DOI:
10.1039/D3RA03259K
(Paper)
RSC Adv., 2023,
13, 21545-21549
The role of electrostatic potential in the translocation of triangulene across membranes†
Received
16th May 2023
, Accepted 30th June 2023
First published on 18th July 2023
Abstract
Triangulene and its derivatives show broad application prospects in the fields of biological imaging and biosensing. However, its interaction with cell membranes is still poorly studied. In this study, classical molecular dynamics simulations were used to adjust the electrostatic potential of triangulene to observe its interactions with cell membranes. We found that electrostatic potential not only affects the behavior as it enters the cell membrane, but also spatial distribution within the cell membrane. The angle distribution of inside-0 and all-0 triangulene when penetrating the membrane is more extensive than that of ESP triangulene. However, inside-0 triangulene could cross the midline of the cell membrane and prefers to stay in the upper leaflet, while all-0 triangulene and ESP triangulene can reach the lower leaflet. These findings can help us regulate the distribution of nanoparticles in cells, so as to design functional nanoparticles that conform to the requirements.
1. Introduction
Recent years have witnessed a spurt of progress in nanotechnology, and nanomaterials have shown increasing potential in biomedical fields such as targeted drug delivery, radiosensitization, and bioimaging.1–4 The cell membrane is the first barrier for the interaction between biological systems against nanoparticles, and its interaction with nanoparticles will be the beginning of a series of subsequent reactions.5–7 Hence, figuring out how nanoparticles interact with the cell membrane is crucial. It has been demonstrated that surface charge of nanoparticles has the potential to regulate the way they interact with cell membranes. For example, the periodically distributed electrostatic potential of C2N nanosheets can prevent it from damaging the cell membrane.8 Moreover, our recent research has found that the more polarized electrostatic potential of graphene quantum dots makes it harder to enter the cell membrane.9 Besides, because of the highly polar nature of the head group of phospholipid molecules of cell membranes, it has been recognized that modulating the electrostatic potential (EP) of nanomaterials is one of the most effective methods to regulate the nanoparticle–cell membrane interactions.10,11 Therefore, it is essential to understand how electrostatic potential (EP) of nanomaterials plays a role in the translocation across membranes. This can help us regulate the location of nanoparticles in the cell and design the required functional nanoparticles.
Triangulene is one of the most famous triplet-ground-state benzenoid hydrocarbons with interesting biological properties.12–14 Yu created a benchmark dataset of 25 magnetic systems with nonlocal spin densities that can more accurately predict electronic ground state for triangulene analogues.15 Shen introduced a new intramolecular radical–radical coupling approach and successfully synthesize two fused triangulene dimers efficiently.16 Arikawa synthesized a kinetically-stabilized nitrogen-doped triangulene cation derivative.17 Besides, atomic force microscopy (AFM) appears as an invaluable experimental technique, allowing the measurement of the mechanical strength of biomolecular complexes to provide a quantitative characterization of their interaction properties from a single molecule perspective.18 The above research indicates that stable triangulene may be synthesized in the future, and tools such as AFM may help us analyze the interaction process between triangulene and cell membrane in experiments.19 Recent research has found that the uniformly distributed electrostatic potential allows the system to have a small permanent dipole moment that blocks the electronic transition in the light excitation such that the electronic transition can only be carried out between adjacent carbon atoms.14 According to the calculation of density functional theory (DFT), the derivatives of triangulene have high affinity for DNA helix.20–23 Nevertheless, there are different mechanisms in healthy tissues and tumours. Unlike the non-toxic buckled form in healthy tissues, in tumours with relatively low pH values, the derivatives can intercalate DNA double stranded base pairs in a planar form to disrupt biological processes, showing their potential anti-cancer properties.13 In addition, the derivatives of triangulene can be used for specifically labeling cell mitochondria, lysosomes and other organelles with cell structures well preserved, thereby allow for various applications in the fields of biosensing and imaging due their excellent photo-physical properties.24,25 However, the interaction mechanism between the electrostatic potential of triangulene and cell membrane is unrevealed, which limits its application in biomedicine.
In this study, we simulated the translocation of triangulene with different electrostatic potential in the cell membrane. Since 1-palmitoyl-2-oleoylphosphatidylcholine (POPC) is among the primary constituents of cellular membranes, it is often used to mimic eukaryote cell membrane.26 Classical molecular dynamics (MD) simulations were employed to illustrate the binding dynamics of triangulene to the 1-palmitoyl-2-oleoylphosphatidylcholine (POPC) membrane. We constructed three groups of triangulene with different potentials, one of which had edges with potential polarity (inside-0), another group of evenly distributed potentials (ESP), and the third group with their potentials ignored (all-0).
2. Models and methods
2.1. System setup
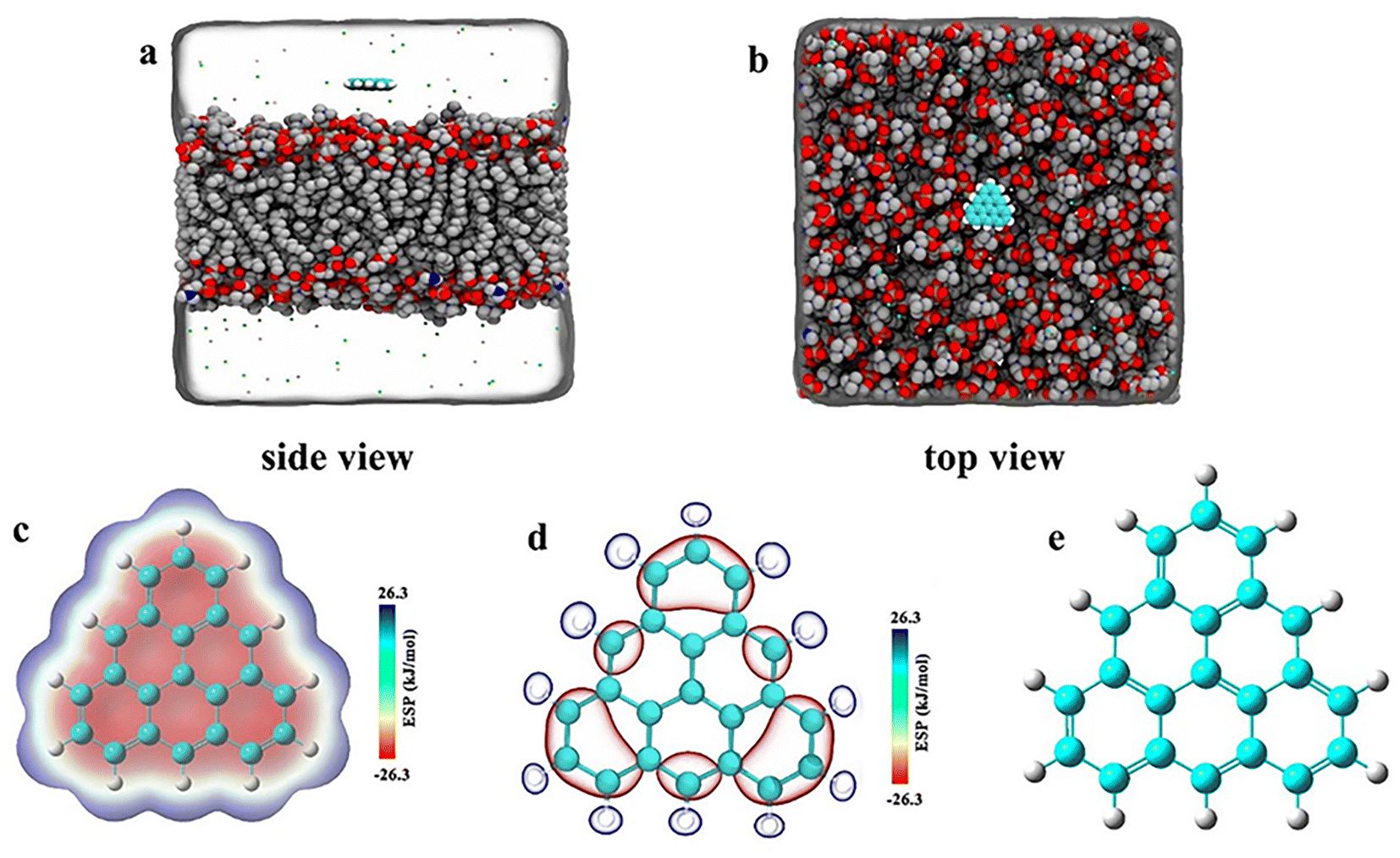
As shown in Fig. 1, we constructed three groups of triangulene with different potentials. The net charge of each triangulene is 0e (shown in the S1†), and the force field parameters are derived from previous studies.27,28 The first group of triangulene atomic charges are calculated by DFT which is called ESP.29 The second group of triangulene atomic charges are acquired from ref. 16, the partial charges of hydrogen atoms and the linked carbon atoms were set to +0.115e and −0.115e, while carbon atoms away from the edge remained neutral.28 The third group of triangulene atomic charges were assigned neutral charge. The electrostatic potential of triangulene are calculated by Adaptive Poisson–Boltzmann Solver (APBS).30–32 The triangulene are placed on a plane parallel to the x–y plane, about 3 nm from the center of the POPC membrane. The side length of triangulene is 0.925 nm. In the MD simulation, 188 POPC lipids were used with 94 lipids in each layer. The POPC membrane was equilibrated at 100 ps in NPT ensemble. The solvent added to the system is the TIP3P-water molecules, and the size of the box is 7.64 nm × 7.64 nm × 10 nm.33 The whole system was filled with 0.15 mol L−1 NaCl (44 Na+ and Cl−) to mimic the physiological environment (shown in the S2†).
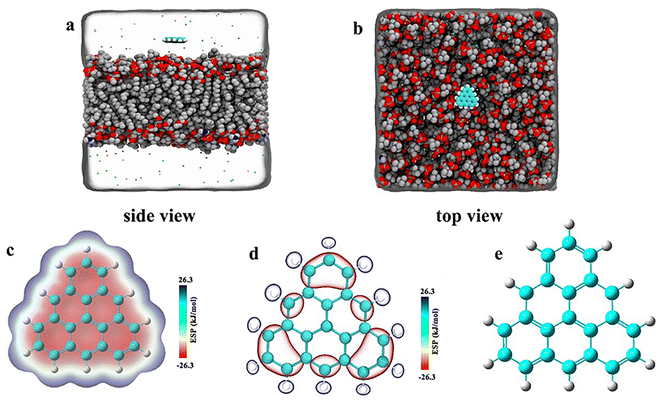 |
| | Fig. 1 The initial placement position of the system. (a) Side view (b) top view. (c), (d), and (e) Corresponding to surface distribution of electrostatic potential of ESP triangulene, inside-0 triangulene and all-0 triangulene, respectively. | |
2.2. Simulation parameters
All MD simulations were performed using the GROMACS package (version 4.6.7) with a time step of 2 fs. The parameters of the POPC lipid membrane were derived from the Charmm36 force field.34 The minimized system by the steepest descent method accepted a 2 ns NPT ensemble pre-equilibration. The v-rescale thermostat and semiisotropic Parrinello–Rahman barostat matained the pressure of the system at 1 atm and the temperature at 310 K.35,36 The short-range electrostatic and van der Waals interaction cut off distance was set to 1.2 nm, and the long-range electrostatic interaction was treated by the particle-mesh Ewald (PME) algorithm.37 The SETTLE algorithm was used for the water model, and all heavy atoms connected to H atoms were constrained using the LINCS algorithm.38,39 More details are available in our previous search.9
2.3. Potential of mean force (PMF) calculation
By analyzing the three different electrostatic potential of the triangulene through the membrane process, we can calculate the free energy, thus explaining why the triangulene will finally stay. To compute the free energy profile along the Z axis we employ an umbrella sampling method.40 Pulling triangulene from 0 nm (center of mass of POPC membrane) to 3.4 nm along the Z axis, with an umbrella sampling window of 0.1 nm. The constraining force constant was 2000 kJ mol−1 nm−2. Notedly, the constraining force was only used for umbrella sampling to calculate PMF, while not imposed on the triangulation during MD simulation. The PMF curve is obtained by g_wham the tool.41
3. Results and discussion
3.1. Angular distribution of triangulene translocation across the POPC membrane
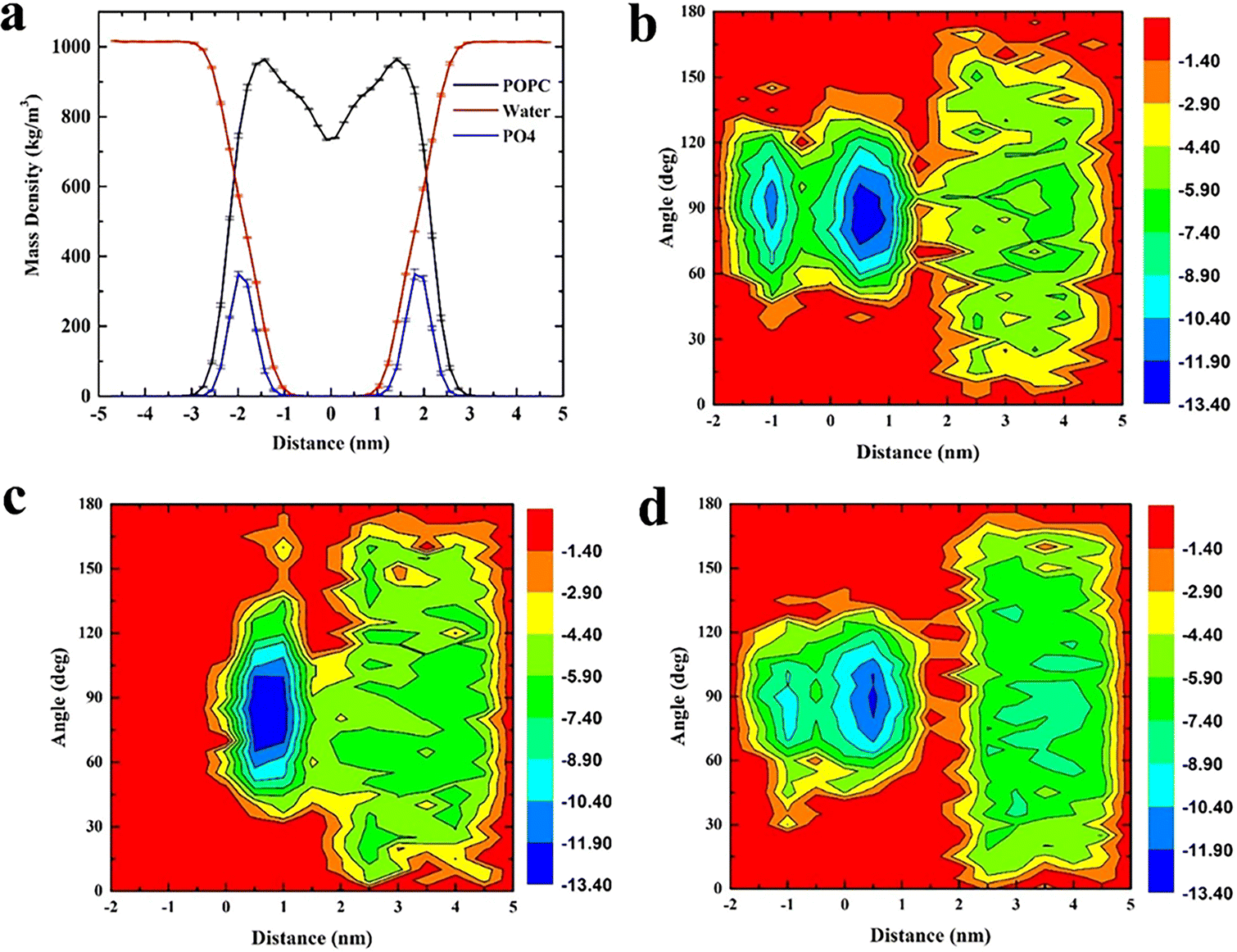
The whole system was divided into three regions along the Z axis, 5 nm to 2 nm was defined as region I (bulk water region), 2 nm to 0 nm was region II (upper leaflet), 0 to −2 nm was region III (lower leaflet). Relationship between angular distribution of triangulene and energy barrier was investigated. We found that ESP triangulene is easier to enter the membrane at an angle of 90° when permeating the membrane (Fig. 2b). However, inside-0 triangulene was more extensive when translocating into the membrane. It had the lowest energy barrier from 45 to 90° for inside-0 triangulene to mitigate from bulk water to upper leaflet (Fig. 2c). The all-0 triangulene was more effortless to enter the membrane at angles of 70, 90, 110 and 120° (Fig. 2d). As shown in Fig. 2, ESP triangulene and all-0 triangulene can get into the membrane and can ulteriorly cross the middle of the membrane to the lower leaflet, with the lowest energy barrier at 0.8 nm and −0.8 nm. While, the inside-0 triangulene can only get the upper leaflet of the membrane and preferred to remain in the upper layer region.
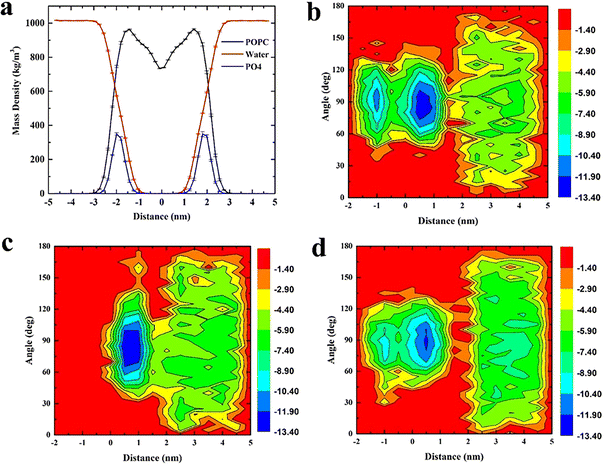 |
| | Fig. 2 (a) Density distribution of POPC membrane, water and phosphate group, defining the boundary of membrane at 2 nm. (b), (c), and (d) respectively show the distribution of angle (formed by ESP, inside-0 and all-0 triangulene surfaces and membrane surfaces) and the energy barrier along the Z axis during translocation. | |
3.2. The spatial distribution of triangulene in the system
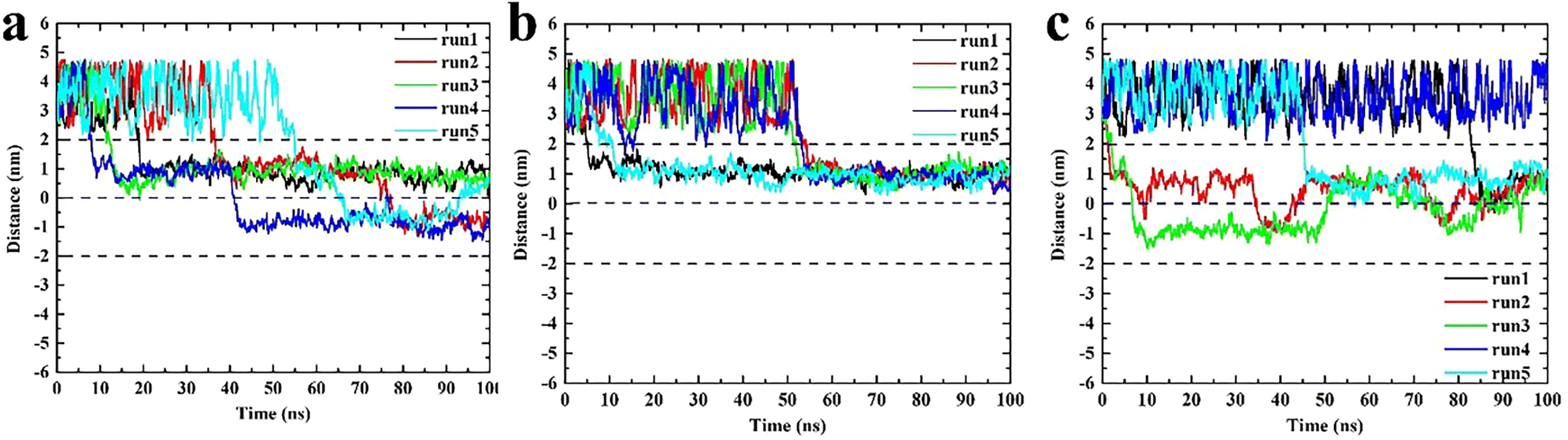
These three types of triangulene moved irregularly outside the membrane, whereas their behaviors changed obviously after entering the membrane (Fig. 3). The inside-0 triangulene had been slightly fluctuated and kept in region II (upper leaflet). ESP triangulene had been stayed mostly in region II, occasionally passing through the midline to the region III (lower leaflet). All-0 triangulene moved back and forth along the POPC membrane's center, and the time between the region II and region III was roughly equal. The results showed that the electrostatic potential of triangulene would have a significant effect on its spatial distribution in the POPC membrane.
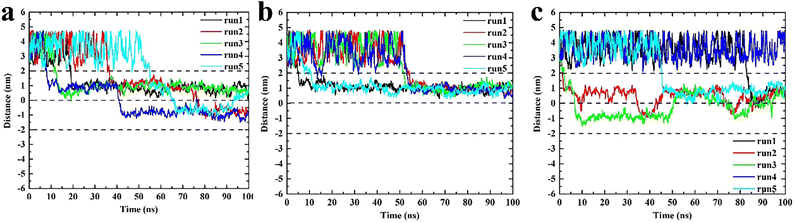 |
| | Fig. 3 The spatial distribution of triangulene in the POPC membrane. (a) ESP triangulene. (b) Inside-0 triangulene. (c) All-0 triangulene. | |
3.3. The potential of mean force (PMF) of triangulene translocating into POPC
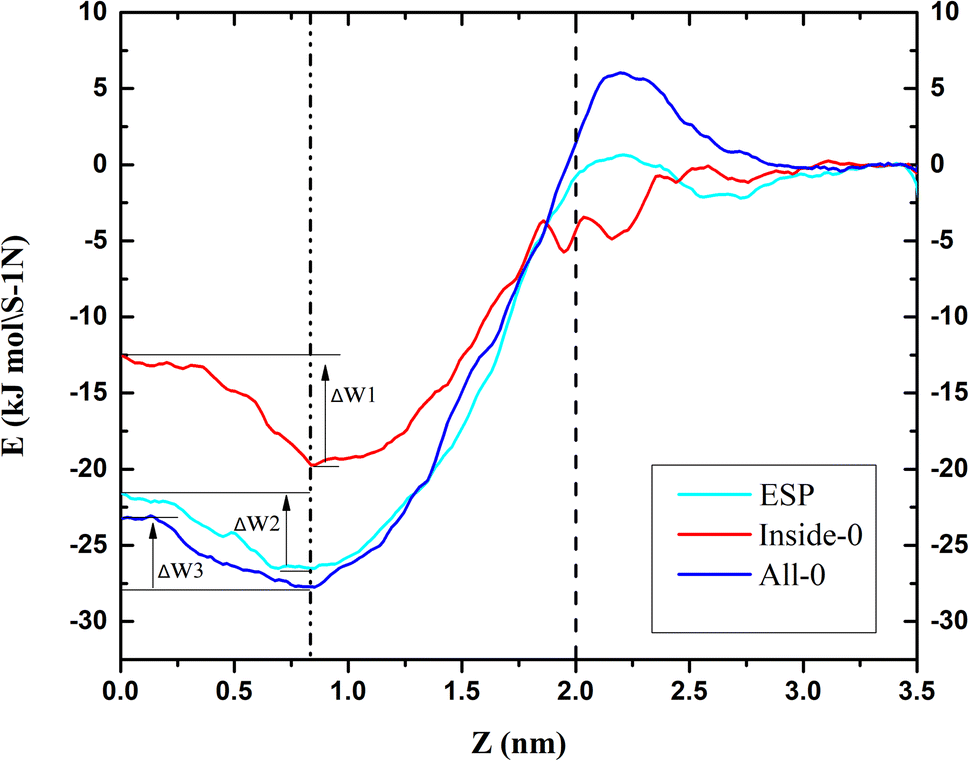
To further explain the reasons above spatial distribution occurs, the mean force potential (PMF) from 5 nm to −2 nm of triangulene was calculated (Fig. 4). The ΔW1, ΔW2 and ΔW3 were defined as the PMF of ESP triangulene, inside-0 triangulene and all-0 triangulene in the midline of the membrane minus the lowest in the region II. The ESP triangulene and all-0 triangulene needed to overcome 4.84 kJ mol−1 (ΔW1), 4.51 kJ mol−1 (ΔW3) to reach the lower leaflet. Inside-0 triangulene needed to overcome 7.15 kJ mol−1 (ΔW2), which explains why the inside-0 triangulene is only stayed in the upper leaflet of the membrane. However, ESP triangulene and all-0 triangulene can reach the lower leaflet of the membrane. From 5 nm to −2 nm, the energy at 0.8 nm is the lowest, so triangulene finally stay at 0.8 nm.
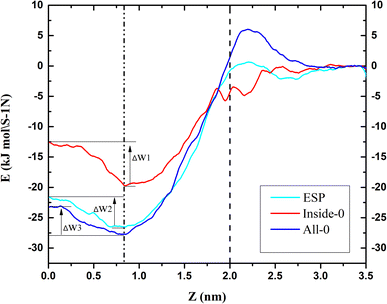 |
| | Fig. 4 The potential of mean force (PMF) of triangulene translocating into POPC along the z-axis direction. | |
4. Conclusion
In this study, the effect of electrostatic potential on the interaction between triangulene and cell membrane were described by setting three kinds of triangulene models with different electrostatic potential distributions. We can conclude that: (1) the angle of ESP triangulene penetrated POPC membrane is mainly 90°, and can cross the midline of the membrane to the opposite side. The angle distribution of inside-0 triangulene penetrated is more extensive than that of ESP. The energy barrier is lower when penetrating the membrane from 45° to 90°, which makes it easier to penetrate the membrane, but it is difficult to cross the central line of the cell membrane. The angle distribution of all-0 triangulene penetrate membrane is more discrete, the energy barrier is lower at 70°, 90°, 110° and 120°, easy to penetrate into the membrane, and can also cross the cell membrane midline to reach the lower leaflet of the POPC membrane; (2) through PMF calculation, it is found that ESP, inside-0 and all-0 triangulene respectively need to overcome 4.84 kJ mol−1 (ΔW1), 7.15 kJ mol−1 (ΔW2) and 4.51 kJ mol−1 (ΔW3). Therefore, inside-0 triangulene can hardly reach the lower leaflet due to the high energy barrier; (3) among these three systems, the energy barrier of the triangulene system is the lowest at 0.8 nm or −0.8 nm, that is the reason they finally stays at there.
Our research suggests the feasibility of regulating the interaction between triangulene and cell membrane through electrostatic potential, which would be helpful for further application of triangulene in biomedicine.
Abbreviations
| MD | Classical molecular dynamics |
| POPC | 1-Palmitoyl-2-oleoylphosphatidylcholine |
| DFT | Density functional theory |
| EP | Electrostatic potential |
| APBS | Adaptive Poisson–Boltzmann solver |
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
We acknowledge financial support by Science, Technology Department of Yunnan Province (202101AY070001-164).
References
- X. Chen and W. Zhang, Chem. Soc. Rev., 2017, 46, 734–760 RSC.
- H. Wang, Q. Chen and S. Zhou, Chem. Soc. Rev., 2018, 47, 4198–4232 RSC.
- H. Kang, J. Gravier, K. Bao, H. Wada, J. H. Lee, Y. Baek, G. El Fakhri, S. Gioux, B. P. Rubin, J. L. Coll and H. S. Choi, Adv. Mater., 2016, 28, 8162–8168 CrossRef CAS PubMed.
- H. M. Xiong, Adv. Mater., 2013, 25, 5329–5335 CrossRef CAS PubMed.
- X. Wang, X. Wang, X. Bai, L. Yan, T. Liu, M. Wang, Y. Song, G. Hu, Z. Gu, Q. Miao and C. Chen, Nano Lett., 2019, 19, 8–18 CrossRef CAS PubMed.
- F. Fontana, H. Lindstedt, A. Correia, J. Chiaro, O. K. Kari, J. Ndika, H. Alenius, J. Buck, S. Sieber, E. Makila, J. Salonen, A. Urtti, V. Cerullo, J. T. Hirvonen and H. A. Santos, Adv. Healthcare Mater., 2020, 9, 2000529 CrossRef CAS.
- D. Zou, Z. Wu, X. Yi, Y. Hui, G. Yang, Y. Liu, Tengjisi, H. Wang, A. Brooks, H. Wang, X. Liu, Z. P. Xu, M. S. Roberts, H. Gao and C. X. Zhao, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2214757120 CrossRef CAS.
- S. Zhang, L. Liu, G. Duan, L. Zhao, S. Liu, B. Zhou and Z. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 34575–34585 CrossRef CAS PubMed.
- X. Tang, S. Zhang, H. Zhou, B. Zhou, S. Liu and Z. Yang, Nanoscale, 2020, 12, 2732–2739 RSC.
- P. Chen, Z. Huang, J. Liang, T. Cui, X. Zhang, B. Miao and L. T. Yan, ACS Nano, 2016, 10, 11541–11547 CrossRef CAS.
- C. Pagnout, S. Jomini, M. Dadhwal, C. Caillet, F. Thomas and P. Bauda, Colloids Surf., B, 2012, 92, 315–321 CrossRef CAS PubMed.
- S. Arikawa, A. Shimizu, D. Shiomi, K. Sato and R. Shintani, J. Am. Chem. Soc., 2021, 143, 19599–19605 CrossRef CAS PubMed.
- E. Leung, L. I. Pilkington, M. M. Naiya, D. Barker, A. Zafar, C. Eurtivong and J. Reynisson, MedChemComm, 2019, 10, 1881–1891 RSC.
- N. Zhang, W. Feng, H. Wen, N. Feng, H. Sheng, Z. Huang and J. Wang, Molecules, 2023, 28, 3744 CrossRef CAS.
- H. Yu, J. Sun and T. Heine, J. Chem. Theory Comput., 2023, 19, 3486–3497 CrossRef CAS.
- T. Shen, D. Dijkstra, A. Farrando-Perez, P. G. Boj, J. M. Villalvilla, J. A. Quintana, Y. Zou, X. Hou, H. Wei, Z. Li, Z. Sun, M. A. Diaz-Garcia and J. Wu, Angew. Chem., Int. Ed., 2023, 62, e202304197 CrossRef CAS PubMed.
- S. Arikawa, A. Shimizu, D. Shiomi, K. Sato, T. Takui, H. Sotome, H. Miyasaka, M. Murai, S. Yamaguchi and R. Shintani, Angew. Chem., Int. Ed., 2023, 62, e202302714 CrossRef CAS PubMed.
- A. Lostao, K. Lim, M. C. Pallares, A. Ptak and C. Marcuello, Int. J. Biol. Macromol., 2023, 238, 124089 CrossRef CAS PubMed.
- E. Escorihuela, A. Concellón, I. Marín, V. J. Kumar, L. Herrer, S. A. Moggach, A. Vezzoli, R. J. Nichols, P. J. Low, P. Cea, J. L. Serrano and S. Martín, Mater. Today Chem., 2022, 26, 101067 CrossRef CAS.
- J. Reynisson, G. B. Schuster, S. B. Howerton, L. D. Williams, R. N. Barnett, C. L. Cleveland, U. Landman, N. Harrit and J. B. Chaires, J. Am. Chem. Soc., 2003, 125, 2072–2083 CrossRef CAS.
- A. Pothukuchy, S. Ellapan, K. R. Gopidas and M. Salazar, Bioorg. Med. Chem. Lett., 2003, 13, 1491–1494 CrossRef CAS PubMed.
- A. Kotar, B. Wang, A. Shivalingam, J. Gonzalez-Garcia, R. Vilar and J. Plavec, Angew. Chem., Int. Ed., 2016, 55, 12508–12511 CrossRef CAS PubMed.
- H. Yu, J. Sun and T. Heine, J. Chem. Theory Comput., 2023, 19, 3486–3497 CrossRef CAS.
- J. Bosson, J. Gouin and J. Lacour, Chem. Soc. Rev., 2014, 43, 2824–2840 RSC.
- C. Bauer, R. Duwald, G. M. Labrador, S. Pascal, P. M. Lorente, J. Bosson, J. Lacour and J. D. Rochaix, Org. Biomol. Chem., 2018, 16, 919–923 RSC.
- M. Mijajlovic, D. Wright, V. Zivkovic, J. X. Bi and M. J. Biggs, Colloids Surf., B, 2013, 104, 276–281 CrossRef CAS PubMed.
- L. Liang, Z. Kong, Z. Kang, H. Wang, L. Zhang and J. W. Shen, ACS Biomater. Sci. Eng., 2016, 2, 1983–1991 CrossRef CAS PubMed.
- D. Cohen-Tanugi and J. C. Grossman, Nano Lett., 2012, 12, 3602–3608 CrossRef CAS PubMed.
- O. A. Gapurenko, A. G. Starikov, R. M. Minyaev and V. I. Minkin, J. Comput. Chem., 2015, 36, 2193–2199 CrossRef CAS PubMed.
- N. A. Baker, D. Sept, S. Joseph, M. J. Holst and J. A. McCammon, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10037–10041 CrossRef CAS.
- T. J. Dolinsky, P. Czodrowski, H. Li, J. E. Nielsen, J. H. Jensen, G. Klebe and N. A. Baker, Nucleic Acids Res., 2007, 35, W522–W525 CrossRef PubMed.
- A. S. Mamidi, A. Ray and N. Surolia, Front. Bioeng. Biotechnol., 2019, 7, 240 CrossRef PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- J. Huang and A. D. Mackerell, J. Comput. Chem., 2013, 34, 2135–2145 CrossRef CAS PubMed.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1998, 52, 7182–7190 CrossRef.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- S. Miyamoto and P. A. Kollman, J. Comput. Chem., 2010, 13, 952–962 CrossRef.
- B. Hess, H. Bekker and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- G. M. Torrie and J. P. Valleau, J. Chem. Phys., 1977, 66, 1402–1408 CrossRef CAS.
- J. S. Hub, J. Chem. Phys., 2015, 6, 3713–3720 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra03259k |
| ‡ Xiaofeng Tang and Youyun Li contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Youyun Li‡b,
Qianyan Lia,
Jinhui Yua and
Han Bai*ac
*a,
Youyun Li‡b,
Qianyan Lia,
Jinhui Yua and
Han Bai*ac