
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis, and docking studies of novel pyrazole-based scaffolds and their evaluation as VEGFR2 inhibitors in the treatment of prostate cancer†
Dalia H. Soliman*ab and
Mohamed S. Nafie *c
*c
aDepartment of Pharmaceutical Medicinal Chemistry and Drug Design, Faculty of Pharmacy (Girls), Al-Azhar University, Egypt
bDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Egyptian Russian University, Badr City, Cairo, Egypt. E-mail: dalia-soliman@eru.edu.eg
cDepartment of Chemistry (Biochemistry Program), Faculty of Science, Suez Canal University, Ismailia 41522, Egypt. E-mail: mohamed_nafie@science.suez.edu.eg
First published on 10th July 2023
Abstract
Since VEGFR-2 plays a crucial role in tumor growth, angiogenesis, and metastasis, it is a prospective target for cancer treatment. In this work, a series of 3-phenyl-4-(2-substituted phenylhydrazono)-1H-pyrazol-5(4H)-ones (3a–l) were synthesized and investigated for their cytotoxicity against the PC-3 human cancer cell line compared to Doxorubicin and Sorafenib as reference drugs. Two compounds 3a and 3i showed comparable cytotoxic activity with IC50 values of 1.22 and 1.24 μM compared to the reference drugs (IC50 = 0.932, 1.13 μM). Compound 3i was found to be the most effective VEGFR-2 inhibitor using in vitro testing of the synthesized compounds, with nearly 3-fold higher activity than Sorafenib (30 nM), with IC50 8.93 nM. Compound 3i significantly stimulated total apoptotic prostate cancer cell death 55.2-fold (34.26% compared to 0.62% for the control) arresting the cell cycle at the S-phase. The genes involved in apoptosis were also impacted, with proapoptotic genes being upregulated and antiapoptotic Bcl-2 being downregulated. These results were supported by docking studies of these two compounds within the active site of the VEGFR2 enzyme. Finally, in vivo, the study revealed the potentiality of compound 3i to inhibit tumor proliferation by 49.8% reducing the tumor weight from 234.6 mg in untreated mice to 83.2 mg. Therefore, 3i could be a promising anti-prostate cancer agent.
1 Introduction
Cancer is an enormous global health overburden that affects almost every region and socioeconomic level. Although many innovative, as well as unconventional, therapies have been introduced to date, a large increase in cancer mortality is estimated in the next few years.1–3 The onset of resistance to therapy still represents a major problem. Therefore, medicinal chemistry researchers are seriously challenged to invent novel small molecules that would act as potent and selective antitumor agents.4 Prostate cancer is one of the most commonly diagnosed cancers and also one of the leading causes of cancer-associated mortality in most developed countries.5 The high mortality rate is mainly attributed to the invasiveness and metastasis of advanced prostate cancer.6 Thus, targeting the molecules involved in metastasis could be an effective mode of prostate cancer treatment. Vascular permeability, survival, and migration of the vascular endothelial cells are controlled by the signaling pathway of vascular endothelial growth factor (VEGFR-2).7,8 It has been proven that VEGF and their kinase receptors have an additional impact on angiogenesis as well as the survival of the newly developed blood vessels in the tumor cells.9–11 It is a new class that is considered to regulate angiogenesis.12,13 One of the advantages of targeting this signaling pathway is that VEGF induction in tumor cells can result from cancer-related changes, the weak expression of VEGFR-2 is observed in the healthy tissue or cells while the over-expression is reported in various types of cancers, one of them is prostate cancer.14 Hence, over-activation of VEGFR signaling positively correlates with poor prognosis and metastasis in the majority of solid tumor patients.15 Consequently, blocking angiogenesis could be a promising strategy to inhibit the growth of tumors with lower adverse effects than other typical chemotherapies.16–18One of the ongoing challenges nowadays would be the quest for new scaffolds, able to act as tyrosine kinase inhibitors.19–22 Interestingly, there are many FDA-approved agents that target VEGFRs (Sorafenib, Sunitinib, Vandetanib, Axitinib, Regorafenib, etc.). However, drug resistance is a major concern associated with the monotherapy of these agents.23,24
The pyrazole-tethered heterocyclic compounds represent an important class in anticancer therapy.25–32 Pyrazoles have been also reported as potent anti-prostate cancer agents, for example, compounds I and II (Fig. 1), comparable to Sorafenib.33,34
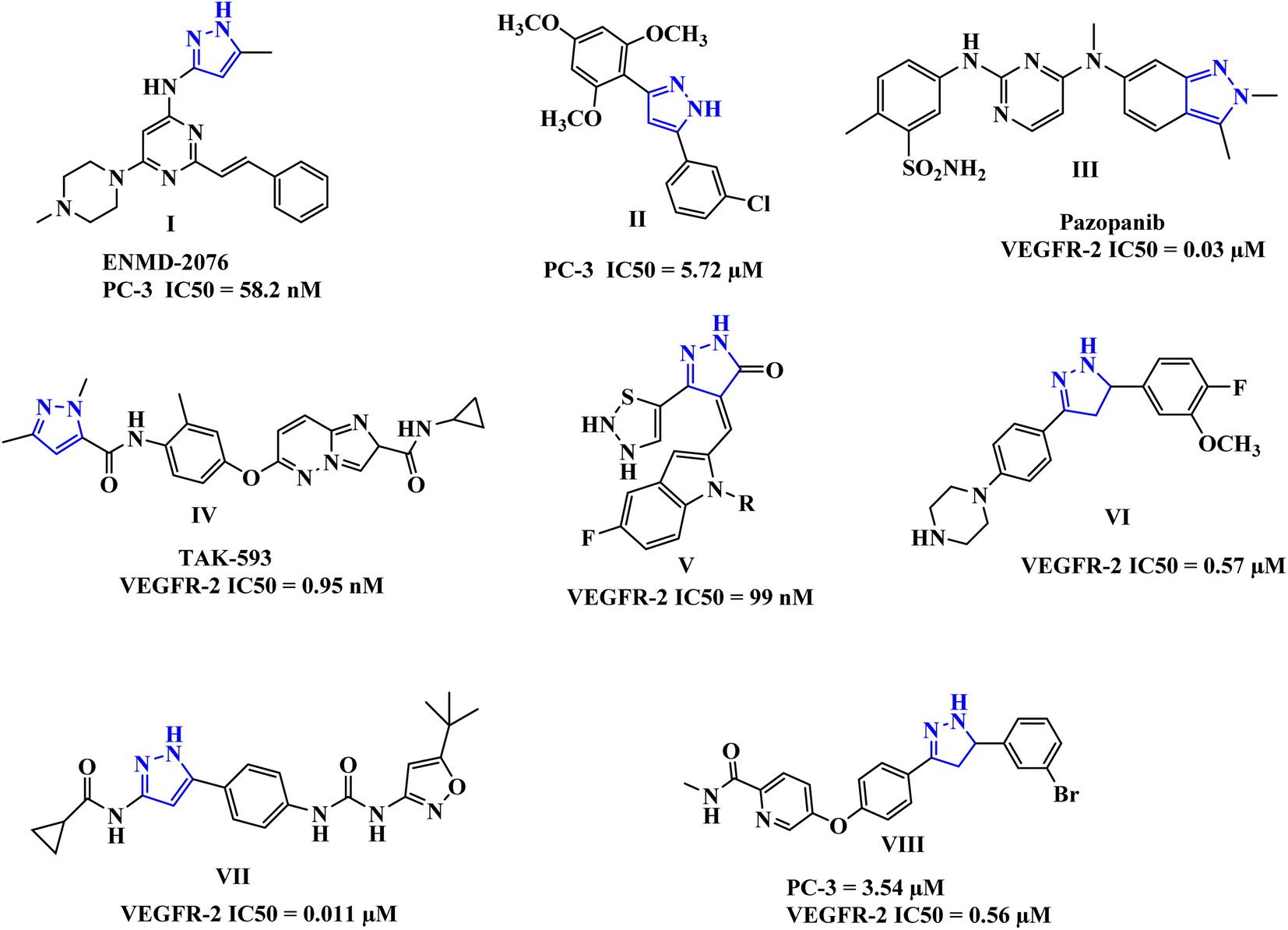 | ||
| Fig. 1 Some representative examples of FDA-approved pyrazole-based drugs, as well as active analogs against VEGFR-2. | ||
Moreover, drugs based on heterocyclic compounds bearing 1,2-diaryl substituted, pyrazole ring have often been on the list of best-selling pharmaceutical products, such as Crizotinib and Ruxolitinib which represent pyrazole-tethered anticancer drugs, currently available in the market.35 Interestingly, the antitumor activity of Celecoxib was reported to be through inhibition of VEGF expression and tumor micro-vessel formation, additionally, several reports emphasize the celecoxib-mediated antiproliferative effects against prostate tumors in experimental models.36–38 Furthermore, these pyrazoline-containing compounds, of which there is the FDA-approved Pazopanib III, and the TAK-593 IV in phase one clinical investigation, have been reported to possess excellent VEGFR-2 inhibition capability along with their antiproliferative potential (Fig. 1).39–46
Accordingly, we aimed in this study to design and synthesize novel pyrazole-based scaffolds as VEGFR-2 inhibitors against prostate cancer using in vitro and in vivo studies.
2 Rationale and design
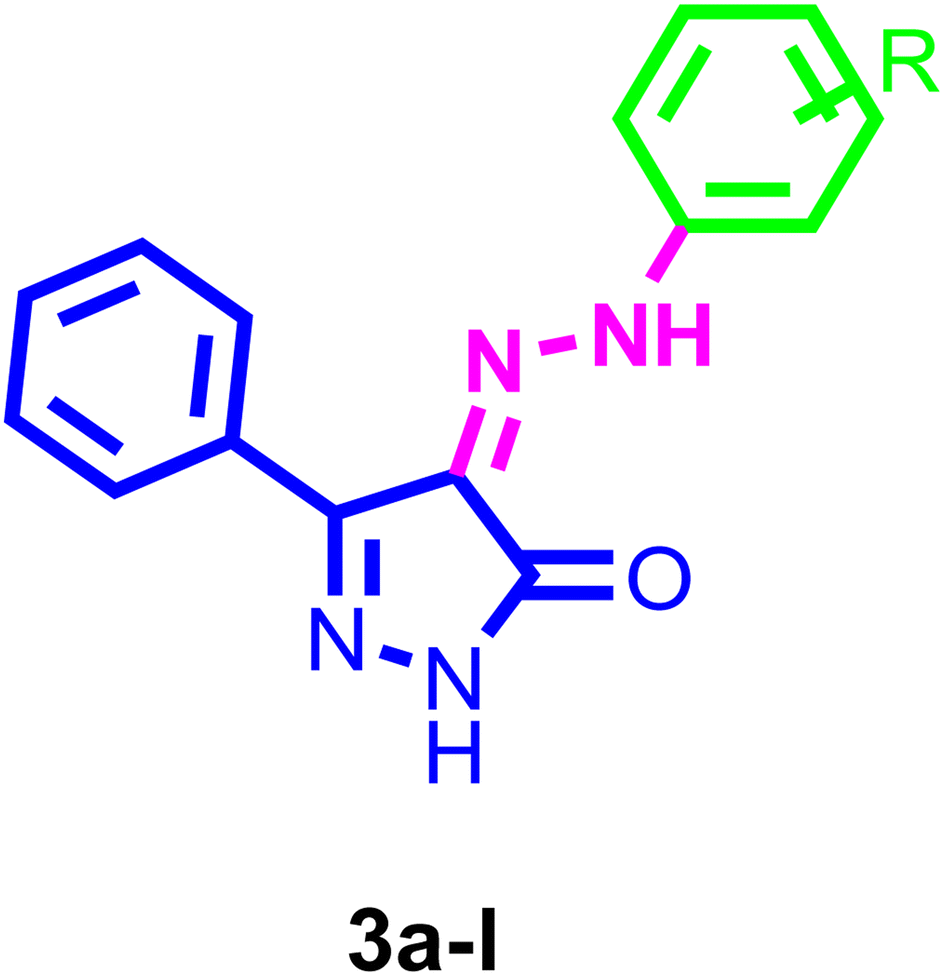
Inhibiting VEGFR-2 was a significant design challenge due to the need for a novel framework to meet the target's binding requirements. Inhibitors of VEGF receptor type 2 (VEGFR-2) need to meet three pharmacophoric criteria: (a) a “hinge-binding” moiety that interacts with the ATP pocket; (b) a “linker” typically consisting of 3–5 bond lengths; and (c) a hydrophobic moiety occupying the allosteric site.4,7,47–51 Thus, we based the structure on appending the bulky substituted diaryl-pyrazoline to an azo-linker, hinge binding moiety for VEGFR-2, that would bind to the gatekeeper, to occlude the basic VEGFR-2 inhibitory activity. Hence, given these findings concerning pyrazole-based VEGFR-2 inhibitors, we aimed to design and synthesize novel 1H-pyrazol-5(4H)-one 3a–l (Fig. 2), these derivatives were subjected to in vitro cytotoxic evaluation against prostate cancer cell line (PC-3), followed by VEGFR-2 inhibitory profile to the most active compounds. To evaluate the binding mechanism and pharmacophoric properties necessary to cause the desired VEGFR-2 inhibition as a potential and efficient target against prostate cancer, molecular docking was done in the active site of VEGFR-2.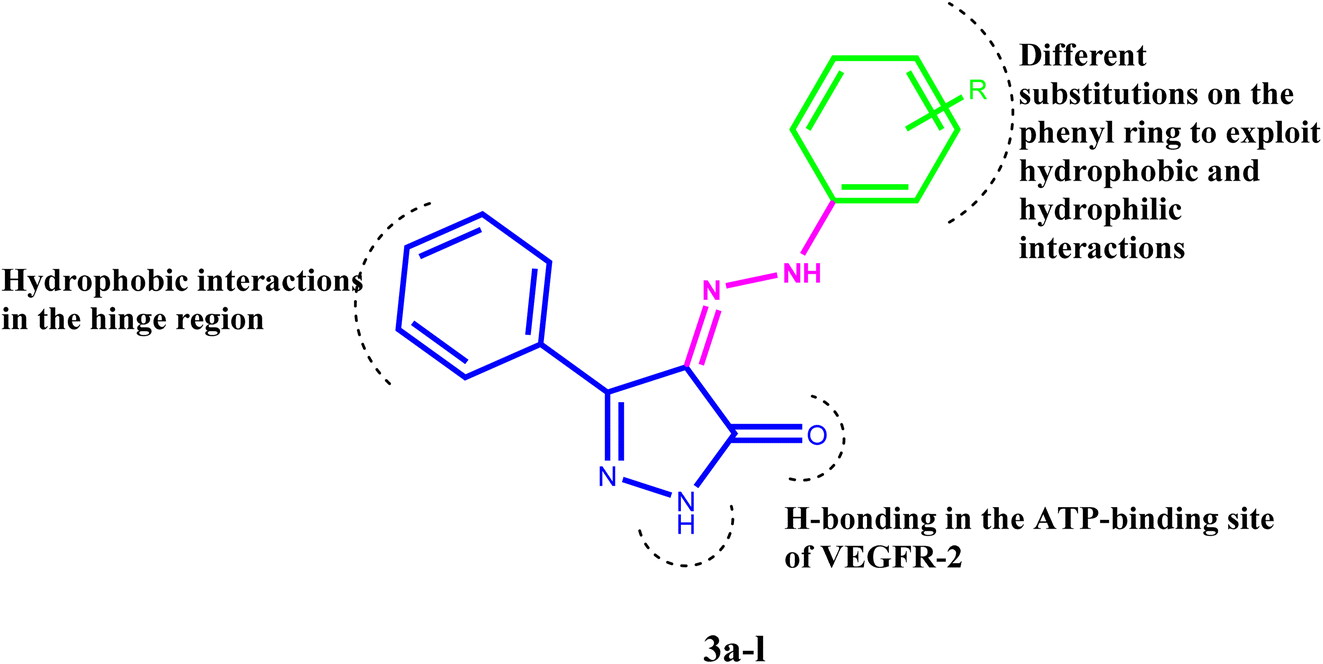 | ||
| Fig. 2 The proposed structures of the new pyrazole derivatives. | ||
3 Results and discussion
3.1. Chemistry
The synthetic pathway adopted for the synthesis of the new target compounds is depicted in Scheme 1. The 3-phenyl-1H-pyrazol-one 2 was obtained by reaction of ethyl benzoylacetate 1 with hydrazine hydrate in ethanol.52,53 The diazonium salts of the corresponding amine were prepared by literature-known procedures, using nitrous acid at 0 °C. Then these salts were further coupled with the pyrazol-5-one 2 at 0 °C to provide the 3-phenyl-4-(2-substituted phenylhydrazono)-1H-pyrazol-5(4H)-one 3a–l in good yields.54 The structures of the newly synthesized compounds were confirmed by spectral and microanalytical data. Infrared spectra showed the characteristics bands at, 1700, 3450, and 1580 cm−1 which authenticated the presence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, NH, and CC groups. IR spectra of these compounds were also characterized by two bands in the region 3552–3142 cm−1 corresponding to the amidic and the hydrazono NHs as well as a band in the range 1658–1710 cm−1 corresponding to the carbonyl groups. The 1H NMR spectra showed two singlets at about δ = 12 and 14 ppm due to the two NH groups. Aromatic protons were also observed in the aromatic region ranging from δ = 7.0–8.1 ppm. Presence of other substituents also authenticated in the 1H NMR spectra at the assigned value. Mass spectra of the synthesized compounds were consistent with their molecular weights.
O, NH, and CC groups. IR spectra of these compounds were also characterized by two bands in the region 3552–3142 cm−1 corresponding to the amidic and the hydrazono NHs as well as a band in the range 1658–1710 cm−1 corresponding to the carbonyl groups. The 1H NMR spectra showed two singlets at about δ = 12 and 14 ppm due to the two NH groups. Aromatic protons were also observed in the aromatic region ranging from δ = 7.0–8.1 ppm. Presence of other substituents also authenticated in the 1H NMR spectra at the assigned value. Mass spectra of the synthesized compounds were consistent with their molecular weights.
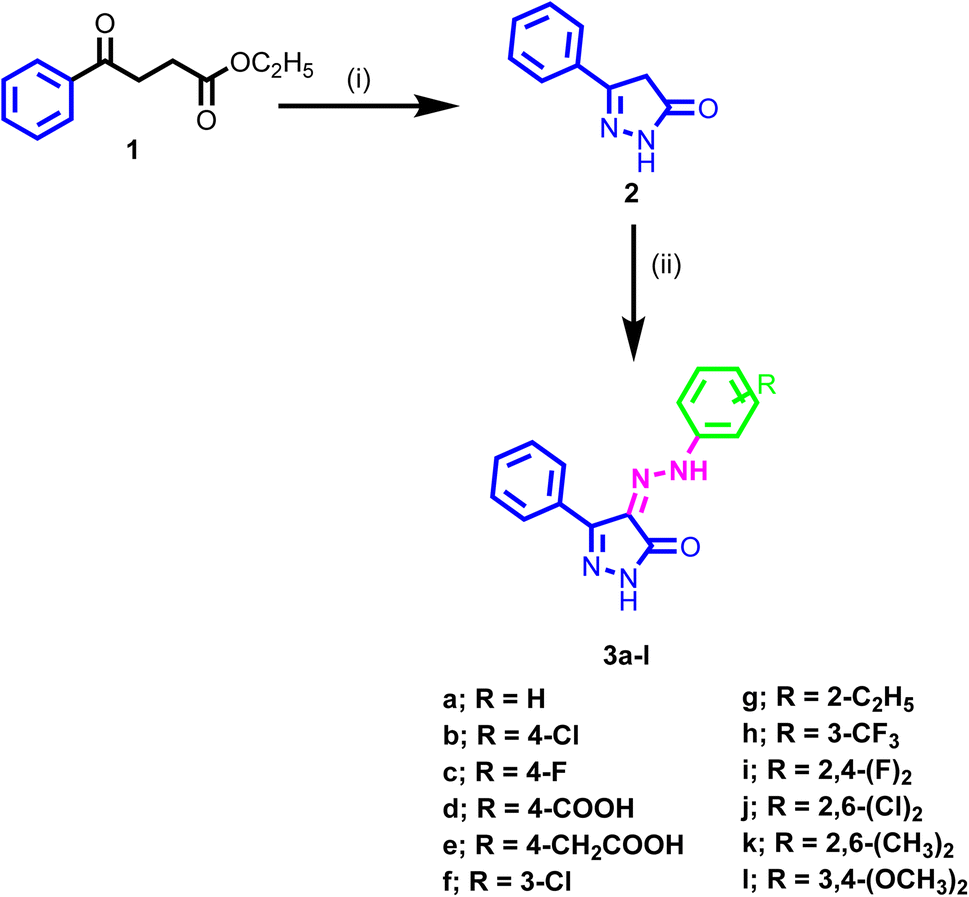 | ||
| Scheme 1 Synthesis of the novel target compounds 3a–l, reagents, and conditions: (i) ethyl benzoylacetate, NH2·NH2 H2O, EtOH, reflux, 60 °C, 6 h. (ii) appropriate amines, HCl/H2O, NaNO2, 0 °C, 2 h, coupling with 3-phenyl-1H-pyrazol-5(4H)-one 2, EtOH, CH3COONa. | ||
3.2 Biological studies
|
||
|---|---|---|
| Compound | R | IC50 (μM) ± SD |
| 3a | H | 1.22 ± 0.3 |
| 3b | 4-Cl | 527.8 ± 61.3 |
| 3c | 4-F | 96.75 ± 6.25 |
| 3d | 4-COOH | 41.68 ± 3.9 |
| 3e | 4-CH2COOH | 178.5 ± 34.6 |
| 3f | 3-Cl | 106.3 ± 6.5 |
| 3g | 2-C2H5 | 93.5 ± 5.1 |
| 3h | 3-CF3 | 22.92 ± 3.4 |
| 3i | 2,4-(F)2 | 1.24 ± 0.15 |
| 3j | 2,6-(Cl)2 | 146.0 ± 8.1 |
| 3k | 2,6-(CH3)2 | 294.4 ± 34.6 |
| 3l | 3,4-(OCH3)2 | 9.32 ± 0.8 |
| Doxorubicin | — | 0.93 ± 0.16 |
| Sorafenib | — | 1.13 ± 0.17 |
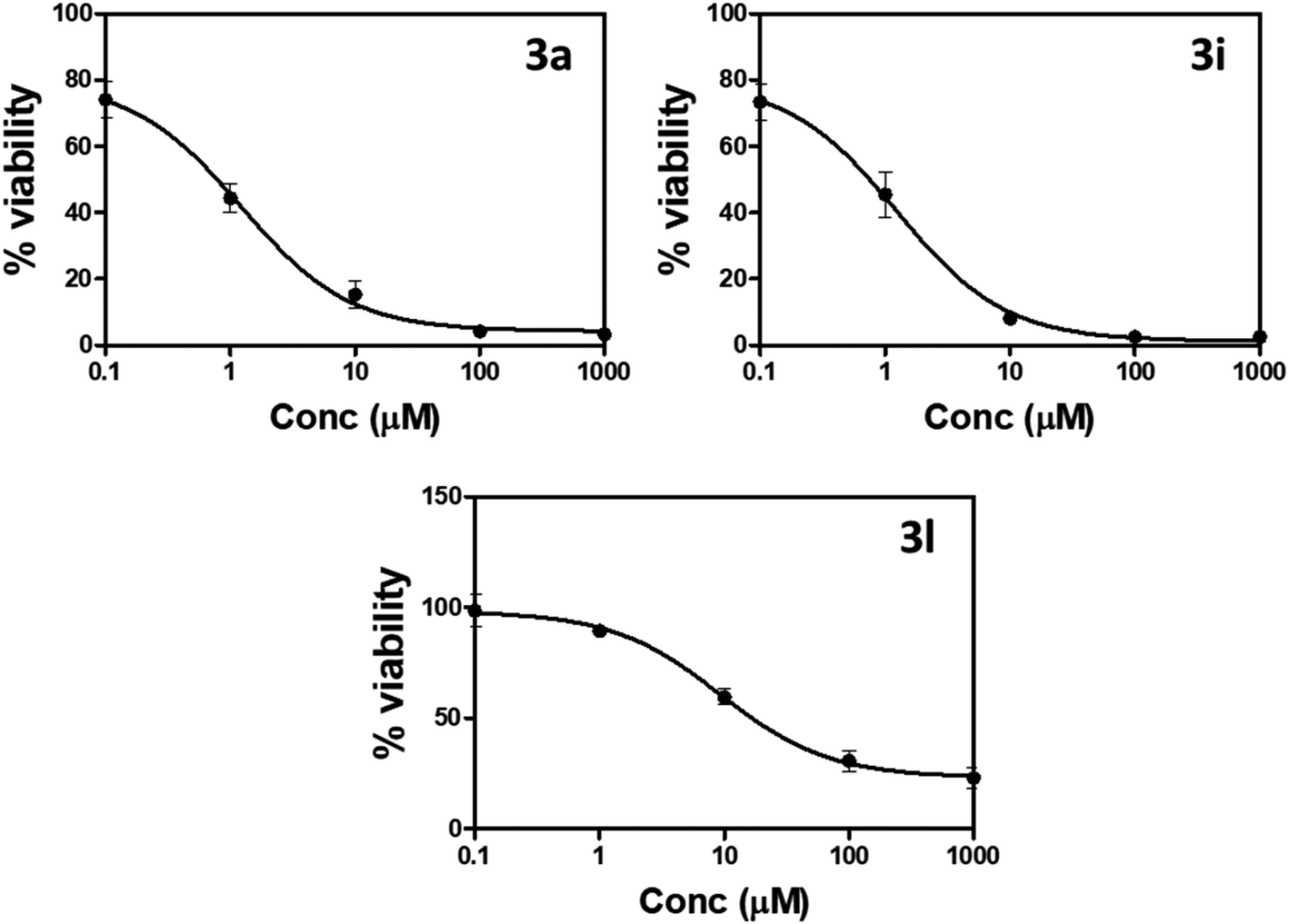 | ||
| Fig. 3 Percentage of cell viability compared to concentrations of compounds 3a, 3i, and 3l against prostate PC-3 cells using MTT assay. | ||
“IC50 values were calculated as the average of three independent trials using the dose–response curve in GraphPad prism”.
| Sample | IC50 nM ± SD |
|---|---|
| a “Values are expressed as the average of three independent replicates”. “IC50 values were calculated via the sigmoidal non-linear regression curve fitting of the percentage inhibition against the concentration of the compound”. | |
| 3a | 38.28 ± 1.79 |
| 3i | 8.93 ± 0.61 |
| 3l | 119 ± 4.1 |
| Sorafenib | 30 ± 1.39 |
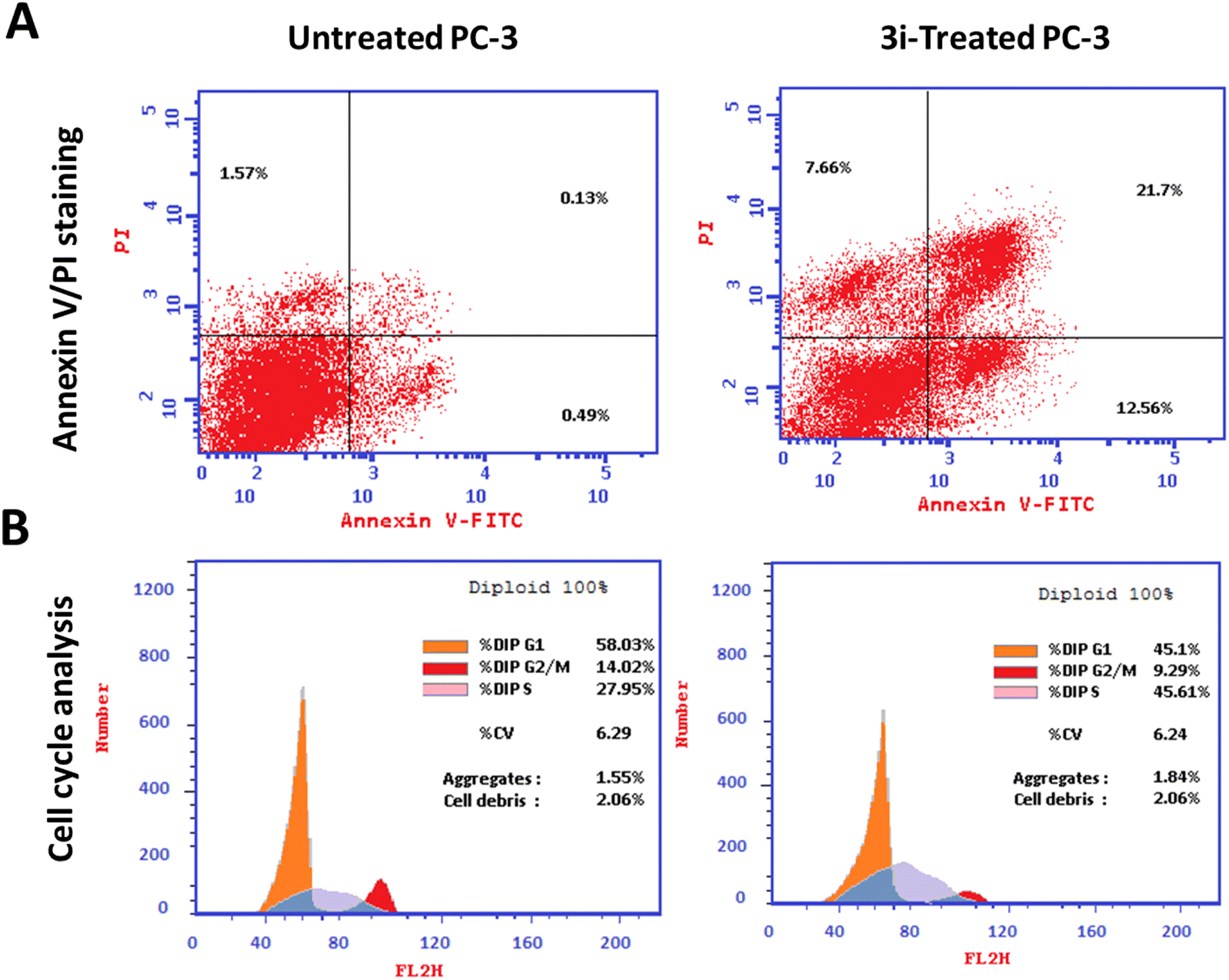 | ||
| Fig. 4 Flow cytometry analysis (A) Annexin V/PI staining for apoptosis-necrosis assessment, “Q1: necrosis, Q2: late apoptosis, Q4: early apoptosis”. (B) Histograms of DNA content at each phase of untreated and 3i-treated PC-3 cells with IC50 values of the IC50 of 1.24 μM, 48 h. | ||
The apoptosis-related gene expression levels were examined by RT-PCR in both untreated and treated cells as further validation of the apoptosis-inducing ability of the tested compound 3i in PC-3 cells. As seen in (Fig. 5), when compared to the untreated control, the levels of the proapoptotic genes P53, Bax, and caspase-3,8,9 were elevated 8.2, 6.3, 7, 2, and 8.2 fold, respectively, by compound 3i treatment, while the level of the anti-apoptotic gene Bcl-2 was decreased by 0.14 fold.
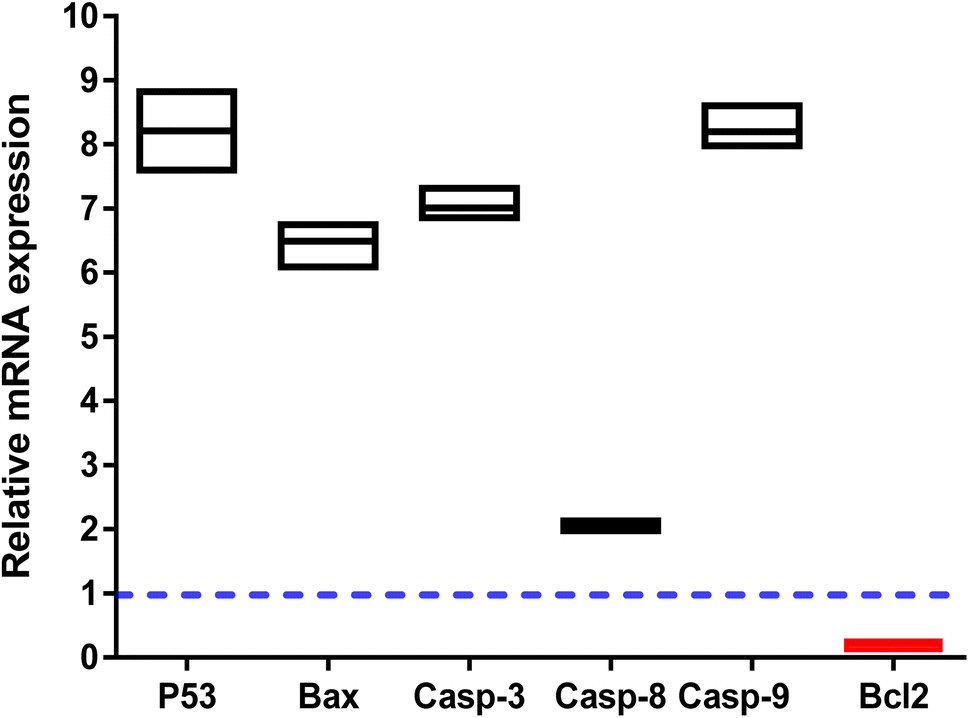 | ||
| Fig. 5 Gene expression analysis in untreated and 3i-treated PC-3 cells at the IC50 of 1.24 μM, 48 h. Values are expressed as “Mean ± SD” for three independent experimental runs. “The housekeeping gene is β-actin. Fold-change = 2−ΔΔCt, where ΔΔCt the difference between mean values of genes CT values in the treated and control groups. Fold of change in the untreated control = 1”. | ||
The apoptosis-initiating gene P53 acts as a tumor suppressor. P53, Bax, caspase 3, and caspase 9 activations were shown to be part of the intrinsic apoptotic pathway. Increased caspase-8 gene expression in treated PC-3 cells also supports the extrinsic apoptotic pathway. Our results were consistent with those of other research that used RT-PCR to draw attention to the apoptotic pathway by increasing the expression of proapoptotic genes and decreasing the expression of anti-apoptotic ones.
| Parameter | Treatments | ||||
|---|---|---|---|---|---|
| Normal control | SEC control | SEC + 3i | SEC + Sorafenib | ||
| a “Mean ± SD values of mice in each group (n = 6)”. “* Values are significantly different (P ≤ 0.05) between SEC control and normal group”, while “# values are significantly different (P ≤ 0.05) between treated SEC and SEC control mice using the un-paired test in GraphPad prism”. TIR% = C − T/C ×100. | |||||
| Tumor potentiality | Tumor weight (mg) | — | 234.6 ± 13.2 | 83.2 ± 3.54 | 79.3 ± 4.63 |
| Tumor volume (mm3) | — | 369.8 ± 17.6 | 185.6 ± 18.9 | 164.3 ± 10.2 | |
| Tumor inhibition ratio (TIR%) | — | — | 49.8 ± 1.12 | 55.5 ± 1.31 | |
As a result, a tumor-potential-relevant increase in solid tumor mass of approximately 234.6 mg was detected by tumor proliferation. The solid tumor mass was reduced to 83.2 mg and 79.3 mg after treatment with 3i and Sorafenib, respectively. Accordingly, treatments with 3i and Sorafenib significantly inhibited tumor proliferation by 49.8% and 55.5%, respectively, by tumor volume reduction from 369.8 mm3 in untreated control to 185.6 mm3 and 164.3 mm3 (Fig. 6).
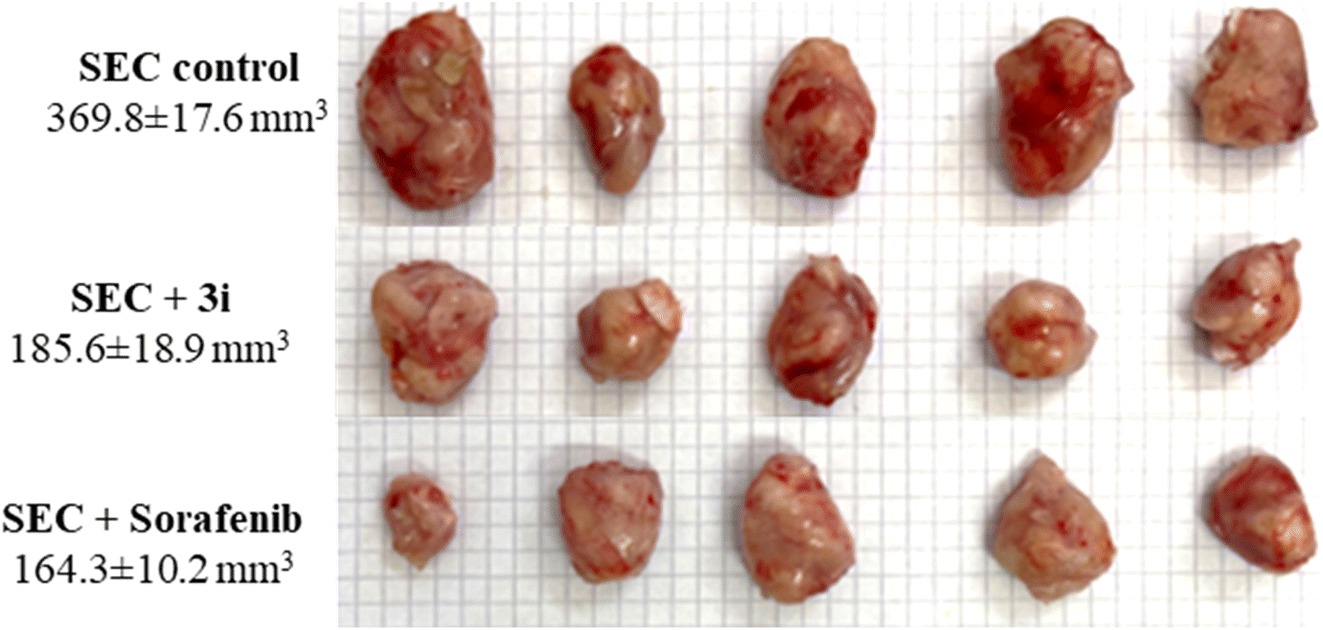 | ||
| Fig. 6 Morphological images of tumor masses and tumor volume in different groups of SEC-bearing mice (n = 5). | ||
As seen in (Fig. 6), with morphological images of tumor masses and tumor volume in different groups of SEC-bearing mice.
Antitumor potentiality of compound 3i-treatment against SEC-bearing mice was validated using histopathological examinations of tumor masses from different groups as seen in (Fig. 7).
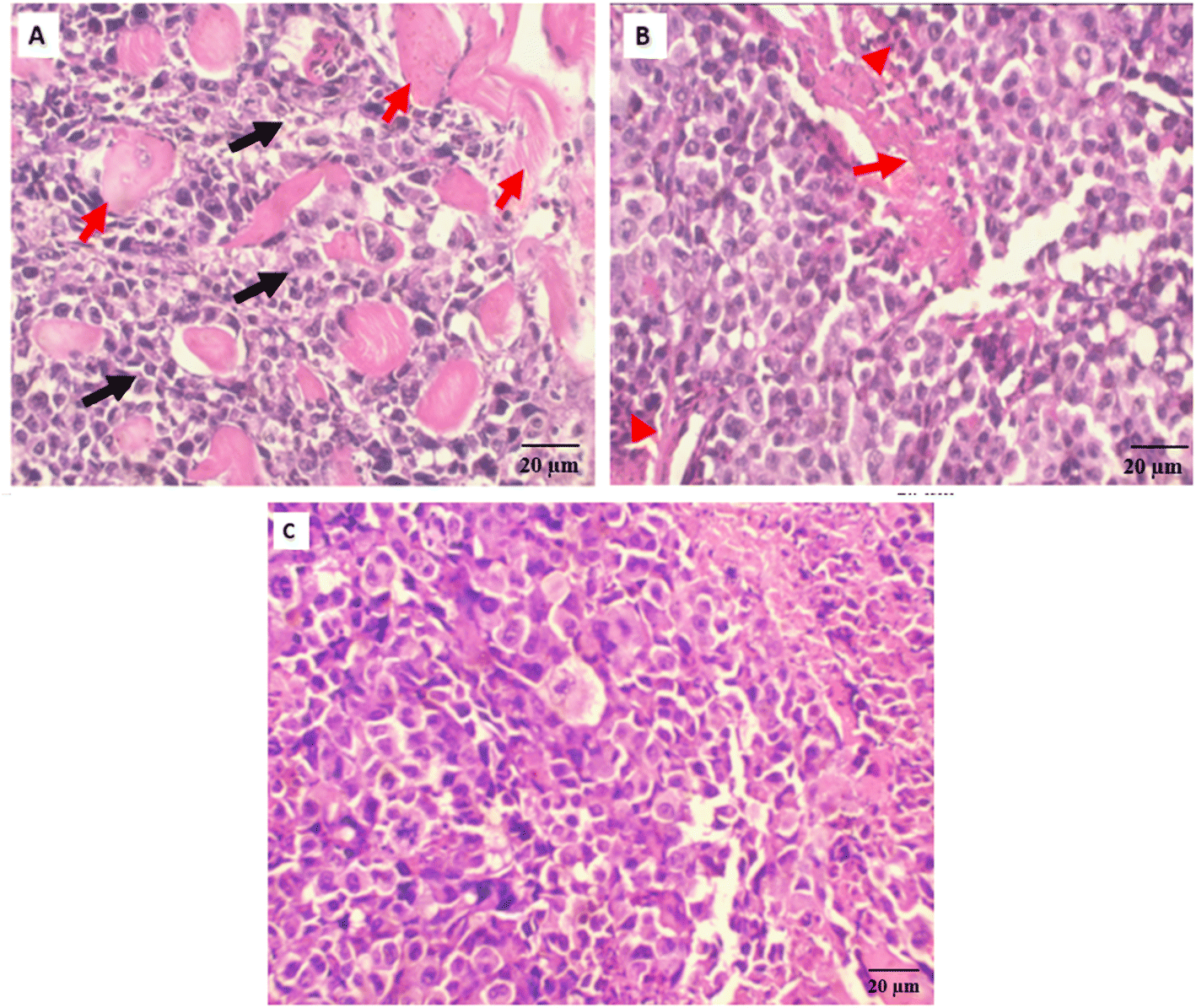 | ||
| Fig. 7 (A) Section of Ehrlich tumor-bearing mice, Tumor tissue is composed of groups and sheets of malignant epithelial cells (black arrows), showing cellular and nuclear pleomorphism, nuclear hyperchromasia, and increased nucleo-cytoplasmic ratio. Tumor cells are infiltrating skeletal muscle bundles (red arrows) (H & E, 10×), (B) section of Ehrlich tumor-bearing mice treated with Sorafenib, Tumor tissue shows multiple areas of necrosis (red arrows) and chronic inflammation (red arrowheads). Tumor cells show decreased cellular and nuclear pleomorphism (H & E, 10×) and (C) section of Ehrlich tumor-bearing mice treated with compound 3i, tumor tissue shows scattered areas of necrosis (red arrows) and chronic inflammation (red arrowheads). Tumor cells show decreased cellular and nuclear pleomorphism (H&E, 10×). | ||
3.3 Molecular modeling
Docking studies were carried out for the pyrazolone derivatives 3a and 3i, which showed excellent VEGFR-2 inhibition, to get insights into the binding mode of these ligands and ascertain the presumed molecular determinants of protein–ligand binding. The di-aryl pyrazolone structures of compounds 3a and 3i, propose its probable inhibitory binding mode which may involve the occupation of the front pocket, the gate area. So, in the present molecular docking simulations, the crystal structure used for VEGFR-2 was PDB ID: 4ASD56 in the DFG-out conformation and complex with a type II PTK inhibitor, Sorafenib, were downloaded from the Protein Data Bank (PDB)[ https://www.rcsb.org/]. First, the molecular docking protocol was validated by performing self-docking of the Sorafenib ligand in the vicinity of the VEGFR-2 active site. The self-docking validation step was able to regenerate the binding pattern of the co-crystallized ligand, indicating that the docking protocol is suitable for the planned docking study. This is indicated by the small RMSD between the docked pose and the co-crystallized ligand (0.21, 1.21 Å); and by the capability of the docking poses to regenerate all the key interactions accomplished by the complexed ligand with the key amino acid in the active site; Glu885, Cys919, and Asp1046.57,58 See ESI (Fig. S1 and S2†).The docking data highlight how well the ligand binds to VEGFR-2's ATP binding site. One possible explanation for why compound 3i is more effective at inhibiting VEGFR-2 than compound 3a is that the shorter bond lengths between the interaction groups and the amino acids allow the chemical to be driven deeper inside the active site. Moreover, compound 3i showed a better docking score (−9.685 kcal mol−1) than compound 3a (−7.520 kcal mol−1) (Fig. 8 and 9). The results elucidated that compound 3i was able to interact with Cys919 through H-bonding with the N1 of the pyrazole (2.08 Å), while the NH-pyrazole was involved in an H-bond interaction with Glu885 (1.83 Å), an arene–π interaction with Lue1035 was also observed for the pyrazole ring altogether with the difloro-phenyl moiety showing stacking interaction with val916 and Gly922 (Fig. 8).
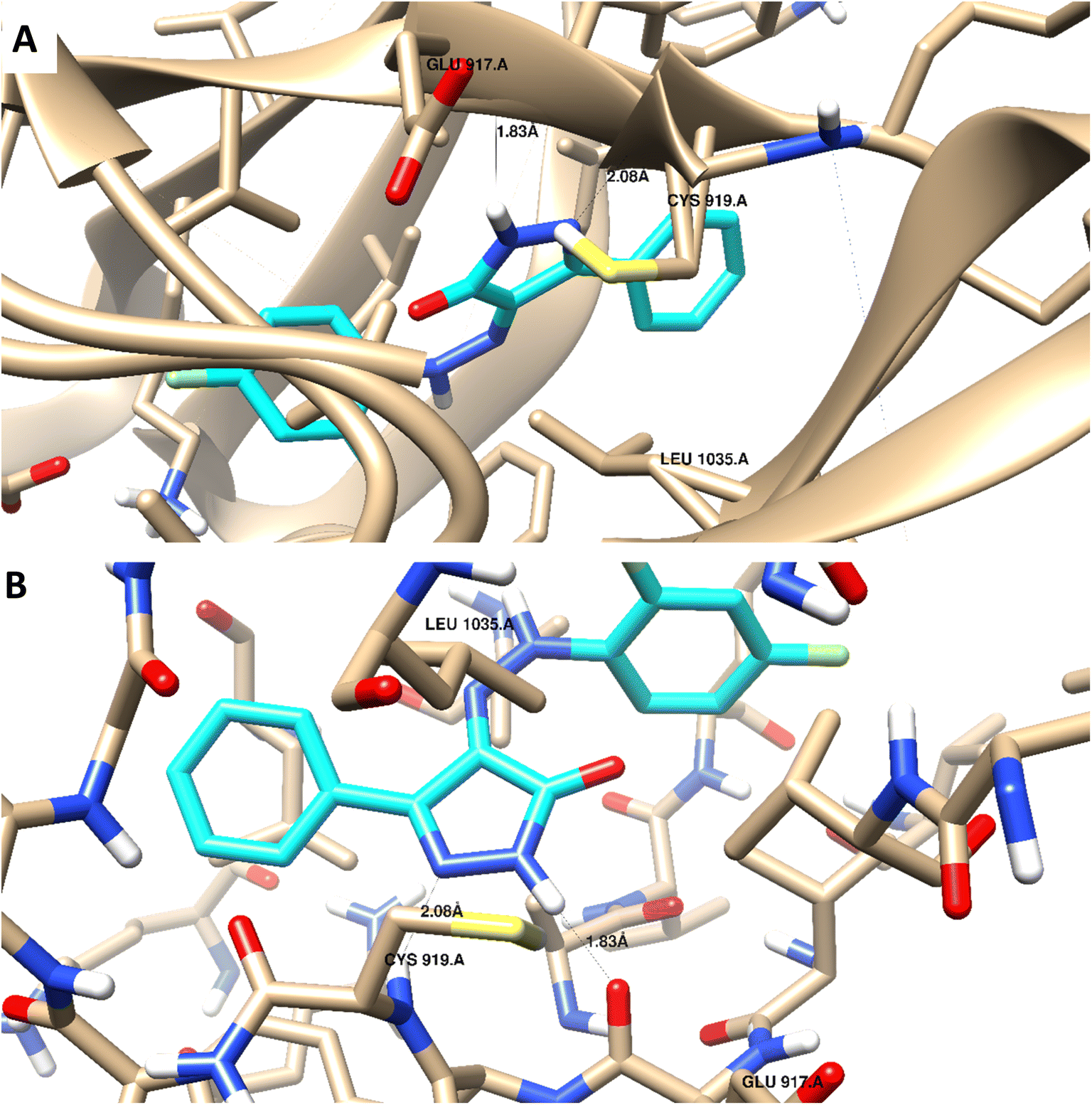 | ||
| Fig. 8 3D interaction diagram with ribbon and binding interaction representation showing compound 3i docking pose interactions with the key amino acids in the VEGFR-2 (PDB = 4ASD) active site (distances in Å). | ||
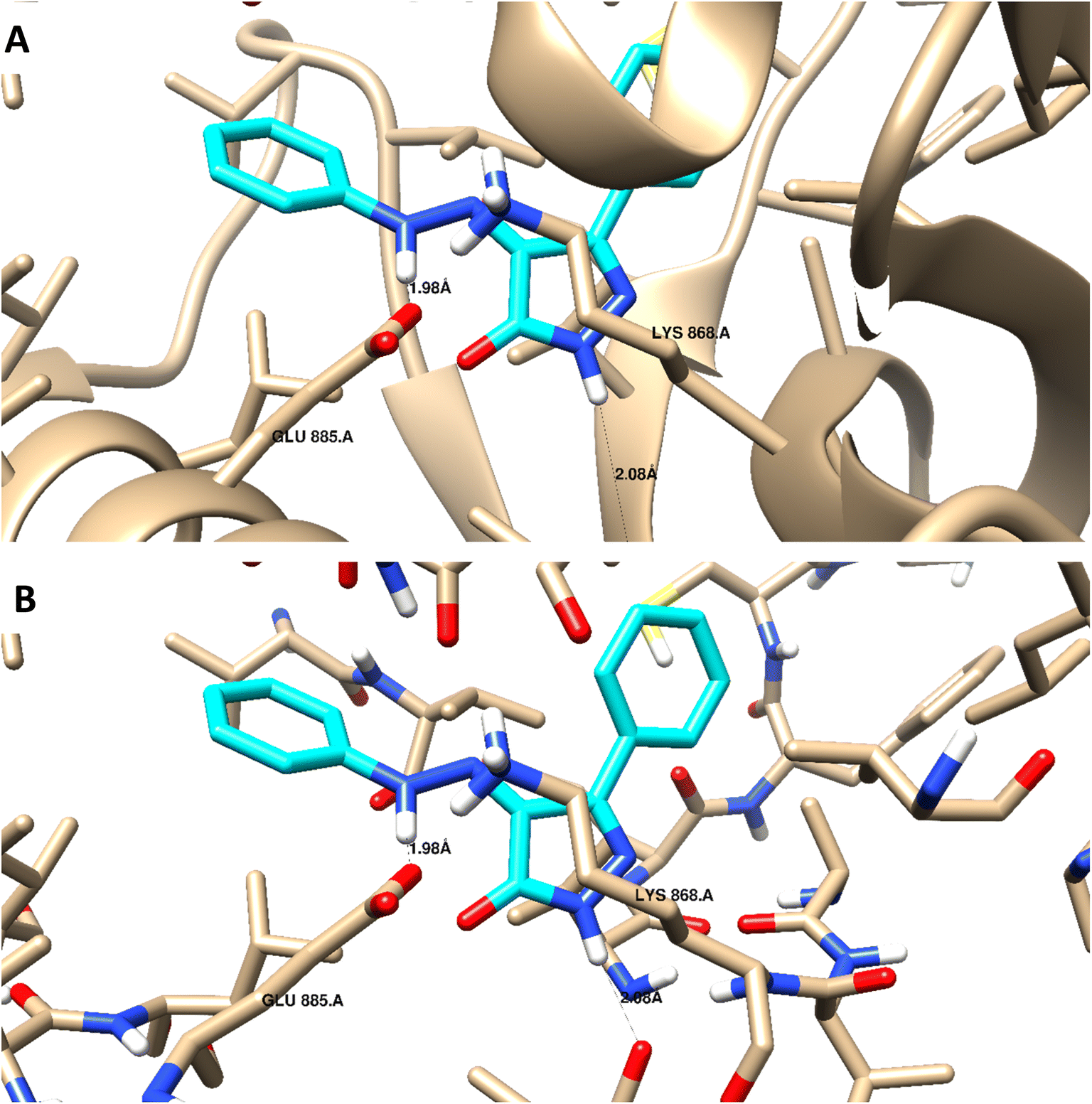 | ||
| Fig. 9 3D interaction diagram with ribbon and binding interaction representation showing compound 3a docking pose interactions with the key amino acids in the VEGFR-2 (PDB = 4ASD) active site (distances in Å). | ||
H-bond interaction with Glu885 through the NH of the azo-linker (1.98 Å), was observed for compound 3a, Val914 also formed a H-bond with NH-pyrazole along with the other hydrophobic interactions with Asp1046 and Lys868. These results are consistent with the literature, which has linked the interaction between these amino acids and effective inhibition.42,49,50 It is worth mentioning that this binding pattern allows the compounds to accommodate, through pyrazole and difluoro-phenyl moiety, into the hydrophobic pocket lined with Leu840, Phe918, Leu889, Lys868, and Ile892 amino acids. These hydrophobic interactions could induce an inactive VEGFR-2 stable conformation of DFG site, which may display improved efficiency and higher potency.59
4 Conclusion
Novel pyrazole derivatives were synthesized and tested for in vitro anticancer activity against the PC-3 human cancer cell line. These compounds' activity was compared to the standard cancer treatments Doxorubicin and Sorafenib. Compounds 3a and 3i showed nearly comparable activities compared to the reference drugs, with IC50 values of 1.22 and 1.24 μM. The compounds were tested for their ability to inhibit VEGFR-2 in vitro, with the results showing that compound 3a is a potent VEGFR-2 inhibitor (IC50 = 38.28 nM), while compound 3i is the most potent VEGFR-2 inhibitor (IC50 = 8.93 nM). Docking of these two compounds within the active site of VEGFR-2 highlighted their mode of action, resulting in potent inhibitory efficacy. According to docking results, compounds 3a and 3i interacted with the catalytic amino acids in this enzyme. Compound 3i significantly stimulated total apoptotic prostate cancer cell death 55.2-fold (34.26% compared to 0.62% for the control) arresting the cell cycle at S-phase. Moreover, it affected the apoptosis-related genes upregulating the proapoptotic genes and downregulating the Bcl-2 as an antiapoptotic one. Finally, in vivo study revealed the potentiality of compound 3i to inhibit tumor proliferation by 49.8% reducing the tumor weight from 234.6 mg in untreated mice to 83.2 mg. Hence, compound 3i may serve as a potential anti-prostate cancer agent through apoptosis-induction.5 Experimental
5.1 Chemistry
Starting materials and reagents were purchased from Sigma-Aldrich (USA), Alfa-Aesar and Loba Chemie Organics and were used without further purification. Solvents were purchased from Fisher scientific or Sigma-Aldrich and used without further purification. Chemical reactions were monitored by analytical thin layer chromatography (TLC), on silica gel 60 F254 packed on Aluminum sheets, 20 × 20 cm (Sigma-Aldrich) and were visualized under U.V. light (254 nm). Dichlomethane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methanol (1:0.1) was the adopted elution system. Melting points were carried out by the open capillary tube method using a Stuart (Stone Staffordshire ST/50SA UK) apparatus and they were uncorrected. Infrared Spectra were performed on Schimadzu FT-IR 8400S spectrophotometer using potassium bromide discs and expressed in wave number (cm−1), at the Faculty of Pharmacy, Cairo University. 1H NMR spectra were recorded on a Bruker 400 MHz spectrometer in δ scale (ppm), using DMSO as solvent at the special unit facility, faculty of pharmacy, Ain-Shams University and Mansoura University. 1H NMR spectra were also recorded on a Gemini 300 MHz spectrometer in δ scale (ppm), using DMSO as solvent at Main Defence Chemical Laboratory. 13C NMR spectra were recorded in δ scale given in ppm on a Bruker 400 (at 101 MHz) spectrophotometer at the special unit facility, Faculty of Pharmacy, Ain-Shams University and Mansoura University. Elemental analyses were carried out using FLASH 2000 CHNS/O analyzer, Thermo Scientific at the Regional Centre for Mycology and Biotechnology (RCMB), Al-Azhar University, Nasr City, Cairo. Mass spectra were carried out on Direct Inlet part to mass analyzer in Thermo Scientific GCMS model ISQ at the Regional Centre for Mycology and Biotechnology (RCMB), Al-Azhar University, Nasr City, Cairo. Mass spectrum was carried out on Direct Probe Controller Inlet part to Single Quadropole mass analyzer in Thermo Scientific GCMS model ISQ LT using Thermo X-Calibur software at the Regional Center for Mycology and Biotechnology (RCMB), Al-Azhar University.
:methanol (1:0.1) was the adopted elution system. Melting points were carried out by the open capillary tube method using a Stuart (Stone Staffordshire ST/50SA UK) apparatus and they were uncorrected. Infrared Spectra were performed on Schimadzu FT-IR 8400S spectrophotometer using potassium bromide discs and expressed in wave number (cm−1), at the Faculty of Pharmacy, Cairo University. 1H NMR spectra were recorded on a Bruker 400 MHz spectrometer in δ scale (ppm), using DMSO as solvent at the special unit facility, faculty of pharmacy, Ain-Shams University and Mansoura University. 1H NMR spectra were also recorded on a Gemini 300 MHz spectrometer in δ scale (ppm), using DMSO as solvent at Main Defence Chemical Laboratory. 13C NMR spectra were recorded in δ scale given in ppm on a Bruker 400 (at 101 MHz) spectrophotometer at the special unit facility, Faculty of Pharmacy, Ain-Shams University and Mansoura University. Elemental analyses were carried out using FLASH 2000 CHNS/O analyzer, Thermo Scientific at the Regional Centre for Mycology and Biotechnology (RCMB), Al-Azhar University, Nasr City, Cairo. Mass spectra were carried out on Direct Inlet part to mass analyzer in Thermo Scientific GCMS model ISQ at the Regional Centre for Mycology and Biotechnology (RCMB), Al-Azhar University, Nasr City, Cairo. Mass spectrum was carried out on Direct Probe Controller Inlet part to Single Quadropole mass analyzer in Thermo Scientific GCMS model ISQ LT using Thermo X-Calibur software at the Regional Center for Mycology and Biotechnology (RCMB), Al-Azhar University.
5.2 General methods
5.2.2.1 (Z)-4-(2-(4-Phenyl)hydrazono)-3-phenyl-1H-pyrazol-5(4H)-one 3a. Yield (%) 75. M.p. 219–221 °C. IR (KBr) (ν, cm−1): 3432, 3191 (NH), 3047 (CH aromatic); 1662 (CO), 1592 (C
N). 1H NMR (400 MHz, DMSO-d6) δ ppm: 7.20–7.23 (t, 1H, H-aromatic); 7.26–7.57 (m, 7H, H-aromatic); 8.06 (d, 2H, H-aromatic, J = 8 Hz); 12.13 (brs, 2H, NH, D2O-exchangeable). 13C NMR (100 MHz, DMSO-d6) δ 116.40, 125.99. 126.98, 127.24, 129.13, 129.68, 130.21, 131.24, 141.75, 145.74, 160.97. MS m/z (%): 264 (8.84), 82 (100). Anal. calcd for C15H12N4O: C, 68.17; H, 4.58; N, 21.20; O, 6.05. Found: C, 67.98; H, 4.66; N, 21.47.
5.2.2.2 (Z)-4-(2-(4-Chlorophenyl)hydrazono)-3-phenyl-1H-pyrazol-5(4H)-one 3b. Yield (%): 84. m.p. 190–191 °C. IR (KBr, ν cm−1): 3543, 3267 (NH), 3007 (CH aromatic); 1671 (CO), 1618 (C
N). 1H NMR (400 MHz, DMSO-d6) δ ppm: 7.40–7.47 (m, 5H, H-aromatic); 7.89–7.92 (m, 4H, H-aromatic); 11.88 (s, 1H, NH, D2O-exchangeable); 11.97 (s, 1H, NH, D2O-exchangeable). 13C NMR (100 MHz, DMSO-d6) δ 127.37, 128.23, 128.96, 130.04, 130.06, 143.81, 145.75, 155.62. MS m/z (%): 299 (12), 297 (31), 283 (93), 112 (100). Anal. calcd for: C15H11ClN4O: C, 60.31; H, 3.71; N, 18.76. Found: C, 60.09; H, 3.84; N, 19.02.
5.2.2.3 (Z)-4-(2-(4-Flourophenyl)hydrazono)-3-phenyl-1H-pyrazol-5(4H)-one 3c. Yield (%): 90. m.p. 176–178 °C. IR (KBr, ν cm−1): 3540, 3268 (NH); 3012 (CH aromatic); 2848 (CH aliphatic); 1671 (CO); 1621 (C
N). 1H NMR (400 MHz, DMSO-d6) δ ppm: 7.32–7.5 (m, 3H, H-aromatic); 7.64–7.66 (t, 2H, 4-F-Ph); 7.88–7.92 (m, 2H, H-aromatic); 8.05 (d, 2H, H-aromatic); 11.87 (s, NH, D2O-exchangeable); 14.52 (s, NH, D2O-exchangeable). 13C NMR (100 MHz, DMSO-d6) δ 127.34, 128.32, 128.77, 129.0, 129.99, 130.69, 143.81, 145.73, 155.67. MS m/z (%): 282 (33), 216 (100). Anal. calcd for: C15H11FN4O: C, 63.83; H, 3.93; N, 19.85. Found: C, 64.12; H, 3.85; N, 20.11.
5.2.2.4 (Z)-4-(2-(5-Oxo-3-phenyl-1H-pyrazol-4(5H)-ylidene)hydrazinyl)benzoic acid 3d. Yield (%): 76. m.p. >300 °C. IR (KBr, ν cm−1): 3552, 3269 (NH); 3010 (CH aromatic); 1709, 1654 (CO); 1590 (C
N). 1H NMR (400 MHz, DMSO-d6) δ ppm: 7.39–7.55 (m, 1H, H-aromatic); 7.62 (d, 2H, 4-COOH-Ph, J = 8.7 Hz); 7.90–7.93 (m, 2H-aromatic); 8.03 (d, 2H, H-aromatic); 8.08 (d, 2H, 4-COOH-Ph, J = 8.7 Hz); 12.15 (brs, 3H, NH, OH, D2O-exchangeable). 13C NMR (100 MHz, DMSO-d6) δ 116.12, 127.05, 127.37, 130.0, 130.74, 130.99, 143.91, 145.35, 155.51, 160.71, 167.21. MS m/z (%): 308 (28), 119 (100). Anal. calcd for: C16H12N4O3: C, 62.33; H, 3.92; N, 18.17. Found: C, 62.19; H, 4.15; N, 18.43.
5.2.2.5 (Z)-2-(4-(2-(5-Oxo-3-phenyl-1H-pyrazol-4(5H)-ylidene)hydrazinyl) phenyl)acetic acid 3e. Yield (%): 83. m.p. 168–170 °C. IR (KBr, ν cm−1): 3421, 3268 (NH, OH), 2973, 2782 (CH); 1709, 1617 (CO), 1470 (C
N). 1H NMR (400 MHz, DMSO-d6) δ ppm: 2.5 (s, 2H, CH2); 7.35–7.45 (m, 5H, H-aromatic); 7.89–7.92 (m, 4H, H-aromatic); 11.86 (s, 1H, NH, D2O-exchangeable); 14.49 (brs, 1H, OH, D2O-exchangeable). 13C NMR (100 MHz, DMSO-d6) δ 34.94, 127.37, 128.28, 128.65, 129.03, 130.15, 130.67, 143.83, 145.71, 155.66. MS m/z (%): 322 (29), 176 (100). Anal. calcd for: C17H14N4O3: C, 63.35; H, 4.38; N, 17.38. Found: C, 63.17; H, 4.50; N, 17.54.
5.2.2.6 (Z)-4-(2-(3-Chlorophenyl)hydrazono)-3-phenyl-1H-pyrazol-5(4H)-one 3f. Yield (%): 70. m.p. 180–181 °C. IR (KBr, ν cm−1): 3552, 3269 (NH), 1709 (CO), 1611 (C
N). 1H NMR (400 MHz, DMSO-d6) δ ppm: 7.39–7.91 (m, 5H, H-aromatic); 7.92 (d, 4H, H-aromatic); 11.81 (s, 1H, NH, D2O-exchangeable); 14.38 (s, 1H, NH, D2O-exchangeable). 13C NMR (100 MHz, DMSO-d6) δ 127.37, 128.24, 128.73, 128.99, 129.66, 130.03, 130.68, 132.35, 143.82, 145.69, 155.58, 163.84.
5.2.2.7 (Z)-4-(2-(2-Ethylphenyl)hydrazono)-3-phenyl-1H-pyrazol-5(4H)-one 3g. Yield (%): 33. M.p. 218–220 °C. IR (KBr, ν cm−1): 3541, 3266 (NH), 3010 (CH aromatic); 2852, 3082 (CH aliphatic); 1676 (CO), 1611 (C
N). 1H NMR (400 MHz, DMSO-d6) δ ppm: 1.26 (t, 3H, CH3); 2.69 (q, 2H, CH2); 7.17–7.55 (m, 6H, H-aromatic); 7.75 (d, 1H, H-aromatic); 8.08 (d, 2H, H-aromatic); 12.18 (s, 1H, NH, D2O-exchangeable); 14.19 (s, 1H, NH, D2O-exchangeable). 13C NMR (100 MHz, DMSO-d6) δ 14.27, 23.62, 114.78, 126.19, 127.03, 128.03, 128.26, 129.33, 129.85, 130.08, 131.30, 131.44, 138.92, 145.60, 161.53. MS m/z (%): 292 (10), 90 (100). Anal. calcd for: C17H16N4O: C, 69.85; H, 5.52; N, 19.17. Found: C, 69.71; H, 5.68; N, 19.41.
5.2.2.8 (Z)-3-Phenyl-4-(2-(3-(trifluoromethyl)phenyl)hydrazono)-1H-pyrazol-5(4H)-one 3h. Yield (%): 60. M.p. 234–235 °C. IR (KBr, ν cm−1): 3380, 3142 (NH), 3043 (CH aromatic); 1675 (CO), 1556 (C
N). 1H NMR (400 MHz, DMSO-d6) δ ppm: 7.41–7.48 (m, 4H, H-aromatic); 7.61–7.66 (m, 2H, H-aromatic); 7.81 (s, 1H, 3-(CF3)-Ph); 7.97 (d, 2H, H-aromatic, J = 8 Hz). 13C NMR (100 MHz, DMSO-d6) δ 112.85, 119.98, 121.73, 122.86, 125.56, 128.20, 129.70, 130.54, 130.87, 131.30, 142.78, 145.91, 160.40. MS m/z (%): 332 (8.45), 93 (100). Anal. calcd for: C16H11F3N4O: C, 57.83; H, 3.34; N, 16.86. Found: C, 58.09; H, 3.47; N, 17.12.
5.2.2.9 (Z)-4-(2-(2,4-Difluorophenyl)hydrazono)-3-phenyl-1H-pyrazol-5(4H)-one 3i. Yield (%) 83. 199–200 °C. IR (KBr, ν cm−1): 3436, 3214 (NH); 3060 (CH aromatic); 1668 (CO), 1604 (C
N). 1H NMR (400 MHz, DMSO-d6) δ ppm: 7.17 (t, 1H, 2,4-(F)2-Ph); 7.52–7.68 (m, 3H, H-aromatic); 7.79–7.87 (m, 2H, 2,4-(F)2-Ph); 8.06 (d, 2H, H-aromatic); 12.24 (brs, 2H, NH, D2O-exchangeable). 13C NMR (100 MHz, DMSO-d6) δ 105.32, 113.47, 117.26, 126.83, 129.22, 129.81, 130.95, 145.55, 150.25, 158.18, 161.31. MS m/z (%): 300.08. Anal. calcd for: C15H10F2N4O. Found: C, 60.00; H, 3.36; F, 12.65; N, 18.66; O, 5.33.
5.2.2.10 (Z)-4-(2-(2,6-Dichlorophenyl)hydrazono)-3-phenyl-1H-pyrazol-5(4H)-one 3j. Yield (%) 90, m.p. 182–183 °C. IR (KBr, ν cm−1): 3542, 3269 (NH), 1670 (CO), 1622 (C
N). 1H NMR (400 MHz, DMSO-d6) δ ppm: 7.39–7.43 (m, 5H, H-aromatic); 7.64–7.67 (m, 1H, H-aromatic); 7.87–7.92 (m, 2H H-aromatic); 11.87 (s, 1H, NH, D2O-exchangeable); 14.50 (s, 1H, NH, D2O-exchangeable). 13C NMR (100 MHz, DMSO-d6) δ 127.36, 128.26, 128.74, 129.01, 129.69, 130.06, 130.64, 132.31, 142.56, 143.78, 145.66, 155.58, 163.83. MS m/z (%): 332 (42), 334 (36), 336 (33). Anal. calcd for: C15H10Cl2N4O: C, 54.08; H, 3.03; N, 16.82. Found: C, 54.29; H, 3.27; N, 17.08.
5.2.2.11 (Z)-4-(2-(2,6-Dimethylphenyl)hydrazono)-3-phenyl-1H-pyrazol-5(4H)-one 3k. Yield (%) 90. M.p. 186–187 °C. IR (KBr, ν cm−1): 3540, 3267 (NH), 3010, 2852 (CH); 1669 (CO), 1622 (C
N). 1H NMR (400 MHz, DMSO-d6) δ ppm: 2.5 (s, 6H, 2CH3); 7.40–7.48 (m, 4H, H-aromatic); 7.64–7.67 (m, 1H, H-aromatic); 7.89–7.92 (m, 3H, H-aromatic); 11.88 (s, 1H, NH, D2O-exchangeable); 14.50 (s, 1H, NH, D2O-exchangeable). 13C NMR (100 MHz, DMSO-d6) δ 127.38, 128.26, 128.74, 129.01, 129.69, 130.06, 130.67, 132.33, 143.79, 145.67, 155.59, 163.89. MS m/z (%): 294 (22), 292 (100). Anal. calcd for: C17H16N4O: C, 69.85; H, 5.52; N, 19.17. Found: C, 70.12; H, 5.68; N, 19.45.
5.2.2.12 (Z)-4-(2-(3,4-Dimethoxyphenyl)hydrazono)-3-phenyl-1H-pyrazol-5(4H)-one 3l. Yield (%) 86. m.p. 244–245 °C. IR (KBr, ν cm−1): 3439, 3325 (NH); 3042 (CH aromatic); 2927 (CH aliphatic); 1658 (CO); 1604 (C
N). 1H NMR (400 MHz, DMSO-d6) δ ppm: 3.78 (s, 3H, OCH3); 3.84 (s, 3H, OCH3); 7.03–7.16 (m, H, 2H-aromatic); 7.25 (s, 1H, 3,4-(OCH3)2-Ph); 7.40–7.50 (m, 3H, H-aromatic); 8.01 (d, 2H, H-aromatic); 11.96 (brs, 2H, NH, D2O-exchangeable). 13C NMR (100 MHz, DMSO-d6) δ 56.02, 56.30, 101.22, 108.53, 113.02, 125.99, 126.99, 129.13, 129.59, 131.47, 145.56, 147.58, 150.11, 160.97. MS m/z (%): 324 (38), 232 (100). Anal. calcd for: C17H16N4O3: C, 62.95; H, 4.97; N, 17.27. Found: C, 62.70; H, 5.13; N, 17.45.
5.3 Biology
PC-3 cells were incubated into 6-well culture plates (3–5× 105 cells per well) overnight, and they were then treated with compound 3i for 48 h. Then, media supernatants and cells were collected the cells were suspended in 100 μL of Annexin binding buffer solution “25 mM CaCl2, 1.4 M NaCl, and 0.1 M Hepes/NaOH, pH 7.4” and incubated with “Annexin V-FITC solution (1:100) and propidium iodide (PI) at a concentration equals 10 μg mL−1 in the dark for 30 min.” The stained cells were then harvested using the Cytoflex FACS machine, and cytExpert software was used to analyze the data.63–65
– Gene expression analysis (RT-PCR) for the selected genes
The apoptotic pathway of compound 3i against PC-3 cells, gene expression of P53, Bax, PUMA, Caspapses-3,8,9 as pro-apoptotic genes and Bcl-2 as the anti-apoptotic gene were investigated; their sequences in forward and reverse direction were supported (Table 4). PC-3 cells were treated with compound 3i at its IC50 value for 48 h. Then, a routine RT-PCR experiment was run, and the data was reported in cycle thresholds (Ct) and Ct to allow comparison of gene expression levels to that of the housekeeping gene, β-actin.63,66,67
| Gene | Forward | Reverse |
|---|---|---|
| P53 | 5′-CCCCTCCTGGCCCCTGTCATCTTC-3′ | 5′-GCAGCGCCTCACAACCTCCGTCAT-3′ |
| BAX | 5′-GTTTCATCCAGGATCGAGCAG-3′ | 5′-CATCTTCTTCCAGATGGTGA-3′ |
| PUMA | 5′-GAGGAGGAACAGTGGGC-3′ | 5′-CTAATTGGGCTCCATCTCGG-3′ |
| CASP-3 | 5′-TGGCCCTGAAATACGAAGTC-3′ | 5′-GGCAGTAGTCGACTCTGAAG-3′ |
| CASP-8 | 5′-AATGTTGGAGGAAAGCAAT-3′ | 5′-CATAGTCGTTGATTATCTTCAGC-3′ |
| CASP-9 | 5′-CGAACTAACAGGCAAGCAGC-3′ | 5′-ACCTCACCAAATCCTCCAGAAC-3′ |
| Bcl-2 | 5′-CCTGTGGATGACTGAGTACC-3′ | 5′-GAGACAGCCAGGAGAAATCA-3′ |
| β-actin | 5′-GTGACATCCACACCCAGAGG-3′ | 5′-ACAGGATGTCAAAACTGCCC-3′ |
Conflicts of interest
There are no conflicts to declare.References
- M. C. Harding, Transitions From Heart Disease to Cancer as the Leading Cause of Death in US States, 1999–2016, Prev. Chronic Dis., 2018, 15, 180151, DOI:10.5888/pcd15.180151.
- M. Montana, F. Mathias, T. Terme and P. Vanelle, Antitumoral activity of quinoxaline derivatives: A systematic review, Eur. J. Med. Chem., 2019, 163, 136–147, DOI:10.1016/j.ejmech.2018.11.059.
- M. Yang, H. Liu, Y. Zhang, X. Wang and Z. Xu, Moxifloxacin-isatin Hybrids Tethered by 1,2,3-triazole and their Anticancer Activities, Curr. Top. Med. Chem., 2020, 20, 1461–1467 CrossRef CAS PubMed.
- L. Zhong, X.-Y. Fu, C. Zou, L.-L. Yang, S. Zhou, J. Yang, Y. Tang, C. Cheng, L.-L. Li, R. Xiang, L.-J. Chen, Y.-Z. Chen, Y.-Q. Wei and S.-Y. Yang, A preclinical evaluation of a novel multikinase inhibitor, SKLB-329, as a therapeutic agent against hepatocellular carcinoma, Int. J. Cancer, 2014, 135, 2972–2983, DOI:10.1002/ijc.28944.
- L. Wang, M. Xu, C.-Y. Kao, S. Y. Tsai and M.-J. Tsai, Small molecule JQ1 promotes prostate cancer invasion via BET-independent inactivation of FOXA1, J. Clin. Invest., 2020, 130, 1782–1792, DOI:10.1172/JCI126327.
- D. P. Petrylak, Future directions in the treatment of androgen-independent prostate cancer, Urology, 2005, 65, 8–12, DOI:10.1016/j.urology.2005.04.020.
- G. McMahon, VEGF Receptor Signaling in Tumor Angiogenesis, Oncologist, 2000, 5, 3–10, DOI:10.1634/theoncologist.5-suppl_1-3.
- D. Shweiki, A. Itin, D. Soffer and E. Keshet, Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis, Nature, 1992, 359, 843–845, DOI:10.1038/359843a0.
- A.-K. Olsson, A. Dimberg, J. Kreuger and L. Claesson-Welsh, VEGF receptor signalling ? In control of vascular function, Nat. Rev. Mol. Cell Biol., 2006, 7, 359–371, DOI:10.1038/nrm1911.
- R. S. Apte, D. S. Chen and N. Ferrara, VEGF in Signaling and Disease: Beyond Discovery and Development, Cell, 2019, 176, 1248–1264, DOI:10.1016/j.cell.2019.01.021.
- M. Shibuya, Vascular endothelial growth factor and its receptor system: physiological functions in angiogenesis and pathological roles in various diseases, J. Biochem., 2013, 153, 13–19, DOI:10.1093/jb/mvs136.
- D. Alferez, R. W. Wilkinson, J. Watkins, R. Poulsom, N. Mandir, S. R. Wedge, I. T. Pyrah, N. R. Smith, L. Jackson, A. J. Ryan and R. A. Goodlad, Dual inhibition of VEGFR and EGFR signaling reduces the incidence and size of intestinal adenomas in ApcMin/+ mice, Mol. Cancer Therapeut., 2008, 7, 590–598, DOI:10.1158/1535-7163.MCT-07-0433.
- A. E.-S. Mghwary, E. M. Gedawy, A. M. Kamal and S. M. Abuel-Maaty, Novel thienopyrimidine derivatives as dual EGFR and VEGFR-2 inhibitors: design, synthesis, anticancer activity and effect on cell cycle profile, J. Enzyme Inhib. Med. Chem., 2019, 34, 838–852, DOI:10.1080/14756366.2019.1593160.
- S. J. Modi and V. M. Kulkarni, Vascular Endothelial Growth Factor Receptor (VEGFR-2)/KDR Inhibitors: Medicinal Chemistry Perspective, Med. Drug Discovery, 2019, 2, 100009, DOI:10.1016/j.medidd.2019.100009.
- R. T.-P. Poon, S.-T. Fan and J. Wong, Clinical Implications of Circulating Angiogenic Factors in Cancer Patients, J. Clin. Oncol., 2001, 19, 1207–1225, DOI:10.1200/JCO.2001.19.4.1207.
- P. Traxler, Tyrosine kinases as targets in cancer therapy – successes and failures, Expert Opin. Ther. Targets, 2003, 7, 215–234, DOI:10.1517/14728222.7.2.215.
- N. Ferrara, VEGF as a Therapeutic Target in Cancer, Oncology, 2005, 69, 11–16, DOI:10.1159/000088479.
- R. S. Kerbel, Tumor angiogenesis: past, present and the near future, Carcinogenesis, 2000, 21, 505–515, DOI:10.1093/carcin/21.3.505.
- P. M. Fischer, Approved and Experimental Small-Molecule Oncology Kinase Inhibitor Drugs: A Mid-2016 Overview, Med. Res. Rev., 2017, 37, 314–367, DOI:10.1002/med.21409.
- N. Berndt, R. M. Karim and E. Schönbrunn, Advances of small molecule targeting of kinases, Curr. Opin. Chem. Biol., 2017, 39, 126–132, DOI:10.1016/j.cbpa.2017.06.015.
- D. Fabbro, 25 Years of Small Molecular Weight Kinase Inhibitors: Potentials and Limitations, Mol. Pharmacol., 2015, 87, 766–775, DOI:10.1124/mol.114.095489.
- M. E. Breen and M. B. Soellner, Small Molecule Substrate Phosphorylation Site Inhibitors of Protein Kinases: Approaches and Challenges, ACS Chem. Biol., 2015, 10, 175–189, DOI:10.1021/cb5008376.
- G. Bergers and D. Hanahan, Modes of resistance to anti-angiogenic therapy, Nat. Rev. Cancer, 2008, 8, 592–603, DOI:10.1038/nrc2442.
- R. N. Gacche and R. J. Meshram, Angiogenic factors as potential drug target: Efficacy and limitations of anti-angiogenic therapy, Biochim. Biophys. Acta, Rev. Cancer, 2014, 1846, 161–179, DOI:10.1016/j.bbcan.2014.05.002.
- İ. Koca, A. Özgür, K. A. Coşkun and Y. Tutar, Synthesis and anticancer activity of acyl thioureas bearing pyrazole moiety, Bioorg. Med. Chem., 2013, 21, 3859–3865, DOI:10.1016/j.bmc.2013.04.021.
- K. M. Dawood, T. M. A. Eldebss, H. S. A. El-Zahabi, M. H. Yousef and P. Metz, Synthesis of some new pyrazole-based 1,3-thiazoles and 1,3,4-thiadiazoles as anticancer agents, Eur. J. Med. Chem., 2013, 70, 740–749, DOI:10.1016/j.ejmech.2013.10.042.
- K. R. A. Abdellatif, E. K. A. Abdelall, M. A. Abdelgawad, R. R. Ahmed and R. B. Bakr, Synthesis and Anticancer Activity of Some New Pyrazolo[3,4-d]pyrimidin-4-one Derivatives, Molecules, 2014, 19, 3297–3309, DOI:10.3390/molecules19033297.
- C.-H. Tu, W.-H. Lin, Y.-H. Peng, T. Hsu, J.-S. Wu, C.-Y. Chang, C.-T. Lu, P.-C. Lyu, C. Shih, W.-T. Jiaang and S.-Y. Wu, Pyrazolylamine Derivatives Reveal the Conformational Switching between Type I and Type II Binding Modes of Anaplastic Lymphoma Kinase (ALK), J. Med. Chem., 2016, 59, 3906–3919, DOI:10.1021/acs.jmedchem.6b00106.
- F. E. Bennani, L. Doudach, Y. Cherrah, Y. Ramli, K. Karrouchi, M. Ansar and M. E. A. Faouzi, Overview of recent developments of pyrazole derivatives as an anticancer agent in different cell line, Bioorg. Chem., 2020, 97, 103470, DOI:10.1016/j.bioorg.2019.103470.
- D. Havrylyuk, B. Zimenkovsky, O. Vasylenko, A. Gzella and R. Lesyk, Synthesis of New 4-Thiazolidinone-, Pyrazoline-, and Isatin-Based Conjugates with Promising Antitumor Activity, J. Med. Chem., 2012, 55, 8630–8641, DOI:10.1021/jm300789g.
- H. H. Fahmy, N. M. Khalifa, M. M. F. Ismail, H. M. El-Sahrawy and E. S. Nossier, Biological Validation of Novel Polysubstituted Pyrazole Candidates with in Vitro Anticancer Activities, Molecules, 2016, 21, 271, DOI:10.3390/molecules21030271.
- G. M. Nitulescu, C. Draghici and O. T. Olaru, New Potential Antitumor Pyrazole Derivatives: Synthesis and Cytotoxic Evaluation, Int. J. Mol. Sci., 2013, 14, 21805–21818, DOI:10.3390/ijms141121805.
- M. Wang, S. Xu, H. Lei, C. Wang, Z. Xiao, S. Jia, J. Zhi, P. Zheng and W. Zhu, Design, synthesis and antitumor activity of Novel Sorafenib derivatives bearing pyrazole scaffold, Bioorg. Med. Chem., 2017, 25, 5754–5763, DOI:10.1016/j.bmc.2017.09.003.
- D. Anil, E. U. Caykoylu, F. Sanli, N. Gambacorta, O. F. Karatas, O. Nicolotti, O. Algul and S. Burmaoglu, Synthesis and biological evaluation of 3,5-diaryl-pyrazole derivatives as potential antiprostate cancer agents, Arch. Pharmazie, 2021, 354, 2100225, DOI:10.1002/ardp.202100225.
- J. Bronson, M. Dhar, W. Ewing, and N. Lonberg, Chapter Thirty-One - To Market, To Market—2011, in Annual Reports in Medicinal Chemistry, ed. M. C. Desai, Academic Press, 2012, pp. 499–569. DOI: DOI:10.1016/B978-0-12-396492-2.00031-X.
- A. L. Hsu, T. T. Ching, D. S. Wang, X. Song, V. M. Rangnekar and C. S. Chen, The cyclooxygenase-2 inhibitor celecoxib induces apoptosis by blocking Akt activation in human prostate cancer cells independently of Bcl-2, J. Biol. Chem., 2000, 275, 11397–11403, DOI:10.1074/jbc.275.15.11397.
- S. K. Kulp, Y.-T. Yang, C.-C. Hung, K.-F. Chen, J.-P. Lai, P.-H. Tseng, J. W. Fowble, P. J. Ward and C.-S. Chen, 3-Phosphoinositide-Dependent Protein Kinase-1/Akt Signaling Represents a Major Cyclooxygenase-2-Independent Target for Celecoxib in Prostate Cancer Cells, Cancer Res., 2004, 64, 1444–1451, DOI:10.1158/0008-5472.CAN-03-2396.
- K. Xu, H. Gao and H.-K. G. Shu, Celecoxib can induce vascular endothelial growth factor expression and tumor angiogenesis, Mol. Cancer Therapeut., 2011, 10, 138–147, DOI:10.1158/1535-7163.MCT-10-0415.
- K. M. Kasiotis, E. N. Tzanetou and S. A. Haroutounian, Pyrazoles as potential anti-angiogenesis agents: a contemporary overview, Front. Chem., 2014, 2, 78, DOI:10.3389/fchem.2014.00078 , accessed August 7, 2022.
- N. Miyamoto, N. Sakai, T. Hirayama, K. Miwa, Y. Oguro, H. Oki, K. Okada, T. Takagi, H. Iwata, Y. Awazu, S. Yamasaki, T. Takeuchi, H. Miki, A. Hori and S. Imamura, Discovery of N-[5-({2-[(cyclopropylcarbonyl)amino]imidazo[1,2-b]pyridazin-6-yl}oxy)-2-methylphenyl]-1,3-dimethyl-1H-pyrazole-5-carboxamide (TAK-593), a highly potent VEGFR2 kinase inhibitor, Bioorg. Med. Chem., 2013, 21, 2333–2345, DOI:10.1016/j.bmc.2013.01.074.
- L.-L. Yang, G.-B. Li, S. Ma, C. Zou, S. Zhou, Q.-Z. Sun, C. Cheng, X. Chen, L.-J. Wang, S. Feng, L.-L. Li and S.-Y. Yang, Structure–Activity Relationship Studies of Pyrazolo[3,4-d]pyrimidine Derivatives Leading to the Discovery of a Novel Multikinase Inhibitor That Potently Inhibits FLT3 and VEGFR2 and Evaluation of Its Activity against Acute Myeloid Leukemia in Vitro and in Vivo, J. Med. Chem., 2013, 56, 1641–1655, DOI:10.1021/jm301537p.
- P. A. Harris, A. Boloor, M. Cheung, R. Kumar, R. M. Crosby, R. G. Davis-Ward, A. H. Epperly, K. W. Hinkle, R. N. Hunter, J. H. Johnson, V. B. Knick, C. P. Laudeman, D. K. Luttrell, R. A. Mook, R. T. Nolte, S. K. Rudolph, J. R. Szewczyk, A. T. Truesdale, J. M. Veal, L. Wang and J. A. Stafford, Discovery of 5-[[4-[(2,3-Dimethyl-2H-indazol-6-yl)methylamino]-2-pyrimidinyl]amino]-2-methyl-benzenesulfonamide (Pazopanib), a Novel and Potent Vascular Endothelial Growth Factor Receptor Inhibitor, J. Med. Chem., 2008, 51, 4632–4640, DOI:10.1021/jm800566m.
- N. H. Metwally, M. S. Mohamed and E. A. Ragb, Design, synthesis, anticancer evaluation, molecular docking and cell cycle analysis of 3-methyl-4,7-dihydropyrazolo[1,5-a]pyrimidine derivatives as potent histone lysine demethylases (KDM) inhibitors and apoptosis inducers, Bioorg. Chem., 2019, 88, 102929, DOI:10.1016/j.bioorg.2019.102929.
- R. Tripathy, A. Ghose, J. Singh, E. R. Bacon, T. S. Angeles, S. X. Yang, M. S. Albom, L. D. Aimone, J. L. Herman and J. P. Mallamo, 1,2,3-Thiadiazole substituted pyrazolones as potent KDR/VEGFR-2 kinase inhibitors, Bioorg. Med. Chem. Lett., 2007, 17, 1793–1798, DOI:10.1016/j.bmcl.2006.12.054.
- N. M. Saleh, M. G. El-Gazzar, H. M. Aly and R. A. Othman, Novel Anticancer Fused Pyrazole Derivatives as EGFR and VEGFR-2 Dual TK Inhibitors, Front. Chem., 2020, 7, 917, DOI:10.3389/fchem.2019.00917 , accessed August 7, 2022.
- P. Hamberg, J. Verweij and S. Sleijfer, (Pre-)clinical pharmacology and activity of pazopanib, a novel multikinase angiogenesis inhibitor, Oncologist, 2010, 15, 539–547, DOI:10.1634/theoncologist.2009-0274.
- Y. Oguro, D. R. Cary, N. Miyamoto, M. Tawada, H. Iwata, H. Miki, A. Hori and S. Imamura, Design, synthesis, and evaluation of novel VEGFR2 kinase inhibitors: Discovery of [1,2,4]triazolo[1,5-a]pyridine derivatives with slow dissociation kinetics, Bioorg. Med. Chem., 2013, 21, 4714–4729, DOI:10.1016/j.bmc.2013.04.042.
- H. Nakamura, Y. Sasaki, M. Uno, T. Yoshikawa, T. Asano, H. S. Ban, H. Fukazawa, M. Shibuya and Y. Uehara, Synthesis and biological evaluation of benzamides and benzamidines as selective inhibitors of VEGFR tyrosine kinases, Bioorg. Med. Chem. Lett., 2006, 16, 5127–5131, DOI:10.1016/j.bmcl.2006.07.075.
- L. Lintnerová, M. García-Caballero, F. Gregáň, M. Melicherčík, A. R. Quesada, J. Dobiaš, J. Lác, M. Sališová and A. Boháč, A development of chimeric VEGFR2 TK inhibitor based on two ligand conformers from PDB: 1Y6A complex – Medicinal chemistry consequences of a TKs analysis, Eur. J. Med. Chem., 2014, 72, 146–159, DOI:10.1016/j.ejmech.2013.11.023.
- U. Bhanushali, S. Rajendran, K. Sarma, P. Kulkarni, K. Chatti, S. Chatterjee and C. S. Ramaa, 5-Benzylidene-2,4-thiazolidenedione derivatives: Design, synthesis and evaluation as inhibitors of angiogenesis targeting VEGR-2, Bioorg. Chem., 2016, 67, 139–147, DOI:10.1016/j.bioorg.2016.06.006.
- R. S. K. Vijayan, P. He, V. Modi, K. C. Duong-Ly, H. Ma, J. R. Peterson, R. L. Dunbrack and R. M. Levy, Conformational Analysis of the DFG-Out Kinase Motif and Biochemical Profiling of Structurally Validated Type II Inhibitors, J. Med. Chem., 2015, 58, 466–479, DOI:10.1021/jm501603h.
- X. Wang, J. Tan and K. Grozinger, Cross-coupling of 1-aryl-5-bromopyrazoles: regioselective synthesis of 3,5-disubstituted 1-arylpyrazoles, Tetrahedron Lett., 2000, 41, 4713–4716, DOI:10.1016/S0040-4039(00)00704-8.
- D. Castagnolo, A. De Logu, M. Radi, B. Bechi, F. Manetti, M. Magnani, S. Supino, R. Meleddu, L. Chisu and M. Botta, Synthesis, biological evaluation and SAR study of novel pyrazole analogues as inhibitors of Mycobacterium tuberculosis, Bioorg. Med. Chem., 2008, 16, 8587–8591, DOI:10.1016/j.bmc.2008.08.016.
- H. Hu, C. Ge, L. Ding and A. Zhang, Synthesis of Novel 1-[(2,6-Dichloro-4-trifluoromethyl)phenyl]-3-aryl-1H-pyrazole-4-carbaldehydes, Molecules, 2010, 15, 7472–7481, DOI:10.3390/molecules15107472.
- N. Ferrara, H.-P. Gerber and J. LeCouter, The biology of VEGF and its receptors, Nat. Med., 2003, 9, 669–676, DOI:10.1038/nm0603-669.
- M. McTigue, B. W. Murray, J. H. Chen, Y.-L. Deng, J. Solowiej and R. S. Kania, Molecular conformations, interactions, and properties associated with drug efficiency and clinical performance among VEGFR TK inhibitors, Proc. Natl. Acad. Sci. U.S.A., 2012, 109, 18281–18289, DOI:10.1073/pnas.1207759109.
- P. Ravula, H. B. Vamaraju, M. Paturi and J. N. G. N. Sharath Chandra, Design, synthesis, in silico and antiproliferative evaluation of novel pyrazole derivatives as VEGFR-2 inhibitors, Arch. Pharmazie, 2018, 351, 1700234, DOI:10.1002/ardp.201700234.
- N. Upadhyay, K. Tilekar, S. Safuan, A. P. Kumar, M. Schweipert, F.-J. Meyer-Almes and C. S. Ramaa, Multi-target weapons: diaryl-pyrazoline thiazolidinediones simultaneously targeting VEGFR-2 and HDAC cancer hallmarks, RSC Med. Chem., 2021, 12, 1540–1554, 10.1039/D1MD00125F.
- F.-W. Peng, J. Xuan, T.-T. Wu, J.-Y. Xue, Z.-W. Ren, D.-K. Liu, X.-Q. Wang, X.-H. Chen, J.-W. Zhang, Y.-G. Xu and L. Shi, Design, synthesis and biological evaluation of N-phenylquinazolin-4-amine hybrids as dual inhibitors of VEGFR-2 and HDAC, Eur. J. Med. Chem., 2016, 109, 1–12, DOI:10.1016/j.ejmech.2015.12.033.
- T. Mosmann, Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays, J. Immunol. Methods, 1983, 65, 55–63, DOI:10.1016/0022-1759(83)90303-4.
- E. S. Tantawy, A. M. Amer, E. K. Mohamed, M. M. Abd Alla and M. S. Nafie, Synthesis, characterization of some pyrazine derivatives as anti-cancer agents: In vitro and in Silico approaches, J. Mol. Struct., 2020, 1210, 128013, DOI:10.1016/j.molstruc.2020.128013.
- M. S. Nafie and A. T. A. Boraei, Exploration of novel VEGFR2 tyrosine kinase inhibitors via design and synthesis of new alkylated indolyl-triazole Schiff bases for targeting breast cancer, Bioorg. Chem., 2022, 122, 105708, DOI:10.1016/j.bioorg.2022.105708.
- M. S. Nafie, K. Arafa, N. K. Sedky, A. A. Alakhdar and R. K. Arafa, Triaryl dicationic DNA minor-groove binders with antioxidant activity display cytotoxicity and induce apoptosis in breast cancer, Chem. Biol. Interact., 2020, 324, 109087, DOI:10.1016/j.cbi.2020.109087.
- M. S. Nafie, A. M. Amer, A. K. Mohamed and E. S. Tantawy, Discovery of novel pyrazolo[3,4-b]pyridine scaffold-based derivatives as potential PIM-1 kinase inhibitors in breast cancer MCF-7 cells, Bioorg. Med. Chem., 2020, 28, 115828, DOI:10.1016/j.bmc.2020.115828.
- E. E. Eltamany, S. S. Elhady, H. A. Ahmed, J. M. Badr, A. O. Noor, S. A. Ahmed and M. S. Nafie, Chemical Profiling, Antioxidant, Cytotoxic Activities and Molecular Docking Simulation of Carrichtera annua DC. (Cruciferae), Antioxidants, 2020, 9, 1286, DOI:10.3390/antiox9121286.
- A. I. Khodair, M. A. Alsafi and M. S. Nafie, Synthesis, molecular modeling and anti-cancer evaluation of a series of quinazoline derivatives, Carbohydr. Res., 2019, 486, 107832, DOI:10.1016/j.carres.2019.107832.
- M. S. Nafie, S. Mahgoub and A. M. Amer, Antimicrobial and antiproliferative activities of novel synthesized 6-(quinolin-2-ylthio) pyridine derivatives with molecular docking study as multi-targeted JAK2/STAT3 inhibitors, Chem. Biol. Drug Des., 2021, 97, 553–564, DOI:10.1111/cbdd.13791.
- M. McTigue, B. W. Murray, J. H. Chen, Y.-L. Deng, J. Solowiej and R. S. Kania, Molecular conformations, interactions, and properties associated with drug efficiency and clinical performance among VEGFR TK inhibitors, Proc. Natl. Acad. Sci. U.S.A., 2012, 109, 18281–18289, DOI:10.1073/pnas.1207759109.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra02579a |
| This journal is © The Royal Society of Chemistry 2023 |