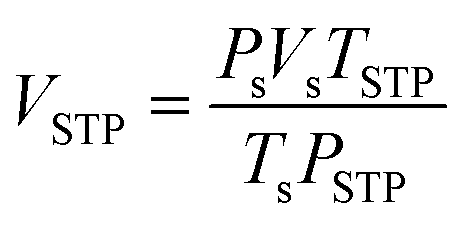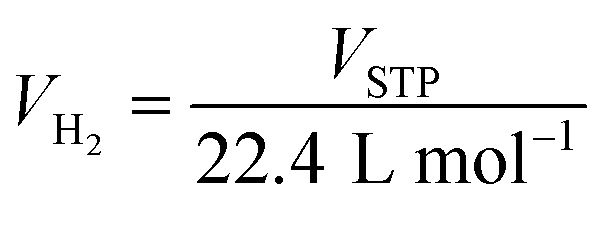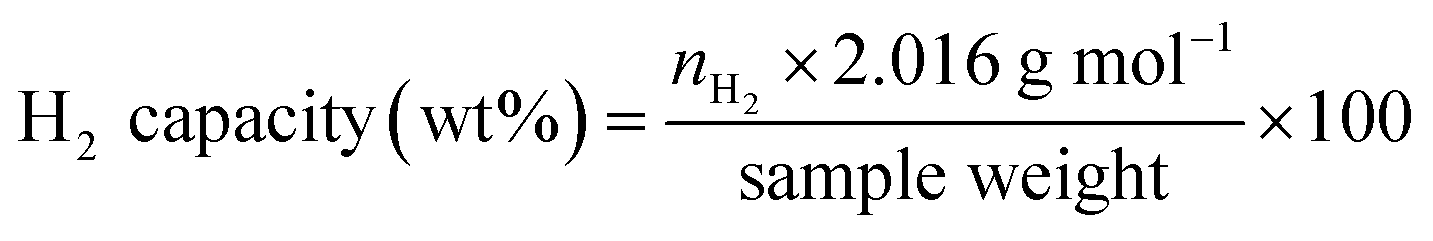DOI:
10.1039/D3RA01914D
(Paper)
RSC Adv., 2023,
13, 16926-16934
Effects of Ni precursors on the formation of Mg–Fe–Ni intermetallic hydrides, kinetics, and reversibility
Received
23rd March 2023
, Accepted 15th May 2023
First published on 5th June 2023
Abstract
This work focuses on the effects of Ni precursors (metallic Ni or Mg2NiH4) on the formation of Mg–Fe–Ni intermetallic hydrides as well as their de/rehydrogenation kinetics and reversibility. After ball milling and sintering, the formation of Mg2FeH6 and Mg2NiH4 are found in both samples, while MgH2 is observed only in the sample with metallic Ni. Both samples show comparable hydrogen capacities of 3.2–3.3 wt% H2 during the 1st dehydrogenation, but the sample with metallic Ni decomposes at a lower temperature (ΔT = 12 °C) and shows faster kinetics. Although phase compositions after dehydrogenation of both samples are comparable, their rehydrogenation mechanisms are different. This affects the kinetic properties upon cycling and reversibility. Reversible capacities of the samples with metallic Ni and Mg2NiH4 during the 2nd dehydrogenation are 3.2 and 2.8 wt% H2, respectively, while those during the 3rd–7th cycles reduce to ∼2.8 and 2.6 wt% H2, respectively. Chemical and microstructural characterizations are carried out to explain de/rehydrogenation pathways.
1. Introduction
The intermetallic hydride of Mg2FeH6 has been considered for hydrogen storage applications due to the highest volumetric hydrogen density (150 kg m−3) and relatively high gravimetric hydrogen density (5.5 wt% H2).1,2 Also, its high reaction enthalpy (∼90 kJ per mol H2) as well as high volumetric and gravimetric energy densities (0.49 kW h L−1 and 0.55 kW h kg−1, respectively) are suitable for the thermochemical energy storage medium.1,3–6 However, Mg2Fe intermetallic alloy is thermodynamically unfavorable and the significant difference in density and melting points of Mg and Fe hinders the formation of homogeneous alloys via metallurgical methods.7,8 Several procedures, such as thermal processes, mechanical milling, cold rolling, and high-pressure compressions have been applied to Mg + Fe or MgH2 + Fe mixtures for Mg2FeH6 syntheses.9–15 Hydrogenation of Mg to MgH2 catalyzed by Fe was first found at ∼200 °C and the obtained MgH2 further reacted with Fe to form Mg2FeH6 only at high temperature (∼350 °C) due to kinetic restriction from solid-solution diffusion processes.16,17 Moreover, reversibility of Mg2FeH6 via the reaction between MgH2 and Fe required high operating temperatures (T = 375–445 °C) to achieve reasonable hydrogen capacity.9,15
Quaternary intermetallic hydrides via partial substitution of transition metals (TMs) for Fe in Mg2FeH6 to form Mg2Fe(1−x)TMxH6 (TM = Cr, Ni, Mn, Co, and Y) have been proposed to enhance kinetics and reversibility. The samples were prepared by (i) milling MgH2 with the plain steel containing TM impurities (e.g., 316L stainless steel and γ-Fe(Ni) nanoparticles)8,18,19 and (ii) compositing TMs in metallic form or compounds with Mg + Fe, MgH2 + Fe, or Mg2FeH6.20–25 These processes increased Mg2FeH6 yield with the improved kinetic properties and reversibility. Immediate reaction between MgH2 and 316L SS via either reactive ball milling under hydrogen pressure or ball milling under Ar atmosphere and annealing under hydrogen pressure resulted in partial substitution of Fe with Cr and Ni to form Mg2(Fe, Cr, Ni)Hx.8,18 Such a faster reactivity with respect to pure iron was induced by martensitic transformation during ball milling and the presence of Ni in the system. Moreover, Mg2Fe(Ni)H6 with tangled nanowire morphology prepared using coarse-grained Mg powder and γ-Fe(Ni) nanoparticles showed lower desorption temperature by 20 °C as compared with Mg2FeH6.19 Catalytic effects on hydrogenation of Ni and Fe as well as comparable fcc lattice of γ-Fe(Ni) and Mg2FeH6, shortening Fe diffusion distance favored the formation of Mg2Fe(Ni)H6. Besides, NiFe-based catalysts favored hydrogen adsorption kinetics, resulting in the enhanced hydrogen evolution capability.26,27 Transition metal complex deuterides of Mg2FexCo(1−x)Dy (x = 0–1 and y = 5–6) prepared by reactive ball milling revealed comparable deuterium desorption temperatures at all compositions, but reversible reaction (T = 400 °C under 30 bar H2) with the enhanced kinetics was detected from Mg2Fe0.5Co0.5H5.5.21 Theoretical studies reported destabilization of Mg2FeH6, i.e., reduction of formation energy and desorption temperature via substitution of Fe with Ni, Co, and Mn.20 The most significant reduction of desorption enthalpy was expected from Mg2Fe0.75Ni0.25H6 (27.7 kJ per mol H).
Among Mg–Fe–TM intermetallic hydrides, Mg–Fe–Ni–H system shows remarkable hydrogen sorption kinetics, meanwhile all metallic compositions (Mg, Fe, and Ni) are inexpensive. From our previous work, Mg2Fe0.75Ni0.25H6 formed during dehydrogenation of 20 wt% Ni-doped Mg2FeH6 showed excellent reversible hydrogen capacities with respect to as-prepared Mg2FeH6, for example, hydrogen reproduction during the 2nd cycle increased from 78 to 85%.23 Besides, Ni-substituted contents in Mg2FeH6 was optimized by varying Mg2FeH6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mg2NiH4 mole ratios to obtain Mg2Fe(1−x)NixH6 with the best kinetics.25 It was found that dehydrogenation kinetics and reversibility were enhanced with Ni-substituted contents, and the most stable composition upon cycling was x ∼ 0.5 (Mg2Fe0.5Ni0.5H6). From these reports, it was found that different starting materials could alter Ni substitution degree in Mg2FeH6, i.e., 25 and 26–47% for the samples prepared from metallic Ni + MgH2 and Mg2FeH6 + Mg2NiH4, respectively. In this work, we would like to extend our study on the effects of Ni precursors on the formation and reversibility of Mg2Fe(1−x)NixH6. Two sample sets with the same stoichiometry of x = 0.25 using MgH2 + Fe + Ni and MgH2 + Fe + Mg2NiH4 mixtures as starting materials are ball milled and sintered under hydrogen pressure. De/rehydrogenation kinetics, reversibility, and hydrogen exchange pathways are investigated. Microstructural analyses are carried out to explain the effects of distribution and contacts among the reactive phases in nanometer range on hydrogen sorption mechanism.
:Mg2NiH4 mole ratios to obtain Mg2Fe(1−x)NixH6 with the best kinetics.25 It was found that dehydrogenation kinetics and reversibility were enhanced with Ni-substituted contents, and the most stable composition upon cycling was x ∼ 0.5 (Mg2Fe0.5Ni0.5H6). From these reports, it was found that different starting materials could alter Ni substitution degree in Mg2FeH6, i.e., 25 and 26–47% for the samples prepared from metallic Ni + MgH2 and Mg2FeH6 + Mg2NiH4, respectively. In this work, we would like to extend our study on the effects of Ni precursors on the formation and reversibility of Mg2Fe(1−x)NixH6. Two sample sets with the same stoichiometry of x = 0.25 using MgH2 + Fe + Ni and MgH2 + Fe + Mg2NiH4 mixtures as starting materials are ball milled and sintered under hydrogen pressure. De/rehydrogenation kinetics, reversibility, and hydrogen exchange pathways are investigated. Microstructural analyses are carried out to explain the effects of distribution and contacts among the reactive phases in nanometer range on hydrogen sorption mechanism.
2. Experimental
2.1 Sample preparation
Mg powder (≥99.0%, Sigma-Aldrich) was hydrogenated at 350 °C under 38–40 bar H2 for 12 h and milled for 1 h 30 min using a Retsch™ PM 100 Model Planetary Ball Mills. The rotational speed and the ball-to-powder weight ratio (BPR) were 500 rpm and 10:1, respectively. Hydrogenation and ball milling under similar conditions were repeatedly carried out until hydrogenation was complete to obtain as-prepared MgH2. Ni powder (99%, Alfa Aesar) was milled with as-prepared MgH2 under 1:2 mole ratio using milling time, BPR, and rotational speed of 5 h, 10:1, and 500 rpm, respectively. Hydrogenation of 2MgH2–Ni mixture was done at 350 °C under 40 bar H2 for 12 h to obtain as-prepared Mg2NiH4. As-prepared MgH2 was milled with the powder samples of Ni and Fe (99.9%, Sigma-Aldrich) with the mole ratio of 8:3:1 (MgH2:Fe:Ni) for 7 h 30 min using BPR and rotational speed of 15:1 and 500 rpm, respectively. The obtained mixture was sintered at 400 °C under 38–40 bar H2 for 48 h to obtain MgH2–Fe–Ni composite, denoted as S1. Fe powder was milled with as-prepared samples of MgH2 and Mg2NiH4 with the mole ratio of 6:3:1 (MgH2:Fe:Mg2NiH4) and the mixture was sintered under similar condition with S1 to produce MgH2–Fe–Mg2NiH4 composite, denoted as S2. The powder samples of S1 and S2 were heated to 500 °C and rehydrogenated at 350 °C under 40 bar H2 for 12 h to obtain S1′ and S2′, respectively.
2.2 Characterizations
Phase compositions of as-prepared and de/rehydrogenated samples were characterized by powder X-ray diffraction (PXRD) at ambient temperature using a Bruker D8 ADVANCE with Cu Kα radiation (λ = 1.5406 Å), a current of 40 mA, and a voltage of 40 kV. The powder sample was packed in an airtight sample holder covered with a poly(methyl methacrylate) dome in a nitrogen-filled glove box. The diffractogram was collected in the 2θ range, scanning step, and acquisition time of 10–80°, 0.02° s−1, and 400 s per step, respectively. Dehydrogenation profiles were investigated by differential scanning calorimetry (DSC) and thermogravimetry (TG) using a Netzsch STA449F3 Jupiter. The powder sample of 20–30 mg was heated from room temperature to 500 °C (5 °C min−1) under N2 flow (50 mL min−1). The relative signal of H2 released from the sample was characterized by mass spectroscopy (MS) using a Netzsch QMS 403C.
X-ray photo-electron spectroscopy (XPS) experiments were carried out at the SUTNANOTEC-SLRI Joint Research Facility, Synchrotron Light Research Institute (Public Organization), Thailand. A PHI5000 Versa Probe II (ULVAC-PHI Inc., Japan) with Al Kα (1.486 keV) radiation as an excitation source was used for characterizations. The powder samples were deposited on the sample holder using carbon glue tape in the glove box. Prior to the measurements, the samples were placed in the high vacuum chamber (1 × 10−8 mbar) for 2 h. The high-resolution scan of each element was collected using a pass energy of 46.95 eV and a step size of 0.05 eV. Dual-beam charge neutralization (low energy electron and ion beam) method was used to minimize sample charging. The binding energy was calibrated with respect to the C 1s peak (284.8 eV). The data was processed and analyzed by using a MultiPak software version 9.6.0 (ULVAC-PHI, Japan). Peak fitting was performed after Shirley background subtraction. Symmetrical Gaussian–Lorentzian function was used to approximate the line shapes of the fitting components.
De/rehydrogenation kinetics and reversibility were studied using a test station automatically controlled by the program developed in a Labview® environment.28,29 Two K-type thermocouples (TCs, −250–1300 °C, SL heater) were used to control and measure the system and sample temperatures during the experiments. Hydrogen release and supply during de/rehydrogenation were controlled by the direct-acting plunger solenoid valves (Type 0255, Bürkert) and the system pressure was detected by a pressure transducer with an operating range of 0–3000 psig (an OMEGA Engineering PX309-3KGI). Hydrogen content desorbed was measured using a mass flow controller (MFC, 0–0.1 standard L min−1 (SLM), a Bronkhorst EL-FLOW selected F-201CV). The signals of temperature, pressure, and mass flow rate were transferred to the computer using the module data loggers (a NI USB-6009, National Instruments and an AI210, Wisco). Hydrogenation was done under isothermal condition at the setting temperature (Tset) of 315 °C under 10–16 bar H2, while dehydrogenation was carried out at Tset = 315 °C by releasing hydrogen through MFC with the flow rate of 0.09 SLM. The volume of hydrogen desorbed was obtained from integrating the peak area of hydrogen flow rate (SLM) versus time (min) plots. The hydrogen storage capacity was calculated by the following equations.
| |
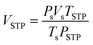 | (1) |
| |
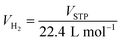 | (2) |
| |
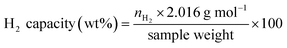 | (3) |
where
VSTP (L) and
Vs (SLM) are the volumes of hydrogen gas at the standard temperature and pressure condition (STP,
TSTP = 273.15 K and
PSTP = 1.0133 bar) and at the standard condition of MFC (
Ts = 296.15 K and
Ps = 1.0156 bar), respectively.
nH2 (mol) is hydrogen moles and standard molar volume is 22.4 L mol
−1.
Morphology and microstructure were characterized by transmission electron microscopy (TEM) technique using a Thermo Scientific TALOS F200X coupled with an energy dispersive X-ray spectroscopy (EDS) micro-analysis. An accelerating voltage of 200 kV was used. Sample preparation was done by ultrasonic dispersion of the powder sample in ethyl alcohol (99% AR grade, RCI Labscan) for 10–15 min and dropping onto a carbon grid.
3. Results and discussion
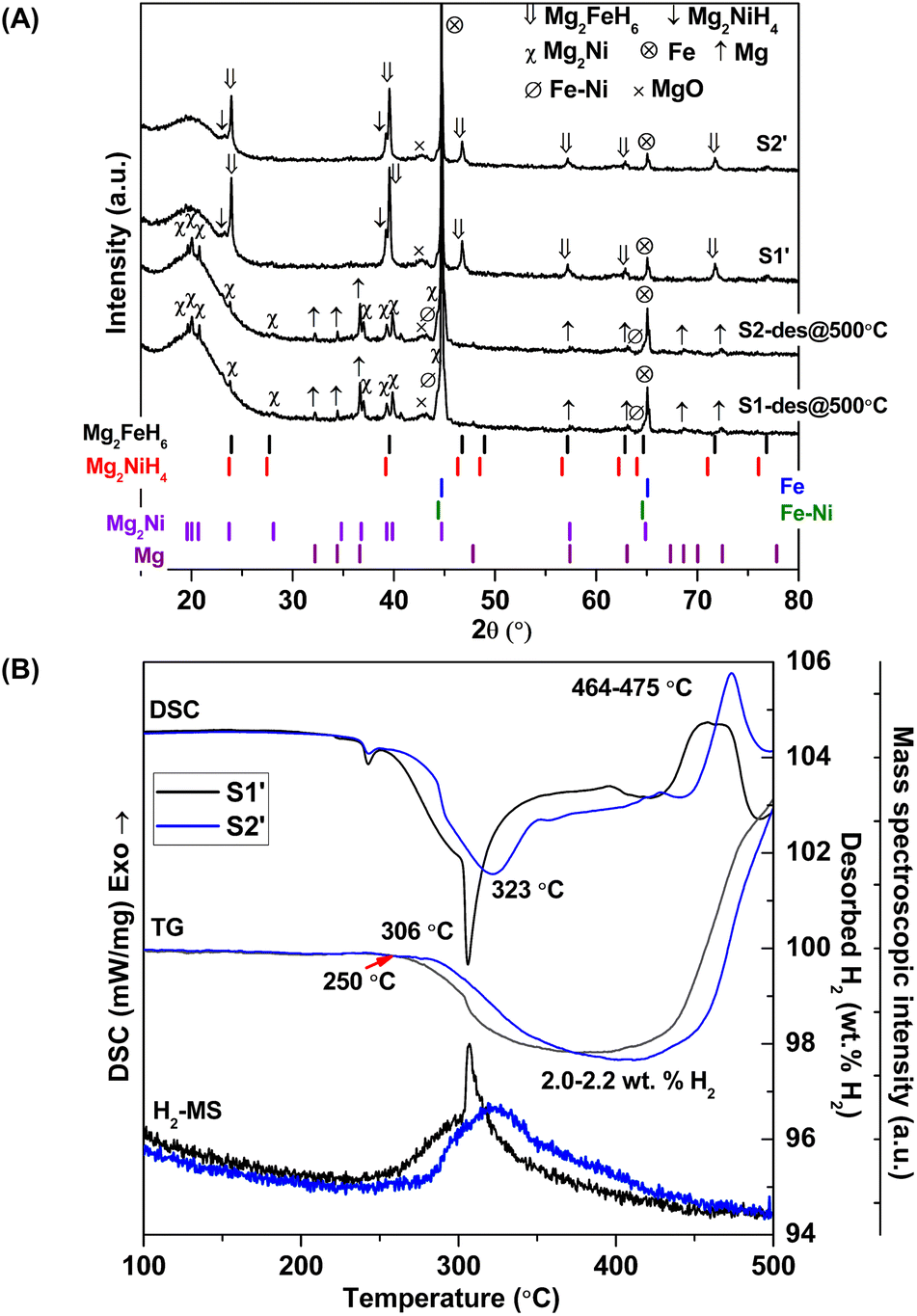
Chemical compositions of as-prepared S1 and S2 are characterized by PXRD technique. From Fig. 1A, PXRD spectra of S1 and S2 show the diffractions of Mg2FeH6, Mg2NiH4, Fe, and MgO as well as MgH2 and Fe–Ni alloy30 from S1 and S2, respectively. Upon milling and sintering, the formations of Mg2FeH6 and Mg2NiH4 confirm hydrogenation of MgH2 + Fe (eqn (4)) and Mg2Ni (eqn (5)), while that of Fe–Ni alloy is solid solution of Fe and Ni.30 MgO is obtained from oxidation of Mg-containing phases with oxygen and/or humidity.| | |
2MgH2(s) + Fe(s) + H2(g) → Mg2FeH6(s)
| (4) |
| | |
Mg2Ni(s) + 2H2(g) → Mg2NiH4(s)
| (5) |
 |
| | Fig. 1 PXRD spectra (A) and simultaneous DSC-TG-MS results (B) of as-prepared S1 and S2. | |
Dehydrogenation of S1 and S2 is investigated by simultaneous DSC-TG-MS experiments. From Fig. 1B, as-prepared S1 and S2 show single-step decomposition at comparable onset dehydrogenation temperatures of ∼250 °C. The main desorption temperatures of S1 and S2 are 304 and 316 °C, respectively. Hydrogen storage capacities of both samples are comparable in the range of 3.2–3.4 wt% H2 (Fig. 1B). Deficient hydrogen capacities with respect to pristine Mg2FeH6 (5.40 wt% H2)23 and Mg2NiH4 (3.4–3.6 wt% H2)31 are described by the formation of unreacted Fe and Fe–Ni alloy in as-prepared samples (Fig. 1A).
Considering DSC and TG profiles of S1 and S2, the exothermic event and the weight-gain signals after 450 °C are observed (Fig. 1B). Chemical compositions of S1 and S2 after dehydrogenation at 500 °C and rehydrogenation (S1′ and S2′) are investigated by PXRD technique. From Fig. 2A, PXRD spectra of desorbed S1 and S2 (T = 500 °C) show comparable diffractions of Mg2Ni, Mg, Fe–Ni alloy, and Fe. Thus, the exothermic peaks at T > 450 °C (Fig. 1B) belong to the formation of Mg2Ni and Fe–Ni alloy. For S1′ and S2′, similar diffractions of Mg2FeH6, Mg2NiH4, and unreacted Fe are observed (Fig. 2A). Dehydrogenation of S1′ and S2′ is characterized by simultaneous DSC-TG-MS experiments. From Fig. 2B, S1′ and S2′ reveal comparable onset and main dehydrogenation temperatures (250 and 306–323 °C, respectively) to those of S1 and S2 (250 and 304–316 °C, respectively) (Fig. 1B). However, storage capacities of S1′ and S2′ (2.0–2.2 wt% H2) are significantly lower than those of S1 and S2 (3.4–3.4 wt% H2). This is because significant amount of unreacted Fe after dehydrogenation at 500 °C is irreversible after rehydrogenation into S1′ and S2′ (Fig. 2A).
 |
| | Fig. 2 PXRD spectra (A) and simultaneous DSC-TG-MS results (B) after dehydrogenation at 500 °C of S1 and S2 as well as S1′ and S2′. | |
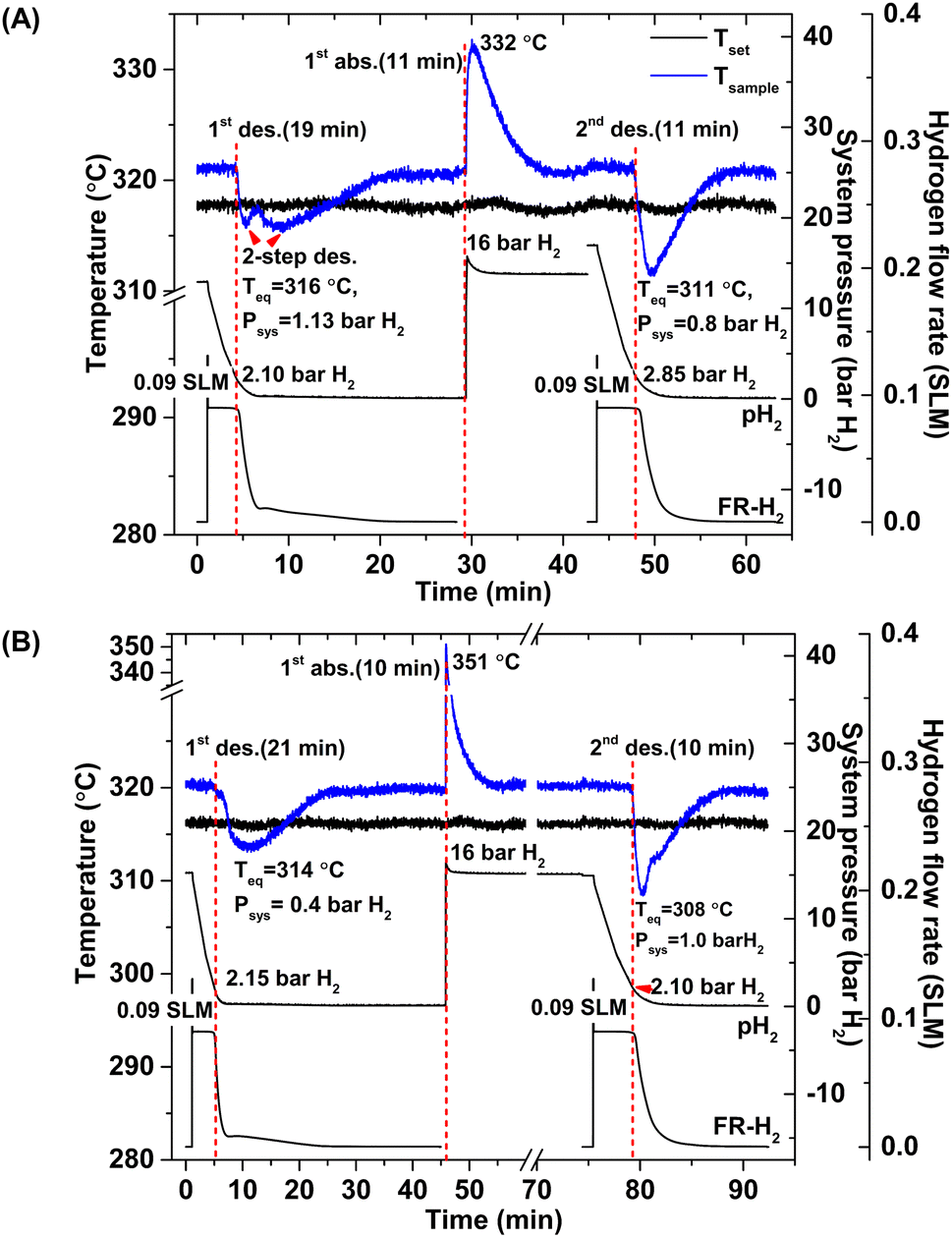
According to greater hydrogen capacities and lower dehydrogenation temperatures, further studies focus on dehydrogenation performance, reversibility, and reaction pathways of S1 and S2. Hydrogen absorption and desorption are carried out at isothermal condition (Tset = 315 °C) under the system pressure (Psys) of 0–16 bar H2. Prior to the measurements, as-prepared samples of S1 and S2 are heated from room temperature to 315 °C under 15 bar H2 to prevent dehydrogenation. Once reaching isothermal condition, dehydrogenation begins with releasing hydrogen through MFC using the constant mass flow rate of 0.09 SLM (Fig. 3). During 0–10 min, the 1st endothermic dehydrogenation of S1 and S2 starts at the system pressure (Psys) of ∼2 bar H2, confirmed by the reduction of sample temperature (Tsample) (Fig. 3). Complete dehydrogenation of both samples is obtained within 19–21 min, shown as the elevated Tsample to the initial temperature. From Fig. 3A, S1 reveals rapid temperature reduction to equilibrium temperature (Teq) of 316 °C under Psys = 1.13 bar H2 with two-step decomposition, possibly belonging to MgH2, Mg2FeH6 and Mg2NiH4. For S2, slow temperature reduction to Teq = 314 °C under Psys = 0.4 bar H2 is found with the single-step dehydrogenation of the mixed Mg2NiH4 + Mg2FeH6 (Fig. 3B). At Teq = 314–316 °C, the equilibrium pressures (Peq) of Mg2FeH6 and Mg2NiH4 are ∼1.5 and 4 bar H2, respectively.32
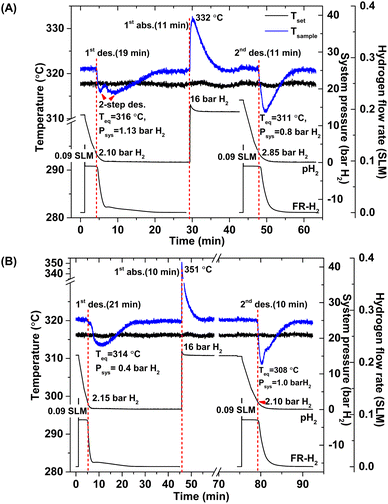 |
| | Fig. 3 Temperature, pressure, and mass flow rate profiles during de/rehydrogenation of S1 (A) and S2 (B). | |
Thus, lower Psys (1.13 and 0.4 bar H2 for S1 and S2, respectively) than Peq at these Teq encourages dehydrogenation of both samples. Afterwards rehydrogenation is carried out at isothermal condition (Tset = 315 °C) under 16 bar H2. By applying hydrogen pressure, Tsample of both S1 and S2 enhance rapidly to Teq = 332 and 351 °C, respectively, due to fast exothermic reaction (Fig. 3). Rehydrogenations of both samples complete within 11 min, assured by the reduction of Tsample to the initial temperature. Under comparable Psys (16 bar H2), different Teq values detected during hydrogenation of S1 and S2 suggest the alteration of reversible phases and reaction pathways. In the case of the 2nd dehydrogenation, S1 and S2 reveal fast temperature reduction to comparable Teq, Psys, and reaction time of 308–311 °C, 0.8–1.0 bar H2, and 10–11 min, respectively (Fig. 3). Afterwards, dehydrogenation kinetics, capacities, and reversibility upon 7 de/rehydrogenation cycles of S1 and S2 are investigated. During the 1st dehydrogenation, hydrogen capacities of S1 and S2 are comparable of 3.2–3.3 wt% H2, but S1 shows faster dehydrogenation rate than S2 (Fig. 4). Considering the 2nd dehydrogenation, kinetic properties of both samples are improved with respect to the 1st cycle. Reversible capacity in the 2nd cycle of S1 is maintained as 3.3 wt% H2, while that of S2 reduces to 2.8 wt% H2 (Fig. 4). Upon the 3rd–7th cycles, kinetic properties of both samples are stable, but their storage capacities reduce to 2.7–2.8 and 2.4–2.6 wt% H2 for S1 and S2, respectively.
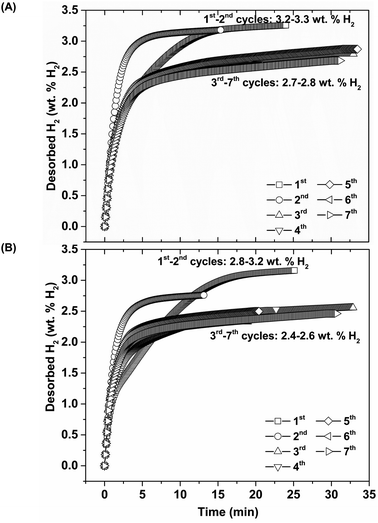 |
| | Fig. 4 Dehydrogenation kinetics and reversible capacities upon 7 hydrogen release and uptake cycles of S1 (A) and S2 (B). | |
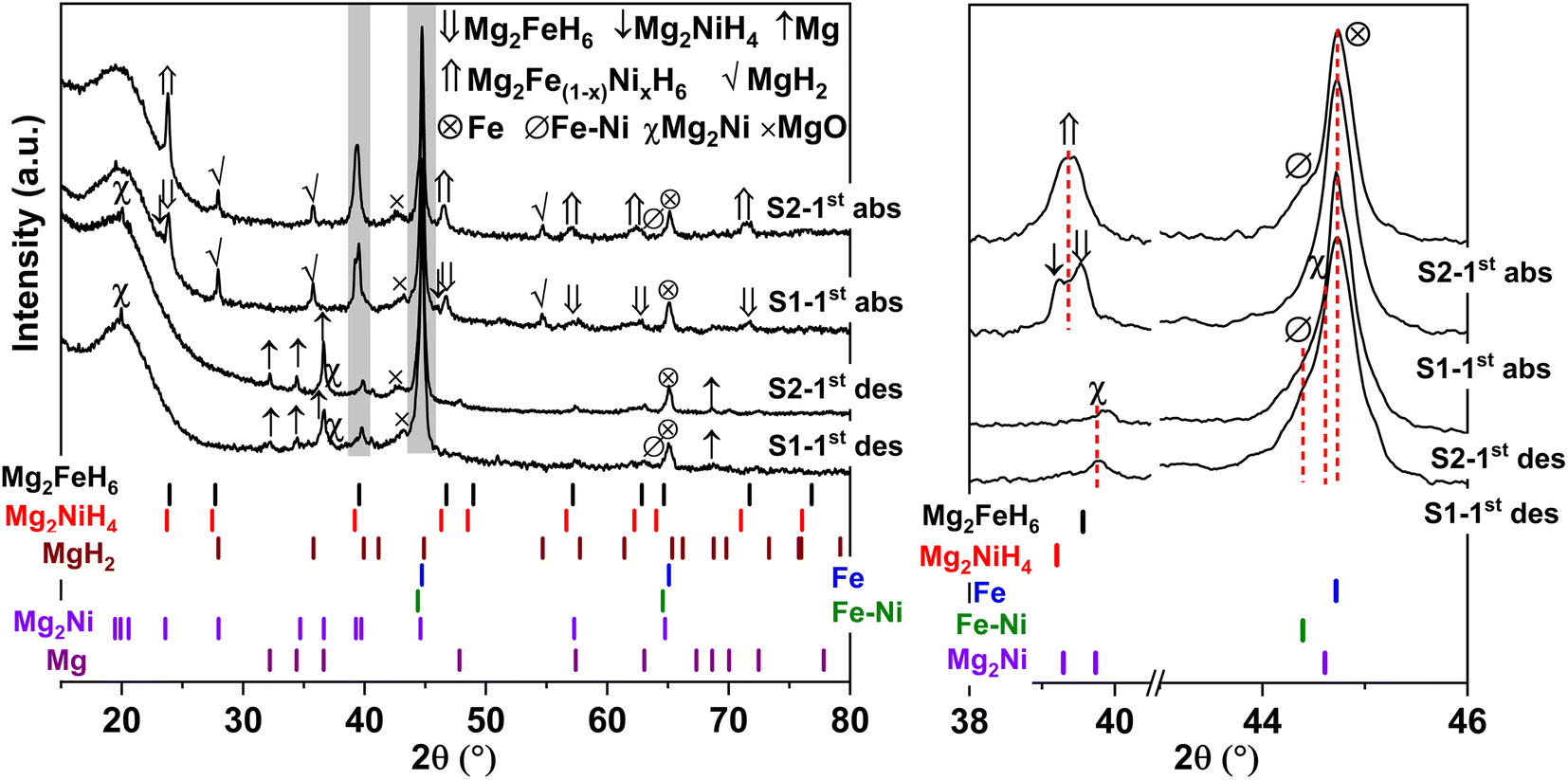
Furthermore, phase compositions of S1 and S2 after the 1st de/rehydrogenation are investigated by PXRD technique. From Fig. 5, the 1st dehydrogenated S1 and S2 reveal comparable diffractions of Mg, Mg2Ni, Fe, MgO, and Fe–Ni alloy. Considering phase compositions of as-prepared and the 1st dehydrogenated samples of S1 and S2, Mg and Fe are obtained from the dehydrogenation of MgH2 and Mg2FeH6 (eqn (6) and (7)), while Mg2Ni is from the decomposition of Mg2NiH4 (reverse reaction of eqn (5)).
| | |
MgH2(S) → Mg(S) + H2(g)
| (6) |
| | |
Mg2FeH6(s) → 2Mg(s) + Fe(s) + 3H2(g)
| (7) |
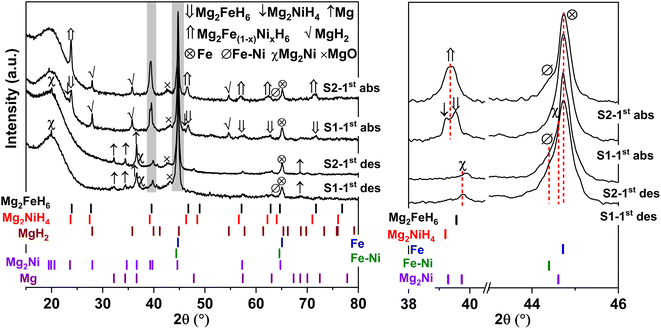 |
| | Fig. 5 PXRD spectra of the 1st de/rehydrogenated S1 and S2. | |
For the 1st rehydrogenation, the formations of MgH2, Mg2NiH4, Mg2FeH6, and MgO are observed in S1. In the case of S2, the 1st rehydrogenated sample reveals the diffractions of MgH2, Fe, Fe–Ni alloy, and MgO as well as Mg2Fe(1−x)NixH6, shown as a new diffraction peak locating between those of Mg2FeH6 and Mg2NiH4.23,25 The formations of MgH2, Mg2FeH6, and Mg2NiH4 in S1 confirm rehydrogenation of Mg, MgH2 + Fe, and Mg2Ni, respectively (reverse reactions of eqn (6) and (7) as well as eqn (5)). Besides, it was reported that Mg2NiH4 was able to be synthesized by hydrogenating the mixture of coarse-grained Mg and Ni(Fe) nanoparticles and most of Ni(Fe) transformed to α-Fe when the reaction completed (eqn (8)).33 Thus, the reduction of Fe–Ni alloy together with the increment of Fe after the 1st rehydrogenation of S1 (Fig. 5) can be explained by the reaction between Fe–Ni alloy and MgH2 to form Mg2NiH4. In the case of the 1st rehydrogenated S2, hydrogenations of Mg into MgH2 (reverse reaction of eqn (6)) and Mg2Ni + Mg2FeH6 into Mg2Fe(1−x)NixH6 (eqn (9))23,25 are observed. Significantly enhanced diffraction of Fe–Ni alloy and irreversibility of Mg2NiH4 upon the 1st hydrogenation of S2 suggest the increase of solid solution of Fe and Ni as well as no reaction between MgH2 and Fe–Ni alloy (eqn (8)). Reaction pathways upon the 1st de/rehydrogenation are summarized in Table 1.
| | |
2MgH2(s) + Ni(Fe)(s) → Mg2NiH4(s) + α-Fe(s)
| (8) |
| | |
xMg2Ni(s) + (1 − x)Mg2FeH6(s) + 3xH2(g) → Mg2Fe(1−x)NixH6(s)
| (9) |
Table 1 Reaction pathways and phase compositions of S1 and S2 during the 1st de/rehydrogenation
| Samples |
Possible reaction pathways and phase compositions |
| S1 |
| As-prepared |
MgH2 + Mg2FeH6 + Mg2NiH4 |
| 1st desorbed |
Mg2FeH6 → 2MgH2 + Fe + H2 |
| MgH2 → Mg + H2 |
| Mg2NiH4 → Mg2Ni + 2H2 |
| Fe + Ni → Fe–Ni |
| 1st absorbed |
Mg + H2 → MgH2 |
| 2MgH2 + Fe + H2 → Mg2FeH6 |
| Mg2Ni + 2H2 → Mg2NiH4 |
| 2MgH2 + Fe–Ni → Mg2NiH4 + Fe33 |
|
| S2 |
| As-prepared |
Mg2FeH6 + Mg2NiH4 + Fe–Ni |
| 1st desorbed |
Mg2FeH6 → 2MgH2 + Fe + H2 |
| MgH2 → Mg + H2 |
| Mg2NiH4 → Mg2Ni + 2H2 |
| Fe–Ni (comparable to as-prepared state) |
| 1st absorbed |
Mg + H2 → MgH2 |
| 2MgH2 + Fe + H2 → Mg2FeH6·xMg2Ni + (1 − x)Mg2FeH6 + 3xH2 → Mg2Fe(1−x)NixH6 |
| Fe + Ni → Fe–Ni |
Due to the changes of reaction pathways and phases formed during the 1st de/rehydrogenation of S1 and S2 (Fig. 5 and Table 1), temperature profiles during the 1st endothermic desorption and exothermic absorption of S1 and S2 are different (Fig. 3). Effective reproducibility of several hydrides in S1 (MgH2 + Mg2FeH6 + Mg2NiH4) probably maintains reversible hydrogen capacities upon 2 cycles (∼3.3 wt% H2) (Fig. 4A). Moreover, phase compositions of the 7th rehydrogenated samples of S1 and S2 are characterized by PXRD technique to describe the reduction of hydrogen capacities upon cycling (Fig. 4). From Fig. 6, both rehydrogenated samples show comparable diffractions of MgH2, Fe, MgO, and unknown phase. Meanwhile, each sample shows different phases of Mg2FeH6 + Mg2NiH4 and Mg2Fe(1−x)NixH6 for the 7th rehydrogenated S1 and S2, respectively. Upon cycling, significant amount of unreacted Fe with respect to the reversible hydrides is observed from both samples. The latter explains the deficient hydrogen capacities of both samples upon the 3rd–7th cycles (Fig. 4).
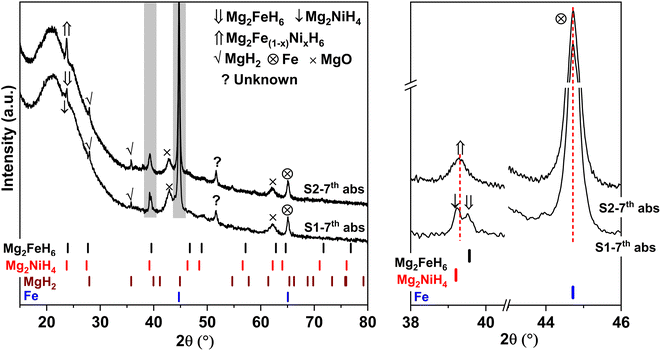 |
| | Fig. 6 PXRD spectra of the 7th rehydrogenated S1 and S2. | |
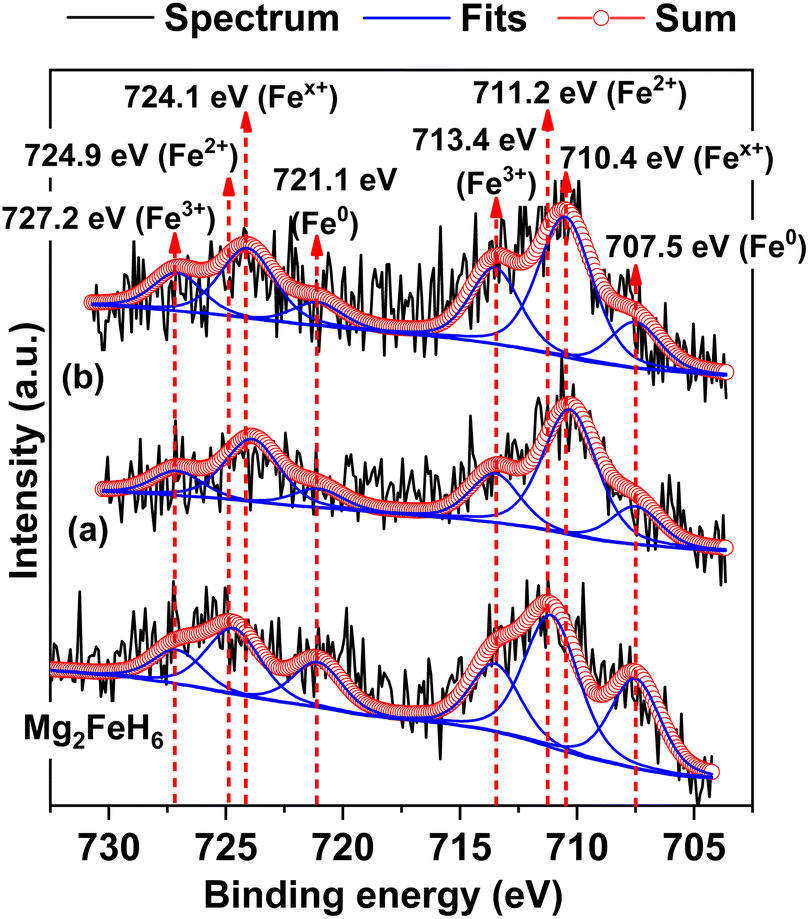
To confirm the formation of Mg2Fe(1−x)NixH6 in the 1st and 7th rehydrogenated S2, Fe 2p XPS experiments are carried out. From Fig. 7, Fe 2p XPS spectrum of as-prepared Mg2FeH6 shows the characteristic peaks of Fe0 (707.5 and 721.1 eV), Fe2+ (711.2 and 724.9 eV), and Fe3+ (713.4 and 727.2 eV), belonging to metallic Fe, Mg2FeH6, and Fe2O3, respectively.34,35 The signal of metallic Fe is attributed to unreacted Fe during Mg2FeH6 preparation, while that of Fe2O3 is likely due to the oxidation of Fe during the measurements. For the 1st and 7th rehydrogenated S2, Fe 2p XPS peaks of Fe0 and Fe3+ of metallic Fe and Fe2O3, respectively, are observed at comparable binding energies with as-prepared Mg2FeH6. Besides, the new peaks of Fex+ (710.4 and 724.1 eV) locating at lower binding energies than Fe2+ are detected (Fig. 7(a) and (b)). This suggests the formation of another Fe-containing phase with lower oxidation state than 2+. Because the energy resolution of XPS measurements is 0.5 eV, the binding energy difference between Fe2+ and Fex+ (∼0.8 eV) is sufficient to imply that the energy shift is due to phase changes. Once partial substitution of Ni for Fe in Mg2FeH6 to form Mg2Fe(1−x)NixH6 occurs, the oxidation state of Fe reduces from Fe2+ to Fex+ (0 < x < 2). Thus, the appearance of Fex+ likely confirms the formation of Mg2Fe(1−x)NixH6 in the 1st and 7th rehydrogenated S2.
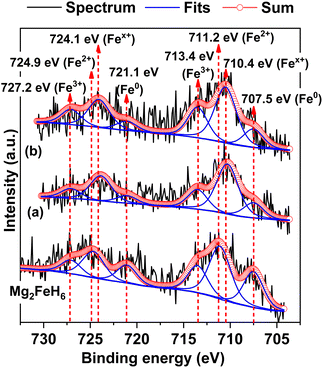 |
| | Fig. 7 Fe 2p XPS spectra of as-prepared Mg2FeH6 as well as the 1st (a) and the 7th (b) rehydrogenated S2. | |
Furthermore, it should be mentioned that phase compositions in the 1st dehydrogenated samples of S1 and S2 are comparable (i.e., Mg, Mg2Ni, Fe–Ni alloy, and Fe) (Fig. 5). However, the reaction pathways during the 1st rehydrogenation of these samples are different, affecting reversible hydrogen capacities (Table 1 and Fig. 4). This might relate to contacts and distribution of the reactive phases in the bulk samples. Therefore, microstructural analyses of the 1st dehydrogenated S1 and S2 are investigated by TEM, electron diffraction, and EDS mapping. TEM image of the 1st dehydrogenated S1 shows that at least two different phases are well distributed in the nanometer scale (Fig. 8A(a)). The corresponding SAED pattern confirms the presence of Mg, Mg2Ni, and Fe–Ni (Fig. 8A(b)), in accordance with PXRD result (Fig. 5). EDS maps reveal excellent distribution of Mg, Fe, and Ni in the sample bulk (Fig. 8A(c) and (e)). These results suggest good contacts among Mg, Fe, Mg2Ni, and Fe–Ni in the 1st dehydrogenated S1. This likely promotes the formation of Mg2FeH6 and Mg2NiH4 upon rehydrogenation (Fig. 5 and Table 1). In the case of the 1st dehydrogenated S2, TEM micrograph shows significant particle agglomeration (Fig. 8B(a)) with comparable phase compositions to S1 (SAED result in Fig. 8B(b)). From EDS maps, Mg and Ni occupying comparable location show well-distributed nanoparticles with partially dense agglomeration (Fig. 8B(c) and (e)), while Fe shows good distribution of sintered particles (Fig. 8B(d)). These distributions either of nanoparticles or sintered particles found in Mg, Ni, and Fe maps lead to the homogeneous reversibility of MgH2, Mg2FeH6, and Fe–Ni alloy all over the sample bulk. The positions with Mg and Ni agglomeration, probably containing high density of Mg2Ni benefit for hydrogenation of Mg2FeH6 + Mg2Ni to form Mg2Fe(1−x)NixH6 (eqn (9)). Thus, using different Ni sources (metallic Ni or Mg2NiH4) as staring material affects the contacts among active phases. S1 using metallic Ni shows better distribution of metal nanoparticles than S2, which Ni is from Mg2NiH4. The 1st dehydrogenated S1 with good metal distribution reproduces individual hydrides (Mg2FeH6 and Mg2NiH4) upon rehydrogenation. For the 1st dehydrogenated S2, agglomeration of Mg2Ni (from direct decomposition of Mg2NiH4), which is in good contacts with Mg and Fe favors the formation of Mg2Fe(1−x)NixH6. Therefore, the distribution and contacts among metal nanoparticles results in different reaction pathways upon de/rehydrogenation and reversible hydrogen capacities.
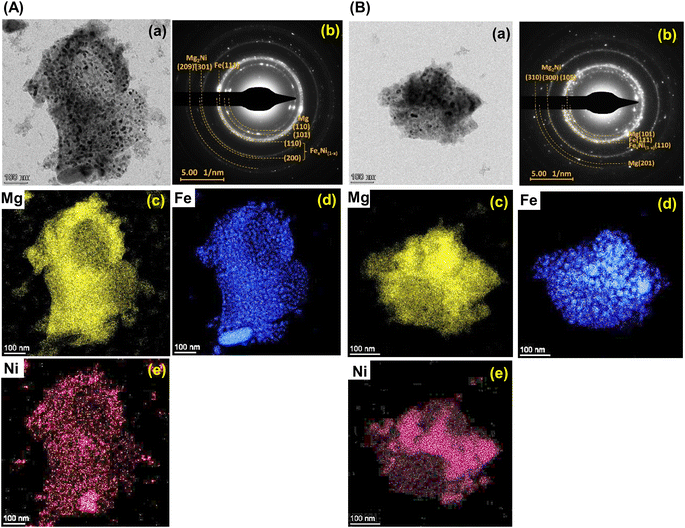 |
| | Fig. 8 The bright field TEM micrographs (a), the corresponding selected area electron diffraction (SAED) pattern (b), and elemental mapping of Mg (c), Fe (d), and Ni (e) of the 1st dehydrogenated samples of S1 (A) and S2 (B). | |
4. Conclusions
The effects of Ni precursors (metallic Ni or Mg2NiH4) on the formation of Mg–Fe–Ni hydrides were studied and the de/rehydrogenation kinetics and reversibility of the obtained samples were investigated. The mixtures of MgH2 + Mg2FeH6 + Mg2NiH4 and Mg2FeH6 + Mg2NiH4 were obtained from the as-prepared samples with metallic Ni and Mg2NiH4, respectively. Although both samples released comparable hydrogen during the 1st cycle (3.2–3.3 wt% H2), the reduction of dehydrogenation temperature (ΔT = 12 °C) and faster kinetics were obtained from the as-prepared sample with metallic Ni. After the 1st dehydrogenation, similar phase compositions of Mg, Mg2Ni, Fe, and Fe–Ni alloy were found in both samples. Nevertheless, different Ni precursors altered phase compositions and reaction pathways during rehydrogenation. The reversible phases of the sample with metallic Ni were MgH2, Mg2FeH6, and Mg2NiH4, while those of the sample with Mg2NiH4 were MgH2 and Mg2Fe(1−x)NixH6. These recovered phases affected reversible capacities. For example, hydrogen capacities during the 2nd–7th cycles of the sample with metallic Ni were 2.7–3.2 wt% H2, while those of the sample with Mg2NiH4 reduced to 2.4–2.8 wt% H2. Deficient reversible capacities of both samples, especially after the 3rd cycles could be described by significant amount of unreacted Fe. Considering microstructural analyses, the sample with metallic Ni contained well-distributed nanoparticles of all metals, benefiting for individual reversibility of MgH2, Mg2FeH6 and Mg2NiH4. For the sample with Mg2NiH4, partial agglomeration of Mg and Ni at comparable location, likely belonging to Mg2Ni favoured the formation of Mg2Fe(1−x)NixH6. Due to the recovery of multiple hydride phases, hydrogen capacities upon cycling of the sample with metallic Ni was significant.
Author contributions
Palmarin Dansirima: sample preparation, characterizations, sharing idea, data analysis, and manuscript writing. Sophida Thiangviriya: sample preparation and characterizations. Praphatsorn Plerdsranoy: characterizations. Narong Chanlek: XPS measurements and analysis. Rapee Utke: conceptualization, supervision, data analysis, manuscript writing, and reviewing and editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research has received the funding support from the National Research Council of Thailand (NRCT) and Suranaree University of Technology (SUT) [grant numbers N42A650323] as well as Thailand Science Research and Innovation (TSRI) and National Science, Research, and Innovation Fund (NSRF) via the Program Management Unit for Human Resources & Institutional Development, Research and Innovation [grant numbers B40G660033 and Fundamental Fund 2566].
References
- B. Bogdanovi, A. Reiser, K. Schlichte, B. Spliethoff and B. Tesche, J. Alloys Compd., 2002, 345, 77–89 CrossRef.
- J. A. Puszkiel, J. J. Andrade-Gamboa and F. C. Gennari, Emerg. Mater. Energy Convers. Storage, 2018, pp. 393–428 Search PubMed.
- A. Reiser, B. Bogdanović and K. Schlichte, Int. J. Hydrogen Energy, 2000, 25, 425–430 CrossRef CAS.
- M. Felderhoff and B. Bogdanović, Int. J. Mol. Sci., 2009, 10, 325 CrossRef CAS PubMed.
- R. Urbanczyk, M. Meggouh, R. Moury, K. Peinecke, S. Peil and M. Felderhoff, Appl. Phys. A: Mater. Sci. Process., 2016, 122, 315–320 CrossRef.
- R. Urbanczyk, K. Peinecke, S. Peil and M. Felderhoff, Int. J. Hydrogen Energy, 2017, 42, 13818–13826 CrossRef CAS.
- A. A. Nayeb-Hashemi, J. B. Clark and L. J. Swartzendruber, Bull. Alloy Phase Diagrams, 1985, 6, 235–238 CrossRef CAS.
- M. Rzeszotarska, T. Czujko and M. Polański, Int. J. Hydrogen Energy, 2020, 45, 19440–19454 CrossRef CAS.
- S. N. Nyamsi, V. Yartys and M. Lototskyy, Mater. Today: Proc., 2018, 5, 10533–10541 Search PubMed.
- A. A. C. Asselli, D. R. Leiva, A. M. Jorge, T. T. Ishikawa and W. J. Botta, J. Alloys Compd., 2012, 536, S250–S254 CrossRef CAS.
- J. O. Fadonougbo, J. Y. Jung, J. Y. Suh, Y. S. Lee, J. H. Shim and Y. W. Cho, Mater. Des., 2017, 135, 239–245 CrossRef CAS.
- J. Y. Jung, J. O. Fadonougbo, J. Y. Suh, Y. S. Lee, J. Y. Huh and Y. W. Cho, Int. J. Hydrogen Energy, 2018, 43, 16758–16765 CrossRef CAS.
- M. Polanski, K. Witek, T. K. Nielsen, L. Jaroszewicz and J. Bystrzycki, Int. J. Hydrogen Energy, 2013, 38, 2785–2789 CrossRef CAS.
- K. Witek, K. Karczewski, M. Karpowicz and M. Polanski, Crystals, 2018, 8, 94 CrossRef.
- D. Khan, S. Panda, Z. Ma, W. Ding and J. Zou, Int. J. Hydrogen Energy, 2020, 45, 21676–21686 CrossRef CAS.
- F. C. Gennari, F. J. Castro and J. J. Andrade Gamboa, J. Alloys Compd., 2002, 339, 261–267 CrossRef CAS.
- J. Puszkiel, F. Gennari, P. A. Larochette, F. Karimi, C. Pistidda, R. Gosalawit-Utke, J. Jepsen, T. R. Jensen, C. Gundlach, J. B. Von Colbe, T. Klassen and M. Dornheim, Int. J. Hydrogen Energy, 2013, 38, 14618–14630 CrossRef CAS.
- M. Rzeszotarska, J. Dworecka-Wójcik, A. Dębski, T. Czujko and M. Polański, J. Alloys Compd., 2022, 901, 163489 CrossRef CAS.
- X. Chen, J. Zou, X. Zeng and W. Ding, Int. J. Hydrogen Energy, 2016, 41, 14795–14806 CrossRef CAS.
- K. Batalović, J. Radaković, J. Belošević-Čavor and V. Koteski, Phys. Chem. Chem. Phys., 2014, 16, 12356–12361 RSC.
- J. Barale, S. Deledda, E. M. Dematteis, M. H. Sørby, M. Baricco and B. C. Hauback, Sci. Rep., 2020, 10, 9000 CrossRef CAS PubMed.
- Y. Zhu, Z. Liu, Y. Yang, H. Gu, L. Li and M. Cai, Int. J. Hydrogen Energy, 2010, 35, 6350–6355 CrossRef CAS.
- S. Thiangviriya, P. Plerdsranoy, A. Hagenah, T. T. Le, P. Kidkhunthod, O. Utke, M. Dornheim, T. Klassen, C. Pistidda and R. Utke, Int. J. Hydrogen Energy, 2021, 46, 32099–32109 CrossRef CAS.
- S. Il Ri, S. Il Hong, J. C. Kim and J. H. Wi, Int. J. Hydrogen Energy, 2021, 46, 3319–3328 CrossRef.
- P. Plerdsranoy, B. Tharat, S. Thiangviriya, P. Kidkhunthod, T. Siritanon, S. Suthirakun and R. Utke, J. Alloys Compd., 2023, 937, 168212 CrossRef CAS.
- T. Xiong, Z. Zhu, Y. He, M. S. Balogun and Y. Huang, Small Methods, 2023, 7, 2201472 CrossRef CAS PubMed.
- Y. Hu, T. Xiong, M. S. J. T. Balogun, Y. Huang, D. Adekoya, S. Zhang and Y. Tong, Mater. Today Phys., 2020, 15, 100267 CrossRef.
- C. Sitthiwet, P. Plerdsranoy, P. Dansirima, P. Eiamlamai, O. Utke and R. Utke, J. Alloys Compd., 2020, 832, 155026 CrossRef CAS.
- P. Dansirima, A. Pangon, O. Utke and R. Utke, Int. J. Hydrogen Energy, 2022, 47, 7351–7361 CrossRef CAS.
- W.-S. Chang, Y. Wei, J.-M. Guo, F.-J. He, W.-S. Chang, Y. Wei, J.-M. Guo and F.-J. He, Open J. Met., 2012, 2, 18–23 CrossRef CAS.
- L. Li, T. Akiyama and J. I. Yagi, J. Alloys Compd., 2001, 316, 118–123 CrossRef CAS.
- A. Züttel, Mater. Today, 2003, 6, 24–33 CrossRef.
- X. Chen, J. Zou, S. Huang, G. He, N. Zhao, X. Zeng and W. Ding, RSC Adv., 2018, 8, 18959–18965 RSC.
- NIST X-ray Photoelectron Spectroscopy (XPS) Database, https://srdata.nist.gov/xps/main_search_menu.aspx, accessed, 9 May 2023 Search PubMed.
- C. Peng, R. Huang, G. Pan, W. Liu and L. Wang, Ionics, 2020, 26, 301–309 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a
*a