DOI:
10.1039/D3RA02650G
(Paper)
RSC Adv., 2023,
13, 15926-15933
Insight into the direct conversion of methane to methanol on modified ZIF-204 from the perspective of DFT-based calculations†
Received
21st April 2023
, Accepted 15th May 2023
First published on 26th May 2023
Abstract
Direct oxidation of methane over oxo-doped ZIF-204, a bio-mimetic metal–organic framework, is investigated under first-principles calculations based on density functional theory. In the pristine ZIF-204, the tetrahedral methane molecule anchors to an open monocopper site via the so-called η2 configuration with a physisorption energy of 0.24 eV. This weak binding arises from an electrostatic interaction between the negative charge of carbon in the methane molecule and the positive Cu2+ cation in the framework. In the modified ZIF-204, the doped oxo species is stabilized at the axial position of a CuN4-base square pyramid at a distance of 2.06 Å. The dative covalent bond between Cu and oxo is responsible for the formation energy of 1.06 eV. With the presence of the oxo group, the presenting of electrons in the O_pz orbital accounts for the adsorption of methane via hydrogen bonding with an adsorption energy of 0.30 eV. The methane oxidation can occur via either a concerted direct oxo insertion mechanism or a hydrogen-atom abstraction radical rebound mechanism. Calculations on transition-state barriers show that reactions via the concerted direct oxo insertion mechanism can happen without energy barriers. Concerning the hydrogen-atom abstraction radical rebound mechanism, the C–H bond dissociation of the CH4 molecule is barrierless, but the C–O bond recombination to form the CH3OH molecule occurs through a low barrier of 0.16 eV. These predictions suggest the modified ZIF-204 is a promising catalyst for methane oxidization.
1. Introduction
Conversion of methane to methanol remains an active research area for diversely beneficial purposes: (1) reducing the amount of greenhouse gas emission as methane, and (2) producing important chemical feedstock as methanol.1–3 However, methane oxidation by chemical methods is challenging due to a prerequisite of high energy for a C–H bond dissociation (4.51 eV) and an overoxidation of the methanol product.4 Traditional transition-metal and oxide-based catalysts have been extensively studied for methane conversion concerning their novel catalytic activities.4–8 Methane molecules actively dissociate on transition metal surfaces. However, the combination of –CH3 and –OH species to form methanol is a multiple-step process that is relatively ineffective and requires high temperatures.4,5 All the above disadvantages also take place for the industrial-scale methanol synthesis through an indirect pathway by using a syngas, catalyzed by metal catalysts at temperatures of above 1000 °C.3,5 Yet that synthesis is expensive and also produces abundant pollution by-products.
On the other hand, biological processes suggest a direct way to convert methane to methanol at low temperatures and high selectivity without releasing any byproducts.2,3 It has been widely known that a class of bacteria so-called methanotrophs can directly oxidize methane to methanol in the presence of oxygen at ambient conditions relying on their soluble methane monooxygenase (sMMO) or particulate methane monooxygenase (pMMO) metalloenzymes.3 Both enzymes are very reactive owing to the presence of containing metal active centers, i.e., iron (Fe) for sMMO or copper (Cu) for pMMO. pMMO enzyme is found to be more popular in the methanotrophic bacteria family and more efficient for methane conversion than sMMO.3,9 Therefore, pMMO attracts much interest in methane oxidation research. Theoretical modeling works on pMMO structures have demonstrated the significant reactivity of copper sites and clarified detailed mechanisms of methane conversion.10,11 However, it is impossible to extract pMMO from bacteria for in vitro studies as well as for practical applications because it tends to lose its original catalytic activities. Therefore, various bioinspired catalytic materials have been proposed to enable direct methane–methanol conversion processes which are environment-friendly.
An attractive family of pMMO-mimetic materials is copper-exchanged zeolites. These materials are of high physical and chemical stability and exhibit significant activity for direct methane–methanol conversion at low temperatures.12–17 Tricopper and dicopper sites in Cu–Na–ZSM-5 and Cu–H–ZSM-5 can activate methane at a high selectivity, low temperature (483–498 K), but low conversion rates.12 Based on first-principles and microkinetic investigations, Engedahl et al. found that dicopper sites in Cu–SSZ-13 can dissociate C–H bonds at lower energy barriers under the influence of H2O molecules.16 Monocopper sites coordinated by four oxygen atoms in Cu1/ZSM-5 exhibit high activity and selectivity for methane oxidation compared to most of the current noble metal catalysts.17 However, monocopper sites in Cu–SSZ-13 are unfavorable for methane activation because of a high activation barrier of 1.14 eV.13
Metal–organic frameworks (MOFs), highly crystalline nanoporous structures, have been proposed as more efficient alternatives for mimicking biocatalysts of methane conversion.1,18–22 MOFs have been utilized as catalysts or hosts for anchoring Cu-based complexes functionalizing as pMMO enzymes.21 Mixed-valent Cu+ and Cu2+ in tricopper18 and dicopper20 oxide clusters were embedded in NU-1000 MOFs. The combination of spectroscopic and activity experiments with DFT calculations demonstrated that such catalyst systems enable the direct methane–methanol conversion at mild conditions with a selectivity up to 70%.18,20 A high selectivity of methanol was also reported for methane oxidation over active bis(μ-oxo) dicopper complexes ligated in MOF-808.19 As opposed to monocopper centers in zeolites which do not activate methane efficiently, monocopper centers in MOFs are more promising. The combined theoretical and experimental study on the single-atom Cu-doped ZIF-7 evidenced a high-rate direct conversion through low activation energy barriers.23 Recent experimental analyses and quantum calculations confirm the dominance of monocopper centers, coordinated by histidine-based imidazole ligands, for methane activation in the pMMO enzymes.11 This finding drives research direction towards single-copper catalysts for direct methane–methanol conversions based on MOF structures.
ZIF-204 is a mixed-metal structure containing intrinsic monocopper active centers. It is stable in water solution and can separate CO2 from CH4 efficiently.24 Additionally, their monocopper sites can coordinate dimethylformamide (DMF) molecules. In this work, ZIF-204 is chemically modified to mimic the pMMO environment by depositing oxo-oxidizing agents to monocopper sites. The oxidation of CH4 by oxo center in ZIF-204 is investigated in terms of reaction paths, energy barriers of C–H bond dissociation and C–O bond recombination, and underlying electronic structure properties by theoretical calculations on the base of density functional theory (DFT). We aim to predict the potential energy surfaces and elucidate the catalytic activity for the methane oxidation over this bioinspired structure.
2. Computational methods
All the results are obtained from the spin-polarized density functional theory (DFT) calculations using the Vienna Ab initio Simulation Package (VASP 5.4.4).25 The generalized gradient approximation functional of Perdew, Burke, and Ernzerhof's (PBE)26,27 is employed with a D3 van der Waals correction.28 The kinetic energy cutoff of 500 eV is used within the projected augmented wave (PAW)29 pseudopotential. The criteria for electronic energy and force convergence are selected to be 10−6 eV and 0.01 eV Å−1, respectively. The k-point mesh is sampled by Monkhorst–Pack grids at the Gamma point. The crystal structure of ZIF-204 is monoclinic with a unit cell containing 170 atoms. Its structural formula is [C15H15N10Cu1.5Zn]4. The electron configurations of the metal ions have been assigned as Cu2+: [Ar] 3d9 4s0 and Zn2+: [Ar] 3d10 4s0. Taking into account strong correlations of electrons in d orbitals, Hubbard U correction of 5.5 eV is properly derived for Cu from the linear response method.30 Detail of the method and U values is provided in Section S1 of the ESI.† For checking the performances of functionals, lattice parameters bench-marked with different functionals including LDA,31 PBE,26,27 revPBE,32 RPBE,33 PBE-D3,26–28 and PBE-D3-U26–28,30 are presented in Section S2 in the ESI.† Among tested functionals, PBE produces the most appropriate lattice constants and corresponding unit-cell volume in comparison to experimental values. Isolated molecules such as CH4 are simulated inside a cubic box with a dimension of 20 Å. Functionalizing Cu center by an oxo group, the binding energy of this oxo ligand is defined| | |
Eb = Ehost + EO − Ehost–O
| (1) |
where Ehost, EO, and Ehost–O are the total energies of the pristine host, the single O atom, and the host with a ligand O bound, respectively.
We formulate the adsorption energy ΔEads of a guest molecule X as following
| | |
Eads = Ehost–X − Ehost − EX
| (2) |
where
Ehost–X,
Ehost, and
EX are the total energies of the host structure with an adsorbed molecule X, the host structure without X, and the isolated X molecule, respectively.
The change of charge density due to the presence of a species X is determined as an electron density difference (EDD) Δρ
| | |
Δρ = ρhost–X − ρhost − ρX
| (3) |
where
ρhost–X,
ρhost, and
ρX are the electron densities of the combined structure with the presence of both the host and X, of the host derived by the elimination of X, and of X derived by the elimination of the host, respectively. On creating individual components from the parent structure, the geometries of derived structures remain unchanged.
A computer program suite named LOBSTER (Local Orbital Basis Suite Towards Electronic-Structure Reconstruction) has been carried out to extract the electronic properties of the involved species.34 Those calculations include the Mulliken charge and projected density of state (PDOS). The climbing image nudged elastic band (CI-NEB) method is also employed to evaluate energy barriers of the transition states along potential energy surfaces of conversion reactions.35 For CI-NEB calculations, only atoms involved in considering reactions are relaxed, while the remaining atoms are frozen to reduce computational cost because they are almost unchanged during reactions. Derived transition states are confirmed by the fact that there has only one imaginary frequency for each transition state found. All atomic structures and EDD plots presented in this work are visualized by VESTA program, version 3.5.7.36
3. Result and discussion
3.1. Optimized structures
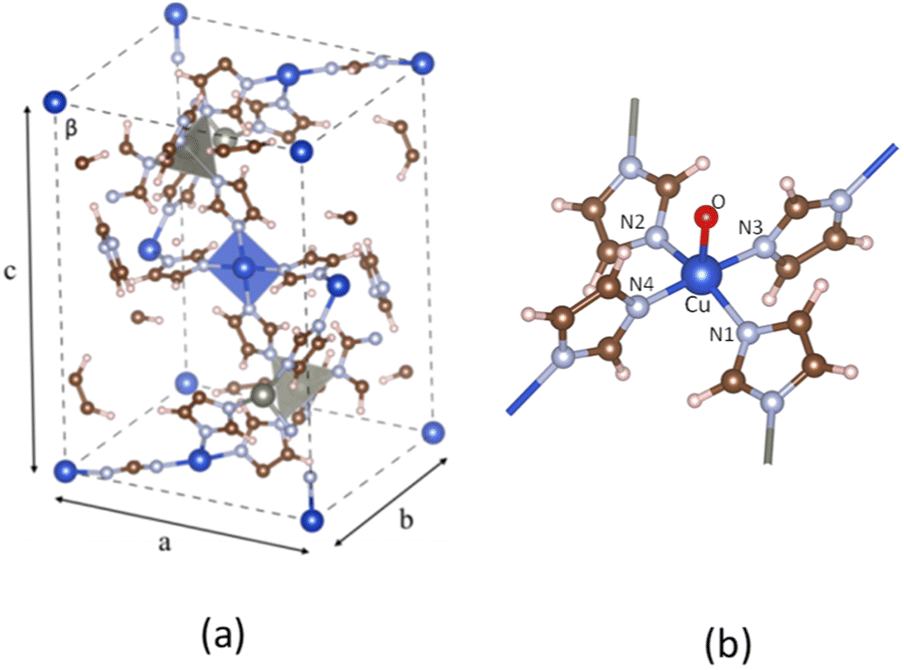
3.1.1. Pristine ZIF-204. The pristine ZIF-204, denoted as ZI hereafter, comprises single-atom transition metal nodes, i.e., either Zn2+ or Cu2+, coordinated by four imidazole linkers. The coordination symmetry at each node is tetrahedral for Zn and square planar for Cu, as shown in Fig. 1(a) The optimized structures show that the bond lengths of Zn and Cu with their imidazole linkers are almost the same, of about 2.00 Å and 1.98 Å, for Zn and Cu, respectively. Structural parameters at the Cu center are provided in Table 1. ZI is not a magnetic MOF, so its total spin is zero. While Zn2+ has a closed-shell configuration, Cu2+ remains to be spin-polarized cations due to their d electrons, as shown by PDOS in Fig. 4(b). Consequently, the antiferromagnetic configuration assigned to six Cu2+ cations in one unit cell is the most stable-optimized structure compared to ferromagnetic ones.
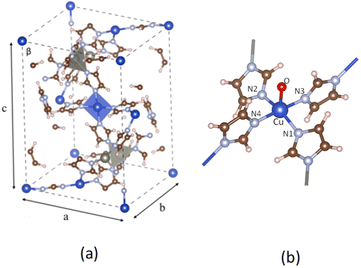 |
| | Fig. 1 The unit cell of the pristine ZI (a), the fragment of ZI_O (b). The spheres colored dark blue, gray, brown, light blue, red, and white represent Cu, Zn, C, N, O, and H atoms, respectively. | |
Table 1 Bond lengths and bond angles related to the Cu center for both the ZI and ZI_O structures
| Structure |
Bond length (Å) |
Bond angle (°) |
| Cu–O |
Cu–N1 |
Cu–N2 |
Cu–N3 |
Cu–N4 |
N1–Cu–N2 |
N3–Cu–N4 |
| ZI |
|
1.98 |
1.98 |
1.97 |
1.97 |
180 |
180 |
| ZI_O |
2.06 |
2.01 |
1.99 |
1.90 |
1.90 |
157 |
174 |
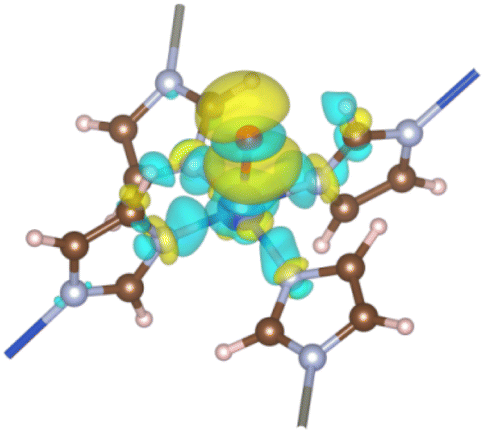
3.1.2. Modified ZIF-204. The oxo group is deposited onto the axial Cu site of the ZI, playing the role of an oxidant for the C–H σ-bond activation. The oxo-doped ZI structure is denoted as ZI_O hereafter which its oxo-containing fragment is shown in Fig. 1(b). Bond lengths and bond angles related to Cu center of the ZI_O are displayed in Table 1. There is a notable distortion of the geometry at the Cu site. Although bond lengths are not much different for ZI against ZI_O, bond angles become smaller in the ZI_O structure. The doped oxo ligand pulls the coordinated Cu atom out of its planar plane. It indicates that the original square planar symmetry turns into square-based pyramidal symmetry in the presence of the oxo group. We found that the doped oxo species locates above CuN4 surface and binds to Cu with a Cu–O bond length of 2.06 Å, and a binding energy of 1.06 eV. Mulliken charge distribution for both the ZI and ZI_O is shown in Table 2. All atoms in the ZI structure lose their negative charges upon the deposition of oxo onto Cu. Cu atom is most affected by losing about 0.17e, while it is only 0.04e lost on N atoms. The oxo has a negative charge of 0.50e induced by the coordination with Cu2+. An EDD plot in Fig. 2 shows negative charge depletion and accommodation regions for Cu, N, and O, respectively. A high negative charge density between Cu and O indicates this is a type of dative covalent bond. PDOS in Fig. 4(c) shows this bond at around −4.0 eV represented by the overlapping of Cu_d and O_p orbitals. Additionally, there is also an extending of the negative charge region above the O atom, outside of the Cu–O bond, as shown in Fig. 2. This orbital corresponds to a crossing of a high density of state of O_p from −2.0 eV to 1.0 eV above the Fermi level, as plotted in Fig. 4(c). The abundance of negative charge in the oxo suggests that this is an active site for arriving adsorbates to adsorb and activate easily.
Table 2 Mulliken charges of involved atoms around the Cu center. The sign (−) or (+) in front of bold numbers represent the loss or gain of negative charges (e) of that atom upon the functionalization with the oxo group, respectively
| Structure |
Mulliken charge |
| Cu |
N1 |
N2 |
N3 |
N4 |
O |
| ZI |
+1.28 |
−0.63 |
−0.63 |
−0.59 |
−0.59 |
|
| ZI_O |
+1.45 |
−0.59 |
−0.59 |
−0.55 |
−0.53 |
−0.50 |
| Lost(−)/gain(+) (e) |
−0.17 |
−0.04 |
−0.04 |
−0.04 |
−0.06 |
+0.50 |
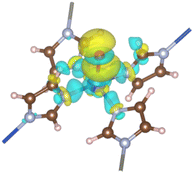 |
| | Fig. 2 EDD for the formation of the oxo ligand. The regions encoded by yellow and cyan stand for electron accumulation and depletion, respectively. The isosurface value is 0.002 |e| Å−3. | |
3.2. Adsorption of CH4 over ZIF-204
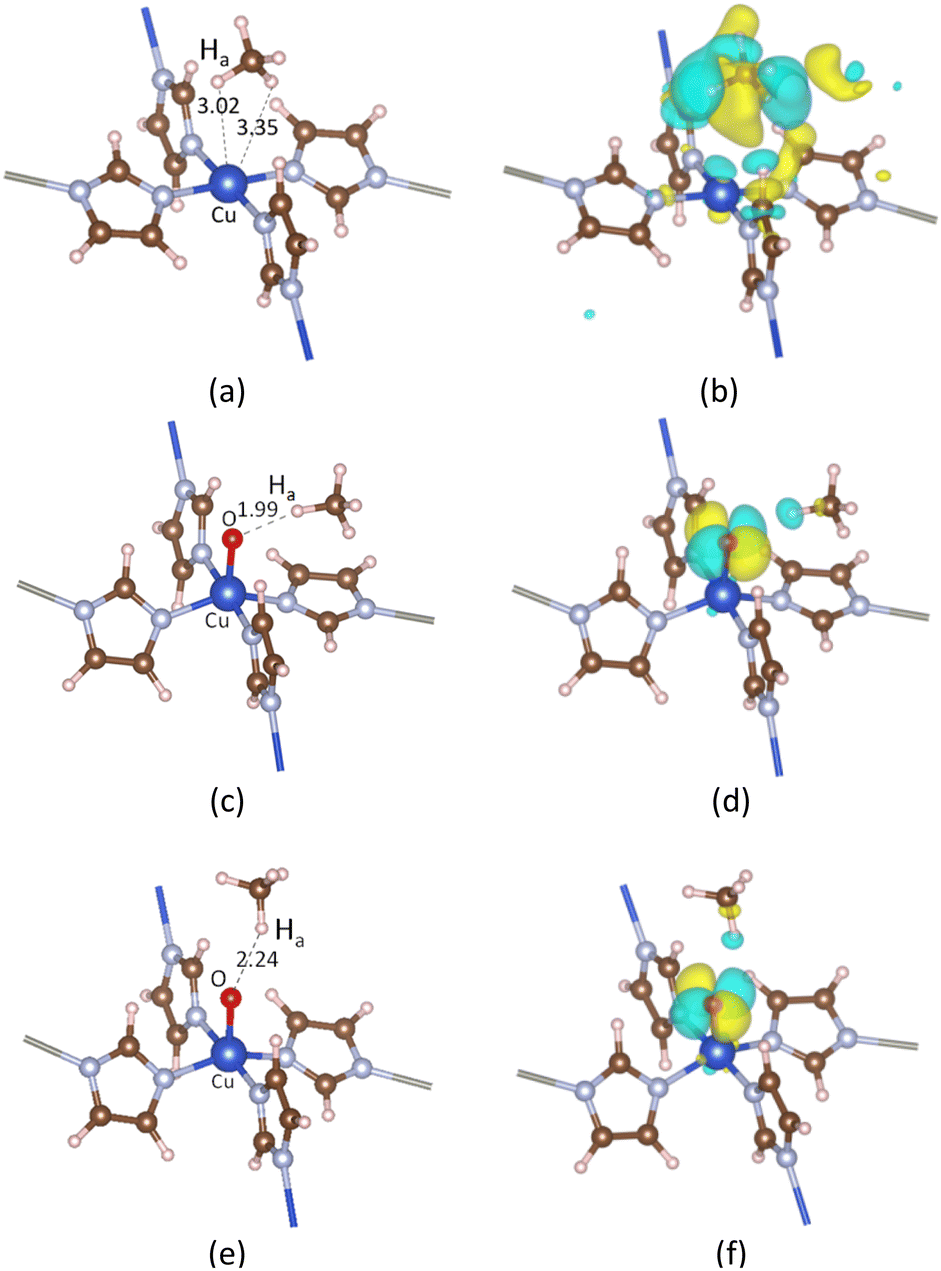
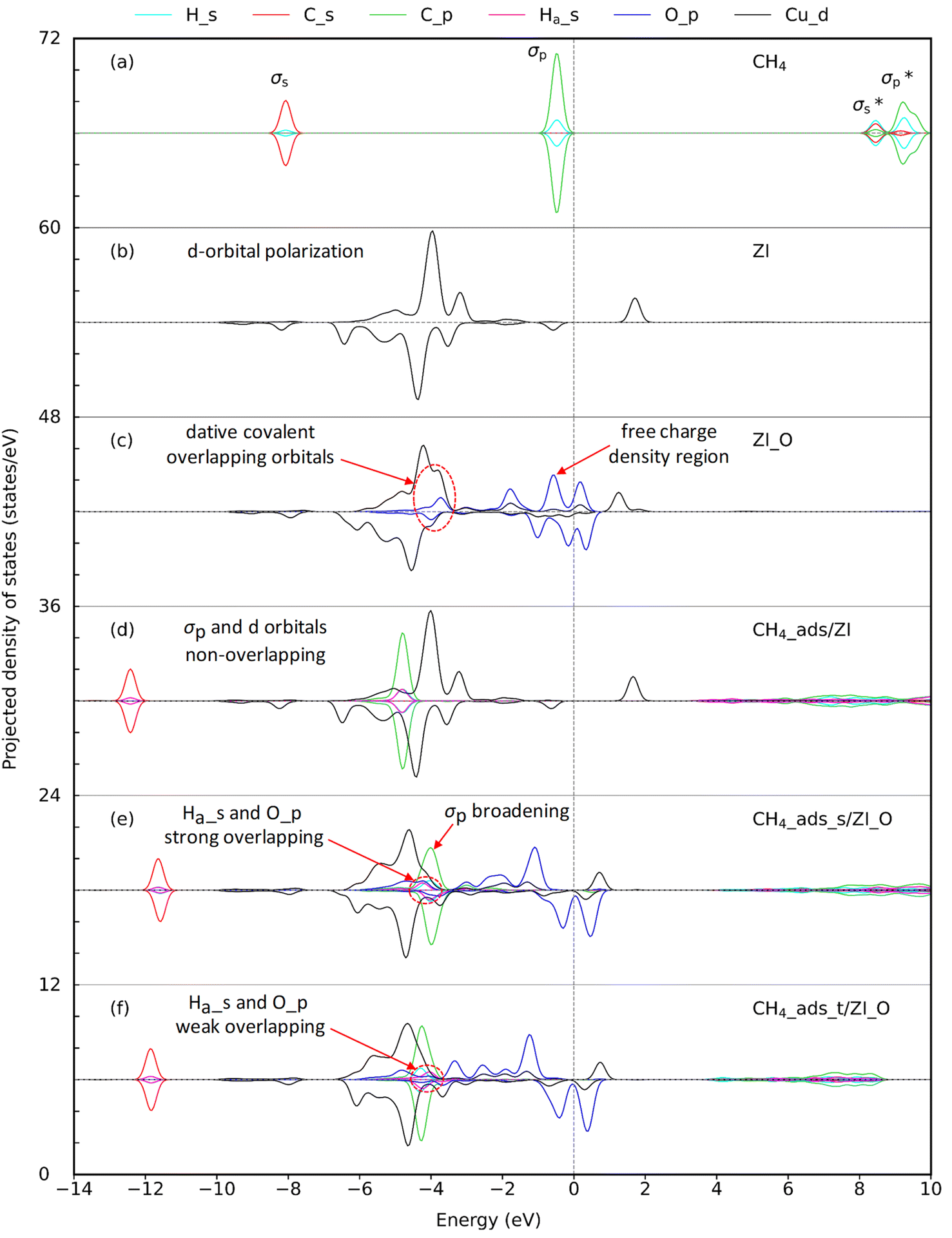
3.2.1. CH4 adsorption on ZI. Breakthrough experiments on methane uptake showed that ZIF-204 can capture methane with an uptake at about 13 cm3 g−1, comparable to 22 cm3 g−1 in BPL activated carbon.24 This demonstrates the fact that ZIF-204 can adsorb methane at a considerable amount, i.e., about half of the uptake of the industrial methane storage materials. The detail of methane adsorption on ZIF-204 has yet been addressed previously. Our calculated results show that methane stabilizes on the Cu site via the η2 configuration, as shown in Fig. 3(a), with the adsorption energy of −0.24 eV, cf. Table 3, entry 1. At this configuration, the two hydrogen atoms are apart from the Cu with similar distances, i.e., 3.02 and 3.35 Å, to minimize the repulsion between positive charges of Cu2+ site and the H atom. The electrostatic attraction mainly arises between the positive Cu2+ and the partial negative charge of the C atom (CH4) indicated by an accommodation of negative charge between two atoms C and Cu2+ plotted in Fig. 3(b). The adsorption configuration at η2 mode found here is reasonable in comparison to the adsorption of CH4 on differently active centers, i.e., transition metal [M(pyridine)n]2+ complexes,37 single-atom Fe-doped graphene and h-BN sheets,38 and Cun+ copper clusters.39 However, in our case, the binding energy of methane on Cu2+ of ZIF-204 is at-most half of the methane binding energies on the structures mentioned above. The smaller interaction energy can be attributed to either the electronic properties of metal centers or the effect of coordinated ligands. Due to a weak electrostatic interaction, the PDOS of CH4_ads/ZI and pristine ZI are almost similar, as shown in Fig. 4(d) and (b), respectively. It is possible for the activation of the C–H σ-bond by the metal active center, which was discussed in the previous work.38 In this study, we found that the activated hydrogen atoms insert into Cu–N bonds, causing the hydrogenation of imidazole ligands. Those hydrogenated structures are unstable and are ignored in our current study.
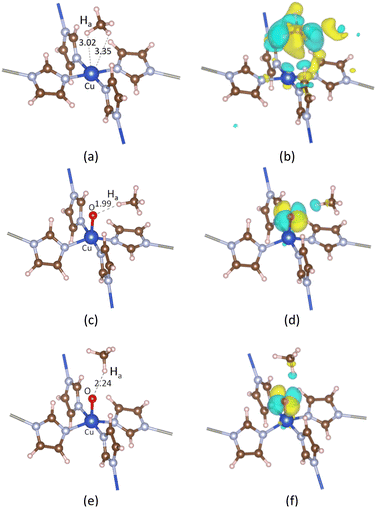 |
| | Fig. 3 Adsorption configurations and the corresponding EDD of CH4 on ZI in (a) and (b), on ZI_O with the most stable in (c) and (d), and the second most stable in (e) and (f). Ha represents the hydrogen atom in CH4 that is closest to the Cu center or the oxo ligand. The isosurface value is 0.002 |e| Å−3. | |
Table 3 Adsorption sites and adsorption energies of CH4 on ZI and ZI_O
| No. |
Structure |
Eads (eV) |
Location |
| 1 |
CH4_ads/ZI |
−0.24 |
Metal-top |
| 2 |
CH4_ads_s/ZI_O |
−0.30 |
Oxo-side |
| 3 |
CH4_ads_t/ZI_O |
−0.22 |
Oxo-top |
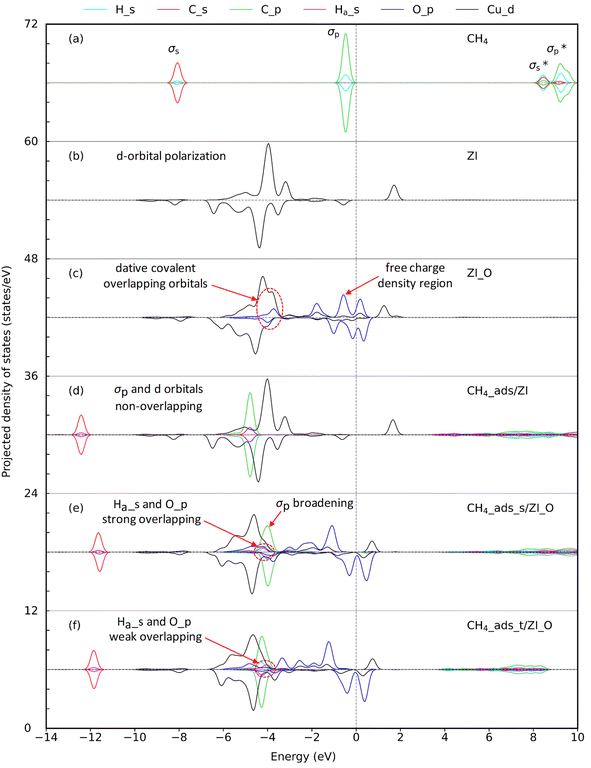 |
| | Fig. 4 Projected density of states (PDOS) for (a) isolated CH4 molecule, (b) pristine ZIF-204 denoted as ZI, (c) oxo-doped ZIF-204 denoted as ZI_O, (d) CH4 molecule adsorbed on ZI, (e) CH4 molecule adsorbed on ZI_O at the side site, and (f) CH4 molecule adsorbed on ZI_O at the top site. Lines in cyan, red, green, pink, blue, and black represent the PDOS of the s-orbital of H, s-orbital of C, and p-orbital of C in CH4, s-orbital of the interacting Ha, p-orbital of oxo, and d-orbital of the copper, respectively. The dashed line crossing at 0 eV represents the Fermi level. | |
3.2.2. CH4 adsorption on ZI_O. There are two distinguished adsorption configurations for the adsorption of CH4 over ZI_O denoted by CH4_ads_s, oxo-side site, see Fig. 3(c), and CH4_ads_t, oxo-top site, see Fig. 3(e). Both the adsorption processes are exothermic at the relative energies of 0.30 and 0.22 eV, as presented in Table 3, entries 2 and 3, respectively. The structural parameters for CH4_ads_s and CH4_ads_t are 1.94, 1.99, 1.10, and 1.95, 2.24, and 1.09(6) Å for CuO, OHa, and CHa, respectively. The bond lengths calculated here can be compared to values obtained for CH4 adsorption on FeO-decorated graphene of 1.63, 2.46, and 1.10 Å for CuO, OHa, and CHa, respectively.38 It can be seen that although the OHa distance is longer for CH4 on FeO than CuO, i.e., 2.46 vs. 1.99 Å, the elongation of CHa is the same with 1.10 Å. This disagreement could be attributed to the difference in electronic properties of Fe and Cu centers. A considerably shorter of OHa distance and an obvious elongation of CHa for CH4 located at the oxo-side position compared to one at the top position demonstrate that the interaction between CH4 and oxo is stronger for CH4 residing on the oxo-side site. In addition, given an elongation of CHa bond lengths, we also observe a decrease, about 200 cm−1, of the stretching mode for CHa bond in CH4_ads_s compared to CH4 in the gas phase, i.e., 2888 cm−1 vs. 3095 cm−1, respectively. The weaker CHa bond in CH4 arising from interacting with oxo is an important feature for the CH4 activation. EDD plots in Fig. 3(d) and (f) show a similar distribution of charge for both adsorption configurations. In the presence of CH4, the p orbital of oxo is split into different regions of the electron accommodation and depletion concerning O_px and O_py, while the electron on O_pz orbital is unchanged. It is obvious that there is a slight loss of electron on the H atom of CH4. This suggests a hydrogen bonding between CH4 and the oxo species. PDOS in Fig. 4(e) shows a strong overlap region of O_p and Ha_s indicated by the same peak at around −4.0 eV demonstrating the existence of such hydrogen bonding. The PDOS in Fig. 4(f) also presents such overlap which is smaller due to weaker hydrogen-bonding interactions.
3.3. Oxidation of CH4 over modified ZI
The overall catalytic reaction is proposed.| | |
CH4_g + ZI_O → CH3OH_g + ZI
| (4) |
The detailed reaction mechanism follows:| | |
CH4_g + ZI_O → CH4_ads + ZI_O
| (5) |
| | |
CH4_ads + ZI_O → CH3_ads + ZI_OH
| (6) |
| | |
CH3_ads + ZI_OH → CH3OH_ads + ZI
| (7) |
| | |
CH3OH_ads + ZI → CH3OH_g + ZI
| (8) |
According to the overall reaction in eqn (4), ZI_O plays the role of a catalyst for transferring its oxo group to CH4 to form CH3OH molecules. The detail mechanism of this transfer is described in the subsequent eqn (5)–(8). The reaction initiates by the attachment of gas-phase molecule CH4 onto the oxo group, shown in eqn (5). The following eqn (6) and (7) describe the activation of the C–H bond and the recombination of the C–O bond, respectively. Eqn (8) presents the desorption of the final product CH3OH_ads into the gas phase.
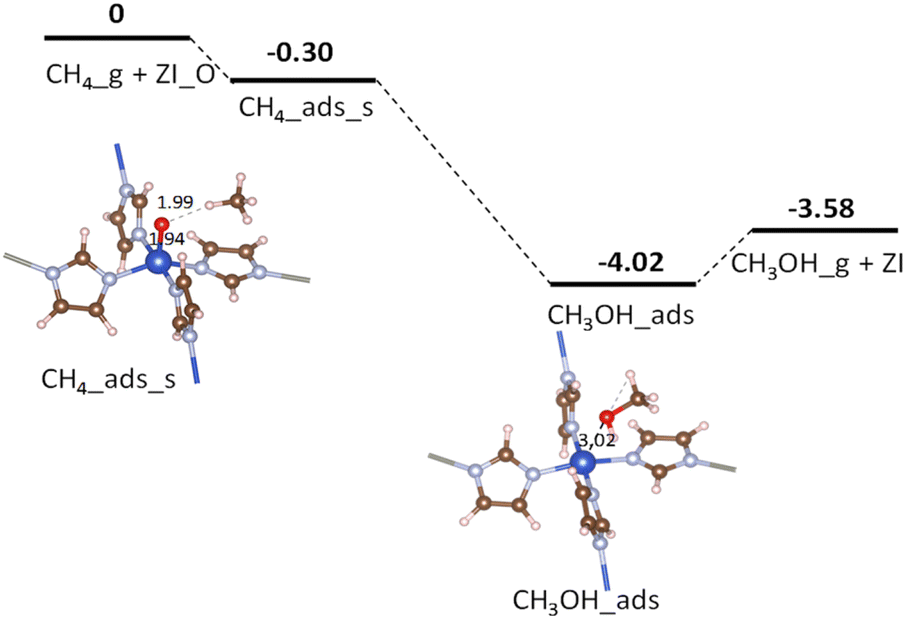
Fig. 5 displays the potential energy surface for the CH4_ads_s pathway. Calculated results from CI-NEB show that none of the proposed reaction steps in eqn (6) or eqn (7) attain intrinsic activation barriers (cf. Table 4, entry 1). Here, the C–H bond is weakened by the presence of the oxo group. After a total transfer of hydrogen atom to the oxo group to become Cu–OH moiety, the remaining induced methyl-like radical directly withdraw the hydroxyl OH group (in Cu–OH) to form the adsorbed CH3OH molecule. The two reaction steps happen simultaneously without the introduction of any transition or intermediate states. It could be due to a strong hydrogen bonding between the oxo and H atom (in CH4) making this bond weaken for the activation with a zero barrier. The broadening of the C–H (in CH4) bonding band around −4.0 eV shown in Fig. 4(e) demonstrates a strong influence of O_p on the C–H σ-bond. Moreover, the short distance between the induced methyl-like radical and the Cu–OH moiety also facilitates its recombination for the final products. The absence of intermediates suggests that the above conversion reactions follow the concerted direct oxo insertion mechanism which has been discussed in early works.40,41
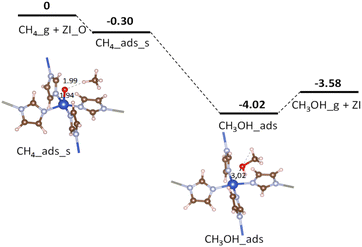 |
| | Fig. 5 Potential energy surface for CH4 oxidation over ZI_O via the CH4_ads_s pathway. Relative energies are given in eV. The zero-reference energy is the sum of the total energies of the isolated CH4 molecule and ZI_O. Zero-point energy correction is not included. | |
Table 4 Activation energies of involved reactions for both CH4_ads_s and CH4_ads_t pathways
| Initial configuration |
No. |
Reaction step |
Bond activation |
Activation energy (eV) |
| CH4_ads_s |
1 |
CH4_ads_s → CH3OH_ads |
C–H/C–O |
0.00 |
| CH4_ads_t |
2 |
CH4_ads_t → Im |
C–H |
0.00 |
| 3 |
Im → CH3OH_ads |
C–O |
0.16 |
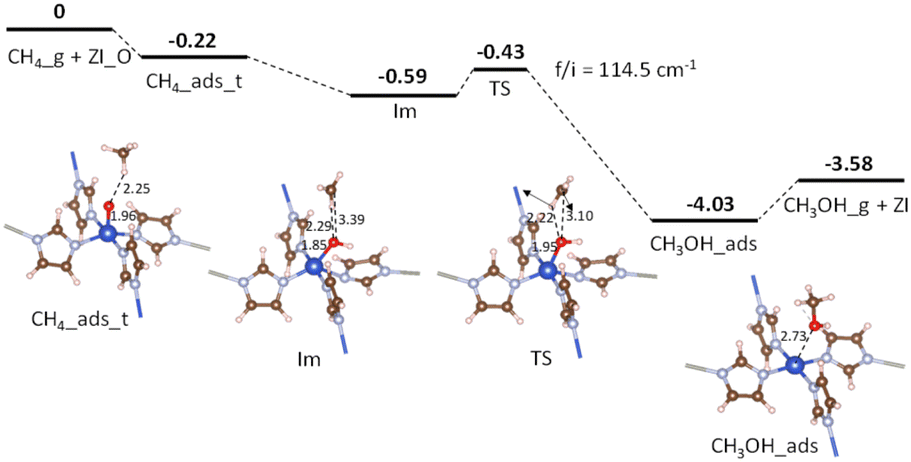
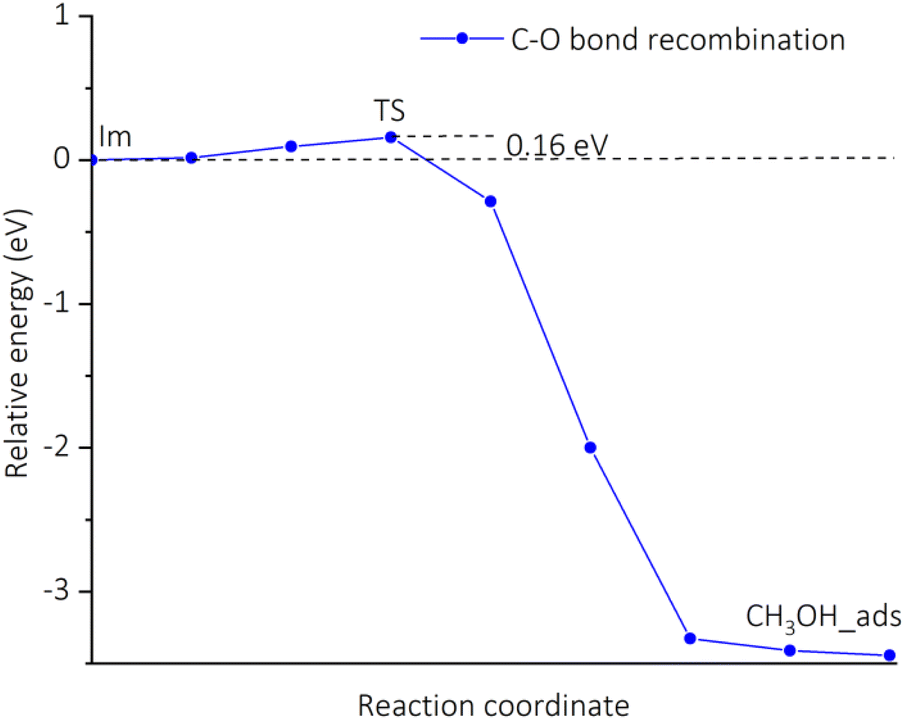
Fig. 6 shows the potential energy surface for the conversion of CH4 into CH3OH starting from the adsorption configuration CH4_ads_t. It has been seen that there is only one transition state (TS) corresponding to the formation of the C–O bond, while the activation of the C–H bond is barrierless (see Table 4, entry 2). The energy barrier is found to be 0.16 eV (see Table 4, entry 3) for the transition from the intermediate Im to TS. For the TS, there is a unique imaginary frequency calculated to be 114.5 cm−1 associated with a concurrent rotation and translation of CH3 radical towards the Cu–OH moiety. The energy profile for a C–O bond recombination is plotted in Fig. 7. The energy deviation is rather smooth with a small activation barrier found, demonstrating an appropriate transition from the initial Im state to TS, and to the final state CH3OH_s. The presence of CH3 radical in the Im state suggests that the CH4_ads_t pathway follows hydrogen-atom abstraction radical rebound mechanism.41,42 For such the mechanism, an early work on methane oxidation on pMMO-inspired trinuclear copper complex, [Cu3(μ-O)2(7-N-Etppz)]+, reported two transition states with moderate barriers of 0.33 and 0.38 eV for C–H bond dissociation and C–O bond recombination, respectively.42 It can be also compared to low barriers of 0.23 eV for H-abstraction and 0.55 eV for methyl radical rebound revealed by Cao et al. for methane oxidation over PMMO-extracted monocopper sites.11 The results found here are also more reasonable by a reference to a recent study on copper-doped ZIF-7, an inspired PMMO structure.23 Accordingly, the active center O–CuN4 moiety can oxidize CH4 by going through two reaction steps similar to our aforementioned mechanism with negligible activation barriers, i.e., 0.08 eV for the C–H bond dissociation and 0.0 eV for the C–O bond recombination.23 It is worth mentioning here that transition barriers are negligible regardless of whether the methane oxidation proceeds via either pathways.
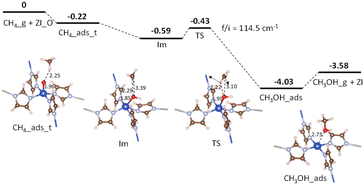 |
| | Fig. 6 Potential energy surface for CH4 conversion over ZI_O via the CH4_ads_t pathway. Relative energies are given in eV. The zero-reference energy is the sum of the total energies of the isolated CH4 molecule and ZI_O. Zero-point energy correction is not included. | |
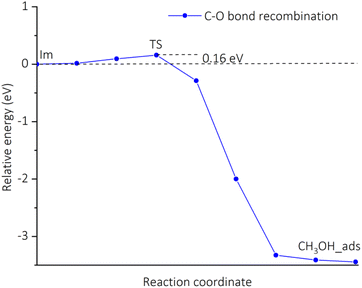 |
| | Fig. 7 Energy changes along the reaction coordinate for a C–O bond recombination via the CH4_ads_t pathway calculated by the CI-NEB method. | |
4. Conclusions
We study the direct conversion of methane to methanol over modified ZIF-204 metal–organic frameworks by means of first-principle DFT-based calculations. The catalytic activity of the modified ZIF-204 is identified by the oxo-deposited species at monocopper sites. In pristine ZIF-204, methane molecules physisorb on the open monocopper sites with the adsorption energy of 0.24 eV. The activation of methane over these copper centers is beyond the interest of the current study due to a weak electrostatic interaction between methane and metal cation and also the instability of the induced hydrogenation structures. In oxo-doped ZIF-204, different reaction mechanisms are found to be affected by the adsorption behavior of methane molecules towards oxo groups through hydrogen bondings with oxo groups. The oxo plays the key role as a reactive center for the activation of methane via either the concerted direct oxo insertion or the hydrogen-atom abstraction radical rebound mechanism. The conversion reactions occur without energy barriers for the concerted pathway or with a negligible transition barrier of 0.16 eV for the C–O bond recombination for the abstraction radical rebound pathway. The reactions happen easily in response to strong catalytic activities of the oxo-oxidizing doped species. Those negligible energy barriers propose a favorable kinetic for the direct oxidation of methane over modified ZIF-204 metal–organic structures.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was funded by Vietnam National University Ho Chi Minh City (VNU-HCM) under grant number C2022-50-03. Y. K. was supported by Suranaree University of Technology (SUT), Thailand Science Research and Innovation (TSRI), and National Science, Research and Innovation Fund (NSRF) (NRIIS Project Number 90465). The authors would like to acknowledge colleagues from Institute for Materials Research, Tohoku University, Japan, Institute of Atomic and Molecular Sciences, Academia Sinica, Taiwan, and Key Laboratory for Multiscale Simulation of Complex Systems, University of Science, Vietnam National University-Hanoi, Hanoi for their help in providing and maintaining high-performance computing resources. T. N. M. L. would also like to thank Dr Jer-Lai Kuo at Institute of Atomic and Molecular Sciences, Academia Sinica, Taiwan and Dr Cheng-Chau Chiu at National Sun Yat-Sen University, Taiwan for assistance and fruitful discussions.
References
- L. S. Andrade, H. H. L. B. Lima, C. T. P. Silva, W. L. N. Amorim, J. G. R. Poço, A. López-Castillo, M. V. Kirillova, W. A. Carvalho, A. M. Kirillov and D. Mandelli, Coord. Chem. Rev., 2023, 481, 215042 CrossRef CAS.
- N. F. Dummer, D. J. Willock, Q. He, M. J. Howard, R. J. Lewis, G. Qi, S. H. Taylor, J. Xu, D. Bethell, C. J. Kiely and G. J. Hutchings, Chem. Rev., 2022, 123(9), 6359–6411 CrossRef PubMed.
- V. C. Wang, S. Maji, P. P. Chen, H. K. Lee, S. S. Yu and S. I. Chan, Chem. Rev., 2017, 117, 8574–8621 CrossRef CAS PubMed.
- A. A. Latimer, A. Kakekhani, A. R. Kulkarni and J. K. Nørskov, ACS Catal., 2018, 8, 6894–6907 CrossRef CAS.
- M. Ravi, M. Ranocchiari and J. A. van Bokhoven, Angew. Chem., Int. Ed., 2017, 56, 16464–16483 CrossRef CAS PubMed.
- Y. Tsuji and K. Yoshizawa, J. Phys. Chem. C, 2018, 122, 15359–15381 CrossRef CAS.
- Z. Liu, E. Huang, I. Orozco, W. Liao, R. M. Palomino, N. Rui, T. Duchon, S. Nemsak, D. C. Grinter, M. Mahapatra, P. Liu, J. A. Rodriguez and S. D. Senanayake, Science, 2020, 368, 513–517 CrossRef CAS PubMed.
- Z. Liang, T. Li, M. Kim, A. Asthagiri and J. F. Weaver, Science, 2017, 356, 299–303 CrossRef CAS.
- M. O. Ross, F. MacMillan, J. Wang, A. Nisthal, T. J. Lawton, B. D. Olafson, S. L. Mayo, A. C. Rosenzweig and B. M. Hoffman, Science, 2019, 364, 566–570 CrossRef CAS PubMed.
- K. Yoshizawa and Y. Shiota, J. Am. Chem. Soc., 2006, 128, 9873–9881 CrossRef CAS PubMed.
- L. Cao, O. Caldararu, A. C. Rosenzweig and U. Ryde, Angew. Chem., Int. Ed., 2018, 57, 162–166 CrossRef CAS PubMed.
- K. Narsimhan, K. Iyoki, K. Dinh and Y. Roman-Leshkov, ACS Cent. Sci., 2016, 2, 424–429 CrossRef CAS PubMed.
- A. R. Kulkarni, Z.-J. Zhao, S. Siahrostami, J. K. Nørskov and F. Studt, ACS Catal., 2016, 6, 6531–6536 CrossRef CAS.
- V. L. Sushkevich and J. A. van Bokhoven, ACS Catal., 2019, 9, 6293–6304 CrossRef CAS.
- M. B. Park, E. D. Park and W. S. Ahn, Front. Chem., 2019, 7, 514 CrossRef CAS PubMed.
- U. Engedahl, A. Boje, H. Ström, H. Grönbeck and A. Hellman, J. Phys. Chem. C, 2021, 125, 14681–14688 CrossRef CAS.
- X. Tang, L. Wang, B. Yang, C. Fei, T. Yao, W. Liu, Y. Lou, Q. Dai, Y. Cai, X.-M. Cao, W. Zhan, Y. Guo, X.-Q. Gong and Y. Guo, Appl. Catal., B, 2021, 285, 119827 CrossRef CAS.
- T. Ikuno, J. Zheng, A. Vjunov, M. Sanchez-Sanchez, M. A. Ortuno, D. R. Pahls, J. L. Fulton, D. M. Camaioni, Z. Li, D. Ray, B. L. Mehdi, N. D. Browning, O. K. Farha, J. T. Hupp, C. J. Cramer, L. Gagliardi and J. A. Lercher, J. Am. Chem. Soc., 2017, 139, 10294–10301 CrossRef CAS PubMed.
- J. Baek, B. Rungtaweevoranit, X. Pei, M. Park, S. C. Fakra, Y. S. Liu, R. Matheu, S. A. Alshmimri, S. Alshehri, C. A. Trickett, G. A. Somorjai and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 18208–18216 CrossRef CAS PubMed.
- J. Zheng, J. Ye, M. A. Ortuno, J. L. Fulton, O. Y. Gutierrez, D. M. Camaioni, R. K. Motkuri, Z. Li, T. E. Webber, B. L. Mehdi, N. D. Browning, R. L. Penn, O. K. Farha, J. T. Hupp, D. G. Truhlar, C. J. Cramer and J. A. Lercher, J. Am. Chem. Soc., 2019, 141, 9292–9304 CrossRef CAS PubMed.
- D. Li, H.-Q. Xu, L. Jiao and H.-L. Jiang, Energy Chem., 2019, 1(1), 100005 CrossRef.
- A. S. Rosen, J. M. Notestein and R. Q. Snurr, ACS Catal., 2019, 9, 3576–3587 CrossRef CAS.
- H. Lee, C. Kwon, C. Keum, H.-E. Kim, H. Lee, B. Han and S.-Y. Lee, Chem. Eng. J., 2022, 450, 138472 CrossRef CAS.
- N. T. Nguyen, T. N. Lo, J. Kim, H. T. Nguyen, T. B. Le, K. E. Cordova and H. Furukawa, Inorg. Chem., 2016, 55, 6201–6207 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- M. Cococcioni and S. de Gironcoli, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71(3), 035105 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- Y. Zhang and W. Yang, Phys. Rev. Lett., 1998, 80, 890 CrossRef CAS.
- B. Hammer, L. B. Hansen and J. K. Nørskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 7413–7421 CrossRef.
- S. Maintz, V. L. Deringer, A. L. Tchougreeff and R. Dronskowski, J. Comput. Chem., 2016, 37, 1030–1035 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- G. W. Roffe and H. Cox, J. Phys. Chem. A, 2013, 117, 3017–3024 CrossRef CAS PubMed.
- S. Impeng, P. Khongpracha, J. Sirijaraensre, B. Jansang, M. Ehara and J. Limtrakul, RSC Adv., 2015, 5, 97918–97927 RSC.
- O. V. Lushchikova, S. Reijmer, P. B. Armentrout and J. M. Bakker, J. Am. Soc. Mass Spectrom., 2022, 33, 1393–1400 CrossRef CAS PubMed.
- D. Balcells, E. Clot and O. Eisenstein, Chem. Rev., 2010, 110, 749–823 CrossRef CAS PubMed.
- P. P. Chen and S. I. Chan, J. Inorg. Biochem., 2006, 100, 801–809 CrossRef CAS PubMed.
- C. H. Yeh, S. S. F. Yu, S. I. Chan and J. C. Jiang, ChemistrySelect, 2018, 3, 5113–5122 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Thu Bao Nguyen Le
*ab,
Thu Bao Nguyen Le