
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-free visible-induced C(sp2)–C(sp2) coupling of quinoxalin-2(H)-ones via oxidative cleavage of the C–N bond†
Mei Wu‡
a,
Nancheng Lian‡b,
Cuimin Wua,
Xinyao Wua,
Houzheng Chena,
Chen Lina,
Sunying Zhou*a and
Fang Ke *a
*a
aInstitute of Materia Medica, School of Pharmacy, Fujian Provincial Key Laboratory of Natural Medicine Pharmacology, Fujian Medical University, Fuzhou 350122, China. E-mail: kefang@mail.fjmu.edu.cn
bDepartment of Spinal Surgery, The First Affiliated Hospital of Fujian Medical University, Fuzhou 350005, China
First published on 16th June 2023
Abstract
A C(sp2)–C(sp2) reaction between aromatic hydrazines and quinoxalines has been developed through a photocatalytic system. The protocol is established for C(sp2)–N bond cleavage and direct C(sp2)–H functionalization for the coupling of C(sp2)–C(sp2) via photocatalysis under mild and ideal air conditions without the presence of a strong base and metal. The mechanistic studies reveal that the generation of a benzene radical via the oxidative cleavage of aromatic hydrazines for the cross-coupling of C(sp2)–C(sp2) with the assistance of a photocatalyst is essential. The process exhibits excellent compatibility with functional groups and provides convenient access to various 3-arylquinoxalin-2(1H)-ones in good to excellent yields.
The C(sp2)–C(sp2) bond1a,b plays an important role in the synthesis of conjugated molecules such as biaryls which are widely used in pharmaceutical chemistry and chemical industry.1c–f Owing to its importance and usefulness, a number of methods for the construction of the C(sp2)–C(sp2) bonds have been developed. The traditional method for the formation of C(sp2)–C(sp2) bonds relies on a phenyl diazonium salt (Scheme 1a).2 However, it is not a stable structure except during pre-preparation due to its explosive disadvantages. During the past decades, coupling reactions catalyzed by various transition metals with C–X(H) (X = halogen) bonds, such as Pd,3 Ru,4 Co,5 Au,6 have had a substantial role in the construction of C(sp2)–C(sp2) bonds. Some of these reaction systems still have certain limitations, such as the need for a large amount of excess base and expensive ligands. In addition, C–X(H) activation is effective but lacks applicability to a wide range of aryl substrates due to the challenging selective synthesis of complex aryl halide starting materials (Scheme 1b).7 To address the issue, the other route for C(sp2)–C(sp2) bond formation is C(sp2)–B bond cleavage, such as the use of boronic acid which is the most useful coupling partner (Scheme 1c).8 Nevertheless, the fracture of the C–B bond in phenylboronic acid requires prefunctionalization under the majority of complex reaction conditions. Therefore, it is still a difficult challenge to develop a mild and simple catalytic method for C(sp2)–C(sp2) formation.
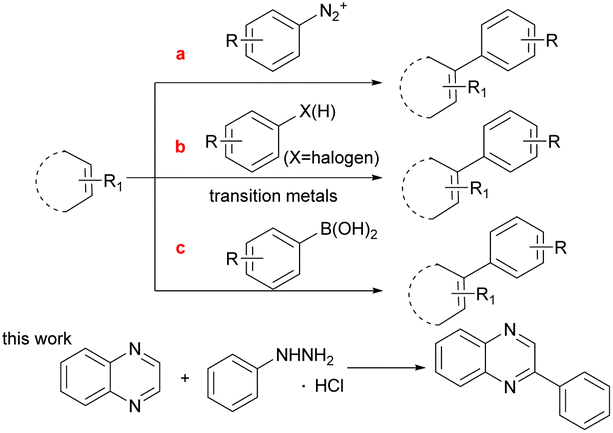 | ||
| Scheme 1 General methods for C(sp2)–C(sp2) bond formation. | ||
Photoinduced C–N bond oxidative cleavage has received more attention because the new methodology for the direct transformation and rapid derivatization of functional molecules containing C–N bonds is a valuable tool in modern organic synthesis. The pioneering work of Ramesh group,9a Hajra group9b et al. in the past few years showed that the method of oxidative cleavage of C(sp2)–N bond can directly construct carbon radical from the free radical path, with high economy and good functional group compatibility. Even more, direct C(sp2)–H functionalization is still an essential route because the C(sp2)–H bond avoids the pre-functionalization of (hetero)arenes as a functional group.10 However, C(sp2)–H bond functionalization remains a huge challenge, which usually use extra noble metal catalysts or excess amounts of strong oxidants.11 The visible-light induced direct C(sp2)–H functionalization has emerged recently in green synthetic chemistry and offered an opportunity to overcome the above shortcomings.12 More recently, Wang and co-workers have reported a HCl-mediated, photocatalytic method for quinoxaline(on)es C3–H arylation with arylhydrazine.12a Different from the method of using acid catalysis, our work employed photocatalyst eosin Y to break the C–N bond and generate benzene free radicals. Hence, continuing our research efforts on developing efficient and straightforward methods,13 we report a photoinduced direct C(sp2)–H functionalization and the C(sp2)–N bond cleavage for the formation of C(sp2)–C(sp2) under air atmosphere without strong base and metal at room temperature.
We began our investigation using quinoxaline (1a) and arylhydrazine hydrochloride (2a) for the screening of the reaction conditions in the presence of eosin Y (10 mol%) in MeOH/H2O (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 12 W blue LED. Several selected photocatalysts such as eosin B, rose bengal, and rhodamine B were investigated in the reactions, but the yields of 3a decreased (Table 1, entries 2–4). We then explored the effects of solvents. Various solvents including MeCN/H2O (v/v = 1/1), and DMSO/H2O (v/v = 1/1) were examined (Table 1, entries 5–6), and none of them was better than MeOH/H2O (v/v = 1/1). Subsequently, performing the reaction under the irradiation of white LED resulted in a 39% yield of 3a (entry 7). Using purple LED as a light source, a 74% yield of 3a was detected (entry 8). When the transformation was conducted under the dark condition or N2 atmosphere (Table 1, entries 9–10), no reaction occurred. Reducing the amount of phenylhydrazine 2a by half resulted in a yield of only 53%, probably because phenylhydrazine was volatile during the reaction.
:1) at 12 W blue LED. Several selected photocatalysts such as eosin B, rose bengal, and rhodamine B were investigated in the reactions, but the yields of 3a decreased (Table 1, entries 2–4). We then explored the effects of solvents. Various solvents including MeCN/H2O (v/v = 1/1), and DMSO/H2O (v/v = 1/1) were examined (Table 1, entries 5–6), and none of them was better than MeOH/H2O (v/v = 1/1). Subsequently, performing the reaction under the irradiation of white LED resulted in a 39% yield of 3a (entry 7). Using purple LED as a light source, a 74% yield of 3a was detected (entry 8). When the transformation was conducted under the dark condition or N2 atmosphere (Table 1, entries 9–10), no reaction occurred. Reducing the amount of phenylhydrazine 2a by half resulted in a yield of only 53%, probably because phenylhydrazine was volatile during the reaction.
|
||
|---|---|---|
| Entry | Variations from the standard conditions | Yieldb (%) |
| a Standard conditions: 1a (0.5 mmol), 2a (1.0 mmol), eosin Y (10.0 mol%), K2CO3 (1.0 mmol), blue LED, 12 W, MeOH/H2O (1/1) = 4 mL, 15 h, under air atmosphere, rt.b Isolated yield.c 1a (10.0 mmol), 2a (20.0 mmol), eosin Y (10.0 mol%), K2CO3 (20.0 mmol), blue LED, 12 W, MeOH/H2O (1/1) = 45 mL, 36 h. | ||
| 1 | None | 87 |
| 2 | Eosin B instead of eosin Y | 71 |
| 3 | Rose bengal instead of eosin Y | 56 |
| 4 | Rhodamine B instead of eosin Y | 43 |
| 5 | MeCN/H2O (1/1) instead of MeOH/H2O (v/v = 1/1) | 64 |
| 6 | DMSO/H2O (1/1) instead of MeOH/H2O (v/v = 1/1) | 13 |
| 7 | White LED instead of blue LED | 39 |
| 8 | Purple LED instead of blue LED | 74 |
| 9 | Under dark condition | Trace |
| 10 | Under N2 atmosphere | Trace |
| 11 | 0.5 mmol 2a | 53 |
| 12 | 5 mol% eosin Y | 64 |
| 13 | 2.0 mmol K2CO3 | 85 |
| 14 | 10 h instead of 15 h | 47 |
| 15 | 18 h instead of 15 h | 87 |
| 16c | Gram-scale experiment | 69 |
When the eosin Y equivalent was reduced to 5 mol%, the yield of 3a was decreased to 64% (Table 1, entry 12). It should be noted that a similar yield was still obtained for 3a when the amount of base was doubled (Table 1, entry 13). In addition, the product was formed only in 47% yield (Table 1, entry 14) when the time of reaction was decreased to 10 h. Moreover, the increase in time to 18 h did not have much effect on the yield (Table 1, entry 15). In order to further demonstrate the practicability of the catalytic system under the similar standard conditions, a gram-scale reaction was implemented (Table 1, entry 16), and the target product 3a was obtained in 69% yield.
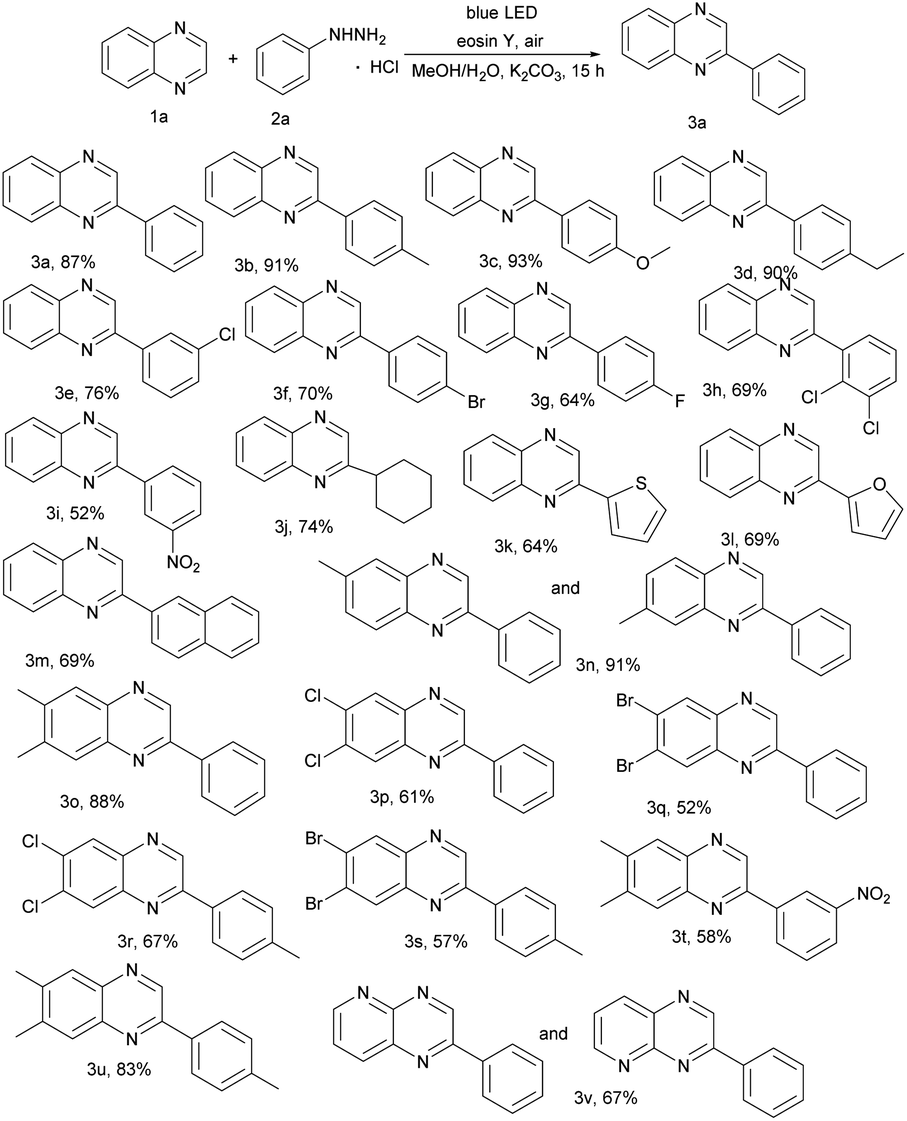
With the optimized reaction conditions in hand, we investigated the scope of the reported reaction by screening a wide range of quinoxalines and arylhydrazine hydrochlorides (Table 2). Generally, electron-donating arylhydrazines gave better product yields than electron withdrawing arylhydrazines. Specifically, various functional groups (e.g., –Me, –OCH3, and –Et) on arenes were well tolerated for the C(sp2)–H arylation and produced corresponding 3b, 3c, and 3d. In addition, halogen atom (–F, –Cl and –Br) substituted arylhydrazine hydrochlorides can also work well and the corresponding products 3e–3h were obtained in moderate yields (64–76%). Moreover, strong electron withdrawing groups such as nitrophenylhydrazine were employed, but gave the relatively low yield. In addition, besides the phenylhydrazines, cyclohexyl group reacted with 1a and provided the C(sp2)–C(sp3) oxidative coupling product 3j in 74% yield (Table 2, 3j). Surprisingly, heterocycles gave the related products 3k–3l in good yields. To our satisfaction, highly catalytic activities were found in transformations of naphthalen-2-ylhydrazine to the corresponding products (Table 2, 3m). We further turned our attention to explore the scope of representative quinoxalines as summarized in Table 2. The catalytic system exhibited a broad substrate scope and high functional group tolerance. Substrates with methyl, halogen groups proved to be compatible with the reaction, furnishing the expected products 3n–3u in 52–91% yields. Moreover, heterocycles afforded the product 3v in 67% yield.
To explore the reaction mechanism, a series of controlled experiments were performed (Scheme 2). Controlled experiments revealed that the blue LED component was essential to the reaction (Scheme 2a). Nevertheless, the yield of 3a was reduced to trace when the photocatalyst was removed (Scheme 2b). Also, we replaced phenylhydrazine hydrochloride with phenylhydrazine hydrochloride and found that the yield of the target product 3a was not reduced, indicating that the effect of base may be used to react with the hydrochloric acid in 2a, thus releasing phenylhydrazine molecules, but it may not be involved in subsequent reactions (Scheme 2c). Additionally, the standard model reaction was conducted in the presence of TEMPO and BHT, respectively. An evident inhibition of these free radical scavengers to the reaction was observed (Scheme 2d, 2e).
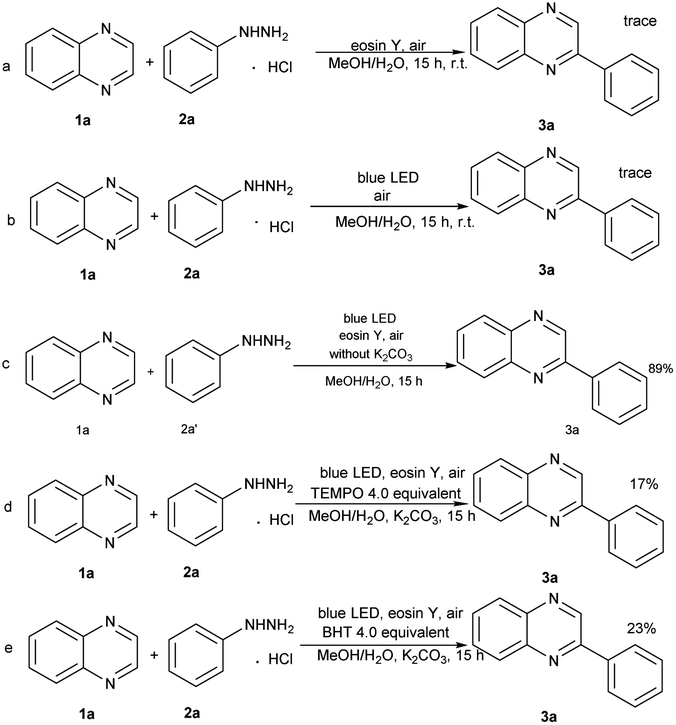 | ||
| Scheme 2 Control experiments for mechanism. | ||
Subsequently, we measured the CV experiment using a solution of the same concentration as the real reaction mixture. The experimental results showed that the curve of eosin Y and phenylhydrazine 2a exhibited oxidative wave from 1.612 V, indicating that 2a may have reacted with eosin Y directly (Fig. 1).
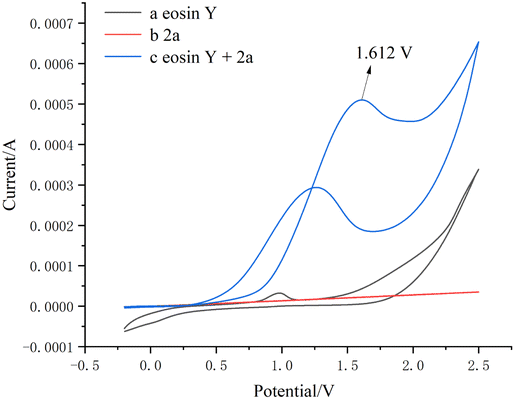 | ||
| Fig. 1 Cyclic voltammetry (CV) studies. | ||
To verify the probable pathway involving 2a′ and the feasibility of this process, DFT calculations were also performed, the results of which are shown in Fig. 3. According to these calculations, the excited eosin Y easily attacked the nitrogen atom of 2a′ to generate A via a single electron transfer (SET) along with the release of 8 kcal mol−1 energy. Similarly, the excited eosin Y promoted the change from Intermediate D to E, and this step needed to surmount the 18.98 kcal mol−1 energy barrier. Afterwards, the resulting radical E spontaneously reacted with an O2˙− to form radical F. The obtained radical F further released N2 into benzene free radicals G. Subsequently, the addition of benzene free-radical G to 1a generated a nitrogen-centered radical H, which undergo a 1,2-hydrogen shift to give a carbon-centered radical I as shown in Fig. 2.14 The intermediate I was further oxidized by eosin Y˙+ radical to give a cationic intermediate J. Finally, the intermediate J followed by deprotonation under the action of a peroxide anion to give the target product 3a.14d,e
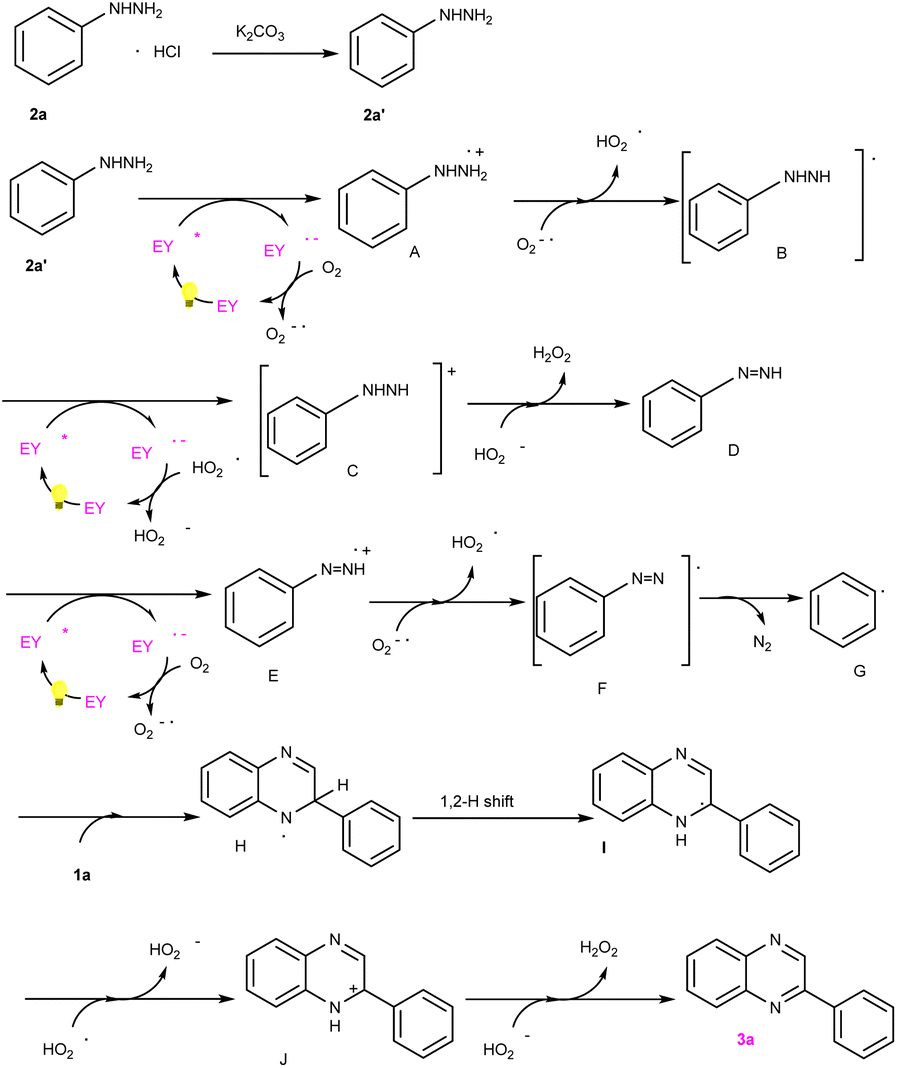 | ||
| Fig. 2 Proposed mechanism for this transformation. | ||
 | ||
| Fig. 3 DFT study of the coupling of C(sp2)–C(sp2). | ||
Conclusions
The C(sp2)–C(sp2) reaction between aromatic hydrazines and quinoxalines has been developed through a photocatalyst. The protocol has been established for the C(sp2)–N bond cleavage and direct C(sp2)–H functionalization for cross-coupling of C(sp2)–C(sp2) under air atmosphere without strong base and metal. Moreover, the protocol has high functional group tolerance and a wide range of substrate scope. The key to the strategy is the generation of benzene radical via oxidative cleavage of aromatic hydrazines for cross-coupling of C(sp2)–C(sp2) with the assistance of photocatalyst.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by the Natural Science Foundation of Fujian Province (No. 2021J01692, 2020J01620, 2020J01627, 2021Y9006); the Research Fund of Fujian Key Laboratory of Oral Diseases (No. 2021kq02).Notes and references
- (a) S. Çakır, S. B. Kavukcu, H. Karabıyık, S. Rethinamcd and H. Türkmen, RSC Adv., 2021, 11, 37684–37699 RSC; (b) Z. Song, X. Huang, S. Jiang, C. He, L. Tang, Q. Ni, M. Ma, B. Chen and Y. Ma, Org. Lett., 2022, 24, 5573–5578 CrossRef CAS PubMed; (c) K. Wu, L.-L. Wu, C.-Y. Zhou and C.-M. Che, Angew. Chem., Int. Ed., 2020, 59, 16202–16208 CrossRef CAS PubMed; (d) S. M. A. Shakoor, S. K. Mandal and R. Sakhuja, Eur. J. Org. Chem., 2017, 2017, 2596–2602 CrossRef; (e) L. Wen, L. Tang, Y. Yang, Z. Zha and Z. Wang, Org. Lett., 2016, 18, 1278–1281 CrossRef CAS PubMed; (f) A. Sharmaab and P. Gogoi, Org. Biomol. Chem., 2019, 17, 9014–9025 RSC.
- J. M. Bhojane, S. A. Sarode and J. M. Nagarkar, New J. Chem., 2016, 40, 1564–1570 RSC.
- (a) C.-H. Park, V. Ryabova, I. V. Seregin, A. W. Sromek and V. Gevorgyan, Org. Lett., 2004, 6, 1159–1162 CrossRef CAS PubMed; (b) J.-P. Leclerc and K. Fagnou, Angew. Chem., Int. Ed., 2006, 45, 7781–7786 CrossRef CAS PubMed; (c) M. P. Huestis and K. Fagnou, Org. Lett., 2009, 11, 1357–1360 CrossRef CAS PubMed.
- A. M. Berman, J. C. Lewis, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2008, 130, 14926–14927 CrossRef CAS.
- (a) D. Zell, M. Bursch, V. Müller, S. Grimme and L. Ackermann, Angew. Chem., 2017, 129, 10514–10518 CrossRef; (b) Y. L. Kim, S. A. Park and J. H. Kim, Eur. J. Org. Chem., 2020, 2020, 4026–4030 CrossRef CAS.
- M. Li and R. Hua, Tetrahedron Lett., 2009, 50, 1478–1481 CrossRef CAS.
- (a) H. Cao, H. Jiang, H. Feng, J. M. C. Kwan, X. Liu and J. Wu, J. Am. Chem. Soc., 2018, 140, 16360–16367 CrossRef CAS PubMed; (b) J. M. Hammann, D. Haas and P. Knochel, Angew. Chem., Int. Ed., 2015, 54, 4478–4481 CrossRef CAS PubMed.
- (a) P. P. Singh, S. K. Aithagani, M. Yadav, V. P. Singh and R. A. Vishwakarma, J. Org. Chem., 2013, 78, 2639–2648 CrossRef CAS PubMed; (b) J. D. Galloway, D. N. Mai and R. D. Baxter, Org. Lett., 2017, 19, 5772–5775 CrossRef CAS PubMed.
- (a) R. Saritha, S. B. Annes, K. Perumal, K. A. Veerappan and S. Ramesh, ChemistrySelect, 2021, 6, 12440–12445 CrossRef CAS; (b) G. Kibriya, S. Mondal and A. Hajra, Org. Lett., 2018, 20, 7740–7743 CrossRef CAS PubMed; (c) S. Liu, W. Pan, S. Wu, X. Bu, S. Xin, J. Yu, H. Xu and X. Yang, Green Chem., 2019, 21, 2905–2910 RSC.
- (a) V. Soni, S. M. Khake and B. Punji, ACS Catal., 2017, 7, 4202–4208 CrossRef CAS; (b) I. Ghosh, L. Marzo, A. Das, R. Shaikh and B. König, Acc. Chem. Res., 2016, 49, 1566–1577 CrossRef CAS; (c) C.-S. Wang, P. H. Dixneuf and J.-F. Soulé, Chem. Rev., 2018, 118, 7532–7585 CrossRef CAS PubMed; (d) B. Chen, L.-Z. Wu and C.-H. Tung, Acc. Chem. Res., 2018, 51, 2512–2523 CrossRef CAS PubMed; (e) Q. Yang, L. Zhang, C. Ye, S. Luo, L.-Z. Wu and C.-H. Tung, Angew. Chem., Int. Ed., 2017, 56, 3694–3698 CrossRef CAS PubMed; (f) Q.-Y. Meng, T.-E. Schirmer, A.-L. Berger, K. Donabauer and B. König, J. Am. Chem. Soc., 2019, 141, 11393–11397 CrossRef CAS PubMed; (g) Q.-Y. Meng, J.-J. Zhong, Q. Liu, X.-W. Gao, H.-H. Zhang, T. Lei, Z.-J. Li, K. Feng, B. Chen, C.-H. Tung and L.-Z. Wu, J. Am. Chem. Soc., 2013, 135, 19052–19055 CrossRef CAS PubMed; (h) A. Hossain, S. Engl, E. Lutsker and O. Reiser, ACS Catal., 2019, 9, 1103–1109 CrossRef CAS; (i) Q. Yang, L. Zhang, C. Ye, S. Luo, L.-Z. Wu and C.-H. Tung, Angew. Chem., Int. Ed., 2017, 56, 3694–3698 CrossRef CAS PubMed; (j) C.-Yu. Huang, J. Li, W. Liu and C.-J. Li, Chem. Sci., 2019, 10, 5018–5024 RSC.
- (a) X.-H. Hu, X.-F. Yang and T.-P. Loh, ACS Catal., 2016, 6, 5930–5934 CrossRef CAS; (b) C. Uyeda, Y. Tan, G. C. Fu and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 9548–9552 CrossRef CAS PubMed; (c) A. Hossain, A. Vidyasagar, C. Eichinger, C. Lankes, J. Phan, J. Rehbein and O. Reiser, Angew. Chem., Int. Ed., 2018, 57, 8288–8292 CrossRef CAS PubMed; (d) T. K. Achar, S. Maiti, S. Jana and D. Maiti, ACS Catal., 2020, 10, 13748–13793 CrossRef CAS; (e) J. Luo and Q. Fu, Adv. Synth. Catal., 2021, 363, 3868–3878 CrossRef CAS; (f) P. Roy, J. R. Bour, J. W. Kampf and M. S. Sanford, J. Am. Chem. Soc., 2019, 141, 17382–17387 CrossRef CAS PubMed; (g) L.-P. Xu, B. E. Haines, M. J. Ajitha, J.-Q. Yu and D. G. Musaev, ACS Catal., 2021, 11, 12620–12631 CrossRef CAS.
- (a) K. Wang, W. Xu, H. Qi, P. Zhang, X. Cao and G. Wang, Catalysts, 2022, 12, 633–642 CrossRef CAS; (b) K. Sun, A. Shi, Y. Liu, X. Chen, P. Xiang, X. Wang, L. Qu and B. Yu, Chem. Sci., 2022, 13, 5659–5666 RSC; (c) S. Paul, J. H. Ha, G. E. Park and Y. R. Lee, Adv. Synth. Catal., 2017, 359, 1515–1521 CrossRef CAS; (d) R. K. Samanta, P. Meher and S. Murarka, J. Org. Chem., 2022, 87, 10947–10957 CrossRef CAS PubMed; (e) K. Sun, F. Xiao, B. Yu and W.-M. He, Chin. J. Catal., 2021, 42, 1921–1943 CrossRef CAS.
- Y. Hu, X. Ma, H. Hou, W. Sun, S. Tu, M. Wu, R. Lin, X. Xu and F. Ke, Catal. Sci. Technol., 2021, 11, 6374–6379 RSC.
- (a) S. P. Pitre, C. D. McTiernan, H. Ismaili and J. C. Scaiano, J. Am. Chem. Soc., 2013, 135, 13286–13289 CrossRef CAS PubMed; (b) T. Taniguchi, Y. Sugiura, H. Zaimoku and H. Ishibashi, Angew. Chem., Int. Ed., 2010, 49, 10154–10157 CrossRef CAS PubMed; (c) T. Taniguchi, A. Idota and H. Ishibashi, Org. Biomol. Chem., 2011, 9, 3151–3153 RSC; (d) T. Xiao, L. Li, G. Lin, Q. Wang, P. Zhang, Z.-W. Mao and L. Zhou, Green Chem., 2014, 16, 2418–2421 RSC; (e) M. Tian, S. Liu, X. Bu, J. Yu and X. Yang, Eur. J. Org. Chem., 2020, 26, 369–373 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra03479h |
| ‡ Mei Wu and Nancheng Lian contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |