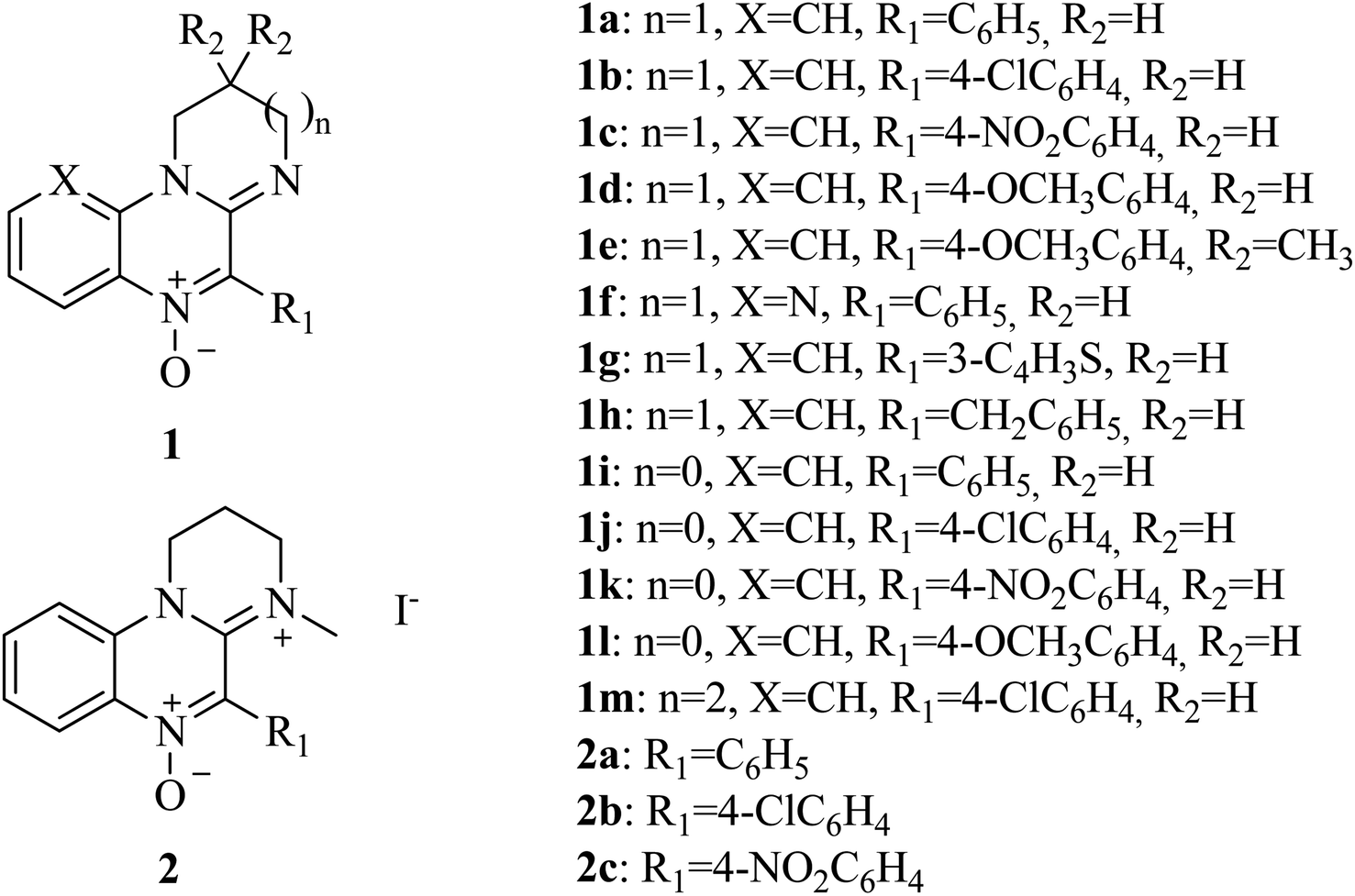 | ||
| Fig. 1 Amidinoquinoxaline N-oxides 1 and their quaternary salts 2. | ||
In this work we have evaluated the activity of the nitrones under study against anaerobic bacteria and analyzed the structural features that influence their bioactivity. We also present the results of cytotoxicity assays and ADME predictions as well as an insight to a general mechanism proposal. The synthetic approach previously reported by our group50 included a spontaneous cyclodehydration step which was too slow for some derivatives, leading in those cases to byproducts and affording low yields. We present herein an improved synthetic procedure that circumvents these problems.
2 Materials and methods
2.1 Synthesis
N-Oxides 1a–h,m were prepared by cyclodehydration of aminoamides 3a–h,m (Scheme 1).45 A mixture of the aminoamide (1 mmol) and ethyl polyphosphate (PPE, 1 mL/0.05 g) was refluxed for 5 h in an oil bath. After reaching room temperature, the resulting solution was extracted with water (5 × 6 mL). The aqueous phases were pooled, filtered and made alkaline with 10% aqueous NaOH. The mixture was extracted with chloroform (3 × 15 mL). The organic phases were washed with water, dried over sodium sulphate and filtered. The crude chloroformic solution of amidines 4a–h,m was left at r.t. until no further conversion to compounds 1a–h,m was evidenced by TLC (silica gel, chloroform![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methanol 9:1). The solvent was then removed in vacuo and the crude product was purified by column chromatography (silica gel, chloroform:methanol 10:0–9:1).
:methanol 9:1). The solvent was then removed in vacuo and the crude product was purified by column chromatography (silica gel, chloroform:methanol 10:0–9:1).
 | ||
| Scheme 1 Synthesis of amidinoquinoxaline N-oxides 1. | ||
Amidinium salts 2a–c were prepared from the corresponding N-oxides 1a–c according to the method developed by our group (Scheme 2).46
 | ||
| Scheme 2 Synthesis of amidinium salts 2a–c. | ||
2.1.2.1 Base-promoted synthesis of 4-aryl-1,2-dihydroimidazo[1,2-a]quinoxaline 5-oxides 1i–l. Imidazolines 4i–l were prepared in the same way as the analogous tetrahydropyrimidines and tetrahydrodiazepines 4a–h,m. The crude imidazolines 4i–l were stirred with 4% KOH in absolute ethanol (10 mL/100 mg of compound) in an ice bath. After 10 min the cold bath was removed and the mixture was stirred at room temperature for 50 min. The mixture was then concentrated in vacuo, diluted with water (5 mL) and extracted with methylene chloride (3 × 15 mL). The organic phases were pooled, dried with anhydrous sodium sulphate, filtered and the solvent was removed in vacuo. The crude compounds were purified by crystallization (EtOH) or column chromatography (Silica gel, chloroform
:methanol 10:0–9:1).Compounds 1a–d,h,m,45 1e,51 2a–c,46 1f,g,48 1i (ref. 51) and 1l (ref. 49) were described in the literature. Yields and analytical data of nitrones 1j,k aminoamides 3j,k and amidines 4j–l are as follows.
2.1.2.1.1 4-(4-Chlorophenyl)-1,2-dihydroimidazo[1,2-a]quinoxaline 5-oxide (1j). This compound was obtained as an orange hygroscopic solid (244 mg, 82% yield), mp = 181–182 °C (from EtOH). Anal. calc. for C16H12ClN3O: C, 64.5; H, 4.1; N, 14.1. Found: C, 64.8; H, 4.1; N, 14.1%. IR (neat)
![[small nu, Greek, macron]](https://www.rsc.org/images/entities/char_e0ce.gif) /cm−1: 1618m, 1578s, 1505s, 1467m, 1373m, 1282s, 1087s, 819s, 548m, 437s. 1H NMR (600 MHz, CDCl3, 25 °C, TMS): δ = 8.27 (1H, dd, J = 8.3, 1.3 Hz), 7.91 (2H, d, J = 8.6 Hz), 7.48–7.52 (1H, m), 7.46 (2H, d, J = 8.6 Hz), 7.10–7.15 (1H, m), 6.82 (1H, dd, J = 8.1, 1.1 Hz), 4.17–4.22 (2H, m), 4.05–4.10 (2H, m). 13C NMR (151 MHz, CDCl3, 25 °C) δ = 152.7, 136.1, 134.8, 133.3, 132.3, 131.7, 131.0, 128.2, 126.8, 121.4, 121.3, 111.9, 54.2, 46.5. HRMS (ESI) m/z: [M + H]+ calcd for C16H13ClN3O: 298.0747. Found: 298.0743.
/cm−1: 1618m, 1578s, 1505s, 1467m, 1373m, 1282s, 1087s, 819s, 548m, 437s. 1H NMR (600 MHz, CDCl3, 25 °C, TMS): δ = 8.27 (1H, dd, J = 8.3, 1.3 Hz), 7.91 (2H, d, J = 8.6 Hz), 7.48–7.52 (1H, m), 7.46 (2H, d, J = 8.6 Hz), 7.10–7.15 (1H, m), 6.82 (1H, dd, J = 8.1, 1.1 Hz), 4.17–4.22 (2H, m), 4.05–4.10 (2H, m). 13C NMR (151 MHz, CDCl3, 25 °C) δ = 152.7, 136.1, 134.8, 133.3, 132.3, 131.7, 131.0, 128.2, 126.8, 121.4, 121.3, 111.9, 54.2, 46.5. HRMS (ESI) m/z: [M + H]+ calcd for C16H13ClN3O: 298.0747. Found: 298.0743.
2.1.2.1.2 4-(4-Nitrophenyl)-1,2-dihydroimidazo[1,2-a]quinoxaline 5-oxide (1k). This compound was obtained as a red hygroscopic solid (246 mg, 80%), mp = 211–212 °C (from EtOH). Anal. calc. for C16H12N4O3: C, 62.3; H, 3.9; N, 18.2. Found: C, 61.4; H, 3.9; N, 17.7%. IR (neat)
/cm−1: 1607m, 1599s, 1577s, 1503s, 1343s, 1282s. 1H NMR (600 MHz, CDCl3, 25 °C, TMS): δ = 8.33 (2H, d, J = 8.9 Hz), 8.26 (1H, dd, J = 8.1, 1.2 Hz), 8.16 (2H, d, J = 8.9 Hz), 7.51–7.56 (1H, m), 7.12–7.17 (1H, m), 6.85 (1H, dd, J = 8.1, 1.0 Hz), 4.18–4.23 (2H, m), 4.07–4.13 (2H, m). 13C NMR (125 MHz, CDCl3, 25 °C): δ = 152.3, 148.1, 134.9, 133.7, 133.5, 132.9, 131.6, 131.0, 123.0, 121.51, 121.48, 112.1, 54.2, 46.5. HRMS (ESI) calcd for C16H13N4O3: 309.0988. Found: 309.0985.
2.1.2.1.3 2-(4-Chlorophenyl)-N-(2-(2-nitrophenylamino)ethyl)acetamide (3j). This compound was obtained as an orange solid (237 mg, 71% yield), mp = 132–134 °C (from hexane/chloroform). 1H NMR (600 MHz, CDCl3, 25 °C, TMS): δ = 8.14 (1H, dd, J = 8.6, 1.0 Hz), 8.08 (1H, bs ex), 7.40–7.44 (1H, m), 7.28 (2H, d, J = 8.3 Hz), 7.15 (2H, d, J = 8.3 Hz), 6.91 (1H, d, J = 8.6 Hz), 6.64–6.69 (1H, m), 5.88 (1H, bs ex), 3.53 (2H, s), 3.46–3.52 (4H, m). 13C NMR (151 MHz, CDCl3, 25 °C) δ = 171.2, 145.3, 136.4, 133.4, 132.9, 132.2, 130.7, 129.1, 126.9, 115.8, 113.7, 42.8, 41.9, 39.0. HRMS (ESI) m/z: [M + H]+ calcd for C16H17ClN3O3: 334.0959. Found: 334.0957.
2.1.2.1.4 2-(4-Nitrophenyl)-N-(2-((2-nitrophenyl)amino)ethyl)acetamide (3k). This compound was obtained as an orange solid (151 mg, 44% yield), mp = 190–192 °C (from hexane/chloroform). 1H NMR (500 MHz, DMSO-d6, 25 °C, TMS): δ = 8.41 (1H, bs), 8.18 (1H, bs), 8.13 (2H, d, J = 8.7 Hz), 8.04 (1H, dd, J = 8.7, 1.6 Hz), 7.48–7.53 (3H, m), 7.11 (1H, dd, J = 8.7, 0.9 Hz), 6.65–6.70 (1H, m), 3.58 (2H, s), 3.44 (2H, c, J = 6.1 Hz), 3.30–3.34 (2H, m). 13C NMR (125 MHz, DMSO-d6, 25 °C): δ = 169.7, 146.3, 145.2, 144.3, 136.6, 131.1, 130.4, 126.3, 123.3, 115.3, 114.4, 41.9, 41.8, 37.8. HRMS (ESI) calcd for C16H17N4O5: 345.1199. Found: 345.1204.
2.1.2.1.5 2-(4-Chlorobenzyl)-1-(2-nitrophenyl)-4,5-dihydro-1H-imidazole (4j). This compound was obtained as an orange oil (246 mg, 78%).1H NMR (600 MHz, CDCl3, 25 °C, TMS): δ = 7.87 (1H, dd, J = 8.1, 1.5 Hz), 7.54 (1H, td, J = 7.7, 1.5 Hz), 7.41–7.44 (1H, m), 7.10–7.14 (3H, m), 6.89 (2H, d, J = 8.5 Hz), 3.98 (2H, t, J = 9.5 Hz), 3.77 (2H, t, J = 9.5 Hz), 3.39 (2H, s). 13C NMR (151 MHz, CDCl3, 25 °C): δ = 162.9, 147.8, 133.7, 133.6, 132.7, 132.2, 130.9, 130.0, 128.5, 128.2, 125.2, 54.0, 53.3, 34.0.
2.1.2.1.6 2-(4-Nitrobenzyl)-1-(2-nitrophenyl)-4,5-dihydro-1H-imidazole (4k). This compound was obtained as an orange oil (231 mg, 71%). 1H NMR (600 MHz, CDCl3, 25 °C, TMS): δ = 8.05 (2H, d, J = 8.5 Hz), 7.88 (1H, dd, J = 8.1, 1.6 Hz), 7.58 (1H, td, J = 7.8, 1.6 Hz), 7.44–7.48 (1H, m), 7.20 (1H, dd, J = 7.8, 1.4 Hz), 7.17 (2H, d, J = 8.5 Hz), 3.98 (2H, t, J = 9.6 Hz), 3.78 (2H, t, J = 9.6 Hz), 3.52 (2H, s). 13C NMR (125 MHz, CDCl3, 25 °C): δ = 162.0, 147.9, 146.9, 142.8, 134.8, 133.9, 130.7, 129.7, 128.5, 125.4, 123.6, 54.1, 53.6, 34.4.
2.1.2.1.7 2-(4-Methoxybenzyl)-1-(2-nitrophenyl)-4,5-dihydro-1H-imidazole (4l). This compound was obtained as a yellow oil (255 mg, 82%). 1H NMR (600 MHz, CDCl3, 25 °C, TMS): δ = 7.88 (1H, dd, J = 8.2, 1.5 Hz), 7.55 (1H, td, J = 7.6, 1.5 Hz), 7.40–7.46 (1H, m), 7.17 (1H, dd, J = 7.6, 1.3 Hz), 6.81 (2H, d, J = 8.6 Hz), 6.68 (2H, d, J = 8.6 Hz), 3.99 (2H, t, J = 9.7 Hz), 3.81 (2H, t, J = 9.7 Hz), 3.74 (3H, s), 3.43 (2H, s). 13C NMR (125 MHz, CDCl3, 25 °C): δ = 164.3, 158.5, 147.5, 134.7, 133.8, 131.98, 131.0, 129.7, 128.3, 125.3, 113.8, 55.2, 54.0, 52.3, 33.6.
2.2 Clinical isolates
A total of 100 bacterial isolates, including ATCC strains and clinical isolates of Gram-positive and Gram-negative anaerobic bacteria were tested. A first panel composed of 84 strains of special clinical relevance was further subdivided into six groups, which include the same or related species (Table 2). Groups 1 to 4, corresponding to Gram-negative bacilli, were assigned to B. fragilis, Bacteroides spp. and Parabacteroides distasonis, Prevotella spp. and Fusobacterium nucleatum, respectively. The species of genus Bacteroides are some of the most frequently isolated anaerobes in clinical laboratories and are also typically more virulent and resistant. Among them, B. fragilis has the greatest clinical relevance and was therefore considered as a separate group. Groups 5 and 6 were assigned to Clostridium perfringens and C. difficile as Gram-positive spore-forming bacilli (Table 2).The remaining isolates were included in Table 3 as representative examples of less common genera and/or species that, together with the previous results, contribute to outline the spectrum of action of this nitrone family.
2.3 Minimal inhibitory concentration (MIC) determinations
MIC determinations were performed using the Agar Dilution Procedure according to CLSI guidelines.52,53 This technique is used to quantitatively measure the in vitro antimicrobial activity and is considered the standard method for anaerobic bacteria. MIC is defined as the minimum antimicrobial concentration that prevents the visible development of a microorganism in a susceptibility test by dilution in broth or agar.54 The culture medium, Brucella agar, was prepared fresh and supplemented with vitamin K (1 μg mL−1), hemin (5 μgmL−1) and laked sheep blood (5% V/V). Clinical isolates from the Microbiology Laboratory of the Hospital Aleman, stored at −70 °C, were subcultured twice or until normal growth rate. The isolate purity was controlled and identification, if needed, was confirmed with a MALDI-TOF BD mass spectrometer.Stock solutions were prepared with 50% DMSO in sterile distilled water and two-fold serial dilutions of the nitrones were made. Compound concentration in the culture plate ranged typically from 64 to 0.06 μg mL−1 standardized bacterial inoculums of ∼1.5 × 108 CFU mL−1 were prepared in BHI broth (0.5 of the McFarland standard). Agar dilution test plates were inoculated with 1 μL (approximately 1.5 × 105 CFU per spot) using a Steers multipoint replicator. Plates were incubated at 37 °C for 48 h in anaerobic conditions using anaerobic atmosphere generation bags (Anaero-Pack, Key Scientific, Mitsubishi).
Positive growth controls were performed at different times of the assay to ensure anaerobes viability. Viability controls with DMSO 5% and 2.5% were also included. Contamination with aerobic bacteria was controlled at the beginning and the end of each assay by culturing the bacterial suspensions in Chocolate Agar under aerobic conditions. Reproducibility was controlled by testing B. fragilis ATCC 25285 strain and metronidazole as inter-assay controls, following CLSI recommendations.58
The results have been reported using population parameters: MIC ranges, MIC50 and MIC90 (minimum concentrations able to inhibit 50 and 90% of the tested isolates, respectively).
2.4 Cell viability assay
Murine macrophage cell line RAW 264.7 was cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 100 U mL−1 penicillin, 100 μg mL−1 streptomycin, and 0.29 mg ml−1 L-glutamine. The cells were grown in plastic flasks in a5% CO2 humidified atmosphere at 37 °C. Medium was changed every 2–3 days until subconfluence was achieved. Then, cells were harvested with an EDTA 0.05% solution and placed thereafter in 96-well plates to perform all assays.The 50% cytotoxic concentration (CC50), defined as the drug concentration that reduces cell viability by 50% when compared to untreated controls, was evaluated using these cells. A preliminary study was conducted to determine the optimal number of cells per well and their tolerance to DMSO, briefly: amounts of 2.5 × 104, 1 × 105 and 4 × 105 cells per well were seeded in duplicate and cultured in DMEM containing serial two-fold dilutions of DMSO. The cells were cultured for 21 h and the cell viability was determined using the Alamar Blue reagent in a fluorometer as described below. In this assay, the optimal amount of cells per well was determined as 4 × 105 and 1% DMSO was tolerated without affecting cell growth.
Afterwards, serial dilutions 1/5 of each nitrone and metronidazole were tested in duplicate, ranging from 0.03 to 100 μg mL−1. Plates were seeded with 4 × 105 cells per well and 100 μL of DMEM supplemented medium with the corresponding nitrone dilution was added. Cells were cultured for 21 h. After that, 20 μL of the Alamar Blue Reagent (Resazurin) were added to each well and incubated for 3 h. The fluorescence of each well was measured using a micro-plate reader (Victor3, PerkinElmer) with excitation/emission 560/590 nm. The resulting data were presented as survival percentage.55
Cell viability controls were performed in every assay in triplicate by culturing the cells with DMEM supplemented medium with and without 1% DMSO. Cell death controls were performed in triplicate with DMSO 10%. Basal fluorescence of each nitrone in a 100 μg mL−1 dilution was measured in duplicate. Metronidazole was used for comparison and as inter-assay control.
2.5 Electron affinity calculations
Density Functional Theory56 calculations were carried out with GAUSSIAN 09.57 Nitrone conformations were systematically screened by means of appropriate relaxed (i.e., with optimization at each point) Potential Energy Surfaces (PES). Scans were performed at the B3LYP/6-31G(d) level to find global minimum energy structures. Imaginary (negative) values were never found in frequency calculations, confirming that the computed geometries were always referred to a minimum. Thermodynamic parameters were computed at 298 K by means of frequency calculations employing the B3LYP functional in conjunction with the 6-31+G(d,p) basis set. Geometries of the corresponding radical anions and their thermodynamic parameters were calculated at the same level. The electron affinity (EA) was calculated as the energy difference (measured as ΔH) between the nitrone and its radical anion in their ground states (Table 4). The EA thus calculated is known as adiabatic electron affinity and is a good general estimation of the experimental EA.582.6 ADME calculations
Qikprop program, version 3.0 (Schrödinger, LLC., New York) was used for this in silico study. Geometry optimization of the input structures was calculated with the Hyperchem program, using the MM+ force field. Since Qikprop is not able to calculate charged species, amidinium salts 2 were excluded of this study, even if numerous works have shown that ionic compounds are capable of crossing lipidic membranes.593 Results and discussion
3.1 Synthesis
In our previously developed synthetic approach,45 the last reaction step (Scheme 1) is a spontaneous heterocyclization that leads from the cyclic amidine 4 to the corresponding N-oxide 1. This reaction was achieved within 1–24 h with high yields for most tetrahydropyrimidines. Imidazolines, in contrast, required significantly longer reaction times (7–15 days) to afford the corresponding N-oxides. As a consequence, these nitrones were isolated in low yields and accompanied by colateral products. Literature on the synthesis of related componds suggested that this step could be base promoted.60 After screening a few basic media (Et3N/DCM; Et3N/MeCN; 10% aq. NaOH/EtOH; 4% KOH/abs. ethanol) we found that treatment of imidazolines 3i–l with an ethanolic solution of KOH 4% resulted in a drastic reduction of the reaction times (from more than one week to one our) and a significant improve in the yields (Table 1).| Comp | Yield 3 → 1 (%) (I) | Yield 3 → 1 (%) (II) |
|---|---|---|
| 1i | 64 | 90 |
| 1j | 59 | 82 |
| 1k | 17 | 80 |
| 1l | 71 | 82 |
In the absence of additional base, the reaction would be autocatalyzed by the amidine moiety with a pKa dependent rate. The striking pKa difference between different cyclic amidines homologues (pKa = 10.51 for 1-phenyl-2-4-nitrophenyl-1,4,5,6-tetrahydropyrimidine vs. 7.65 for its imidazoline homologue)61 would account for the different conversion times. A plausible reaction mechanism involves the semistabilized carbanion I as the intermediate (Scheme 3).
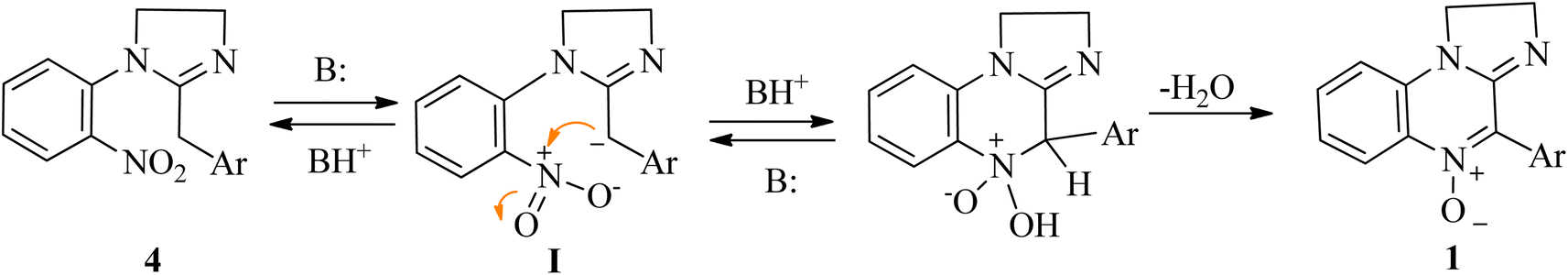 | ||
| Scheme 3 Probable base-promoted cyclodehydration mechanism. | ||
3.2 Antimicrobial activity
Results, expressed as MIC50 and MIC90 are shown in Tables 2 and 3 (a complete version including the MIC ranges is included in the ESI, Table S1†). The N-oxides under study showed great efficiency against anaerobic bacteria. Among them, compounds 1c,f,i–l exhibited significantly lower MIC90 than those determined for metronidazole.‡ Exceptions to the high activity of these heterocycles are non-spore-forming Gram-positive bacilli, against which these compounds, as well as metronidazole, were not active.| Organism and compounds | MIC50 (μg mL−1)/MIC90 (μg mL−1)/(n° of isolates) | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1a | 1b | 1c | 1d | 1e | 1f | 1g | 1h | 1i | 1j | 1k | 1l | 1m | 2a | 2b | 2c | Mtz | |
| a Includes 3 isolates of Parabacteroides distasonis and 10 corresponding to species of Bacteroides: 6 Bacteroides thetaiotaomicron/ovatus, 2 Bacteroides uniformis, 1 Bacteroides vulgatus and 1 Bacteroides caccae.b Includes 16 isolates of Prevotella intermedia/nigrescens, 2 of Prevotella oralis group, 2 of Prevotella buccae and 1 of Prevotella bivia.c Includes 5 isolates of Prevotella intermedia/nigrescens, 1 of Prevotella corporis, 1 of Prevotella oralis group, 1 of Prevotella oris, 1 of Prevotella baroniae, 2 of Prevotella buccae, 2 of Prevotella bivia and 1 of Prevotella dentalis.d MTZ = metronidazole.e ND: not determined. | |||||||||||||||||
| Gram negative bacilli | |||||||||||||||||
| Bacteroides fragilis (ATCC 25285) | 1 | 2 | ≤0.06 | 2 | 0.5 | 0.125 | 0.5 | 1 | 0.125 | 0.125 | ≤0.06 | ≤0.06 | 2 | 4 | 4 | 1 | 0.25 |
| Bacteroides fragilis | 0.5 | 1 | ≤0.06 | 1 | 0.25 | ≤0.06 | 0.25 | 1 | ≤0.06 | ≤0.06 | ≤0.06 | ≤0.06 | 1 | 2 | 4 | 0.5 | 0.5 |
| 1 (19) | 2 (19) | ≤0.06 (19) | 2 (19) | 0.5 (16) | ≤0.06 (16) | 0.5 (16) | 0.4 (16) | ≤0.06 (16) | 0.125 (16) | ≤0.06 (16) | ≤0.06 (16) | 2 (19) | 2 (16) | 8 (16) | 1 (16) | 1 (16) | |
| Bacteroides thetaiotaomicron (ATCC 29148) | 0.5 | 1 | 0.125 | 1 | 0.5 | ≤0.06 | 0.5 | 2 | ≤0.06 | 0.125 | ≤0.06 | ≤0.06 | 1 | 2 | 4 | 0.5 | 1 |
| Bacteroides ovatus (ATCC 84834) | ND | ND | ND | ND | 0.5 | 0.125 | 1 | 4 | ≤0.06 | 0.125 | ≤0.06 | ≤0.06 | ND | 4 | 8 | 0.5 | 1 |
| Other Bacteroides spp. and Parabacteroidesa | 0.5 | 1 | ≤0.06 | 1 | 0.25 | ≤0.06 | 0.25 | 1 | ≤0.06 | ≤0.06 | ≤0.06 | ≤0.06 | 1 | 2 | 4 | 0.25 | 1 |
| 1 (13) | 1 (13) | ≤0.06 (13) | 2 (13) | 0.5 (13) | ≤0.06 (13) | 0.5 (13) | 4 (13) | ≤0.06 (13) | 0.125 (13) | ≤0.06 (13) | ≤0.06 (13) | 2 (13) | 4 (13) | 8 (13) | 1 (13) | 1 (13) | |
| Prevotella intermedia/nigres-cens (ATCC 25611) | 0.5 | 1 | ≤0.06 | 1 | 0.5 | 0.125 | 0.25 | 1 | ≤0.06 | ≤0.06 | ≤0.06 | ≤0.06 | 0.5 | 8 | 16 | 2 | 1 |
| Prevotella spp. | 0.25 | 0.5 | ≤0.06 | 0.5 | 0.25 | ≤0.06 | 0.125 | 1 | ≤0.06 | ≤0.06 | ≤0.06 | ≤0.06 | 0.5 | 4 | 4 | 0.5 | 0.5 |
| 1 (21)b | 1 (21)b | 0.125 (21)b | 1 (21)b | 0.5 (14)c | 0.125 (14)c | 0.5 (14)c | 4 (14)c | ≤0.06 (14)c | ≤0.06 (14)c | ≤0.06 (14)c | ≤0.06 (14)c | 0.5 (21)b | 8 (14)c | 8 (14)c | 1 (14)c | 1 (21) b | |
| Fusobacterium nucleatum (ATCC 25586) | 0.125 | 0.25 | ≤0.06 | 0.5 | 0.25 | 0.125 | 0.125 | 0.5 | ≤0.06 | ≤0.06 | ≤0.06 | ≤0.06 | 0.5 | 2 | 4 | 0.25 | 0.125 |
| Fusobacterium nucleatum | 0.25 | 0.25 | ≤0.06 | 0.5 | 0.25 | 0.125 | 0.25 | 1 | ≤0.06 | ≤0.06 | ≤0.06 | ≤0.06 | 0.5 | 4 | 4 | 0.25 | ≤0.06 |
| 1 (7) | 1 (7) | ≤0.06 (7) | 2 (7) | 0.25 (7) | 0.5 (7) | 0.5 (7) | 4 (7) | 0.125 (7) | 0.125 (7) | ≤0.06 (7) | ≤0.06 (7) | 0.5 (7) | 4 (7) | 8 (7) | 1 (7) | 0.25 (7) | |
|
|||||||||||||||||
| Gram positive bacilli | |||||||||||||||||
| Clostridium difficile (ATCC 43255) | 4 | 8 | ≤0.06 | 8 | 4 | 0.5 | 4 | 32 | 1 | 0.25 | ≤0.06 | 0.5 | 4 | 32 | 16 | 0.5 | 0.25 |
| Clostridium difficile | 2 | 8 | 0.25 | 4 | 2 | 0.5 | 2 | 16 | 0.5 | 0.25 | ≤0.06 | 0.25 | 2 | 16 | 32 | 0.25 | 0.25 |
| 4 (9) | 16 (9) | 0.5 (9) | 8 (9) | 8 (14) | 0.5 (14) | 2 (14) | 16 (14) | 0.5 (14) | 0.25 (14) | ≤0.06 (14) | 0.5 (14) | 4 (9) | 32 (14) | 32 (14) | 0.5 (14) | 0.25 (14) | |
| Clostridium perfringens | 8 | 32 | 0.5 | 16 | 4 | 0.5 | 4 | <32 | 0.5 | 0.5 | ≤0.06 | 0.5 | 16 | 32 | 32 | 0.25 | 0.5 |
| 16 (10) | 64 (10) | 2 (10) | 32 (10) | 8 (10) | 1 (10) | 8 (10) | <32 (10) | 1 (10) | 1 (10) | 0.125 (10) | 1 (10) | 32 (10) | 32 (10) | 32 (10) | 1 (10) | 1 (10) | |
| Organism | MIC (μg mL−1) | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1a | 1b | 1c | 1d | 1e | 1f | 1g | 1h | 1i | 1j | 1k | 1l | 1m | 2a | 2b | 2c | Mtz | |
| a ND: not determined.b Mtz: metronidazole. | |||||||||||||||||
| Gram negative bacilli | |||||||||||||||||
| Porphyromonas gingivalis (ATCC 33277) | ≤0.06 | 0.125 | ≤0.06 | ≤0.06 | ND | ND | ND | ND | ND | ≤0.06 | ND | ND | 0.25 | ND | ND | ND | ≤0.06 |
| Porphyromonas asaccharolytica (CI) | ND | ND | ND | ND | 0.25 | ≤0.06 | 0.125 | 2 | ≤0.06 | ≤0.06 | ≤0.06 | ≤0.06 | ND | 4 | 4 | 0.25 | 0.25 |
| Porphyromonas sp. (CI) | 0.125 | 0.5 | ≤0.06 | 0.5 | 0.25 | ≤0.06 | 0.125 | 1 | ≤0.06 | ≤0.06 | ≤0.06 | ≤0.06 | 0.25 | 4 | 4 | 0.5 | 0.125 |
| Fusobacterium mortiferum (CI 1) | ND | ND | ND | ND | 0.5 | 0.125 | 0.25 | 2 | ≤0.06 | ≤0.06 | ≤0.06 | ≤0.06 | ND | 4 | 8 | 1 | 0.25 |
| Fusobacterium mortiferum (CI 2) | ND | ND | ND | ND | 2 | 0.25 | 1 | 8 | 0.25 | 0.25 | 0.125 | 0.25 | ND | 8 | 16 | 2 | 0.25 |
|
|||||||||||||||||
| Gram positive bacilli | |||||||||||||||||
| Spore-forming | |||||||||||||||||
| Clostridium sporogenes (ATCC 3584) | 0.5 | 2 | ≤0.06 | 1 | 1 | 0.25 | 1 | 8 | 0.25 | 0.25 | ≤0.06 | 0.125 | 1 | 8 | 8 | 0.125 | 0.25 |
| Eggerthella lenta (ATCC 43055) | 0.5 | 0.5 | ≤0.06 | 0.5 | 0.5 | ≤0.06 | 0.25 | 2 | ≤0.06 | ≤0.06 | ≤0.06 | ≤0.06 | 1 | 4 | 8 | 0.5 | 0.25 |
| Clostridium sordellii (CI) | 16 | 64 | 0.5 | 32 | ND | ND | ND | ND | ND | 4 | ND | ND | 64 | ND | ND | ND | 1 |
| Clostridium butyricum (CI) | ND | ND | ND | ND | 0.25 | ≤0.06 | 0.25 | 8 | ≤0.06 | ≤0.06 | ≤0.06 | ≤0.06 | ND | 8 | 4 | ≤0.06 | ≤0.06 |
| Non-spore-forming | |||||||||||||||||
| Actinomyces odontolyticus (CI) | 32 | 32 | >64 | 64 | ND | ND | ND | ND | ND | 64 | ND | ND | >64 | ND | ND | ND | 64 |
| Cutibacterium acnes (CI 1) | 64 | 64 | >64 | >64 | ND | ND | ND | ND | ND | 64 | ND | ND | >64 | ND | ND | ND | >64 |
| Cutibacterium acnes (CI 2) | ND | ND | ND | ND | 16 | 16 | 32 | >32 | 16 | 8 | 32 | 8 | ND | 16 | 8 | >16 | >64 |
| Cutibacterium acnes (CI 3) | >64 | >64 | >64 | >64 | ND | ND | ND | ND | ND | 32 | ND | ND | >64 | ND | ND | ND | >64 |
|
|||||||||||||||||
| Gram positive cocci | |||||||||||||||||
| Parvimonas micra (ATCC 3870) | 0.125 | 0.25 | ≤0.06 | 0.25 | ≤0.06 | ≤0.06 | ≤0.06 | 0.125 | ≤0.06 | ≤0.06 | ≤0.06 | ≤0.06 | 0.25 | 0.5 | 1 | ≤0.06 | 0.125 |
| Finegoldia magna (CI) | 2 | 2 | 1 | 1 | 4 | 0.5 | 0.25 | 8 | 0.125 | 0.25 | 1 | 0.25 | 0.25 | 32 | >16 | 1 | 0.125 |
| Peptostreptococcus anaerobius (CI) | ND | ND | ND | ND | 1 | 0.25 | 1 | 8 | 0.5 | 0.25 | ≤0.06 | 0.25 | ND | 16 | 16 | 0.125 | 0.125 |
Several nitrones had MIC50 and MIC90 values against B. fragilis 8 and 16 fold lower than MTZ and related compounds,35 and their activity also compared favourably to other reference drugs like clindamycin and amoxicillin-clavulanate.38 MICs of 1c,k,l were lower than those reported for these compounds also for F. nucleatum.35,38
Regarding clostridia, compound 1k had MIC values significantly lower than MTZ and some analogues as well as other reference drugs for C. difficile.36–39 It is striking that most compounds show a lower MIC90 against C. difficile than that observed with C. perfringens. This is an interesting feature, since C. difficile has high levels of resistance62–64 to many antibacterial drugs (β-lactams including carbapenems, quinolones, clindamycin and rifampicin, among others) except for vancomycin and metronidazole, while C. perfringens remains susceptible to a large number of antimicrobials, including penicillin.
The activity of each nitrone did not vary significantly against C. difficile isolates. This is important since C. difficile has a highly mobile, mosaic genome and there is wide strain diversity,65 resulting sometimes in activity variations among new drug candidates.29
3.3 Structure–activity relationship (SAR)
General trends are presented in this section. Non-sporulated Gram-positive bacilli, against which this family of compounds was only slightly active, were excluded from the analysis.In order to analyze the influence of some model electronic variations in the aryl moiety in position α− to the N-oxide function we compared compounds 1a–d, 1i–1l, 2a–c. Derivatives with a 4-nitro group were considerably more active, while 4-H, 4-Cl, and 4-OCH3 substitution did not lead to significant differences, although 4-H compounds were generally slightly more active. Additionally, replacement of the phenyl moiety in 1a by a thienyl ring (1g) subtly enhanced the activity, while a benzyl substituent (1h) had a negative influence. In summary, besides p-nitrophenyl substitution, the remaining electronic variations did not significantly improve the antimicrobial activity.
Introduction of a gem-dimethyl group in the methylene chain of a pyrimido derivative (1e) resulted in a slightly more active compound than its counterpart 1d. On the contrary, quaternization of the amidine nitrogen was generally not favourable for antianaerobic activity since amidinium salts 2a–c were in almost all cases considerably less active than the corresponding N-oxides 1a–c. Concerning the fused ring, replacement of the phenyl ring in compound 1a by a pyridine core (1f) improved the activity. This suggests that EWGs in the fused ring enhance the antianaerobic activity of these heterocycles.
When comparing compounds 1b, 1j and 1m, no significant differences in the activity between 6-and 7-membered homologues was observed, although 1m was slightly more active against C. difficile and C. perfringens than 1b. On the other hand, imidazoquinoxaline 1j was notoriously more active than its six- and seven-membered homologues. The trend was confirmed when comparing the remaining derivatives 1i,k–l with their counterparts 1a,c–d: in every case, the 5-membered derivatives were more active than their higher homologues. Among imidazoquinoxalines 1i–l, the 4-nitrophenyl derivative (1k) was again the most active compound.
From the previous analysis it turns out that the most significant structural variations are the presence of a 5-membered amidine ring, a fused pyridine core and a p-nitrophenyl substituent α− to the N-oxide function. Compound 1k, containing two out of the three favourable structural features, namely the five-membered amidine ring and the p-nitrophenyl substituent, was the most active in the series. Although the nitro group is generally not sought for during drug discovery due to safety issues, there are many therapeutic agents that include it in their composition such as antibacterials and antiparasitics, among others.66
3.4 Analysis of the probable mechanism of action
The activity spectrum of the new molecules is analogous to that of metronidazole. Aerobic bacteria and non-sporulated Gram positive bacilli show resistance both to metronidazole and amidinoquinoxaline N-oxides, which would in principle suggest a similar mechanism of action. Metronidazole enters the cell as a prodrug by passive diffusion and is reduced to the corresponding nitro radical anion by electron carriers in an anaerobic environment.67,68 The active form of the drug interacts with DNA by a mechanism not fully elucidated, causing DNA damage and non specific macromolecular alterations leading to cell death.69,70 Similarly, many N-oxides have been described as bioreducible drugs, i.e. they are inactive per se, but become cytotoxic after a reduction step which in many cases requires hypoxic conditions.71 According to these facts, it would be reasonable that nitrones 1 and 2 would not exert their action as such but after activation through a bioreduction step. To investigate this mechanistic hypothesis, we calculated the electron affinity (EA) of the compounds (Table 4), as an indicator of their redox potential, and related it to their antimicrobial activity as logCIM90 (μM).| Compounds | EAcalc − ΔH (kcal mol−1) |
|---|---|
| 1a | 24.5 |
| 1b | 29.2 |
| 1c | 43.2 |
| 1d | 22.7 |
| 1e | 23.8 |
| 1f | 28.4 |
| 1g | 25.6 |
| 1h | 21.7 |
| 1i | 28.1 |
| 1j | 32.2 |
| 1k | 46.9 |
| 1l | 26.0 |
| 1m | 29.0 |
| 2a | 117.6 |
| 2b | 120.2 |
| 2c | 126.1 |
Fig. 2 shows the relationship between the calculated EA and logCIM90 (with CIM values expressed as μM) for C. difficile, chosen as a representative example due to its clinical relevance. Gram negative bacilli were extremely susceptible and the MIC90 was in many cases less than the minimum concentration tested (Table 2, values ≤0.06 μg mL−1). Even so, results presented in Fig. 2 were consistent for the 6 groups of bacteria classified according to Table 2.
 | ||
| Fig. 2 Relationship between calculated electron affinity and log CIM90 for C. difficile. Six-membered ring amidines are shown in blue, five-membered analogues in orange and the 7-membered ring nitrone in violet; quaternary salts are in green. | ||
Fig. 2 shows that compounds with the highest EA are the most active within each group. The behavior is not linear since, as expected, antibacterial activity does not depend exclusively on a single parameter such as electronic affinity. Imidazoquinoxaline derivatives 1i–l always show higher electron affinities and are more active than the homologous pyrimidoquinoxalines 1a–d. Additionally, pyrimidoquinoxaline 1b and diazepinoquinoxaline N-oxides 1m have comparable electron affinity values and display similar activities.
The relationship between the electron affinity and antibacterial activity supports the hypothesis that the mechanism of action of the N-oxides would include a reduction step to transform the compound into its active form. Compounds 1c,k and 2c, where both the N-oxide and the nitro functional groups may undergo reduction, are more complex to analyze, although it is worth highlighting that these compounds were the most active within each series.
3.5 Cytotoxicity
Cell viability data at 100 μg mL−1 (the maximum concentrations tested) are shown in Table 5. In all cases CC50 were higher than 100 μg mL−1. Considering that values of CC50 > 50 μg mL−1 correspond to non-cytotoxic compounds,72 the results of these tests are excellent and show the low cytotoxicity of the compounds in the eukaryotic cell model RAW 264.7.| Comp. | Cell viability (%) | Selectivity index (>100 μg mL−1/MIC90) | |||||
|---|---|---|---|---|---|---|---|
| B. fragilis | Other Bacteroides spp. and Parabacteroides | Prevotella spp. | Fusobacterium nucleatum | C. difficile | C. perfringens | ||
| a Mtz = metronidazole.b ND: not determined.c Compound 1m was not tested in this study. | |||||||
| 1a | 98 | >100 | >100 | >100 | >100 | >25 | >6 |
| 1b | 94 | >50 | >100 | >100 | >100 | >6 | >2 |
| 1c | 74 | >1667 | >1667 | >800 | >1667 | >200 | >50 |
| 1d | 94 | >50 | >50 | >100 | >50 | >13 | >3 |
| 1e | 94 | >200 | >200 | >200 | >400 | >13 | >13 |
| 1f | 83 | >1667 | >1667 | >800 | >800 | >200 | >100 |
| 1g | 88 | >200 | >200 | >200 | >200 | >50 | >8 |
| 1h | 61 | >25 | >25 | >25 | >25 | >6 | ND |
| 1i | 91 | >1667 | >1667 | >1667 | >800 | >200 | >100 |
| 1j | 75 | >800 | >800 | >1667 | >800 | >400 | >100 |
| 1k | 63 | >1667 | >1667 | >1667 | >1667 | >1667 | >800 |
| 1l | 59 | >1667 | >1667 | >1667 | >1667 | >200 | >100 |
| 2a | 98 | >50 | >25 | >12.5 | >25 | >3 | >3 |
| 2b | 67 | >13 | >13 | >13 | >13 | >3 | >3 |
| 2c | 59 | >100 | >100 | >100 | >100 | >200 | >100 |
The Selectivity Index (SI), defined as SI = CC50/MIC, allows to relate the MIC value with the cytotoxicity. The greater the SI, the higher is the cytotoxic concentration (represented by the CC50) with respect to the active concentration (symbolized by the MIC), and the greater the probability that the compounds will not be toxic to host cells in vivo. According to literature reports73 values of IS ≥ 10 are considered suitable. SI values calculated as SI > 100 μg mL−1/CIM90 for the six bacteria groups presented in Table 2, show very promising results (Table 5). Even if the most favorable structural variations in terms of antibacterial activity seem to be associated with higher cytotoxicity of the compounds (Table 5, second column), the increase in antibacterial activity is so important that the Selectivity Indexes still show very favourable results. Interestingly, the most active compounds of the series 1k, shows IS > 800 for all groups of bacteria. Results presented in Table 5 demonstrate the low toxicity of the compounds in the eukaryotic cell model, in comparison to their antianaerobic activity.
3.6 ADME properties
The predominant role of pharmacokinetics in drug discovery has been recognized for years. Ideally, a new drug should be target-specific, orally-absorbed, cause minimal or no adverse effects and be distributed and excreted in a way that allows its administration once a day.74,75 Many drug candidates fail in clinical trials due to their pharmacokinetic properties, causing enormous loss of time and funds. Therefore in silico ADME (absorption, distribution, metabolism, and excretion) predictions are nowadays an important tool to identify problematic compounds at an early stage and to rationalize the overall development progression. The drug-like properties of the N-oxides 1 were calculated as a first approximation to investigate the pharmacokinetic features (ADME) of the new compounds. Selected molecular descriptors are presented in Table 6, which also includes the experimental logP values of some derivatives.50| Comp | CNS | MW | SASA | Vol | DHB | AHB | logP | logS | PCaco | logBB | PMDCK | nM | HOA | %HOA | PSA | VR5 | VR3 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a MW: molecular weight (recommended range 130–725 Da); SASA: total solvent-accessible surface area (recommended range 300.0–1000.0); Vol: total solvent-accessible volume (recommended range 500.0–2000.0); DHB: estimated number of hydrogen bond donors (recommended range 0.0–6.0); AHB: estimated number of hydrogen bond acceptors (recommended range 2.0–20.0); logP: experimental log of the octanol/water partition coefficient determined in PBS buffer (data taken from ref. 47) (recommended range −2.0 – 6.5); logS: predicted aqueous solubility (recommended range −6.5 – 0.5); PCaco: predicted apparent Caco-2 cell permeability (<25 poor, >500 great); logBB: predicted log of the brain/blood partition coefficient (recommended range −3.0 – 1.2); PMDCK: predicted apparent MDCK cell permeability (<25 poor, >500 great); nM: number of likely metabolic reactions (recommended range 1 – 8); HOA: qualitative human oral absorption – 1, 2, or 3 for low, medium, or high; % HOA: predicted human oral absorption (>80% is high <25% is poor); PSA: polar (N and O) van der Waals surface area (recommended range 7.0–200); VR5: number of violations for Lipinski's rule of five (MW < 500, logP < 5, DHB ≤ 5, accptHB ≤ 10); VR3: number of violations for Jörgensen's rule of three: logS > −5.7; PCaco > 22 nm s−1; nM < 7. The Qikprop ranges/recommended values were determined with 95% of known drugs. | |||||||||||||||||
| 1a | 1 | 277.325 | 523.783 | 902.771 | 0 | 2.500 | −0.149 | −4.671 | 4160.303 | 0.147 | 2309.675 | 3 | 3 | 100.000 | 32.038 | 0 | 0 |
| 1b | 1 | 311.770 | 545.805 | 944.637 | 0 | 2.500 | 0.836 | −5.400 | 4205.739 | 0.319 | 5764.794 | 3 | 3 | 100.000 | 31.973 | 0 | 0 |
| 1c | −1 | 322.323 | 562.589 | 977.280 | 0 | 3.500 | 0.657 | −4.720 | 466.578 | −0.859 | 217.018 | 3 | 3 | 94.215 | 78.769 | 0 | 0 |
| 1d | 1 | 307.351 | 558.782 | 975.816 | 0 | 3.250 | −0.128 | −4.779 | 4179.554 | 0.080 | 2321.229 | 4 | 3 | 100.000 | 40.297 | 0 | 0 |
| 1e | 1 | 335.405 | 598.020 | 1071.044 | 0 | 3.250 | — | −5.522 | 4378.159 | 0.095 | 2440.677 | 4 | 3 | 100.000 | 39.808 | 0 | 0 |
| 1f | 1 | 278.313 | 516.286 | 889.391 | 0 | 3.500 | 0.311 | −4.005 | 3398.734 | 0.068 | 1856.269 | 3 | 3 | 100.000 | 41.993 | 0 | 0 |
| 1g | 1 | 283.347 | 500.778 | 868.766 | 0 | 2.500 | −0.010 | −4.636 | 4678.076 | 0.401 | 5214.276 | 4 | 3 | 100.000 | 31.116 | 0 | 0 |
| 1h | 0 | 291.352 | 591.760 | 1000.602 | 0 | 2.500 | — | −5.796 | 3341.608 | −0.053 | 1822.568 | 3 | 3 | 100.000 | 33.186 | 0 | 1 |
| 1i | 1 | 263.298 | 502.438 | 854.774 | 0 | 2.500 | — | −4.267 | 4124.038 | 0.147 | 2287.921 | 2 | 3 | 100.000 | 33.198 | 0 | 0 |
| 1j | 1 | 297.743 | 525.850 | 898.135 | 0 | 2.500 | 1.292 | −5.073 | 4093.646 | 0.311 | 5601.908 | 1 | 3 | 100.000 | 33.222 | 0 | 0 |
| 1k | −1 | 308.296 | 543.140 | 931.304 | 0 | 3.500 | — | −4.352 | 458.238 | −0.859 | 212.828 | 2 | 3 | 92.378 | 80.021 | 0 | 0 |
| 1l | 1 | 293.324 | 540.213 | 930.486 | 0 | 3.250 | 1.247 | −4.427 | 4111.646 | 0.074 | 2280.490 | 3 | 3 | 100.000 | 41.477 | 0 | 0 |
| m | 1 | 325.797 | 563.828 | 986.954 | 0 | 2.500 | 1.247 | −4.427 | 4111.646 | 0.074 | 2280.490 | 3 | 3 | 100.000 | 41.477 | 0 | 0 |
Nitrones 1 comply with Lipinski's rule of five,76 Jorgensen's rule of three (except for 1h whose logS is too low),77 indicating that the new compounds are drug-like and, according to their predicted bioavailability, could be orally administered. Other individual parameters like logS, logP, PCaco and the predicted HOA are also very encouraging concerning the gut-blood barrier penetration, making these nitrones good candidates for oral absorption. This is a fundamental feature in a potential drug, since the oral route facilitates its administration and contributes to patient's compliance.
4 Conclusions
The activity of 13 amidinoquinoxaline N-oxides 1 and 3 related quaternary salts with selected structural variations against Gram-positive and Gram-negative anaerobic bacteria was evaluated, as well as their cytotoxicity and ADME (calculated) properties. Six and seven membered derivatives were synthesized by a method previously developed by our group, which was not efficient for imidazoquinoxaline N-oxides. Such compounds were prepared in high yields by base-promoted ring closure of the quinoxaline ring.The majority of the N-oxides under study showed high to excellent antianaerobic activity together with low cytotoxicity and suitable selectivity indexes, with an activity spectrum similar to that of metronidazole. Among them, six out of 13 compounds (1c,f,i–l) exhibited MIC90 values significantly lower than metronidazole, its analogues and other reference drugs. It is also remarkable that these compounds are very active against C. difficile, a multidrug-resistant anaerobe which can cause severe intestinal disease. Regarding their structure–activity relationship, the dominating factor was the ring size of the amidine ring: derivatives containing imidazoline rings were the most active within each series. Substitution with strong electron withdrawing groups either in the aryl moiety adjacent to the N-oxide function or in the benzofused ring significantly improved the activity. A combination of both relevant structural features led to the most active compound 1k. The N-oxides also showed favourable drug likeness profiles.
It is known that metronidazole, a first choice antianaerobic agent, is a prodrug which needs a bioreductive step to yield the active species. A preliminary study on the mechanism suggested that the N-oxides 1 would also undergo reductive activation in the biological medium in order to exert their activity. In fact, compounds with the highest electron affinities were the most active within each group. Taken together with the ease of preparation of the compounds (3–4 steps with high overall yields), all these features make amidinoquinoxalines N-oxides attractive candidates for further studies on their therapeutic potential.
Author contributions
Synthesis: NG, NBK, JAB, MBG, LROMIC determinations: LFC, NG Cytotoxicity: DGM, MLP, NG. Experimental planning: NG, LFC, DGM, LRO. DFT calculations: PS, NG. Paper writing: NG, LRO.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the Universidad de Buenos Aires [Grant 20020170100189BA] and by CONICET [Grant 11220200101694CO].References
- H. Jousimies-Somer, P. Summanen, D. M. Citron, E. J. Baron, H. M. Wexler and S. M. Finegold, Wadsworth-KTL Anaerobic Bacteriology Manual, Star, Corea, 6th edn, 2002 Search PubMed.
- I. Brook, Antimicrobial Resistance of Anaerobic Bacteria, in Antimicrobial Drug Resistance, ed. D.L. Mayers, J. D. Sobel, M. Ouellette, K. S. Kaye and D. Marchaim, Springer International Publishing, 2017, p. 1007 Search PubMed.
- S. Löfmark, C. Edlund and C. E. Nor, Clin. Infect. Dis., 2010, 50, 16 CrossRef PubMed.
- (a) M. A. Pence, Clin. Microbiol. Newsl., 2019, 41, 1 CrossRef; (b) L. Fernández-Canigia, M. Litterio, M. C. Legaria, L. Castello, S. C. Predari, A. Di Martino, A. Rossetti, R. Rollet, G. Carloni, H. Bianchini, D. Cejas, M. Radice and G. Gutkind, Anaerobe Surveillance Team, Antimicrob. Agents Chemother., 2012, 56(3), 1309, DOI:10.1128/AAC.05622-11.
- H. R. Ingham, S. Eaton, C. W. Venables and P. C. Adams, Lancet, 1978, 1, 214 CrossRef CAS PubMed.
- J. S. Brazier, S. L. Stubbs and B. I. Duerden, J. Antimicrob. Chemother., 1999, 44, 580 CrossRef CAS PubMed.
- V. O. Rotimi, M. Khoursheed, J. S. Brazier, W. Y. Jamal and F. B. Khodakhast, Clin. Microbiol. Infect., 1999, 5, 166 CrossRef PubMed.
- S. P. Sadarangani, S. A. Cunningham, P. R. Jeraldo, J. W. Wilson, R. Khare and R. Patel, Antimicrob. Agents Chemother., 2015, 59, 4157, DOI:10.1128/AAC.00677-15.
- A. A. Elsaghier, J. S. Brazier and E. A. James, J. Antimicrob. Chemother., 2003, 51(6), 1436, DOI:10.1093/jac/dkg265.
- C. Alauzet, A. Lozniewski and H. Marchandin, Anaerobe, 2019, 55, 40 CrossRef CAS PubMed.
- S. Sethi, R. Shukla, K. Bala, V. Gautam, A. Angrup and P. Ray, J. Global Antimicrob. Resist., 2019, 16, 210 CrossRef PubMed.
- (a) A. N. Schuetz, Anaerobic Bacteria: Antimicrobial Susceptibility Testing and Resistance Patterns, in Antimicrobial Resistance in the 21st Century, Emerging Infectious Diseases of the 21st Century, ed. I. W. Fong, D. Shlaes and K. Drlica, Springer International Publishing AG, 2018, DOI:10.1007/978-3-319-78538-7_6; (b) A. C. M. Veloo, H. B. Tokman, H. Jean-Pierre, Y. Dumont, S. Jeverica, R. Lienhard, A. Novak, A. Rodloff, V. Rotimi, I. Wybo and E. Nagy, ESGAI study group, Anaerobe, 2020, 61, 102111, DOI:10.1016/j.anaerobe.2019.102111; (c) B. Fox, M. A. Berger, M. Roncallo, L. Pinoche, M. E. Ibáñez, S. González-Fraga and L. Fernández-Canigia, Anaerobe, 2018, 54, 267, DOI:10.1016/j.anaerobe.2018.04.003; (d) S. C. Predari, A. N. de Paulis, E. Bertona, D. Guevara Núñez, J. P. Suárez and L. Castello, Rev. Argent. Microbiol., 2017, 49(2), 146, DOI:10.1016/j.ram.2016.12.008.
- K. M. Land and P. J. Johnson, Drug Resistance Updates, 1999, 2, 289 CrossRef CAS PubMed.
- L. Pumbwe, A. Chang, R. L. Smith and H. M. Wexler, Microb. Drug Resist., 2007, 13, 96, DOI:10.1089/mdr.2007.719.
- R. Ghotaslou, M. Yekani and M. Y. Memar, Microbiol. Res., 2018, 210, 1 CrossRef CAS PubMed.
- R. Ghotaslou, H. B. Baghi, N. Alizadeh, M. Yekani, S. Arbabi and M. Y. Memar, Infect., Genet. Evol., 2018, 64, 156 CrossRef CAS PubMed.
- H. K. Leiros, S. Kozielski-Stuhrmann, U. Kapp, L. Terradot, G. A. Leonard and S. M. McSweeney, J. Biol. Chem., 2004, 279, 55840 CrossRef CAS PubMed.
- G. Reysset, Anaerobe, 1996, 2, 59 CrossRef CAS PubMed.
- M. M. Theron, M. N. Janse Van Rensburg and L. J. Chalkley, J. Antimicrob. Chemother., 2004, 54, 240 CrossRef CAS PubMed.
- S. Lofmark, H. Fang, M. Hedberg and C. Edlund, Antimicrob. Agents Chemother., 2005, 49, 1253 CrossRef PubMed.
- E. Otte, H. L. Nielsen, H. Hasman and D. Fuglsang-Damgaard, Anaerobe, 2017, 43, 91 CrossRef PubMed.
- M. M. Theron, M. N. Janse van Rensburg and J. Chalkley, J. Antimicrob. Chemother., 2004, 54, 240 CrossRef CAS PubMed.
- (a) F. C. Lessa, Y. Mu, W. M. Bamberg, Z. G. Beldavs, G. K. Dumyati, J. R. Dunn, M. M. Farley, S. M. Holzbauer, J. I. Meek, E. Phipps, L. E. Wilson, L. G. Winston, J. A. Cohen, B. M. Limbago, S. K. Fridkin, D. N. Gerding and L. C. McDonald, N. Engl. J. Med., 2015, 372(9), 825, DOI:10.1056/NEJMoa1408913; (b) A. Cheknis, S. Johnson, L. Chesnel, L. Petrella, S. Sambol, S. E. Dale, J. Nary, P. Sears, D. M. Citron, E. J. C. Goldstein and D. N. Gerding, Anaerobe, 2018, 53, 38, DOI:10.1016/j.anaerobe.2018.05.009.
- https://www.cdc.gov/hai/organisms/cdiff/cdiff_infect.html.
- K. Z. Vardakas, K. A. Polyzos, K. Patouni, P. I. Rafailidis, G. Samonis and M. E. Falagas, Int. J. Antimicrob. Agents, 2012, 40, 1 CrossRef CAS PubMed.
- Z. Peng, D. Jin, H. B. Kim, W. Stratton, B. Wu, Y.-W. Tang and X. Sun, J. Clin. Microbiol., 2017, 55(7), 1998, DOI:10.1128/JCM.02250-16.
- A. M. Jarrad, T. Karoli, M. A. T. Blaskovich, D. Lyras and M. A. Cooper, J. Med. Chem., 2015, 58(13), 5164, DOI:10.1021/jm5016846.
- A. Malath Al-Qtaitat, A. Haythem, A. Saadeh, A. G. Al-Bakri, K. Hargobinder, G. Kapil, R. Sehgal and M. S. Mubarak, Monatsh. Chem., 2015, 146, 705, DOI:10.1007/s00706-014-1352-0.
- Y. Miyamoto, J. Kalisiak, K. Korthals, T. Lauwaet, D. Young Cheung, R. Lozano, E. R. Cobo, P. Upcroft, J. A. Upcroft, D. E. Berg, F. D. Gillin, V. V. Fokin, K. B. Sharpless and L. Eckmann, Proc. Natl. Acad. Sci. U.S.A., 2013, 110(43), 17564 CrossRef CAS PubMed.
- H. A. Saadeh, I. M. Mosleh, A. G. Al-Bakri and M. S. Mubarak, Monatsh. Chem., 2010, 141, 471, DOI:10.1007/s00706-010-0281-9.
- M. M. Bkhaitan, M. Alarjah, A. Z. Mirza, A. N. Abdalla, H. M. El-Said and H. S. Faidah, Chem. Biol. Drug Des., 2018, 92, 1954, DOI:10.1111/cbdd.13366.
- A. A. Oliveira, A. P. A. Oliveira, L. L. Franco, M. O. Ferencs, J. F. G. Ferreira, S. M. P. S. Bachi, N. L. Speziali, L. M. Farias, P. P. Magalhães and H. Beraldo Alexandre, BioMetals, 2018, 31, 571, DOI:10.1111/cbdd.13366.
- B. R. Prajapati and A. K. Seth, Pharma Sci. Monit., 2018, 9(1), 318 CAS.
- T. E. Ballard, X. Wang, I. Olekhnovich, T. Koerner, C. Seymour, P. S. Hoffman and T. L. Macdonald, Bioorg. Med. Chem. Lett., 2010, 20, 3537, DOI:10.1016/j.bmcl.2010.04.126.
- T. E. Ballard, X. Wang, I. Olekhnovich, T. Koerner, C. Seymour, J. Salamoun, M. Warthan, P. S. Hoffman and T. L. Macdonald, ChemMedChem, 2011, 6, 362, DOI:10.1002/cmdc.201000475.
- G. A. Pankuch and P. C. Appelbaum, Antimicrob. Agents Chemother., 2006, 50, 1112, DOI:10.1128/AAC.50.3.1112–1117.2006.
- C. Costello, T. Karpanen, P. A. Lambert, P. Mistry, K. J. Parker, D. L. Rathbone, J. Ren, L. Wheeldon and T. Worthington, Bioorg. Med. Chem. Lett., 2008, 18, 1708, DOI:10.1016/j.bmcl.2008.01.041.
- Antibacterial agents in clinical development: an analysis of the antibacterial clinical development pipeline, including tuberculosis, World Health Organization, Geneva, 2017, WHO/EMP/IAU/2017.11 Search PubMed.
- G. E. Adams, E. M. Fielden, M. A. Naylor and I. J. Stratford, UK Patent Application No: GB2257360, 1993.
- M. A. Naylor, M. A. Stephens, J. Nolan, B. Sutton, J. H. Tocher, E. M. Fielden, G. E. Adams and I. J. Stratford, Anti Cancer Drug Des., 1993, 8, 439 CAS.
- (a) M. A. Naylor, Oncol. Res., 1994, 6, 483 CAS; (b) M. A. Naylor, G. E. Adams, A. Haigh, S. Cole, T. Jenner, N. Robertson, D. Siemann, M. A. Stephens and I. J. Stratford, Anti Cancer Drugs, 1995, 6, 259 CrossRef CAS PubMed; (c) H. M. Barham and I. J Stratford, Biochem. Pharmacol., 1996, 51, 829 CrossRef CAS PubMed; (d) B. M. Sutton, N. J. Reeves, M. A. Naylor, E. M. Fielden, S. Cole, G. E. Adams and I. J. Stratford, Int. J. Radiat. Oncol., Biol., Phys., 1994, 29, 339 CrossRef CAS PubMed.
- P. C. Parthasarathy, B. S. Joshi, M. R. Chaphekar, D. H. Gawad, L. Anandan, M. A. Likhate, M. Hendi, S. Mudaliar, S. Iyer, D. K. Ray and V. B. Srivastava, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1983, 22, 1250 Search PubMed.
- G. J. Ellames and A. A. Jaxa-Chamiec, US Pat., 4696928, 1987.
- G. J. Ellames, K. R. Lawson, A. A. Jaxa-Chamiec and R. M. Upton, European Pat., EP0256545, 1988.
- G. J. Ellames, R. M. Upton, A. A. Jaxa-Chamiec and P. L. Myers, US Pat., 4761414, 1988.
- J. Segreti, L. J. Goodman and G. M. Trenholme, Diagn. Microbiol. Infect. Dis., 1993, 17, 177 CrossRef CAS PubMed.
- N. Gruber, L. Orelli, C. Minnelli, L. Mangano, E. Laudadio, G. Mobbili and P. Stipa, Antioxidants, 2021, 10, 1185, DOI:10.3390/antiox10081185.
- N. Gruber, L. L. Piehl, E. Rubin de Celis, J. E. Díaz, M. B. García, P. Stipa and L. R. Orelli, RSC Adv., 2015, 5, 2724 RSC.
- N. Gruber, L. R. Orelli, R. Cipolletti and P. Stipa, Org. Biomol. Chem., 2017, 15, 7685 RSC.
- M. B. García, L. R. Orelli, M. L. Magri and I. A. Perillo, Synthesis, 2002, 2687 Search PubMed.
- N. A. Isley, R. T. H. Linstadt, S. M. Kelly, F. Gallou and B. H. Lipshutz, Org. Lett., 2015, 17(19), 4734 CrossRef CAS PubMed.
- Clinical and Laboratory Standards Institute (CLSI), Methods for antimicrobial susceptibility testing of anaerobic bacteria, Approved standard M11-A7, CLSI, Wayne, PA, 7th edn, 2007 Search PubMed.
- Clinical and Laboratory Standards Institute (CLSI), Performance standards for antimicrobial susceptibility testing; 24th informational supplement, CLSI, Wayne, PA, 2012, CLSI M100-S22 Search PubMed.
- Manual of clinical microbiology, ed. J. H. Jorgensen, M. Pfaller and K. C. Carroll, ASM press, Washington, DC, 11th edn, 2015, vol. 1 Search PubMed.
- J. O'Brien, I. Wilson, T. Orton and F. Pognan, Eur. J. Biochem., 2000, 267, 5421, DOI:10.1046/j.1432-1327.2000.01606.x.
- (a) R. G. Parr and W. Yang, Density Functional Theory of Atoms and Molecules, Oxford University, New York, NY, 1998 Search PubMed; (b) W. Koch and M. C. Holthausen, A Chemist's Guide to Density Functional Theory, Wiley-VCH, Weinheim, Germany, 2000 Search PubMed.
- M. J. Frisch, et al., Gaussian 09 (Revision D.01), Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- J. C. Rienstra-Kiracofe, G. S. Tschumper and H. F. Schaefer, Chem. Rev., 2002, 102, 231 CrossRef CAS PubMed.
- H. van de Waterbeemd, D. A. Smith, K. Beaumont and D. K. Walker, J. Med. Chem., 2001, 44, 1313 CrossRef CAS PubMed.
- (a) C. W. Muth, J. C. Ellers and O. F. Folmer, J. Am. Chem. Soc., 1957, 79, 6500 CrossRef CAS; (b) R. P. Barnes, J. H. Graham and M. A. Salim Qureshi, J. Org. Chem., 1963, 28, 2890 CrossRef CAS; (c) G. Tennant, J. Chem. Soc., 1963, 2428 RSC; (d) R. Fusco and S. Rossi, Gazz. Chim. Ital., 1964, 94, 3 CAS.
- I. Perillo, B. Fernandez and S. Lamdan, J. Chem. Soc., Perkin Trans. 1, 1977, 2068 RSC.
- O. Aspevall, A. Lundberg, L. G. Burman, T. Åkerlund and B. Svenungsson, Antimicrob. Agents Chemother., 2006, 50, 1890 CrossRef CAS PubMed.
- J. Freeman, J. Vernon, K. Morris, S. Nicholson, S. Todhunter, C. Longshaw and M. H. Wilcox, Clin. Microbiol. Infect., 2015, 21, 248 Search PubMed.
- J. Arca-Suárez, F. Galán-Sánchez, F. Cano-Cano, G. García-Santos and M. A. Rodríguez-Iglesias, Anaerobe, 2018, 54, 146 CrossRef PubMed.
- M. Sebaihia, B. W. Wren, P. Mullany, N. F. Fairweather, N. Minton and R. Stabler, et al., Nat. Genet., 2006, 38, 779, DOI:10.1038/ng1830.
- K. Nepali, H.-Y. Lee and J.-P. Liou, J. Med. Chem., 2019, 62(6), 2851 CrossRef CAS PubMed.
- K. M. Land and P. J. Johnson, Drug Resistance Updates, 1999, 2, 289 CrossRef CAS PubMed.
- H. K. Leiros, S. Kozielski-Stuhrmann, U. Kapp, L. Terradot, G. A. Leonard and S. M. McSweeney, J. Biol. Chem., 2004, 279, 55840 CrossRef CAS PubMed.
- J. Muller, P. Schildknecht and N. Muller, J. Antimicrob. Chemother., 2013, 68, 1781, DOI:10.1093/jac/dkt106.
- C. G. Diniz, S. G. Santos, A. C. Pestana, L. M. Farias and M. A. Carvalho, Anaerobe, 2000, 6, 149 CrossRef CAS.
- H. Cerecetto and M. González, Mini-Rev. Med. Chem., 2001, 1, 219 CrossRef CAS PubMed.
- A. B. Patel, K. H. Chikhalia and P. Kumari, Eur. J. Med. Chem., 2014, 79, 57 CrossRef CAS PubMed.
- (a) R. Yendapally, J. G. Hurdle, E. I. Carson, R. B. Lee and R. E. Lee, J. Med. Chem., 2008, 51, 1487 CrossRef CAS PubMed; (b) T. Yempala, J. P. Sridevi, P. Yogeeswari, D. Sriram and S. Kantevari, Eur. J. Med. Chem., 2014, 71, 160 CrossRef CAS PubMed.
- K. M. Merz and J. J. Baldwin, J. Med. Chem., 2000, 43, 3867 CrossRef PubMed.
- U. Norinder and C. A. S. Bergström, ChemMedChem, 2006, 1, 920 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3 CrossRef CAS.
- W. L. Jorgensen, Acc. Chem. Res., 2009, 42, 724 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra01184d |
| ‡ Since metronidazole has a molecular weight of 171 g mol−1 and compounds 1 and 2 range between 263–464 g mol−1, this difference is accentuated if the results are expressed in molar concentration instead of μg mL−1 units. |
| This journal is © The Royal Society of Chemistry 2023 |