
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Introduction of a new and safe synthesis procedure for Ni-MOF-I in aqueous solution and its application for the extraction of some pesticides from different beverages†
Mir Ali Farajzadeh *ab,
Nastaran Khoshnavaza,
Sakha Pezhhanfara and
Mohammad Reza Afshar Mogaddamcd
*ab,
Nastaran Khoshnavaza,
Sakha Pezhhanfara and
Mohammad Reza Afshar Mogaddamcd
aDepartment of Analytical Chemistry, Faculty of Chemistry, University of Tabriz, Tabriz, Iran. E-mail: mafarajzadeh@yahoo.com; mafarajzadeh@tabrizu.ac.ir; Fax: +98 41 33340191; Tel: +98 41 33393084
bEngineering Faculty, Near East University, Mersin 10, 99138 Nicosia, North Cyprus, Turkey
cFood and Drug Safety Research Center, Tabriz University of Medical Sciences, Tabriz, Iran
dPharmaceutical Analysis Research Center, Tabriz University of Medical Sciences, Tabriz, Iran
First published on 19th July 2023
Abstract
For the first time, this research introduces an analytical application of Ni-MOF-I, which was used as an adsorbent in a dispersive micro solid phase extraction procedure followed by dispersive liquid–liquid microextraction for the extraction and preconcentration of seven pesticides from different fruit juices. Also, Ni-MOF-I was synthesized by a new and green method with many advantages over the previously published synthesis procedures. For example, effortless and green synthesis, no need for autoclaves and ovens, and elimination of organic solvent usage are the main highlights. The synthesized Ni-MOF-I was characterized by applying nitrogen adsorption/desorption, energy-dispersive X-ray, scanning electron microscopy, Fourier transform infrared spectrophotometry, and X-ray diffraction analyses. The studied pesticides were extracted and preconcentrated by the proposed method. Then, the extracted analytes in the sedimented organic phase were injected into a gas chromatography-flame ionization detector. Acceptable analytical results such as low limits of detection (0.15–0.60 μg L−1) and quantification (0.50–2.0 μg L−1), reasonable extraction recoveries (51–80%), high enrichment factors (255–400), satisfactory relative standard deviation values of 4.8–7.2% (intra-day precision, n = 6) and 5.3–7.5% (inter-day precision, n = 4), and wide linear ranges were obtained. The proposed method can be introduced as an effective analytical technique based on Ni-MOF-I for the analysis of different pesticides in fruit beverages.
1. Introduction
The request for fruit-based beverages in the juice market is considerably increasing over recent years due to them containing an abundance of different phytochemicals and having valuable potential for strengthening human health.1–3 In addition to the mentioned positive factors, juices can be harmful for human health because of various pesticides that might be present and abundance of additive sugar.4 Pesticides are one of the main inputs, which are utilized to eradicate various pests in households and agriculture, heighten the output and quality of crops, decrease the energy cost, and restrict many vector-borne diseases. Despite the mentioned advantages, pesticides may put the human body at considerable danger of diseases, cause irrecoverable harm, and persist in the environment and cause the contamination of fruits, vegetables, and surface waters because of their environmental stability, capability to bioaccumulate, and toxicity.5,6 The residue of pesticides is out of the consumer's control.7 Therefore, based on the aforementioned information, it becomes imperative to develop highly sensitive, reliable, and selective analytical methods to analyze pesticide residues in foods to decrease the possible dangers for human society. These days, various types of analytical techniques such as gas chromatography (GC),8,9 and high performance liquid chromatography (HPLC)10,11 are used to determine pesticide residues in food samples including juices. Usually, these methods have great selectivity with low detection limits and high sensitivity,12 but most samples cannot be injected directly into them because of some limitations like the low concentration of analytes, having a complex matrix, and the intrinsic limitation of aqueous solutions in injection into GC instrument.3 The amounts of pesticide residues in various environments are usually under the limit of detection (LOD) of many analytical instruments, so it is necessary to integrate sample preparation steps with instrumental analysis in order to concentrate the pesticides, reduce the LODs, and acquire a reliable signal while utilizing different analytical instruments for analyzing pesticides.13 More than 80% of the analysis time is dedicated to the extraction, preconcentration, and preparation steps. For this reason, the critical part of most analytical procedures is sample preparation step.14 Methods of sample preparation should be able to isolate the matrix from the analytes15 and transfer them into a phase which is suitable for injection into the analysis system by extracting the targeted analytes from the sample matrix.16 Moreover, they should be eco-friendly, time-saving, inexpensive, and simple.13 The two coventional sample preparation methods which are applied for determining the pesticide residues in fruit juices are solid phase extraction (SPE) and liquid–liquid extraction (LLE).17 Beside the advantages they contain, they have some serious disadvantages such as high-volume use of organic solvents in the case of LLE and cartridge obstruction in the case of SPE.18,19 To overcome these issues, Anastassiades et al.20 introduced an alternative method over conventional SPE named dispersive solid phase extraction (DSPE) in which the adsorbent is straightly dispersed into the sample solution instead of being loaded as the cartridge. Various kinds of materials have been utilized as solid adsorbents like graphene, carbon nanotubes, metal–organic frameworks (MOFs), silica nanoparticles, mesoporous silica, etc.21 Recently, dispersive micro solid phase extraction (DμSPE) which is the miniaturized modification of DSPE has been widely developed22 in which it needs less than 500 mg of solid adsorbent.4 In comparison to DSPE, DμSPE has simpler operation, and is more economic due to the utilization of lower sorbent weights. These make the approach economical and environmentally-friendly.22,23 However, the main drawback of this method is the low value of enrichment factors (EFs) because of consuming mL-level of elution solvent. To solve this issue a combination of DμSPE with dispersive liquid–liquid microextraction (DLLME) is required which is an affordable and simple extraction method and results in high EFs.24–26 The main drawback of DLLME is the limitation of its direct application in complex matrices due to the extraction of the co-extractives that interfere on the analysis process.27 Considering the advantages of DμSPE and DLLME, simultaneous use of these two methods can make more benefits like the achievement of high EFs, efficient sample clean up, and low LODs and limits of quantification (LOQs).25 MOFs are a novel class of hybrid porous materials and a new type of three-dimensional coordination polymers formed by organic ligands and metallic clusters through coordination bonds.28,29 Owing to their tailorable polarity, structure flexibility, high and tunable porosity, good thermal stability, uniform cavities, adsorption capacity, and high surface area, MOFs have been used in drug delivery, photovoltaic materials, catalysis, gas purification and separation, gas storage systems, biomedicine, as adsorbents in analytical sample preparation methods, sensors in electroanalytical and spectroscopy methods, and the stationary phases of chromatographic columns.29–31Many papers were reported in literature regards pesticide residues analysis in different matrices. Barrek et al.32 separated residual pesticides from olive oil by size-exclusion chromatography. After the extraction, eleven pesticides were separated and analyzed by HPLC-mass spectrometry (MS) and 20 others by GC-MS. Ma et al.33 extracted and preconcentrated four pesticides including fenpyroximate, chlorfenapyr, fipronil, and flusilazole in environmental water samples using magnetic MIL-101 in magnetic solid phase extraction procedure and analyzed by HPLC-diode array detector (DAD). Chahkandi et al.34 extracted some organophosphorus pesticides including diazinon, phosalone, fenthion, fenitrothion, and profenofos from fruit juices and water samples applying magnetic potassium substituted hydroxyapatite and analyzed by GC-flame ionization detector (FID). Michel et al.35 extracted ten fungicide, insecticide, and herbicide residues in vegetables, fruits, and cereal using LLE, SPE, and matrix solid phase dispersion. Determination was performed by reversed-HPLC-DAD. Shamsipur et al.36 used SPE coupled with DLLME and GC-MS for the extraction and determination of 20 pesticide residues from fruit juice, honey, milk, and water. Wang et al.37 developed an ultrasound-assisted DLLME method based on solidification of floating organic droplets followed by GC-FID for the extraction and determination of some pesticides including pyridaben, triazophos, es-fenvalerate, buprofezin, and λ-cyhalothrin in water samples.
This research has two main novel aspects. The first one is the introduction of a new, facile, and safe synthesis procedure for Ni-MOF-I which is done in aqueous solution. This approach is effortless, green, and needs no special synthesis instruments such as autoclaves, ovens, and toxic solvents such as N,N-dimethylformamide. The second aspect is the utilization of Ni-MOF-I for the first time in a sample preparation process for the extraction of different pesticides from juices through DμSPE-DLLME procedure and their analysis using GC-FID.
2. Experimental
2.1. Chemicals and solutions
All the target compounds (chlorpyrifos, haloxyfop-R-methyl, oxadiazon, diniconazole, clodinafop-propargyl, fenpropathrin, and fenoxprop-P-ethyl) with purity >98% were procured from Dr Ehrenstorfer (Agsburg, Germany). Sodium sulfate, potassium chloride, sodium chloride, sodium hydroxide, acetonitrile (ACN), acetone, hydrochloric acid (37%, w/w), and methanol (analytical grade) were obtained from Merck (Darmstadt, Germany). 2-Propanol was acquired from Caledon (Georgetown, Canada). Deionized water was supplied from Ghazi Co. (Tabriz, Iran). Extraction solvents including 1,1,2-trichloroethane (1,1,2-TCE), 1,1,1-trichloroethane (1,1,1-TCE), 1,2-dibromoethane (1,2-DBE), and carbon tetrachloride were purchased from Janssen (Beerse, Belgium). To synthesize the adsorbent, nickel(II) chloride hexahydrate (NiCl2·6H2O), 1,4-benzenedicarboxylic acid (1,4-BDCA), and concentrated ammonia solution (25%, w/w) were purchased from Merck (Darmstadt, Germany). A mixture standard solution including 250 mg L−1 of each pesticide was provided in methanol and diluted using deionized water for daily-used solutions of interest.2.2. Samples
Four commercial fruit juices including mango (the content of 200 mL juice includes sodium (13 mg), potassium (75 mg), total carbohydrates (28 g), sugar (26.25 g), vitamin A, vitamin C, calcium, iron, and natural mango puree), pineapple (the content of 200 mL juice includes sodium (22 mg), potassium (71 mg), total carbohydrates (27.5 g), sugar (27.5 g), vitamin A, vitamin C, calcium, iron, and natural pineapple puree), apple (the content of 200 mL juice includes sodium (23 mg), potassium (201 mg), total carbohydrate (26 g), vitamin A, vitamin C, calcium, iron, and natural apple puree), and peach (the contents of 200 mL juice includes sodium (22 mg), potassium (125 mg), total carbohydrate (28 g), sugars (26.25 g), vitamin A, vitamin C, calcium, iron, and natural mango puree) were purchased from a local store in Tabriz (Iran). The brand of all the commercial fruit juices used in this study Sun Ich. In addition, in order to evaluate the juices of fresh fruits, two fresh fruits including apple and orange were purchased. A juicer was used to extract the juices of these fruits. For separating the juice from its scum, centrifugation was done at 6000 rpm for 5 min. All the mentioned juices were diluted at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 with deionized water prior to carrying out the extraction method on them.
:1 with deionized water prior to carrying out the extraction method on them.
2.3. Apparatus
Chromatographic analysis and separation of the pesticides were carried out by a Shimadzu 2014 gas chromatograph (Kyoto, Japan) equipped with a split/splitless injector and an FID. A split ratio of 1:10 and a sampling time of 1 min were adjusted. The temperatures of the injection port and FID were set at 300 °C during the analysis. The initial temperature of the column oven was held at 60 °C for 1 min, then programmed at a rate of 18 °C min−1 to 300 °C, and held for 3 min to complete the separation run. Chromatographic separation was attained on a Zebron capillary column (95% dimethyl, 5% diphenyl polysiloxane) (30 m × 0.25 mm i.d, a film thickness of 0.25 μm) (Phenomenex, Torrance, CA, USA). Helium (99.999%, Gulf Cryo, Dubai, United Arab Emirates) was used as the make up and carrier gases. A flow rate of 30 mL min−1 and linear velocity of 30 cm s−1 were adjusted for the make up and carrier gases, respectively. The required hydrogen as the fuel of FID was provided by a hydrogen generator (OPGU-1500S, Shimadzu, Japan) which was set at the flow rate of 30 mL min−1. Additionally, the air flow rate was set to be 300 mL min−1. For injection of the standards and extracted samples into GC-FID, a 1.0 μL microsyringe (zero dead volume, Hamilton, Switzerland) was utilized. A Metrohm pH meter, model 654 (Herisau, Switzerland) was utilized for pH adjustment. Phase separation during the extraction process was accomplished by a Hettich centrifuge, model D-7200 (Kirchlengern, Germany). A magnetic heater-stirrer (Heidolph MR 3001 K, Germany) was applied in providing the MOF. An L46 vortex (Labinco, Breda, the Netherlands) was utilized for vortexing. The synthesized MOF's X-ray diffraction (XRD) pattern was attained by applying a Siemens D500 diffractometer (Siemens AG, Karlsruhe, Germany) at a voltage of 35 kV. It was performed at the rate and scan range of 1° min−1 and 4–73°, respectively. Surface characterization of the synthesized MOF was also accessed by energy-dispersive X-ray (EDX) and scanning electron microscopy (SEM) analyses utilizing a Mira 3 microscope (Tescan, Czech Republic). A Fourier transform infrared (FTIR) spectrophotometer (Bruker, Billerica, USA) was utilized in the range of 400–4000 cm−1 for obtaining the FTIR spectrum of the adsorbent applied in the present investigation. A BELSORP-mini II (BEL, Japan) analyzer was used to carry out nitrogen adsorption/desorption analysis for getting information about average pores diameter, pores size, and surface area of the MOF.
2.4. Synthesis of Ni-MOF-I
In this study, Ni-MOF-I was synthesized through a new green procedure. In comparison to the previously reported synthesis method38 the developed approach is more economic, time-saving, and needs no expensive and special synthesis instruments. Also, this synthesis process is environmentally friendly and not harmful for human health because unlike the previous synthesis method in which dimethylformamide was utilized as the reaction solvent, deionized water was used instead. This synthesis method entails the subsequent steps:(a) Initially, 0.80 g of 1,4-BDCA was added as the ligand to 5.0 mL of concentrated ammonia solution and put on a stirrer. Deionized water (20 mL) was slowly added until the solution became clear. Then, the solution was poured into a burette.
(b) 1.14 g of NiCl2·6H2O was dissolved in 100 mL of deionized water in an Erlen mayer flask. Afterward, the flask was put into a water bath set at 80 °C.
(c) The solution inside the burette was added dropwisely into the flask under stirring at 300 rpm. After finishing the content of the burette, a lid was put on the flask and continued stirring for 1 h. Then, the flask was let to cool down at room temperature. The formed light green precipitate was Ni-MOF-I. Then, the content of flask was transferred into glass tubes and then were centrifuged at 6000 rpm for 5 min. Afterwards, the supernatant was decanted. The product remaining at the bottom of the tubes was vortexed with 5 mL of deionized water for 5 min, centrifuged at 6000 rpm for 5 min, and the supernatant was decanted. This process was repeated for 5 times until the product was completely washed and the decanted phase was neutralized. At the end, the obtained product was poured on a watch glass to dry at room temperature. The yield of the MOF according to the adopted synthesis method was 68.41%.
Eventually, the synthesized Ni-MOF-I was subjected to FTIR, XRD, nitrogen adsorption/desorption, EDX, and SEM analyses and after that it was applied as an adsorbent in the extraction procedure.
2.5. Extraction procedure
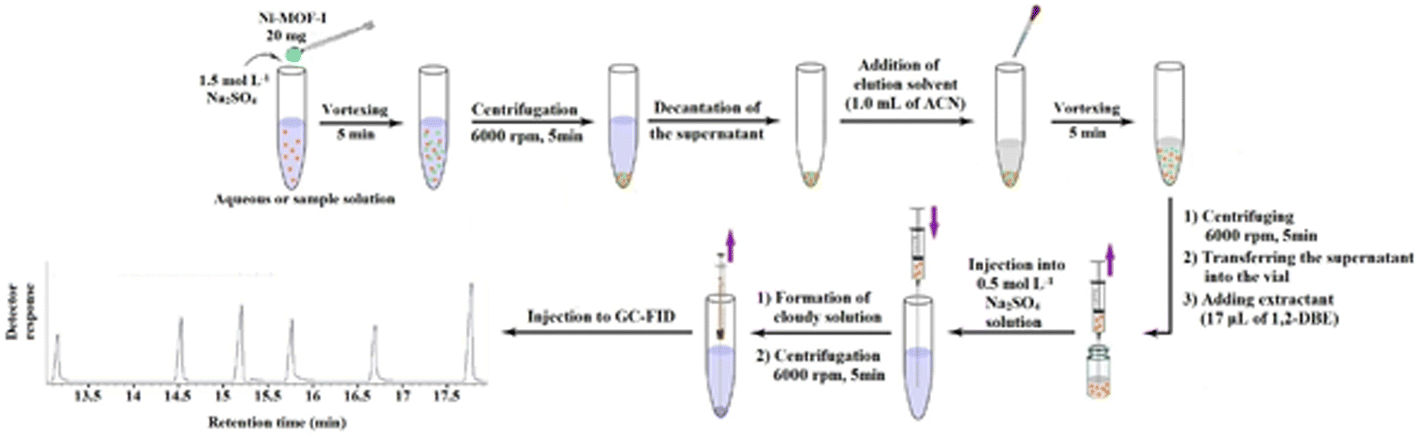 | ||
| Fig. 1 Schematic figure of the developed extraction procedure. | ||
2.6. Calculation of EF and extraction recovery (ER)
ER and EF were utilized to appraise efficiency of the extraction in various experimental conditions. The equations used to calculate their values are written below.
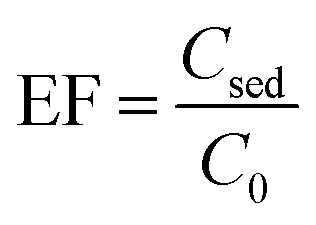 | (1) |
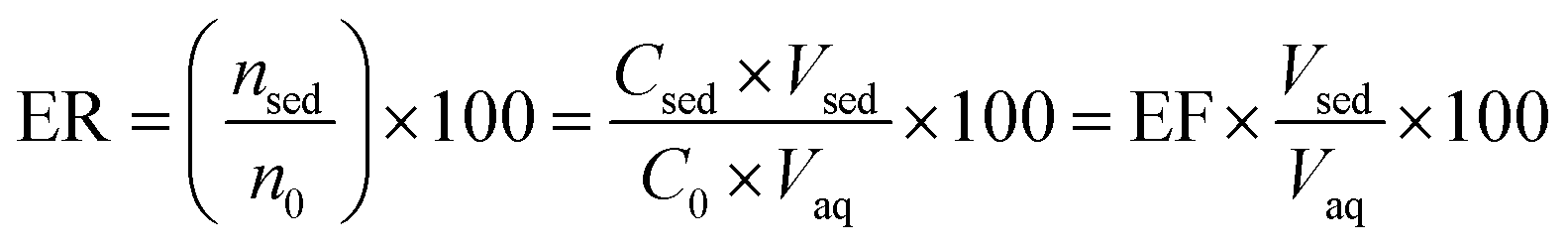 | (2) |
The ratio of pesticide concentration in the extraction solvent (Csed) to the pesticide concentration in the initial sample solution (C0) is described as EF. The ER is the ratio of the extracted pesticide amount (nsed) to its initial amount (no) multiplied by 100. Moreover, the term Vsed indicates volume of the settled organic phase in DLLME and Vaq shows the initial aqueous sample volume.
3. Results and discussion
3.1. Characterization of Ni-MOF-I
To identify the synthesized MOF, some characterization analyses were performed such as FTIR, XRD, nitrogen adsorption/desorption, EDX, and SEM. By comparing the obtained results with the results reported previously,38,39 it was concluded that the synthesized adsorbent is Ni-MOF-I.XRD is an influential and essential analysis for diagnosing and validating the creation of the required crystalline compound. For this purpose, XRD analysis was done in the range of 4–73°. The outcome XRD pattern of Ni-MOF-I is shown in Fig. 2a. Same peaks are observed at 2θ values of around 10, 12, 16, 18, 19, 24, 28, and 29°. Based on the attained XRD pattern, some acute and distinct peaks are observed which is the reason for the crystalline structure and successful formation of Ni-MOF-I. Furthermore, the perfect match between the obtained pattern and the previously reported pattern32 verifies the successful synthesis of Ni-MOF-I in this research.
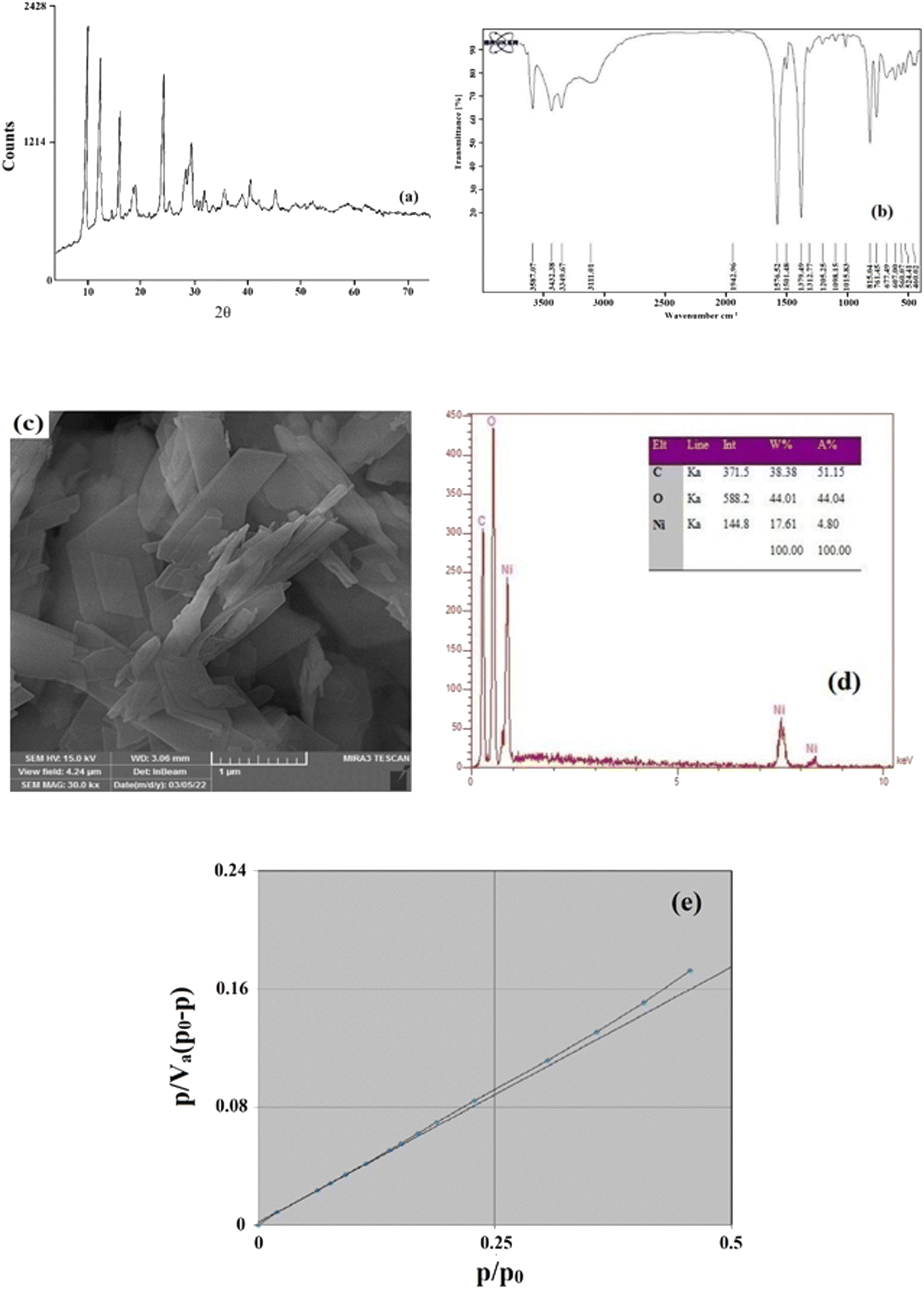 | ||
| Fig. 2 XRD pattern (a), FTIR spectrum (b), SEM image (c), EDX data (d), and BET plot (e) of Ni-MOF-I. | ||
FTIR spectrometry was also applied to identify the formation of the intended MOF. Fig. 2b shows the FTIR spectrum of the synthesized Ni-MOF-I. Two absorption peaks at 1576.52 and 1379.49 cm−1 are related to the symmetric and asymmetric stretching vibration of the –COO– functional group of the ligand coordinated to the metallic center. The presence of peaks at 460.02, 607.00, and 677.49 cm−1 are ascribed to the formation of the metal–oxygen bond between the carboxylic group of 1,4-BDCA and Ni (Ni–O). The peaks at 1501.48 and 3432.38 cm−1 are attributed to the stretching vibration of CH and OH, respectively. Also, the peaks at 761.05 and 815.05 cm−1 are related to the C–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds present in the ligand section of the synthesized MOF. According to the above-mentioned results and their perfect agreement with the previously published FTIR spectrum of the desired MOF,33 it reveals that Ni-MOF-I synthesis was done correctly.
C bonds present in the ligand section of the synthesized MOF. According to the above-mentioned results and their perfect agreement with the previously published FTIR spectrum of the desired MOF,33 it reveals that Ni-MOF-I synthesis was done correctly.
The morphology of the surface of the synthesized MOF can be investigated by SEM. So, high resolution image of the MOF which is shown in Fig. 2c, was obtained to attain the surface morphology of Ni-MOF-I. As can be observed, the Ni-MOF-I shows a layer-cuboid morphology with plain surface which provides an appropriate adsorption surface for the investigated pesticides.
EDX analysis was applied to reveal the presence of elements in the structure of the synthesized MOF and also to obtain information about the MOF's purity. The results are shown in Fig. 2d. As can be clearly noticed, there are some main and sharp peaks corresponding to C, O, and Ni which confirm the presence of composing elements in Ni-MOF-I. No appearance of peaks related to other elements is the proof of the purity of the synthesized MOF. The EDX analysis reveals that the surface of Ni-MOF-I contains 17.61% nickel, 44.01% oxygen, and 38.38% carbon.
To get information regarding the surface area, average pores diameter, and total pores volume of Ni-MOF-I, nitrogen adsorption/desorption analysis was applied (Fig. 2e). The results of the analysis were reported as follows: 0.055 cm3 g−1 for total pores volume, 17.641 nm for average pores diameter, and 12.503 m2 g−1 for the surface area of the MOF. The mentioned data were obtained from the BET data.
3.2. Optimization of parameters in DμSPE
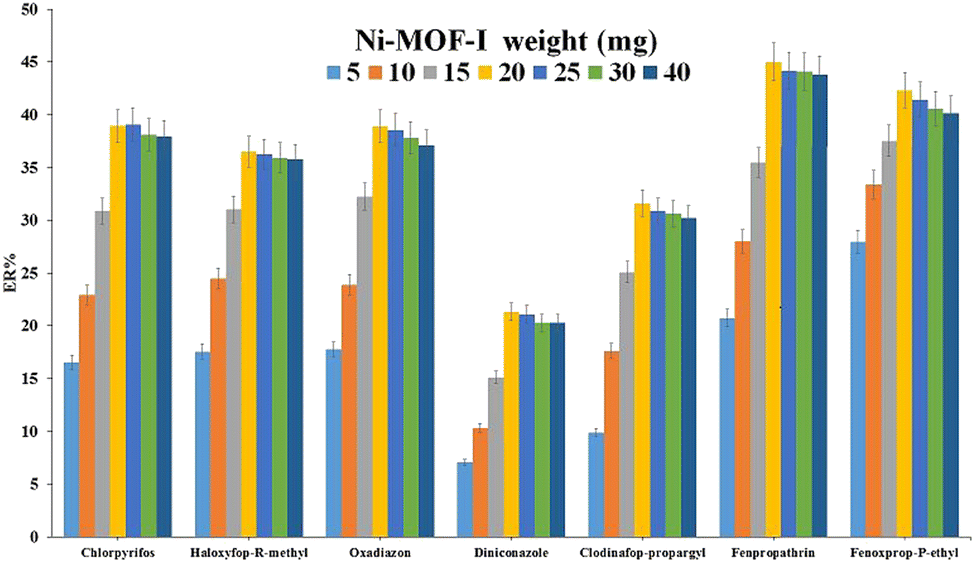 | ||
| Fig. 3 Evaluation of Ni-MOF-I weight. Extraction conditions: DμSPE procedure: aqueous solution, 5 mL deionized water containing 1.0 mol L−1 Na2SO4 spiked with 150 μg L−1 of each analyte without pH adjustment; vortex time in adsorption step, 5 min; desorption solvent (volume), ACN (1.0 mL); vortex time in desorption step, 5 min; and centrifugation speed and time, 6000 rpm and 5 min, respectively. DLLME procedure: aqueous phase, 5 mL deionized water without pH adjustment and salt addition; extraction solvent (volume), 1,2-DBE (31 μL); centrifugation rate, 6000 rpm; and centrifugation time, 5 min. The error bars show the minimum and maximum of three repeated determinations. | ||
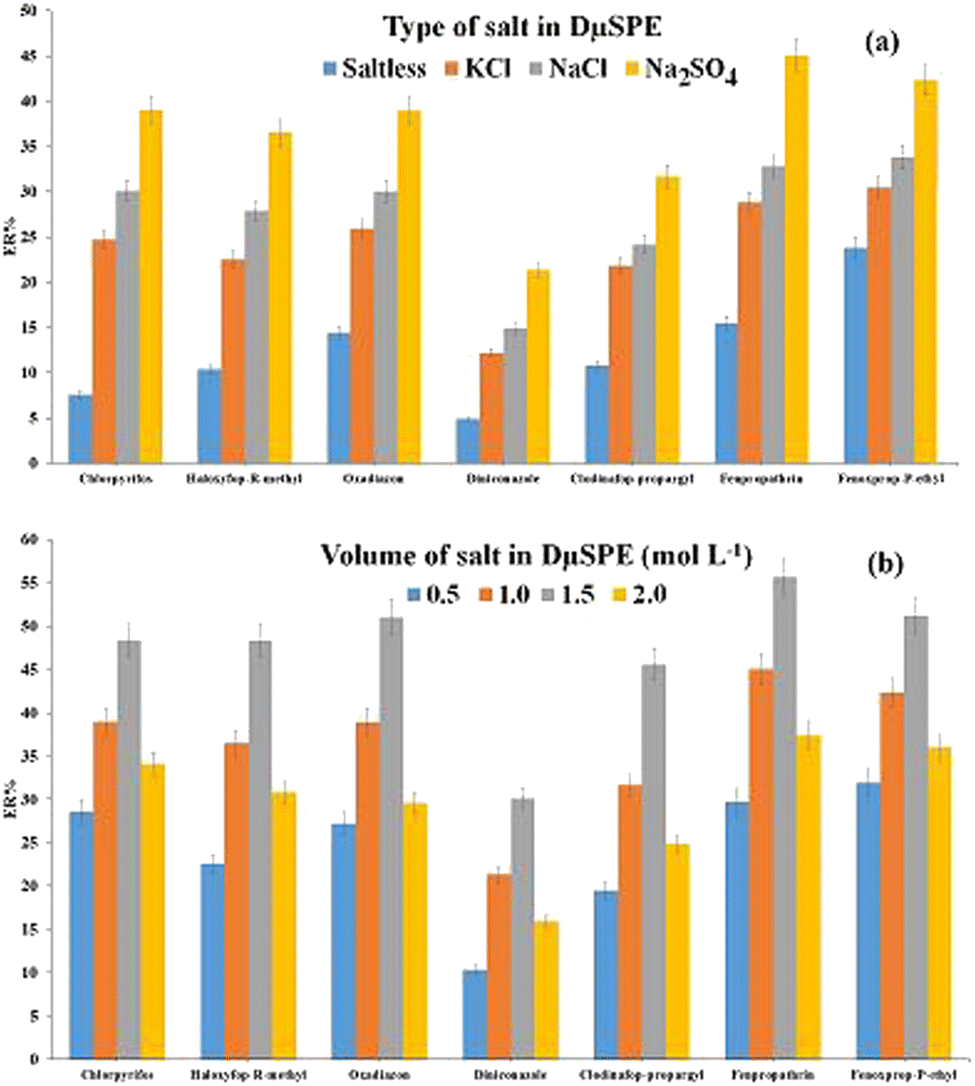 | ||
| Fig. 4 Study of ionic strength in DμSPE. (a) Salt type and (b) concentration of Na2SO4. Extraction conditions: are the same as those used in Fig. 3, except 20 mg of Ni-MOF-I was used. | ||
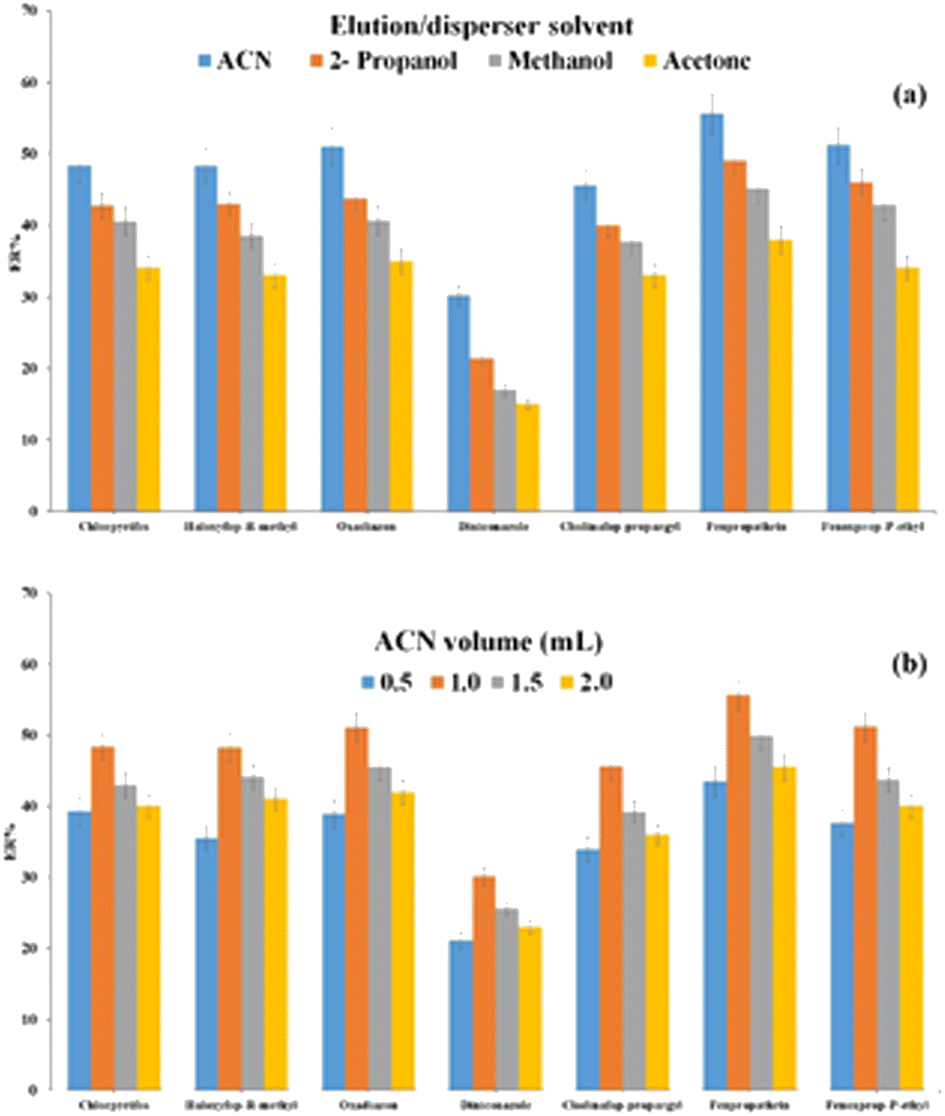 | ||
| Fig. 5 Optimization of type (a) and volume (b) of elution/disperser solvent. Extraction conditions: are the same as those used in Fig. 4, except 1.5 mol L−1 Na2SO4 was used. | ||
3.3. Optimization of parameters in DLLME
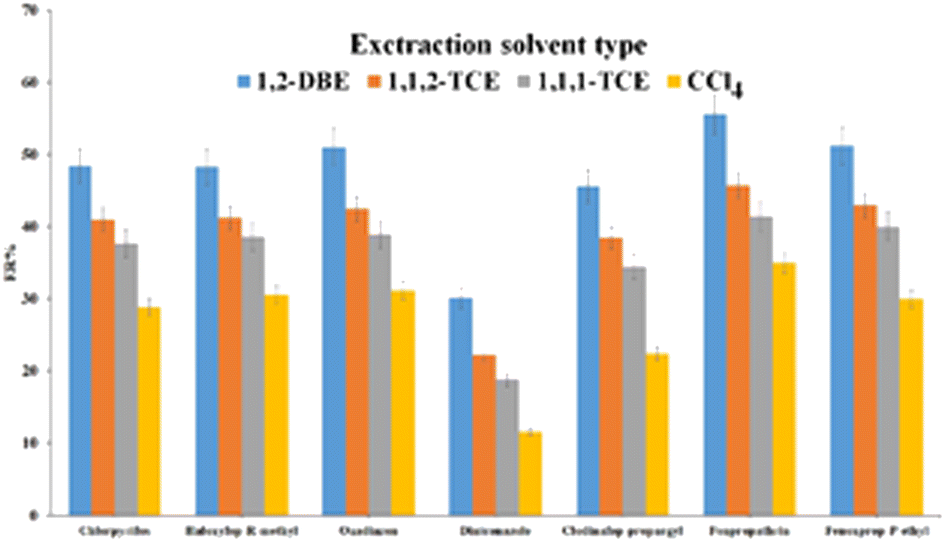 | ||
| Fig. 6 Study of extraction solvent type. Extraction conditions: are the same as those used in Fig. 5, except 1.0 mL ACN was used as the elution solvent. | ||
3.4. The reusability of Ni-MOF-I
One of the effective factors in evaluating the efficiency of an adsorbent is its reusability in the repetitive extraction processes. In order to study this factor, the MOF was subjected to repetitive extraction cycles. Before repeating the extraction, the used adsorbent was eluted twice (each time by 0.5 mL of ACN) and vortexing for 5 min to prevent carry-over. Based on the obtained data, the adsorbent showed proper adsorption ability of the analytes after three cycles without significant variations in the ERs. The results showed that Ni-MOF-I is a sufficient adsorbent for utilization in repetitive extraction cycles.3.5. The interaction mechanisms among the pesticides and Ni-MOF-I
The adsorption of the pesticides on the MOF with appreciable ERs is due to the adsorptive interactions among the pesticides and Ni-MOF-I. By considering the chemical structures of Ni-MOF-I and studied pesticides, it can be concluded that a noncovalent interaction called π–π stacking happens among the MOF's ligand and the cyclic sections of fenoxaprop-P-ethyl, fenpropathrin, clodinafop propargyl, oxadiazon, haloxyfop-R-methyl, and chlorpyrifos. Halogen bond is another significant intermolecular interaction which is based on the noncovalent interaction among a Lewis base, π-system, or an anion and the halogen atom in another compound. In this research, halogen bonds emerge between the oxygen atoms of Ni-MOF-I and chlorine in chlorpyrifos, oxadiazon, and fenoxaprop-P-ethyl, and also fluoride and chlorine in clodinafop propargyl and haloxyfop-R-methyl. Based on the descriptions mentioned above, efficient adsorptive intermolecular bonds are formed among Ni-MOF-I and the studied pesticides.3.6. Method validation
A sign of the success of a new method is its obtained analytical figures of merit such as LOQ, LOD, EF, ER, relative standard deviation (RSD), linear range (LR), and coefficient of determination (r2). The abstract of the above-mentioned quantitative parameters is given in Table 1. The EFs and ERs of this approach were in the ranges of 255–400 and 51–80%, respectively. The RSD values were computed at the concentration of 50 μg L−1 of each pesticide. The RSDs for intra- (n = 6) and inter-day (n = 4) precisions were in the ranges of 4.8–7.2 and 5.3–7.5%, respectively. The LOQ and LOD calculations were based on signal-to-noise ratios of 10 and 3, respectively. The LOQ and LOD values of this method were in the ranges of 0.50–2.0 and 0.15–0.60 μg L−1, respectively. Calibration curves were plotted at different concentrations of pesticides utilizing standard solutions. The r2 values for this method were greater than or equal to 0.995 which indicate the good linearity of the method. The LRs were in the range of 2.0–500 μg L−1. Low LOD, LOQ, and RSD values, wide LRs, and applying little amounts of organic solvents and Ni-MOF-I are the main privileges of this current procedure.| Analyte | LODa | LOQb | LRc | r2 d | RSDe (%) | EF ± SDf | ER ± SDg | |
|---|---|---|---|---|---|---|---|---|
| Intra-day | Inter-day | |||||||
| a Limit of detection (S/N = 3) (μg L−1).b Limit of quantification (S/N = 10) (μg L−1).c Linear range (μg L−1).d Coefficient of determination.e Relative standard deviation at a concentration of 50 μg L−1 of each analyte for intra- (n = 6) and inter-day (n = 4) precisions.f Enrichment factor ± standard deviation (n = 3).g Extraction recovery ± standard deviation (n = 3). | ||||||||
| Chlorpyrifos | 0.60 | 2.0 | 2.0–500 | 0.996 | 5.6 | 6.0 | 375 ± 25 | 75 ± 5 |
| Haloxyfop-R-methyl | 0.30 | 1.0 | 1.0–500 | 0.996 | 5.1 | 5.9 | 380 ± 15 | 76 ± 3 |
| Oxadiazon | 0.30 | 1.0 | 1.0–500 | 0.995 | 4.8 | 5.3 | 390 ± 15 | 78 ± 3 |
| Clodinafop-propargyl | 0.15 | 0.50 | 0.50–500 | 0.998 | 7.2 | 7.5 | 255 ± 20 | 51 ± 4 |
| Clodinafop propargyl | 0.25 | 0.85 | 0.85–500 | 0.997 | 6.2 | 7.2 | 350 ± 10 | 70 ± 2 |
| Fenpropathrin | 0.25 | 0.85 | 0.85–500 | 0.995 | 5.9 | 7.1 | 400 ± 15 | 80 ± 3 |
| Fenoxaprop-P-ethyl | 0.30 | 1.0 | 1.0–500 | 0.997 | 5.0 | 5.8 | 360 ± 20 | 72 ± 4 |
3.7. Analysis of real samples
In this research, the applicability of the method was evaluated by analyzing four commercial fruit juices including mango, pineapple, apple, and peach juices and two fresh orange and apple juices. For this purpose, deionized water and the fruit juices were spiked at 30 and 60 μg L−1 of each pesticides, extracted in the optimum conditions, and eventually the obtained sedimented phase injected into GC-FID. The achieved relative recoveries of the samples compared with deionized water are summarized in Table 2. As it can be deduced from the obtained results, the analyzed juices matrices have no significant effect on the present method. Therefore, this method can be used as an efficient and appropriate method for the extracting and analyzing the studied pesticides in the aforementioned samples. Fig. 7 indicates the GC-FID chromatograms of a standard solution (250 mg L−1 of each pesticide) which was directly injected, the extracted standard aqueous solution (containing 150 μg L−1 of each pesticide), and the extracted unspiked juices under the optimum conditions. According to the chromatograms, it can be seen that the chromatograms of the real samples do not show traces of the presence of any of the surveyed pesticides.:1
| Analytes | Mean relative recovery ± standard deviation (n = 3) | |||||
|---|---|---|---|---|---|---|
| Fresh apple juice | Peach juice | Pineapple juice | Apple juice | Mango juice | Fresh orange juice | |
| All samples were spiked with each analyte at a concentration of 30 μg L−1 | ||||||
| Chlorpyrifos | 95 ± 2 | 97 ± 2 | 94 ± 3 | 97 ± 2 | 92 ± 2 | 96 ± 2 |
| Haloxyfop-R-methyl | 97 ± 2 | 93 ± 3 | 96 ± 3 | 92 ± 2 | 91 ± 2 | 91 ± 2 |
| Oxadiazon | 96 ± 3 | 90 ± 3 | 90 ± 3 | 90 ± 4 | 93 ± 4 | 92 ± 2 |
| Diniconazole | 91 ± 2 | 92 ± 4 | 91 ± 3 | 91 ± 3 | 90 ± 3 | 97 ± 2 |
| Clodinafop-propargyl | 94 ± 3 | 88 ± 4 | 93 ± 2 | 92 ± 2 | 95 ± 2 | 96 ± 2 |
| Fenpropathrin | 93 ± 2 | 98 ± 4 | 94 ± 3 | 93 ± 3 | 92 ± 4 | 93 ± 2 |
| Fenaxaprop-P-ethyl | 90 ± 4 | 94 ± 3 | 92 ± 4 | 95 ± 3 | 89 ± 3 | 90 ± 3 |
|
||||||
| All samples were spiked with each analyte at a concentration of 60 μg L−1 | ||||||
| Chlorpyrifos | 95 ± 3 | 93 ± 2 | 94 ± 2 | 97 ± 3 | 92 ± 3 | 96 ± 2 |
| Haloxyfop-R-methyl | 90 ± 4 | 90 ± 3 | 91 ± 2 | 92 ± 2 | 89 ± 2 | 91 ± 3 |
| Oxadiazon | 91 ± 2 | 96 ± 2 | 92 ± 2 | 87 ± 3 | 93 ± 2 | 89 ± 3 |
| Diniconazole | 93 ± 3 | 92 ± 3 | 96 ± 2 | 95 ± 2 | 85 ± 3 | 98 ± 3 |
| Clodinafop-propargyl | 92 ± 3 | 87 ± 4 | 89 ± 2 | 89 ± 3 | 94 ± 4 | 99 ± 2 |
| Fenpropathrin | 94 ± 4 | 98 ± 3 | 93 ± 2 | 93 ± 3 | 92 ± 4 | 94 ± 2 |
| Fenaxaprop-P-ethyl | 97 ± 3 | 89 ± 2 | 95 ± 2 | 91 ± 2 | 90 ± 3 | 97 ± 3 |
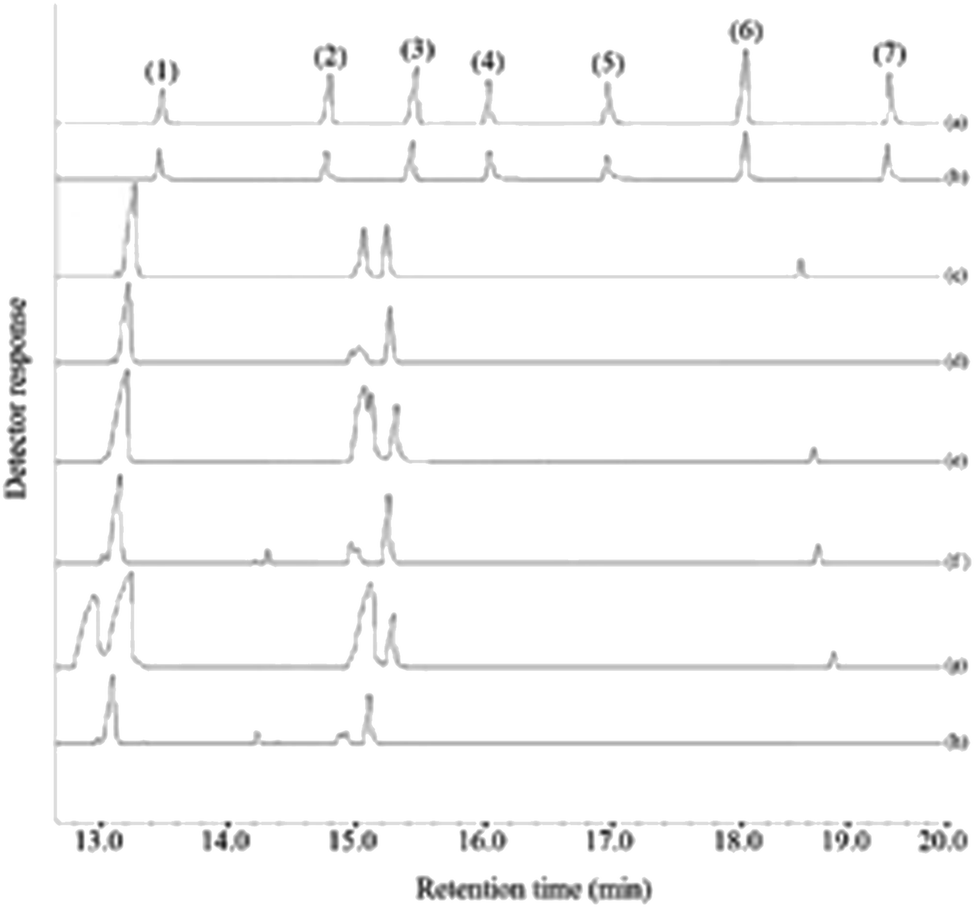 | ||
| Fig. 7 Typical GC-FID chromatograms of: (a) standard solution (250 mg L−1 of each pesticide in methanol), (b) aqueous standard solution at a concentration of 150 μg L−1 of each pesticide, (c) pineapple juice, (d) fresh orange juice, (e) peach juice, (f) fresh apple juice, (g) mango juice, and (h) commercial apple juice. In all cases, except chromatogram (a) the proposed method was performed and 1 μL of the sedimented phase was injected into the separation system. Peaks identification: (1) chlorpyrifos, (2) haloxyfop-R-methyl, (3) oxadiazon, (4) clodinafop propargyl, (5) fenpropathrin, and (6) fenoxaprop-P-ethyl. | ||
3.8. Method comparison
Table 3 contains comparison of the analytical characteristics (EF, ER, LOD, RSD, and LR) of the proposed method in this research and other previously reported methods for analyzing the mentioned pesticides. According to this table, the present method has comparable LRs with most of the other methods. Also, the RSDs are comparable with or lower than those attained for the mentioned methods. Moreover, the EFs and ERs of the studied approach are comparable with those of the other mentioned techniques. In addition, the proposed method LOQs and LODs are lower than or comparable with the reported methods data, except for one method in which mass spectrometry has been used as the detector instead of FID which is intrinsically more sensitive than FID. Eventually, it can be said that this method is an effective and efficient analytical approach due to high EFs, reasonable ERs, low LODs and LOQs, applying little amount of Ni-MOF-I as the adsorbent, and green synthetic procedure of the MOF.| Method | Sample | LODa | LOQb | LRc | r2 d | RSDe (%) | EFf | ERg (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a Limit of detection (μg L−1).b Limit of quantification (μg L−1).c Linear range (μg L−1).d Coefficient of determination.e Relative standard deviation.f Enrichment factor.g Extraction recovery.h Dispersive solid phase extraction-dispersive liquid–liquid microextraction-gas chromatography-flame ionization detection.i Hollow fiber-liquid phase microextraction-gas chromatography-mass spectrometry.j Stir bar sorptive extraction-solidification of floating organic droplet-gas chromatography-mass spectrometry.k Headspace-solid phase microextraction-gas chromatography-mass spectrometry.l Dispersive micro solid phase extraction-dispersive liquid–liquid microextraction-gas chromatography-flame ionization detection. | |||||||||
| DSPE-DLLME-GC-FIDh | Fruit juices | 0.32–0.76 | 1.10–2.60 | 1.1–2000 | 0.994–0.999 | 4.6–8.4 | 680–840 | 68–84 | 40 |
| HF-LPME-GC-MSi | Water | 0.3–0.8 | 1.0 | 1–5000 | 0.997–0.999 | 6–9 | 134–240 | — | 41 |
| SBSE-SFOD-GC-MSj | Tomato juice | 0.007–0.014 | 0.023–0.047 | 0.037–2000 | 0.998 | 6–9 | — | 76–90 | 42 |
| HS-SPME-GC-MSk | Wine | 0.1 | 0.4 | 0.5–150 | 0.995 | 13.5 | — | — | 43 |
| DμSPE-DLLME-GC-FIDl | Fruit beverages | 0.15–0.60 | 0.50–2.0 | 2.0–500 | 0.995–0.997 | 4.8–7.2 | 255–400 | 51–80 | Present method |
4. Conclusions
For the first time in this research, Ni-MOF-1 was synthesized through a new, facile, and safe process which was done in deionized water. The approach was green, economic, and time-saving. The MOF was also applied as an efficient adsorbent in DμSPE-DLLME-GC-FID method for the extraction, preconcentration, and determination of some pesticides in fruit juice samples. Furthermore, characterization of the synthesized MOF was done by applying nitrogen adsorption/desorption, SEM, EDX, FTIR, and XRD techniques. The proposed analytical method showed satisfactory analytical figures of merit such as low LODs (0.15–0.60 μg L−1), and LOQs (0.50–2.0 μg L−1), high EFs (255–400), acceptable ERs (51–80%), good repeatability (RSD ≤ 7.2%), and wide LRs (2.0–500 μg L−1). Green synthesis of the sorbent and applying low weight of MOF during the analytical approach, were the other important advantages of the proposed procedure. Also, no critical matrix effect was detected in the real samples analysis. Based on the obtained results, it can be concluded that this method can be utilized in the extraction, preconcentration, and analyzing low concentrations of the studied pesticides in different fruit juices with high reliability.Author contributions
Mir Ali Farajzadeh: analytical methodology, synthesis methodology, and editing the manuscript. Nastaran Khoshnavaz: analytical methodology, synthesis methodology, MOF synthesis, analytical analysis, and writing the manuscript. Sakha Pezhhanfar: analytical methodology, synthesis methodology, MOF synthesis, and editing the manuscript. Mohammad Reza Afshar Mogaddam: analytical methodology.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
The authors are thankful to the University of Tabriz for financial support.References
- L. G. Hassan, S. M. Dangoggo, S. W. Hassan, S. Muhammad and K. J. Umar, Nutritional and antinutritional composition of Sclerocarya birrea fruit juice, Nig. J. Basic Appl. Sci., 2010, 18, 222–228 Search PubMed.
- S. Pezhhanfar, M. A. Farajzadeh, S. A. Hosseini-Yazdi and M. R. Afshar Mogaddam, Development of a sample preparation procedure using an iron-based metal-organic framework for the extraction of pesticides from fruit juices by dispersive micro solid-phase extraction followed by their preconcentration by dispersive liquid-liquid microextraction, Sep. Sci. plus, 2022, 5, 184–192 CrossRef CAS.
- S. Pezhhanfar, M. A. Farajzadeh, S. A. Hosseini-Yazdi and M. R. Afshar Mogaddam, Synthesis of MOF-70 based on diffusion method; microgram amount application as a highly efficient sorbent in dispersive micro solid phase extraction prior to dispersive liquid–liquid microextraction for the preconcentration and extraction of pesticides from fruit juices, J. Iran. Chem. Soc., 2022, 19, 2407–2419 CrossRef CAS.
- S. Williamson, A. Ball and J. Pretty, Trends in pesticide use and drivers for safer pest management in four African countries, Crop Prot., 2008, 27, 1327–1334 CrossRef.
- J. Fenik, M. Tankiewicz and M. Biziuk, Properties and determination of pesticides in fruits and vegetables, Trends Anal. Chem., 2011, 30, 814–826 CrossRef CAS.
- M. A. Farajzadeh, R. Safi and A. Yadeghari, Combination of QuEChERS extraction with magnetic solid phase extraction followed by dispersive liquid–liquid microextraction as an efficient procedure for the extraction of pesticides from vegetable, fruit, and nectar samples having high content of solids, Microchem. J., 2019, 147, 571–581 CrossRef CAS.
- U. Bajwa and K. S. Sandhu, Effect of handling and processing on pesticide residues in food-a review, J. Food Sci. Technol., 2014, 51, 201–220 CrossRef CAS PubMed.
- M. A. Farajzadeh, M. Sattari Dabbagh and A. Yadeghari, Deep eutectic solvent based gas-assisted dispersive liquid-phase microextraction combined with gas chromatography and flame ionization detection for the determination of some pesticide residues in fruit and vegetable samples, J. Sep. Sci., 2017, 40, 2253–2260 CrossRef CAS.
- M. A. Farajzadeh, M. R. Mogaddam, S. R. Aghdam, N. Nouri and M. Bamorrowat, Application of elevated temperature-dispersive liquid-liquid microextraction for determination of organophosphorus pesticides residues in aqueous samples followed by gas chromatography-flame ionization detection, Food Chem., 2016, 212, 198–204 CrossRef CAS PubMed.
- L. Velkoska-Markovska, B. Petanovska-Ilievska and A. Markovski, Application of high performance liquid chromatography to the analysis of pesticide residues in apple juice, Contemporary Agriculture, 2018, 67, 93–102 CrossRef.
- Y. Song, R. Ma, L. Hao, X. Yang, C. Wang, Q. Wu and Z. Wang, Application of covalent organic framework as the adsorbent for solid-phase extraction of trace levels of pesticide residues prior to high-performance liquid chromatography-ultraviolet detection, J. Chromatogr. A, 2018, 1572, 20–26 CrossRef CAS PubMed.
- A. Samsidar, S. Siddiquee and S. M. Shaarani, A review of extraction, analytical and advanced methods for determination of pesticides in environment and foodstuffs, Trends Food Sci. Technol., 2018, 71, 188–201 CrossRef CAS.
- M. Nasiri, H. Ahmadzadeh and A. Amiri, Sample preparation and extraction methods for pesticides in aquatic environments: A review, Trends Anal. Chem., 2020, 123, 115772 CrossRef CAS.
- M. A. Farajzadeh, A. Yadeghari and L. Khoshmaram, Magnetic solid phase extraction using Fe3O4@SiO2@C8 nanoparticles performed in a narrow-bore tube followed by dispersive liquid–liquid microextraction for extraction and preconcentration of nine pesticides, New J. Chem., 2018, 42, 6215–6224 RSC.
- M. A. Habila, Z. A. ALOthman, E. R. Yilmaz, E. A. Alabdullkarem and M. U. Soylak, A new amine based microextraction of lead (II) in real water samples using flame atomic absorption spectrometry, Microchem. J., 2019, 148, 214–219 CrossRef CAS.
- B. Feriduni, A. Mohebbi, M. A. Farajzadeh and M. Namvar, Magnetic graphene oxide–based solid-phase extraction combined with dispersive liquid–liquid microextraction for the simultaneous preconcentration of four typical pesticide residues in fruit juice and pulp, Food Anal. Methods, 2019, 12, 2742–2752 CrossRef.
- M. A. Farajzadeh, A. Mohebbi, M. R. Mogaddam, M. Davaran and M. Norouzi, Development of salt-induced homogenous liquid–liquid microextraction based on iso-propanol/sodium sulfate system for extraction of some pesticides in fruit juices, Food Anal. Methods, 2018, 11, 2497–2507 CrossRef.
- K. Gorynski, A critical review of solid-phase microextraction applied in drugs of abuse determinations and potential applications for targeted doping testing, Trends Anal. Chem., 2019, 112, 135–146 CrossRef CAS.
- A. Mohebbi, S. Yaripour, M. A. Farajzadeh and M. R. Mogaddam, Combination of dispersive solid phase extraction and deep eutectic solvent–based air–assisted liquid–liquid microextraction followed by gas chromatography–mass spectrometry as an efficient analytical method for the quantification of some tricyclic antidepressant drugs in biological fluids, J. Chromatogr. A, 2018, 1571, 84–93 CrossRef CAS.
- M. Anastassiades, S. J. Lehotay, D. Stajnbaher and F. J. Schenck, Fast and easy multiresidue method employing acetonitrile extraction/partitioning and “dispersive solid-phase extraction” for the determination of pesticide residues in produce, J. AOAC Int., 2003, 86, 412–431 CrossRef CAS.
- F. J. Ruiz-Espinar, L. Ripoll-Seguer, M. Hidalgo and A. Canals, Dispersive micro solid-phase extraction (DμSPE) with graphene oxide as adsorbent for sensitive elemental analysis of aqueous samples by laser induced breakdown spectroscopy (LIBS), Talanta, 2019, 191, 162–170 CrossRef.
- T. Khezeli and A. Daneshfar, Development of dispersive micro-solid phase extraction based on micro and nano sorbents, Trends Anal. Chem., 2017, 89, 99–118 CrossRef CAS.
- Z. Moradi, E. A. Dil and A. Asfaram, Dispersive micro-solid phase extraction based on Fe3O4@ SiO2@ TI-MOF as a magnetic nanocomposite sorbent for the trace analysis of caffeic acid in the medical extracts of plants and water samples prior to HPLC-UV analysis, Analyst, 2019, 144, 4351–4361 RSC.
- M. A. Farajzadeh, A. Pirmohamadlou and M. Sattari Dabbagh, Extraction and preconcentration of some pesticides in vegetable and fruit juice samples using SA@CaCO3 sorbent combined with dispersive liquid-liquid microextraction, Food Anal. Methods, 2021, 14, 2395–2407 CrossRef.
- M. A. Farajzadeh, A. Pirmohamadlou and M. Sattari Dabbagh, Combining dispersive solid phase extraction using an inexpensive sorbent with dispersive liquid–liquid microextraction for the efficient extraction of some plasticisers from plastic-packed liquids, J. Environ. Anal. Chem., 2021, 1–7 Search PubMed.
- I. Rykowska, J. Ziemblińska and I. Nowak, Modern approaches in dispersive liquid-liquid microextraction (DLLME) based on ionic liquids: a review, J. Mol. Liq., 2018, 259, 319–339 CrossRef CAS.
- M. A. Farajzadeh, H. Sohrabi and A. Mohebbi, Combination of modified QuEChERS extraction method and dispersive liquid–liquid microextraction as an efficient sample preparation approach for extraction and preconcentration of pesticides from fruit and vegetable samples, Food Anal. Methods, 2019, 12, 534–543 CrossRef.
- W. Ma, X. Li, Y. Bai and H. Liu, Applications of metal-organic frameworks as advanced sorbents in biomacromolecules sample preparation, Trends Anal. Chem., 2018, 109, 154–162 CrossRef CAS.
- P. Rocío-Bautista, I. Taima-Mancera, J. Pasán and V. Pino, Metal-organic frameworks in green analytical chemistry, Separations, 2019, 6, 1–21 CrossRef.
- G. Liu, L. Li, X. Huang, S. Zheng, D. Xu, X. Xu, Y. Zhang and H. Lin, Determination of triazole pesticides in aqueous solution based on magnetic graphene oxide functionalized MOF-199 as solid phase extraction sorbents, Mesoporous Mater., 2018, 270, 258–264 CrossRef CAS.
- M. Faraji, Y. Yamini and M. Gholami, Recent advances and trends in applications of solid-phase extraction techniques in food and environmental analysis, Chromatographia, 2019, 82, 1207–1249 CrossRef CAS.
- S. Barrek, O. Paisse and M. F. Grenier-Loustalot, Determination of residual pesticides in olive oil by GC-MS and HPLC-MS after extraction by size-exclusion chromatography, Anal. Bioanal. Chem., 2003, 376, 355–359 CrossRef CAS PubMed.
- J. Ma, Z. Yao, L. Hou, W. Lu, Q. Yang, J. Li and L. Chen, Metal organic frameworks (MOFs) for magnetic solid-phase extraction of pyrazole/pyrrole pesticides in environmental water samples followed by HPLC-DAD determination, Talanta, 2016, 161, 686–692 CrossRef CAS PubMed.
- M. Chahkandi, A. Amiri and S. R. Arami, Extraction and preconcentration of organophosphorus pesticides from water samples and fruit juices utilizing hydroxyapatite/Fe3O4 nanocomposite, Microchem. J., 2019, 144, 261–269 CrossRef CAS.
- M. Michel and B. Buszewski, HPLC determination of pesticide residue isolated from food matrices, J. Liq. Chromatogr. Relat. Technol., 2002, 25, 2293–2306 CrossRef CAS.
- M. Shamsipur, N. Yazdanfar and M. Ghambarian, Combination of solid-phase extraction with dispersive liquid–liquid microextraction followed by GC-MS for determination of pesticide residues from water, milk, honey and fruit juice, Food Chem., 2016, 204, 289–297 CrossRef CAS.
- W. Wang, H. Zhu, S. Cui, J. Miao and J. Chen, Ultrasound-assisted dispersive liquid–liquid microextraction based on solidification of floating organic droplets coupled with gas chromatography for the determination of pesticide residues in water samples, Anal. Methods, 2014, 6, 3388–3394 RSC.
- S. Gao, Y. Sui, F. Wei, J. Qi, Q. Meng and Y. He, Facile synthesis of cuboid Ni-MOF for high-performance supercapacitors, J. Mater. Sci., 2018, 53, 6807–6818 CrossRef CAS.
- P. Thangasamy, S. Shanmuganathan and V. Subramanian, A NiCo-MOF nanosheet array based electrocatalyst for the oxygen evolution reaction, Nanoscale Adv., 2020, 2, 2073–2079 RSC.
- A. Mohebbi, M. A. Farajzadeh, A. Mahmoudzadeh and A. Etemady, Combination of poly (ε–caprolactone) grafted graphene quantum dots–based dispersive solid phase extraction followed by dispersive liquid–liquid microextraction for extraction of some pesticides from fruit juices prior to their quantification by gas chromatography, Microchem. J., 2020, 153, 104328 CrossRef CAS.
- A. Sarafraz-Yazdi, H. Assadi and W. A. Wan Ibrahim, Determination of triazole fungicides using hollow fiber liquid phase microextraction prior to gas chromatography–mass spectrometry analysis, Ind. Eng. Chem. Res., 2012, 51, 3101–3107 CrossRef CAS.
- M. Nemati, M. A. Farajzadeh, A. Mohebbi, F. Khodadadeian and M. R. Afshar Mogaddam, Development of a stir bar sorptive extraction method coupled to solidification of floating droplets dispersive liquid-liquid microextraction based on deep eutectic solvents for the extraction of acidic pesticides from tomato samples, J. Sep. Sci., 2020, 43, 1119–1127 CrossRef CAS.
- A. Navalon, A. Prieto, L. Araujo and J. L. Vilchez, Determination of oxadiazon residues by headspace solid-phase microextraction and gas chromatography–mass spectrometry, J. Chromatogr. A, 2002, 946, 239–245 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra03441k |
| This journal is © The Royal Society of Chemistry 2023 |