
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of new class of indole acetic acid sulfonate derivatives as ectonucleotidases inhibitors†
Muhammad Siraj Khan Jadoonab,
Julie Pelletierc,
Jean Sévigny cd and
Jamshed Iqbal*abe
cd and
Jamshed Iqbal*abe
aDepartment of Pharmacy, COMSATS University Islamabad, Abbottabad Campus, Abbottabad, 22060, Pakistan. E-mail: drjamshed@cuiatd.edu.pk; jamshediqb@googlemail.com
bCentre for Advanced Drug Research, COMSATS University Islamabad, Abbottabad Campus, Abbottabad, 22060, Pakistan
cCentre de Recherche du CHU de Québec-Université Laval, Québec G1V 4G2, Canada
dDépartement de microbiologie-infectiologie et d'immunologie, Faculté de Médecine, Université Laval, Québec G1V 0A6, Canada
eDepartment of Chemistry, COMSATS University Islamabad, Abbottabad Campus, Abbottabad, 22060, Pakistan
First published on 10th October 2023
Abstract
Ectonucleotidases inhibitors (ENPPs, e5′NT (CD73) and h-TNAP) are potential therapeutic candidates for the treatment of cancer. Adenosine, the cancer-developing, and growth moiety is the resultant product of these enzymes. The synthesis of small molecules that can increase the acidic and ionizable structure of adenosine 5-monophosphate (AMP) has been used in traditional attempts to inhibit ENPPs, ecto-5′-nucleotidase and h-TNAP. In this article, we present a short and interesting method for developing substituted indole acetic acid sulfonate derivatives (5a–5o), which are non-nucleotide based small molecules, and investigated their inhibitory potential against recombinant h-ENPP1, h-ENPP3, h-TNAP, h-e5′NT and r-e5′NT. Their overexpression in the tumor environment leads to high adenosine level that results in tumor development as well as immune evasion. Therefore, selective, and potent inhibitors of these enzymes would be expected to decrease adenosine levels and manage tumor development and progression. Our intended outcome led to the discovery of new potent inhibitors like' 5e (IC50 against h-ENPP1 = 0.32 ± 0.01 μM, 58 folds increased with respect to suramin), 5j (IC50 against h-ENPP3 = 0.62 ± 0.003 μM, 21 folds increase with respect to suramin), 5c (IC50 against h-e5′NT = 0.37 ± 0.03 μM, 115 folds increase with respect to sulfamic acid), 5i (IC50 against r-e5′NT = 0.81 ± 0.05 μM, 95 folds increase with respect to sulfamic acid), and 5g (IC50 against h-TNAP = 0.59 ± 0.08 μM, 36 folds increase with respect to Levamisole). Molecular docking studies revealed that inhibitors of these selected target enzymes induced favorable interactions with the key amino acids of the active site, including Lys255, Lys278, Asn277, Gly533, Lys528, Tyr451, Phe257, Tyr340, Gln465, Gln434, Lys437, Glu830, Cys818, Asn499, Arg40, Phe417, Phe500, Asn503, Asn599, Tyr281, Arg397, Asp526, Phe419 and Tyr502. Enzyme kinetic studies revealed that potent compounds such as 5j and 5e blocked these ectonucleotidases competitively while compounds 5e and 5c presented an un-competitive binding mode. 5g revealed a non-competitive mode of inhibition.
Introduction
The proliferation of tumors is perhaps dependent on immune cell infiltration and the release of inflammatory mediators by these infiltrating immune cells, according to currently available data.1 Tumors use various immunosuppressive mechanisms that aid non-responders avert the immune system and modulate the tumor microenvironment and angiogenesis.2,3 In order to enhance clinical outcomes, combination treatments that aim to target a variety of immunosuppressive pathways may be necessary for improved cancer treatment.4 One way that tumors avoid the immune system is through reducing adenosine-like small molecule signaling chemicals in the tumor microenvironment. Adenosine, has been demonstrated to accumulate in tumors and have potent immunosuppressive effects on a variety of tumor-infiltrating leukocytes (TILs) through the A2A/A2B adenosine receptors.5 In fact, elevated levels of ATP, AMP and adenosine in the tumor microenvironment are emerging as important modulators of cancer-related inflammation, supporting tumor development as well as immune evasion.6The members of ectonucleotidases family such as e5′NT (ecto-5′-nucleotidase, CD73), ectonucleotide pyrophosphatases/phosphodiesterases (ENPPs) and human tissue nonspecific alkaline phosphatase (h-TNAP) which changes the extracellular level of adenosine triphosphate (ATP) into adenosine monophosphate (AMP) and finally to adenosine (as shown in Fig. 1), that controls the intratumoral synthesis of adenosine.7–15 Adenosine, which is produced by the hydrolysis of the ATP and AMP via ENPPs, e5′NT and h-TNAP, is crucial for many therapeutically essential processes, including anti-inflammatory,16–18 vasodilatation, and antidiuretic effects,19 immunosuppression,20,21 and antinociceptive effects.22 Numerous human malignancies have been discovered to overexpressed NPPs, e5′NT and, h-TNAP, which is expressed on a variety of cell surfaces including myocardial ischemia, insulin resistance, carcinogenesis and metastasis of tumor cell, and hyper-mineralization like calcific aortic valve disease.21–23 Moreover, a weak clinical prognosis is linked to increased e5′NT expression.24–26 It is anticipated that these ectonucleotidases inhibition can reverse adenosine buildup in the tumor microenvironment, including adenosine mediated immunity suppression by obstructing the primary tumor microenvironment-based adenosine synthesis mechanism.5,27,28
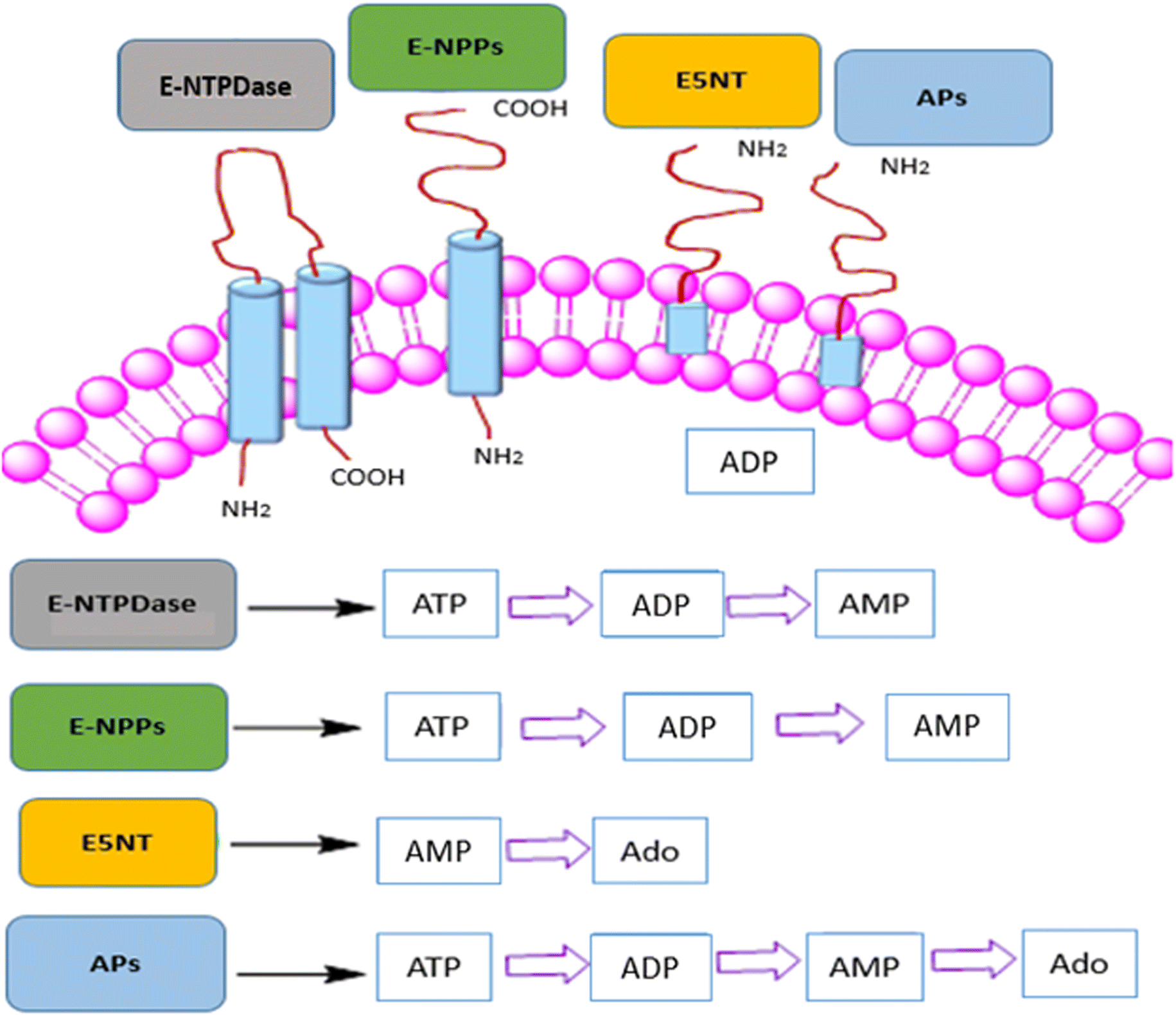 | ||
| Fig. 1 Ectonucleotidases family presented with catalytic transformation of adenosine triphosphate (ATP) into adenosine monophosphate (AMP) and adenosine (Ado). | ||
These ectonucleotidases, belong to the metalloprotease family of enzymes. e5′NT has been found to be significantly expressed by melanoma cells, while levels of e5′NT have been linked to the propensity of these cells to metastasis.29,30 Adenosine and e5′NT play roles in immunological responses, including those that involves T cells and B cells,31 as well as in the development of tumors.32 e5′NT inhibitors have the potential to be used in illness treatments that aim to lower adenosine levels.30,33 Additionally, specific e5′NT inhibitors may be helpful pharmacologically as well as for purposes of diagnosis. ENPPs are present as transmembrane bound glycoproteins and soluble proteins, constituting a family of seven members (NPP1-7).34,35 Among these, NPP1-3 are well characterized and h-ENPP1 and 3 (membrane bound glycoproteins) have superior nucleotide hydrolyzing activity than NPP2 (soluble protein). h-ENPP1 and h-ENPP3 are found on the surface of basophil cells, mast cells,36 mucosal and epithelial cell surfaces.37 The overexpression of h-ENPP1 suppresses the tyrosine kinase based activity of insulin receptors23 including human stem cells38 and brain cancer39 whereas h-ENPP3 act as diagnostic marker for allergen (IgE) sensitivity36,37 and tumor marker.21,40 Since both h-ENPP1 and h-ENPP3 are isozymes that play a role in a number of clinical illnesses, finding biologically active chemicals to change how they operate has been a popular area of research to discover novel drugs. In case of APs the four different isozymes that make up the tissue-specific and tissue-nonspecific alkaline phosphatases have been encoded in separate gene loci.41 Nonspecific alkaline phosphates are found in the kidney, liver, and bones while tissue-specific APs are further classified into intestinal, placental, and germ cell alkaline phosphatases.42 Numerous diseases, including carcinoma of the breast, behavioral disorders, and severe medical conditions including osteoarthritis, ankyloses, and calcification of the vascular system are brought on by the overproduction of these enzymes.43 Alkaline phosphatase (h-TNAP) upregulation and many cancer types are closely related. Most often, cells originating from the breast cancer region exhibit placental APs overexpression. Likewise, to hepatocellular carcinoma, increased saturation of intestine and germ cell APs was observed in the case of choriocarcinoma.44 In addition to the biological application already mentioned, h-TNAP is crucial for purinergic signaling in a number of malignancies (Fig. 2).
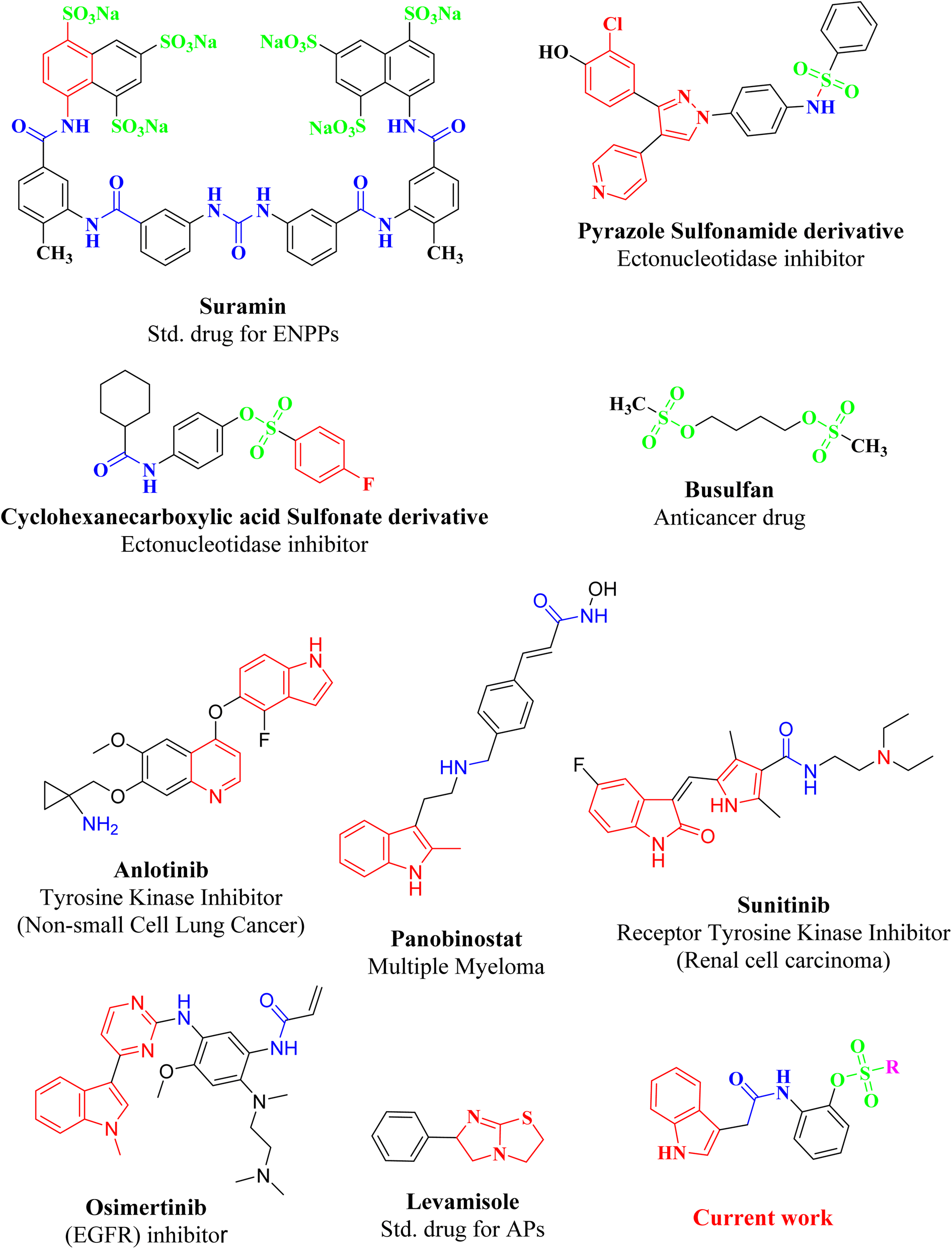 | ||
| Fig. 2 Molecules from literature, reported for exhibiting ectononucleotidases inhibitory and anticancer activity, having either sulfonate, carboxamide or indole moiety in major nucleus of scaffolds. | ||
Affirming the role of ENPPs, e5′NT and h-TNAP in numerous pathophysiological conditions including cancer, various therapeutic interventions have been employed. By inhibiting these enzymes, the adenosine moiety, one of the causes of cancer development and progression could be limited. One of the most important sources of bioactive molecules is heterocyclic compounds, which can be produced in a laboratory or derived from natural sources.45–47 Because these heterocyclic compounds contain one or more heteroatoms with the potential to donate and accept H-bonds, such as N, O, and S, they can easily attach to a variety of therapeutic targets, producing a wide range of biological effects.48 Variety of heterocyclic molecules with ectonucleotidases inhibitory potential either nucleotide or non-nucleotide49–54 have been synthesized till date including inhibitors of natural origin to target cancer using different modes.55,56 A nucleotide-based ectonucleotidase inhibitor drug, Quemliclustat (AB680), is under phase 1 clinical trials for the treatment of tumors.52 One such non-nucleotide class of inhibitors is a variety of sulfonates reported active against the ectonucleotidases with Ki values in low micro-molar ranges.57–61 Sulfonates have been reported to be anticancer agents with additional therapeutic potentials. Since 1959, persistent myeloid leukaemia, refractory lymphoma and myeloproliferative ailments have all been treated with busulfan, one of the alkyl sulfonate.62 The antiproliferative effects of a number of sulfonate esters were tested on the human breast cancer cell line MCF-7, among these compounds p-methoxyphenyl p-toluenesulfonate and 3-methoxyphenyl p-toluenesulfonate significantly decreased cell viability, with lowest IC50 values of 125.9 μM and 89.8 μM, respectively.63 Similarly ectonucleotidase inhibitors, pyrazole sulfonate derivatives expressed good activity against (MCF-7) breast cancer cell line, cervical cancer cell line (HeLa), bone marrow cancer cell line (K-562) and brain astrocytoma cell line (1321N1).64 In the present study indole acetic acid sulfonate derivatives (5a–5o) were synthesized and investigated for in vitro activity against selected ectonucleotidases (ENPPs, e5′NT and h-TNAP). The newly synthesized sulfonate derivatives exhibited noticeable inhibitory potential against the investigated enzymes. The inhibitory potential of compounds 5c, 5e, 5g, 5i, and 5j showed IC50 values in the sub micromolar range of 0.32 to 0.81 μM as shown in Fig. 4.
Among the heterocyclic compounds, nitrogen heterocycles, particularly indole, are thought to be among the most significant nitrogen heterocyclic cores that have attracted significant research in recent years due to its numerous bioactivities.65 The indole moiety, which is made up of a combination of aromatic and heterocyclic compounds, serves as a useful framework for the synthesis of new leads.66 Various indole based newly synthesized compounds showed remarkable activity against numerous cancers. This is the reason that indole based drugs have been in common practice for the treatment of various cancers since long. Such clinically applied indole-based anticancer medications include panobinostat,67 alectinib,68 sunitinib,69 osimertinib,70 anlotinib, and nintedanib.71 Keeping in consideration, indole based drugs role in cancer treatment via different mechanism the newly synthesized compounds with indole, carboxamide and sulfonate substituents are focused to target cancer via ectonucleotidases inhibition, involved in the synthesis of adenosine that develop and promotes different cancers.
Our current work expresses indole acetic acid derivatives with sulfonate, carboxamide, and indole moiety unified in newly synthesized molecules important for ectonucleotidases inhibitory activity as shown in Fig. 3. These compounds were found potentially very active against all the targeted enzymes which contribute to the high level of adenosine formation in tumor microenvironment. Exhibition of excellent inhibitory activities by the synthesized indole acetic acid sulfonate derivatives against ENPPs, e5′NT and h-TNAP pose their importance as a candidate in tumors management strategies with improved IC50 values than the existing compounds of this class.
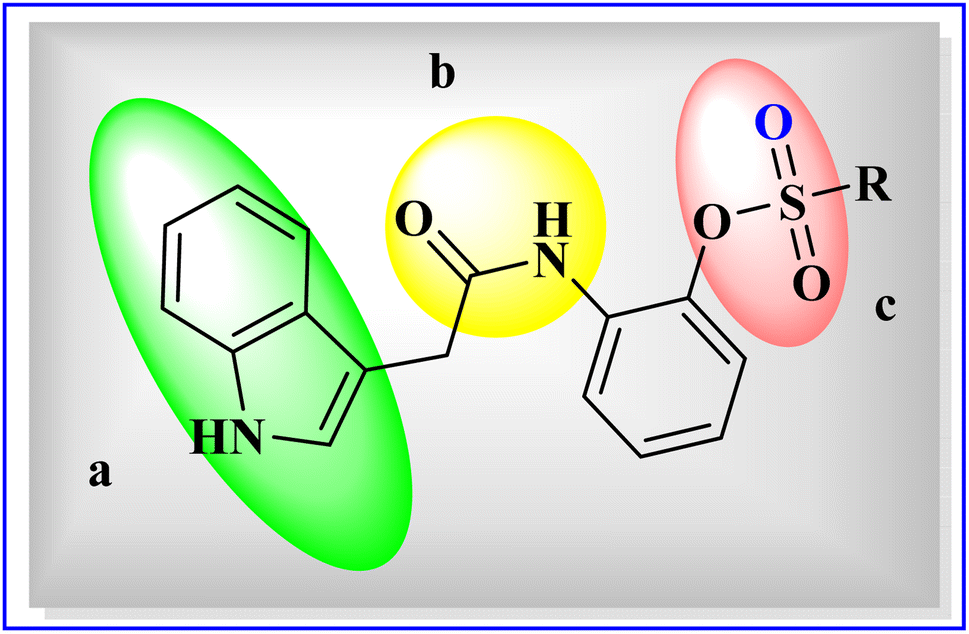 | ||
| Fig. 3 General structure of indole acetic acid sulfonate derivatives (5a–5o), (a) indole ring, (b) carboxamide group, and (c) sulfonate group. | ||
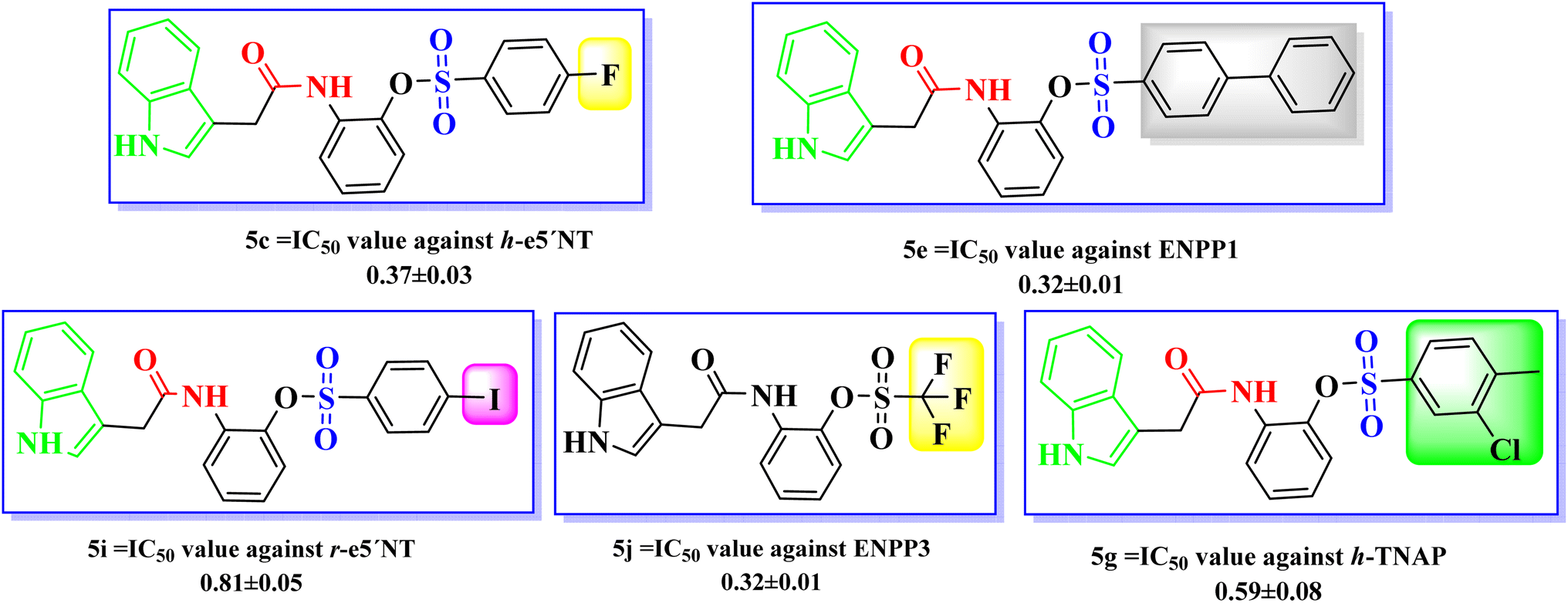 | ||
| Fig. 4 Most potent compounds among indole acetic acid sulfonate derivatives (5a–5o). | ||
Results and discussion
Chemistry
The indole acetic acid sulfonate derivatives (5a–5o) were synthesized by facile in situ synthesis as shown in Scheme 1. In first step the N-(2-hydroxyphenyl)-2-(1H-indol-3-yl)acetamide (3a) was synthesized by treating 2-aminophenol (1a) with 2-indole acetic acid (2a) in the presence of 4-(dimethylamino)pyridine (DMAP) and cabonyldiimidazole (CDI). In the next step the synthesized compound 3a, was treated with substituted sulfonyl chlorides (4a) in the presence of triethylamine (TEA) and acetonitrile as a solvent. The reaction was allowed for 24 hours followed by workup of the reaction resulting in the formation of indole acetic acid sulfonate derivatives (5a–5o) in a good to excellent yield. | ||
| Scheme 1 Synthesis of indole acetic acid sulfonate derivatives (5a–5o). | ||
Physical properties like melting points and color of final products were also determined. Structure elucidation of indole acetic acid sulfonate derivatives (5a–5o) was done using FTIR, 1H NMR, 13C NMR and elemental analysis. In FTIR spectra, absorbance band appeared at 3340–3300, 3150–3194 (free and associated –NH), 2900–2990 (–CH), 1600–1685 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and 1040–1098 (SO). In 1H NMR, two characteristic broad singlets were observed in the range of δ 10.50–12.0 ppm, δ 9.5–8.5 ppm for CONH and NH respectively. In 13C NMR, indole ring carbons appear in the region of δ 111.39–136.58 ppm, while carbonyl carbon attached to the NH of carboxamide gives a peak at δ 171.61 ppm and that of carbon atoms attached to sulfonyl groups present a peak at δ 132.87 ppm and δ 140.11 ppm. These characteristic peaks confirmed the formation of indole acetic acid sulfonate derivatives. Purity of compounds was determined through elemental analysis that were found to be within 0.4% of the calculated results.
O) and 1040–1098 (SO). In 1H NMR, two characteristic broad singlets were observed in the range of δ 10.50–12.0 ppm, δ 9.5–8.5 ppm for CONH and NH respectively. In 13C NMR, indole ring carbons appear in the region of δ 111.39–136.58 ppm, while carbonyl carbon attached to the NH of carboxamide gives a peak at δ 171.61 ppm and that of carbon atoms attached to sulfonyl groups present a peak at δ 132.87 ppm and δ 140.11 ppm. These characteristic peaks confirmed the formation of indole acetic acid sulfonate derivatives. Purity of compounds was determined through elemental analysis that were found to be within 0.4% of the calculated results.
Enzymatic assays
The synthetic compounds' ability to inhibit the hydrolytic activity of the h-ENPP1 and h-ENPP3 isozymes, h-e5′NT and r-e5′NT along with h-TNAP was assessed. The activity of these selected enzymes was potentially and significantly inhibited by newly developed compounds. The results of the enzymatic investigation of compounds (5a–5o) showed highly potent inhibitors with IC50 values ranging from 0.32 to 0.80 μM as shown in Table 1. The main scaffold features include the presence of an indole moiety, carboxamide and a sulfonate group, whereas R representing aryl or alkyl moieties attached to the main scaffold as shown in Fig. 3.| Compound codes | R | IC50 ± SEM (μM)a or inhibition%b | ||||
|---|---|---|---|---|---|---|
| h-ENPP1 | h-ENPP3 | h-e5′NT | r-e5′NT | h-TNAP | ||
| a Results are presented as an average value of two independent experiments, which were conducted in triplicate (only compounds with more than 50% inhibitory concentration were subjected to evaluate their IC50 values).b Percentage inhibition at 100 μM per well inhibitor concentration. (Compounds with less than 50% inhibition are present with inhibition% values). | ||||||
| 5a | C6H4Cl | 2.07 ± 0.22 | 1.66 ± 0.007 | 1.19 ± 0.02 | (22.72)% | 1.6 ± 0.11 |
| 5b | C7H7 | 1.35 ± 0.02 | 1.71 ± 0.11 | 13.22 ± 3.14 | (38.52)% | (42.39)% |
| 5c | C6H4F | 1.12 ± 0.12 | 1.37 ± 0.05 | 0.37 ± 0.03 | 1.66 ± 0.04 | 1.48 ± 0.02 |
| 5d | C7H7O | 1.09 ± 0.01 | 2.05 ± 0.25 | 3.46 ± 0.12 | 1.77 ± 0.12 | 5.18 ± 0.46 |
| 5e | C12H9 | 0.32 ± 0.01 | 4.51 ± 0.38 | 0.56 ± 0.016 | 0.98 ± 0.02 | 1.06 ± 0.03 |
| 5f | C6H4Br | 1.14 ± 0.002 | 1.23 ± 0.01 | 0.69 ± 0.001 | 1.64 ± 0.02 | 6.58 ± 0.25 |
| 5g | C7H6Cl | 1.69 ± 0.05 | 1.31 ± 0.11 | 4.41 ± 0.19 | 1.85 ± 0.11 | 0.59 ± 0.08 |
| 5h | C10H8N | 0.91 ± 0.02 | 1.02 ± 0.004 | 1.17 ± 0.13 | 0.91 ± 0.04 | 2.86 ± 0.06 |
| 5i | C6H4I | 1.07 ± 0.06 | 3.26 ± 0.15 | 4.85 ± 0.55 | 0.81 ± 0.05 | 2.27 ± 0.24 |
| 5j | CF3 | 0.59 ± 0.003 | 0.62 ± 0.003 | 0.89 ± 0.04 | 5.12 ± 0.61 | 1.68 ± 0.07 |
| 5k | C3H7 | 1.74 ± 0.05 | 0.89 ± 0.04 | (37.52)% | (28.65)% | (31.75)% |
| 5l | C6H4Cl | 0.85 ± 0.03 | 0.77 ± 0.03 | 0.43 ± 0.03 | 5.67 ± 0.11 | 1.69 ± 0.18 |
| 5m | C9H6N | 1.36 ± 0.09 | 1.22 ± 0.01 | 6.53 ± 0.67 | (30.03)% | 1.3 ± 0.38 |
| 5n | C4H3 | (38.03)% | 2.55 ± 0.07 | (44.68)% | (46.64)% | (28.88)% |
| 5o | C6H7 | (28.32)% | 2.67 ± 0.46 | 3.21 ± 0.83 | 4.86 ± 0.04 | 3.37 ± 0.05 |
| Suramin | 18.54 ± 1.14 | 12.83 ± 0.23 | — | — | — | |
| Sulfamic acid | — | — | 42.1 ± 7.80 | 77.3 ± 7.0 | — | |
| Levamisole | — | — | — | — | 21.38 ± 4.17 | |
Structure activity relationships (SARs)
A new class of indole acetic acid sulfonate derivatives were synthesized and evaluated against human h-ENPP1, h-ENPP3, h-e5′NT, r-e5′NT and h-TNAP via in vitro assay. Compounds presented noticeable inhibitory activity against these selected targets. Among all the synthesized compounds 5c, 5e, 5g, 5i, and 5j were found to be more potent against the target enzymes. The pharmacophore structure including indole ring, carboxamide group and sulfonate group are responsible for the primary inhibitory activity of the indole acetic acid sulfonate derivatives (5a–5o), whereas substitution to the main scaffold resulted in the increase in inhibitory potential of newly synthesized compounds (as shown in Fig. 5). In case of compound 5c, the substitution of fluorine at para position enhanced the activity of the main scaffold with IC50 against h-e5′NT = 0.37 μM. Electronegative effect of the fluorine resulted in an increased potency by reducing the hydrolytic activity of the enzyme. It was concluded from the in vitro results that fluorine substitution adds to the good inhibitory activity for all targeted enzymes ranging IC50 between 0.37 to 1.67 μM.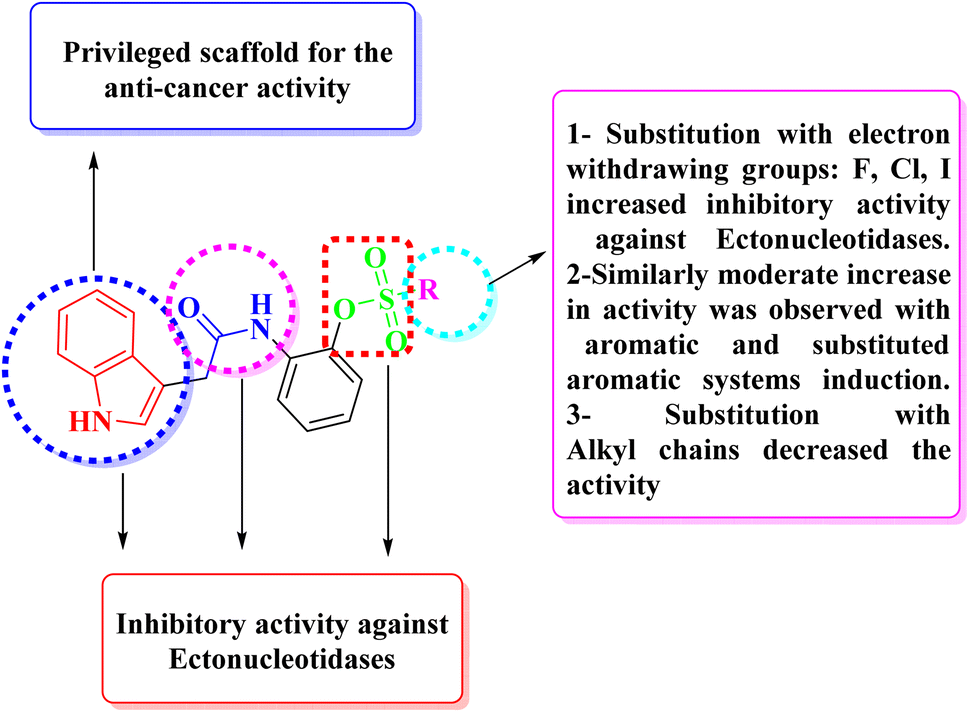 | ||
| Fig. 5 Illustration of SAR for newly synthesized indole acetic acid sulfonate derivatives (5a–5o) highlighting the role of pharmacophoric core including indole ring, carboxamide, and sulfonate group in main scaffold structure with different substitutions. | ||
Biphenyl ring substitution to the main scaffold developed 5e and resulted in very good activity that increase the activity by 57 folds against h-ENPP1 as compared to standard inhibitor suramin. Similarly, it increased the activity against h-ENPP3, h-e5′NT, r-e5′NT and h-TNAP by 3, 75, 79 and 20 folds, respectively. Compound 5g, when substituted with chloro and methyl groups at ortho and para position respectively, resulted in an increase in its inhibitory potential against h-TNAP with an IC50 value of 0.59 μM. Electronegative effect of chlorine and presence of methyl group impart increase in inhibitory potential of this compound against all target enzymes as concluded from the in vitro activity assay. Additionally, the methyl ring contributes to better pharmacokinetic profile of the compound by enhancing its bioavailability and excretion of the drug from the body. Substitution of iodo group in compound 5i at para position of the scaffold resulted in an increase inhibitory potential of the compound against r-e5′NT with an IC50 value of 0.81 μM contributing 95 folds increase compared to reference standard sulfamic acid. Compound 5j having a substitution of three strong electronegative fluorine groups resulted in increase in in vitro determined inhibitory potential of h-ENPP3. The incorporation of substitution with strong electronegative group contributed to increase inhibitory potential against target enzymes with IC50 values ranging from 0.59 to 5 μM.
Some compounds showed selectivity and good potency between same enzymes from different species, such as 5a, 5b and 5m expressed good potency and selectivity for h-e5′NT as compared to the r-e5′NT. Similarly compounds expressed selectivity and high potency between different isozymes such as h-ENPP1 and h-ENPP3. Substitution of more electronegative chlorine group in compound 5a, phenyl group in compound 5b and quinolone moiety in compound 5m resulted in selectivity of the compounds towards h-e5′NT. In a similar way, compound 5n and 5o, which have a thiophene moiety substitution and a para methyl substitution to the primary scaffold consisting of an indole, a carboxamide, and sulfonate moieties, respectively, were found to be more effective and selective for h-ENPP3 between the two h-ENPP isozymes (Table 2).
| Ligands | Binding energy (kcal mol−1) | ||||
|---|---|---|---|---|---|
| h-ENPP1 | h-ENPP3 | h-e5′NT | r-e5′NT | h-TNAP | |
| 5a | −6.3 | −5.9 | −6.4 | −8.0 | −5.1 |
| 5b | −6.6 | −5.9 | −6.4 | −8.1 | −4.7 |
| 5c | −6.6 | −5.7 | −6.1 | −7.7 | −5.0 |
| 5d | −6.4 | −6.1 | −6.3 | −8.0 | −5.1 |
| 5e | −8.3 | −6.6 | −7.6 | −8.3 | −5.3 |
| 5f | −6.7 | −6.6 | −6.4 | −8.0 | −5.2 |
| 5g | −7.2 | −6.3 | −6.3 | −8.3 | −5.0 |
| 5h | −7 | −6.2 | −6.6 | −7.7 | −5.2 |
| 5i | −6.9 | −5.9 | −5.6 | −7.9 | −4.9 |
| 5j | −5.6 | −5.2 | −6.0 | −6.8 | −4.4 |
| 5k | −5.9 | −5.4 | −6.2 | −7.1 | −4.5 |
| 5l | −6.6 | −6.1 | −6.7 | −7.7 | −4.9 |
| 5m | −6.8 | −5.9 | −6.7 | −8.0 | −5.1 |
| 5n | −6.5 | −5.6 | −6.2 | −7.5 | −4.9 |
| 5o | −6.3 | −6.1 | −6.2 | −7.8 | −5.1 |
| Ref. | −5.8 | −5.4 | −11.1 | −11.7 | −3.5 |
Enzyme kinetic studies for h-ENPPs, e5′NT and h-TNAP
Enzyme kinetic studies were performed for the most potent inhibitors against h-ENPP1, h-ENPP3, h-e5′NT, r-e5′NT and h-TNAP. For h-ENPP1and h-ENPP3 the potent inhibitors were 5e and 5j, while compound 5c and 5i showed good potential for inhibition of h-e5′NT and r-e5′NT, whereas, 5g expressed good inhibitory potential against h-TNAP. Compound 5j and 5i exhibited competitive mode of inhibition. Inhibitors 5e and 5c presented uncompetitive mode of inhibition as shown in Fig. 6 and 7, whereas, competitive inhibition mode was shown by compound 5g as shown in Fig. 8. | ||
| Fig. 6 Illustration of enzyme kinetics for compounds 5e and 5j against human h-ENPP1 and h-ENPP3, respectively. | ||
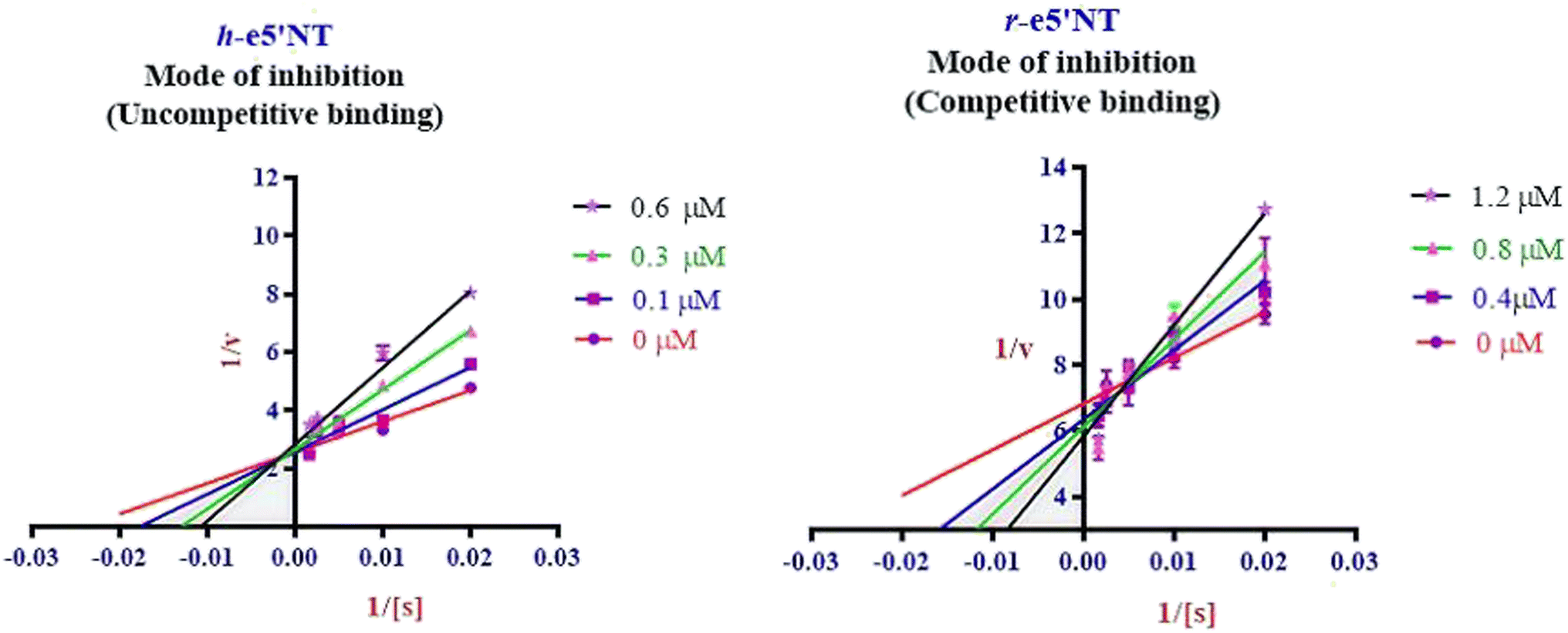 | ||
| Fig. 7 Illustration of enzyme kinetics for compounds 5c and 5i against h-e5′NT and r-e5′NT respectively. | ||
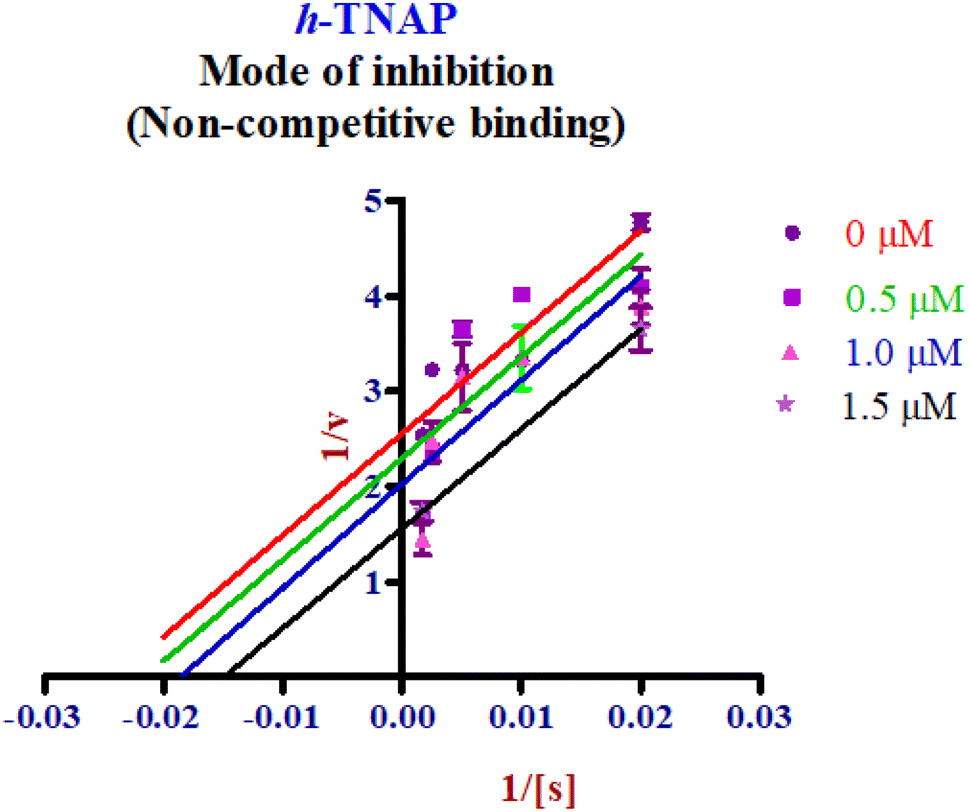 | ||
| Fig. 8 Enzyme kinetics graphs presenting non-competitive mode of inhibition for compound 5g against h-TNAP. | ||
Molecular docking studies analysis
Based upon the binding energy values and results of in vitro studies, 3D interactions of potent compounds are visualized in the following figures.Human ectonucleotide pyrophosphatase/phosphodiesterase 1(h-ENPP1)
Active site amino acids residues involved in the binding interactions of h-ENPP1 are;(1) (Asp218 Lys255 Thr256 Phe257 Asp276 Asn277 Lys278 Ser289 Leu290 Lys291 Lys295 Phe321 Trp322 Pro323 Ser325 Asp326 Lys338 Met339 Tyr340 Tyr371 Glu373 Asp376 Ser377 His380 Ser381 Tyr434 Ile450 Tyr451 Gly452 Pro453 Gln519 Arg527 Lys528 Tyr529 Cys530 Gly531 Ser532 Gly533 Phe534 His535 Po41001 Zn1002).
Protein–ligand 5j interaction showed hydrogen bonding between Lys255 and carbonyl oxygen atom, Lys278 and oxygen atom of sulfonyl group, Asn277 and oxygen atom of sulfonyl group, and hydrogen atom of indole ring and Gly533. One hydrogen bond was also observed between phosphate group and fluorine atom.
In case of ligand 5e Amino acid residues Lys528 and Tyr451 are making the hydrogen bonds with the oxygen atom of carbamide group as shown in (Fig. 8). Metal–acceptor interaction was observed between Zn atom and oxygen atom of sulfonyl group. Phe257 and Tyr340 formed π–π stacked and π–π T shaped interaction with the terminal phenyl ring respectively.
Human ectonucleotide pyrophosphatase/phosphodiesterase 3 (h-ENPP3)
Amino acids residues in the active site, involved in the active site of h-ENPP3 are;(1): (Lys382 Leu427 Ser428 Cys429 Lys431 Pro432 Asp433 Gln434 His435 Phe436 Lys437 Pro438 Phe462 Val463 Asp464 Gln465 Trp467 Phe489 Val815 Glu816 Ser817 Cys818 Lys822 Trp827 Glu830 Arg831 Ala834).
Binding interaction analysis of 5j revealed that amino acid residue Gln465 made two hydrogen bonds with oxygen atom of sulfonyl group and fluorine atom of trifluoro group (Fig. 9). The hydrogen atom of indole ring formed hydrogen bond with Gln434. In case of 5e, oxygen atom of sulfonyl group made hydrogen bond with Gln465. Gln434 is forming hydrogen bond with hydrogen atom of indole ring. π–cation, π–anion and π–sulphur interactions were observed with amino acid residues Lys437, Glu830 and Cys818 respectively.
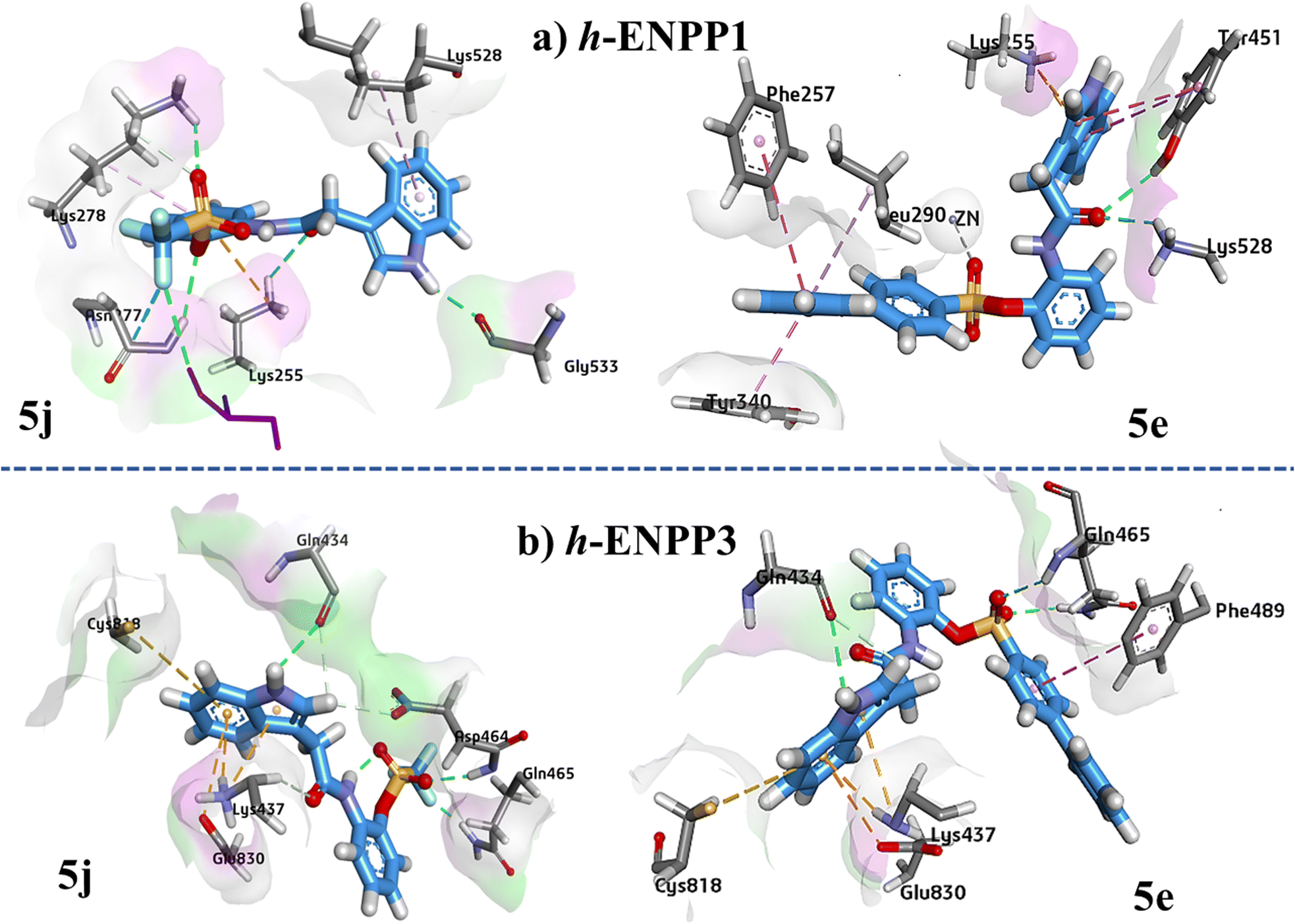 | ||
| Fig. 9 Illustration of 3D ligand–protein interactions conformations of 5j and 5e against (a) h-ENPP1, (b) h-ENPP3. | ||
Human ecto-5′-nucleotidase (h-e5′NT)
Active site amino acids residues involved in the active site of h-e5′NT are; (Arg40 Glu46 Asp47 Ser48 Ser49 His243 Ser244 Asn245 Glu257 Tyr281 Phe283 Leu389 Asn390 Gly392 Gly393 Arg395 Leu415 Phe417 Gly418 Gly419 Thr420 Phe421 Asp422 Pro498 Asn499 Phe500 Asn503 Asp506 His518 Asp519 Ser520 Asp524).In case of compound 5j, amino acid residues Asn499 and Arg40 developed hydrogen bonds with the fluorine atoms of tri-fluorine group while π–π stacking interaction was found with amino acids Phe417 and Phe500 (Fig. 10). Docking study of protein–ligand interactions of (5c) revealed that amino acid residue Asn503 and Asn599 is involved in hydrogen bonding with both oxygen atoms of sulfonate group. π–π stacking is observed involving indole ring and Phe500 and Phe417 amino acids. π–donor hydrogen bond was observed between terminal ring attached to fluorine and amino acid Tyr281.
 | ||
| Fig. 10 Presentation of 3D binding modes of inhibitors, 5c and 5j against (a) h-e5′NT residues and 5e and 5l against (b) r-e5′NT residues. | ||
Rat ecto-5′-nucleotidase (r-e5′NT)
Active site amino acids residues involved in the active site of r-e5′NT are; (Gln90 Phe95 Thr96 Lys99 His120 Asp123 Asn124 Ser180 Lys181 Glu182 Phe185 Leu186 Ser187 Asn188 Glu198 Ser223 Gly224 Phe225 Glu226 Met227 His245 Thr246 Asn247 Arg356 Val391 Asn392 Gly394 Gly395 Arg397 Leu417 Pro418 Phe419 Gly420 Gly421 Thr422 Phe423 Arg443 Gln446 Ser447 Thr448 Gly449 Glu450 Pro500 Ser501 Tyr502 Gly506 Gly507 Asp508 Gly509 Asp526).Study of binding interaction of 5e showed that pyrrole ring is involved in forming the hydrogen bond with Asp526 While oxygen atom of sulfonyl moiety made hydrogen bond with amino acid residue His120. π–cation and π–anion interaction were observed with Asp508 and Arg397 respectively. Phe419 developed π–π stacking with 5e (as shown in Fig. 10). Synthesized compound 5l showed maximum inhibition potential against r-e5′NT. Binding interaction analysis of 5l showed that amino acid residues Asn392 and Arg397 are responsible for hydrogen bonding with the oxygen atoms of carboxamide group. Amino acid Asp526 developed hydrogen bond with the hydrogen atom of pyrrole ring. π–π stacking was observed with amino acid residues Phe419 and Tyr502.
Human tissue nonspecific alkaline phosphatase (h-TNAP)
Active site amino acid residues involved in the active site of h-TNAP are;(1) (Ala188 Leu191 Met192 His193 Ile195 Arg196 Asp197 Ile198 Ile201 Met209 Leu235 Trp239 Pro244 Arg245 Tyr246 Lys247 Tyr268 Leu270).
Analysis of indole acetic acid sulfonate derivative 5g presents that amino acid residue Arg196 is responsible for the formation of hydrogen bond with oxygen atom of sulfonyl group while Arg245 developed hydrogen bond with carbonyl oxygen. π–alkyl interactions were observed with amino acid residues Tyr246, Lys247 and His248. In case of 5e, oxygen atom of carboxamide is responsible for hydrogen bonding with Arg245. π–cation interactions were formed with Arg196 (as shown in Fig. 11).
 | ||
| Fig. 11 3D ligand–protein interactions docked conformations presentation for compound 5e and 5g against h-TNAP active pocket. | ||
Experimental part
General
All the chemicals and solvents needed for this study were procured from commercial sources and used without additional purification. Analytical grade solvents were used for synthesis while lab grade solvents were employed for purification of newly synthesized compounds. Using the Gallenkamp melting point equipment, the melting point was identified. The thin layer chromatography was carried out using aluminium plates coated with silica gel (0.20 mm, 60 A), while the spots were detected using a UV absorbance lamp 254–366 nm. For 1H NMR and 13C NMR Brucker Avance III HD (400 MHz spectrometer) was employed. The multiplicities observed were given as s for singlet, d for doublet, t for triplet, and q for quintet. The coupling constant (J) was estimated in Hz. Elemental analysis of the test compounds was carried out using LECO 630-200-200 TruSpec CHNS microanalyzer.General procedure for the synthesis (5a–5o)
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4), colour: off-white solid, mp: 181–183 °C, FT-IR (
:4), colour: off-white solid, mp: 181–183 °C, FT-IR (![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) , cm−1): 3404 (N–H, stretching), 2958 (C–H, stretching), 1613 (CO, stretching), 1085 (SO, stretching); 1H NMR (400 MHz, DMSO, δ = ppm): 10.98 (s, 1H), 9.44 (s, 1H), 7.75 (d, J = 8.1 Hz, 1H), 7.64 (d, J = 8.3 Hz, 2H), 7.56 (d, J = 8.0 Hz, 1H), 7.50 (d, J = 8.3 Hz, 2H), 7.37 (d, J = 8.1 Hz, 1H), 7.25 (q, J = 8.0 Hz, 3H), 7.11 (m, 2H), 6.99 (t, J = 7.5 Hz, 1H), 3.60 (s, 2H). 13C NMR (101 MHz, DMSO, δ = ppm): 169.90, 140.14, 136.55, 133.84, 133.12, 131.42, 130.45, 129.87, 128.13, 127.71, 125.25, 124.70, 124.52, 123.25, 121.54, 119.22, 118.93, 111.86, 108.57, 33.38. Anal. calcd. for C22H17ClN2O4S: C, 59.87; H, 3.89; N, 6.35; S, 7.27%. Found: C, 59.87; H, 3.88; N, 6.38; S, 7.34%.:4), colour: off-white solid, mp: 134–137 °C, FT-IR (, cm−1): 3172 (N–H, stretching), 2931 (C–H, stretching), 1613 (CO, stretching), 1085 (SO, stretching); 1H NMR (400 MHz, DMSO, δ = ppm): 10.99 (s, 1H), 9.36 (s, 1H), 7.91 (d, J = 8.1 Hz, 1H), 7.60 (d, J = 7.9 Hz, 1H), 7.39 (m, 3H), 7.39–7.25 (m, 5H), 7.22 (dd, J = 8.2, 1.7 Hz, 1H), 7.20–7.11 (m, 1H), 7.05 (t, J = 7.5 Hz, 1H), 6.97 (t, J = 7.4 Hz, 1H), 4.66 (s, 2H), 3.82 (s, 2H). 13C NMR (101 MHz, DMSO, δ = ppm): 170.44, 136.60, 131.59, 131.50, 129.38, 129.10, 127.96, 127.73, 127.61, 125.55, 124.93, 124.80, 123.06, 121.57, 119.07, 119.01, 111.90, 108.40, 56.52, 33.76. Anal. calcd. for C23H20N2O4S: C, 65.70; H, 4.79; N, 6.66; S, 7.62%. Found: C, 65.75; H, 4.73; N, 6.58; S, 7.60%.:4), colour: white solid, mp: 161–163 °C, FT-IR (, cm−1): 3453 (N–H, stretching), 2937 (C–H, stretching), 1608 (CO, stretching), 1092 (SO, stretching); 1H NMR (400 MHz, DMSO, δ = ppm): 10.98 (s, 1H), 9.42 (s, 1H), 7.83 (d, J = 8.1 Hz, 2H), 7.76 (d, J = 8.0 Hz, 1H), 7.57 (d, J = 7.8 Hz, 1H), 7.37 (m, 3H), 7.29–7.20 (m, 3H), 7.11 (m, 2H), 7.00 (t, J = 7.4 Hz, 1H), 3.59 (s, 2H). 13C NMR (101 MHz, DMSO, δ = ppm): 169.89, 140.14, 138.94, 136.56, 134.21, 131.43, 129.91, 128.09, 127.72, 125.22, 124.65, 124.51, 123.22, 121.54, 119.24, 118.95, 111.86, 108.60, 104.88, 33.41. Anal. calcd. for C22H17FN2O4S: C, 62.26; H, 4.04; N, 6.60; S, 7.55%. Found: C, 62.25; H, 4.13; N, 6.68; S, 7.62%.:4), colour: white to off-white solid, mp: 200–203 °C, FT-IR (, cm−1): 3438 (N–H, stretching), 2963 (C–H, stretching), 1655 (CO, stretching), 1071 (SO, stretching); 1H NMR (400 MHz, DMSO, δ = ppm): 11.01 (s, 1H), 9.32 (s, 1H), 7.80 (dd, J = 8.0, 1.6 Hz, 1H), 7.55 (t, J = 9.0 Hz, 3H), 7.38 (d, J = 8.1 Hz, 1H), 7.30 (d, J = 2.4 Hz, 1H), 7.22 (m, 2H), 7.10 (m, 2H), 7.00 (t, J = 7.4 Hz, 1H), 6.93–6.85 (m, 2H), 3.74 (s, 3H), 3.66 (s, 2H). 13C NMR (101 MHz, DMSO, δ = ppm): 169.93, 164.45, 136.58, 131.53, 131.04, 127.80, 127.74, 125.69, 124.92, 124.62, 124.11, 123.05, 121.56, 119.22, 118.93, 115.10, 111.86, 108.59, 56.32, 33.55. Anal. calcd. for C23H20N2O5S: C, 63.29; H, 4.62; N, 6.42; S, 7.35%. Found: C, 63.22; H, 4.55; N, 6.38; S, 7.29%.:4), colour: off-white solid, mp: 193–195 °C, FT-IR (, cm−1): 3395 (N–H, stretching), 2954 (C–H, stretching), 1601 (CO, stretching), 1076 (SO, stretching); 1H NMR (400 MHz, DMSO, δ = ppm): 10.99 (d, J = 2.5 Hz, 1H), 9.44 (s, 1H), 7.77 (dd, J = 8.2, 1.7 Hz, 1H), 7.70 (d, J = 8.6 Hz, 2H), 7.66 (d, J = 8.6 Hz, 2H), 7.62–7.40 (m, 6H), 7.40–7.29 (m, 2H), 7.28–7.19 (m, 2H), 7.20–7.04 (m, 2H), 6.96 (ddd, J = 7.9, 7.0, 1.0 Hz, 1H), 3.62 (s, 2H). 13C NMR (101 MHz, DMSO, δ = ppm): 169.86, 146.65, 140.12, 136.56, 133.16, 131.49, 129.60, 129.43, 129.27, 127.96, 127.92, 127.74, 127.65, 125.09, 124.54, 124.45, 123.21, 121.55, 119.25, 118.92, 111.87, 108.61, 33.44. Anal. calcd. for C28H22N2O4S: C, 69.69; H, 4.60; N, 5.81; S, 6.64%. Found: C, 69.73; H, 4.64; N, 5.78; S, 6.56%.:4), colour: off-white solid, mp: 183–186 °C, FT-IR (, cm−1): 3449 (N–H, stretching), 2963 (C–H, stretching), 1613 (CO, stretching), 1085 (SO, stretching); 1H NMR (400 MHz, DMSO, δ = ppm): 10.98 (s, 1H), 9.43 (s, 1H), 7.75 (d, J = 8.5 Hz, 1H), 7.65 (d, J = 8.5 Hz, 2H), 7.59–7.52 (m, 3H), 7.37 (d, J = 8.4 Hz, 1H), 7.25 (d, J = 8.6 Hz, 3H), 7.11 (m, 2H), 7.01 (d, J = 7.8 Hz, 1H), 3.59 (s, 2H). 13C NMR (101 MHz, DMSO, δ = ppm):169.90, 140.14, 136.55, 133.84, 133.12, 131.42, 130.45, 129.87, 128.13, 127.71, 125.25, 124.70, 124.52, 123.25, 121.54, 119.22, 118.93, 111.86, 108.57, 33.38. Anal. calcd. for C22H17BrN2O4S: C, 54.44; H, 3.53; N, 5.77; S, 6.61%. Found: C, 54.73; H, 4.64; N, 5.78; S, 6.56%.:4), colour: off-white solid, mp: 115–117 °C, FT-IR (, cm−1): 3438 (N–H, stretching), 2948 (C–H, stretching), 1688 (CO, stretching), 1086 (SO, stretching); 1H NMR (400 MHz, DMSO, δ = ppm): 10.95 (s, 1H), 9.47 (s, 1H), 7.82–7.73 (m, 2H), 7.53 (dd, J = 8.1, 1.7 Hz, 2H), 7.42 (d, J = 8.1 Hz, 1H), 7.36 (d, J = 8.1 Hz, 1H), 7.32–7.21 (m, 3H), 7.15 (td, J = 7.8, 1.7 Hz, 1H), 7.08 (m, 1H), 6.98 (m, 1H), 3.58 (s, 2H), 2.35 (s, 3H). 13C NMR (101 MHz, DMSO, δ = ppm): 169.93, 144.17, 140.15, 136.55, 134.67, 133.76, 132.69, 131.46, 128.58, 128.13, 127.70, 127.25, 125.23, 124.42, 123.40, 121.49, 119.15, 118.86, 111.83, 108.58, 33.30, 20.36. Anal. calcd. for C23H19ClN2O4S: C, 60.72; H, 4.21; N, 6.16; S, 7.05%. Found: C, 60.66; H, 4.17; N, 6.05; S, 7.13%.:4), colour: white to off-white solid, mp: 116–118 °C, FT-IR (, cm−1): 3438 (N–H, stretching), 2927 (C–H, stretching), 1618 (CO, stretching), 1081 (SO, stretching); 1H NMR (400 MHz, DMSO, δ = ppm): 10.93 (s, 1H), 9.40 (s, 1H), 8.68 (dd, J = 8.6, 1.1 Hz, 1H), 8.42 (d, J = 8.2 Hz, 1H), 8.20 (dd, J = 8.3, 1.4 Hz, 1H), 8.10 (dd, J = 7.5, 1.2 Hz, 1H), 7.83 (m, 1H), 7.75 (m, 1H), 7.72–7.62 (m, 2H), 7.49 (d, J = 7.9 Hz, 1H), 7.38–7.31 (m, 1H), 7.24–7.15 (m, 2H), 7.11–6.92 (m, 4H), 6.88 (dd, J = 8.2, 1.5 Hz, 1H), 3.37 (s, 2H). 13C NMR (101 MHz, DMSO, δ = ppm): 169.96, 141.04, 136.98, 136.54, 134.29, 131.79, 130.55, 129.88, 129.71, 128.02, 127.84, 127.65, 125.10, 124.68, 124.46, 122.40, 121.46, 119.08, 118.85, 111.79, 108.47, 33.08. Anal. calcd. for C26H21N3O4S: C, 66.23; H, 4.49; N, 8.91; S, 6.80%. Found: C, 66.16; H, 4.57; N, 8.79; S, 6.76%.:4), colour: white to off-white solid, mp: 169–172 °C, FT-IR (, cm−1): 3282 (N–H, stretching), 2947 (C–H, stretching), 1613 (CO, stretching), 1097 (SO, stretching); 1H NMR (400 MHz, DMSO, δ = ppm): 10.98 (s, 1H), 9.42 (s, 1H), 7.83 (d, J = 8.1 Hz, 2H), 7.76 (d, J = 8.0 Hz, 1H), 7.57 (d, J = 7.8 Hz, 1H), 7.37 (dd, J = 8.1, 3.4 Hz, 3H), 7.29–7.20 (m, 3H), 7.11 (dt, J = 15.3, 7.8 Hz, 2H), 7.00 (t, J = 7.4 Hz, 1H), 3.59 (s, 2H). 13C NMR (101 MHz, DMSO, δ = ppm): 169.89, 140.14, 138.94, 136.56, 134.21, 131.43, 129.91, 128.09, 127.72, 125.22, 124.65, 124.51, 123.22, 121.54, 119.24, 118.95, 111.86, 108.60, 104.88, 33.41. Anal. calcd. for C22H17IN2O4S: C, 49.64; H, 3.22; N, 5.26; S, 6.02%. Found: C, 49.76; H, 3.31; N, 5.29; S, 6.11%.:4), colour: off-white solid, mp: 114–116 °C, FT-IR (, cm−1): 3407 (N–H, stretching), 2922 (C–H, stretching), 1618 (CO, stretching), 1066 (SO, stretching); 1H NMR (400 MHz, DMSO, δ = ppm): 10.93 (s, 1H), 10.13 (s, 1H), 7.65–7.54 (m, 2H), 7.53–7.39 (m, 2H), 7.38–7.29 (m, 2H), 7.26 (d, J = 2.4 Hz, 1H), 7.07 (m, 1H), 6.98 (m, 1H), 3.77 (s, 2H). 13C NMR (101 MHz, DMSO, δ = ppm): 170.38, 143.06, 136.55, 131.08, 129.49, 127.75, 127.60, 127.10, 124.53, 122.52, 121.44, 119.04, 118.83, 111.78, 108.32, 32.99. Anal. calcd. for C17H13F3N2O4S: C, 51.26; H, 3.29; N, 7.03; S, 8.05%. Found: C, 51.22; H, 3.35; N, 7.08; S, 8.14%.:4), colour: off-white solid, mp: 139–141 °C, FT-IR (, cm−1): 3412 (N–H, stretching), 2958 (C–H, stretching), 1605 (CO, stretching), 1076 (SO, stretching); 1H NMR (400 MHz, DMSO, δ = ppm): 11.01 (s, 1H), 9.29 (s, 1H), 7.88–7.81 (m, 1H), 7.58 (d, J = 7.9 Hz, 1H), 7.37 (d, J = 8.1 Hz, 1H), 7.34–7.25 (m, 3H), 7.22–7.14 (m, 1H), 7.13–7.04 (m, 1H), 6.99 (t, J = 7.4 Hz, 1H), 3.80 (s, 2H), 1.21 (d, J = 6.8 Hz, 6H). 13C NMR (101 MHz, DMSO, δ = ppm): 170.28, 140.52, 136.63, 131.26, 127.58, 127.51, 125.68, 125.23, 124.92, 123.02, 121.60, 119.05, 119.02, 111.88, 108.25, 53.26, 33.81, 16.56. Anal. calcd. for C19H20N2O4S: C, 61.27; H, 5.41; N, 7.52; S, 8.61%. Found: C, 61.19; H, 5.36; N, 7.43; S, 8.73%.:4), colour: off-white solid, mp: 186–188 °C, FT-IR (, cm−1): 3376 (N–H, stretching), 2948 (C–H, stretching), 1598 (CO, stretching), 1071 (SO, stretching); 1H NMR (400 MHz, DMSO, δ = ppm): 10.98 (s, 1H), 9.44 (s, 1H), 7.75 (d, J = 8.1 Hz, 1H), 7.64 (d, J = 8.3 Hz, 2H), 7.56 (d, J = 8.0 Hz, 1H), 7.50 (d, J = 8.3 Hz, 2H), 7.37 (d, J = 8.1 Hz, 1H), 7.25 (q, J = 8.0 Hz, 3H), 7.11 (m, 2H), 6.99 (t, J = 7.5 Hz, 1H), 3.60 (s, 2H). 13C NMR (101 MHz, DMSO, δ = ppm): 169.91, 140.63, 140.15, 136.55, 133.39, 131.43, 130.50, 130.16, 128.14, 127.72, 125.26, 124.70, 124.53, 123.25, 121.54, 119.21, 118.92, 111.86, 108.56, 33.37. Anal. calcd. for C22H17ClN2O4S: C, 59.93; H, 3.89; N, 6.35; S, 7.27%. Found: C, 61.01; H, 3.86; N, 6.39; S, 7.25%.:4), colour: off-white solid, mp: 178–180 °C, FT-IR (, cm−1): 3428 (N–H, stretching), 2922 (C–H, stretching), 1613 (CO, stretching), 1073 (SO, stretching); 1H NMR (400 MHz, DMSO, δ = ppm): 10.93 (s, 1H), 9.53 (s, 1H), 9.19 (dd, J = 4.3, 1.7 Hz, 1H), 8.65 (dd, J = 8.4, 1.8 Hz, 1H), 8.49 (m, 2H), 8.05–7.98 (m, 1H), 7.84 (t, J = 7.8 Hz, 1H), 7.77 (dd, J = 8.3, 4.3 Hz, 1H), 7.48 (d, J = 7.9 Hz, 1H), 7.33 (dt, J = 8.1, 0.8 Hz, 1H), 7.25 (m, 2H), 7.14 (dd, J = 8.2, 1.6 Hz, 1H), 7.06 (m, 2H), 6.93 (m, 1H), 3.67 (s, 2H). 13C NMR (101 MHz, DMSO, δ = ppm): 170.16, 153.03, 143.46, 139.53, 138.03, 137.06, 136.55, 134.34, 132.25, 131.93, 129.42, 127.88, 127.63, 126.30, 124.84, 124.58, 123.64 (d, J = 6.2 Hz), 123.09, 121.49, 118.98, 118.89, 111.81, 108.27, 33.77. Anal. calcd. for C25H19N3O4S: C, 65.63; H, 4.19; N, 9.18; S, 7.01%. Found: C, 65.57; H, 4.24; N, 9.39; S, 7.25%.:4), colour: off-white solid, mp: 180–183 °C, FT-IR (, cm−1): 3431 (N–H, stretching), 2925 (C–H, stretching), 1649 (CO, stretching), 1083 (SO, stretching); 1H NMR 1H NMR (400 MHz, DMSO, δ = ppm): 10.96 (s, 1H), 9.42 (s, 1H), 8.06 (d, J = 5.0 Hz, 1H), 7.83 (d, J = 8.1 Hz, 1H), 7.56 (d, J = 8.1 Hz, 2H), 7.36 (d, J = 8.1 Hz, 1H), 7.29–7.18 (m, 3H), 7.17–7.03 (m, 3H), 6.99 (t, J = 7.6 Hz, 1H), 3.66 (s, 2H). 13C NMR (101 MHz, DMSO, δ = ppm): 170.01, 140.20, 137.76, 136.75, 136.57, 133.40, 131.72, 128.80, 128.15, 127.75, 125.05, 124.61, 124.37, 123.13, 121.51, 119.25, 118.90, 111.83, 108.61, 33.50. Anal. calcd. for C20H16N2O4S2: C, 58.24; H, 3.91; N, 6.79; S, 15.55%. Found: C, 58.31; H, 3.84; N, 6.70; S, 15.58%.:4), colour: white solid, mp: 160–162 °C, FT-IR (, cm−1): 3404 (N–H, stretching), 2955 (C–H, stretching), 1519 (CO, stretching), 1373 (SO, stretching); 1H NMR (400 MHz, DMSO, δ = ppm): 10.95 (s, 1H), 9.47 (s, 1H), 7.77 (m, 2H), 7.53 (dd = 8.06, 1.71 Hz, 2H), 7.42 (d = 8.11 Hz, 1H), 7.36 (d = 8.07 Hz, 1H), 7.26 (m, 3H), 7.15 (td = 7.79, 1.69 Hz, 1H), 7.08 (m, 1H), 6.98 (m, 1H), 3.58 (s, 2H), 2.35 (s, 3H).·13C NMR (101 MHz, DMSO, δ = ppm): 169.91, 140.63, 140.15, 136.55, 133.39, 131.43, 130.50, 130.16, 128.14, 127.72, 125.26, 124.70, 124.53, 123.25, 121.54, 119.21, 118.92, 111.86, 108.56, 33.37, 20.36. Anal. calcd. for C23H20N2O4S: C, 65.70; H, 4.79; N, 6.66; S, 7.62%. Found: C, 65.53; H, 4.84; N, 6.72; S, 7.55%.
, cm−1): 3404 (N–H, stretching), 2958 (C–H, stretching), 1613 (CO, stretching), 1085 (SO, stretching); 1H NMR (400 MHz, DMSO, δ = ppm): 10.98 (s, 1H), 9.44 (s, 1H), 7.75 (d, J = 8.1 Hz, 1H), 7.64 (d, J = 8.3 Hz, 2H), 7.56 (d, J = 8.0 Hz, 1H), 7.50 (d, J = 8.3 Hz, 2H), 7.37 (d, J = 8.1 Hz, 1H), 7.25 (q, J = 8.0 Hz, 3H), 7.11 (m, 2H), 6.99 (t, J = 7.5 Hz, 1H), 3.60 (s, 2H). 13C NMR (101 MHz, DMSO, δ = ppm): 169.90, 140.14, 136.55, 133.84, 133.12, 131.42, 130.45, 129.87, 128.13, 127.71, 125.25, 124.70, 124.52, 123.25, 121.54, 119.22, 118.93, 111.86, 108.57, 33.38. Anal. calcd. for C22H17ClN2O4S: C, 59.87; H, 3.89; N, 6.35; S, 7.27%. Found: C, 59.87; H, 3.88; N, 6.38; S, 7.34%.:4), colour: off-white solid, mp: 134–137 °C, FT-IR (, cm−1): 3172 (N–H, stretching), 2931 (C–H, stretching), 1613 (CO, stretching), 1085 (SO, stretching); 1H NMR (400 MHz, DMSO, δ = ppm): 10.99 (s, 1H), 9.36 (s, 1H), 7.91 (d, J = 8.1 Hz, 1H), 7.60 (d, J = 7.9 Hz, 1H), 7.39 (m, 3H), 7.39–7.25 (m, 5H), 7.22 (dd, J = 8.2, 1.7 Hz, 1H), 7.20–7.11 (m, 1H), 7.05 (t, J = 7.5 Hz, 1H), 6.97 (t, J = 7.4 Hz, 1H), 4.66 (s, 2H), 3.82 (s, 2H). 13C NMR (101 MHz, DMSO, δ = ppm): 170.44, 136.60, 131.59, 131.50, 129.38, 129.10, 127.96, 127.73, 127.61, 125.55, 124.93, 124.80, 123.06, 121.57, 119.07, 119.01, 111.90, 108.40, 56.52, 33.76. Anal. calcd. for C23H20N2O4S: C, 65.70; H, 4.79; N, 6.66; S, 7.62%. Found: C, 65.75; H, 4.73; N, 6.58; S, 7.60%.:4), colour: white solid, mp: 161–163 °C, FT-IR (, cm−1): 3453 (N–H, stretching), 2937 (C–H, stretching), 1608 (CO, stretching), 1092 (SO, stretching); 1H NMR (400 MHz, DMSO, δ = ppm): 10.98 (s, 1H), 9.42 (s, 1H), 7.83 (d, J = 8.1 Hz, 2H), 7.76 (d, J = 8.0 Hz, 1H), 7.57 (d, J = 7.8 Hz, 1H), 7.37 (m, 3H), 7.29–7.20 (m, 3H), 7.11 (m, 2H), 7.00 (t, J = 7.4 Hz, 1H), 3.59 (s, 2H). 13C NMR (101 MHz, DMSO, δ = ppm): 169.89, 140.14, 138.94, 136.56, 134.21, 131.43, 129.91, 128.09, 127.72, 125.22, 124.65, 124.51, 123.22, 121.54, 119.24, 118.95, 111.86, 108.60, 104.88, 33.41. Anal. calcd. for C22H17FN2O4S: C, 62.26; H, 4.04; N, 6.60; S, 7.55%. Found: C, 62.25; H, 4.13; N, 6.68; S, 7.62%.:4), colour: white to off-white solid, mp: 200–203 °C, FT-IR (, cm−1): 3438 (N–H, stretching), 2963 (C–H, stretching), 1655 (CO, stretching), 1071 (SO, stretching); 1H NMR (400 MHz, DMSO, δ = ppm): 11.01 (s, 1H), 9.32 (s, 1H), 7.80 (dd, J = 8.0, 1.6 Hz, 1H), 7.55 (t, J = 9.0 Hz, 3H), 7.38 (d, J = 8.1 Hz, 1H), 7.30 (d, J = 2.4 Hz, 1H), 7.22 (m, 2H), 7.10 (m, 2H), 7.00 (t, J = 7.4 Hz, 1H), 6.93–6.85 (m, 2H), 3.74 (s, 3H), 3.66 (s, 2H). 13C NMR (101 MHz, DMSO, δ = ppm): 169.93, 164.45, 136.58, 131.53, 131.04, 127.80, 127.74, 125.69, 124.92, 124.62, 124.11, 123.05, 121.56, 119.22, 118.93, 115.10, 111.86, 108.59, 56.32, 33.55. Anal. calcd. for C23H20N2O5S: C, 63.29; H, 4.62; N, 6.42; S, 7.35%. Found: C, 63.22; H, 4.55; N, 6.38; S, 7.29%.:4), colour: off-white solid, mp: 193–195 °C, FT-IR (, cm−1): 3395 (N–H, stretching), 2954 (C–H, stretching), 1601 (CO, stretching), 1076 (SO, stretching); 1H NMR (400 MHz, DMSO, δ = ppm): 10.99 (d, J = 2.5 Hz, 1H), 9.44 (s, 1H), 7.77 (dd, J = 8.2, 1.7 Hz, 1H), 7.70 (d, J = 8.6 Hz, 2H), 7.66 (d, J = 8.6 Hz, 2H), 7.62–7.40 (m, 6H), 7.40–7.29 (m, 2H), 7.28–7.19 (m, 2H), 7.20–7.04 (m, 2H), 6.96 (ddd, J = 7.9, 7.0, 1.0 Hz, 1H), 3.62 (s, 2H). 13C NMR (101 MHz, DMSO, δ = ppm): 169.86, 146.65, 140.12, 136.56, 133.16, 131.49, 129.60, 129.43, 129.27, 127.96, 127.92, 127.74, 127.65, 125.09, 124.54, 124.45, 123.21, 121.55, 119.25, 118.92, 111.87, 108.61, 33.44. Anal. calcd. for C28H22N2O4S: C, 69.69; H, 4.60; N, 5.81; S, 6.64%. Found: C, 69.73; H, 4.64; N, 5.78; S, 6.56%.:4), colour: off-white solid, mp: 183–186 °C, FT-IR (, cm−1): 3449 (N–H, stretching), 2963 (C–H, stretching), 1613 (CO, stretching), 1085 (SO, stretching); 1H NMR (400 MHz, DMSO, δ = ppm): 10.98 (s, 1H), 9.43 (s, 1H), 7.75 (d, J = 8.5 Hz, 1H), 7.65 (d, J = 8.5 Hz, 2H), 7.59–7.52 (m, 3H), 7.37 (d, J = 8.4 Hz, 1H), 7.25 (d, J = 8.6 Hz, 3H), 7.11 (m, 2H), 7.01 (d, J = 7.8 Hz, 1H), 3.59 (s, 2H). 13C NMR (101 MHz, DMSO, δ = ppm):169.90, 140.14, 136.55, 133.84, 133.12, 131.42, 130.45, 129.87, 128.13, 127.71, 125.25, 124.70, 124.52, 123.25, 121.54, 119.22, 118.93, 111.86, 108.57, 33.38. Anal. calcd. for C22H17BrN2O4S: C, 54.44; H, 3.53; N, 5.77; S, 6.61%. Found: C, 54.73; H, 4.64; N, 5.78; S, 6.56%.:4), colour: off-white solid, mp: 115–117 °C, FT-IR (, cm−1): 3438 (N–H, stretching), 2948 (C–H, stretching), 1688 (CO, stretching), 1086 (SO, stretching); 1H NMR (400 MHz, DMSO, δ = ppm): 10.95 (s, 1H), 9.47 (s, 1H), 7.82–7.73 (m, 2H), 7.53 (dd, J = 8.1, 1.7 Hz, 2H), 7.42 (d, J = 8.1 Hz, 1H), 7.36 (d, J = 8.1 Hz, 1H), 7.32–7.21 (m, 3H), 7.15 (td, J = 7.8, 1.7 Hz, 1H), 7.08 (m, 1H), 6.98 (m, 1H), 3.58 (s, 2H), 2.35 (s, 3H). 13C NMR (101 MHz, DMSO, δ = ppm): 169.93, 144.17, 140.15, 136.55, 134.67, 133.76, 132.69, 131.46, 128.58, 128.13, 127.70, 127.25, 125.23, 124.42, 123.40, 121.49, 119.15, 118.86, 111.83, 108.58, 33.30, 20.36. Anal. calcd. for C23H19ClN2O4S: C, 60.72; H, 4.21; N, 6.16; S, 7.05%. Found: C, 60.66; H, 4.17; N, 6.05; S, 7.13%.:4), colour: white to off-white solid, mp: 116–118 °C, FT-IR (, cm−1): 3438 (N–H, stretching), 2927 (C–H, stretching), 1618 (CO, stretching), 1081 (SO, stretching); 1H NMR (400 MHz, DMSO, δ = ppm): 10.93 (s, 1H), 9.40 (s, 1H), 8.68 (dd, J = 8.6, 1.1 Hz, 1H), 8.42 (d, J = 8.2 Hz, 1H), 8.20 (dd, J = 8.3, 1.4 Hz, 1H), 8.10 (dd, J = 7.5, 1.2 Hz, 1H), 7.83 (m, 1H), 7.75 (m, 1H), 7.72–7.62 (m, 2H), 7.49 (d, J = 7.9 Hz, 1H), 7.38–7.31 (m, 1H), 7.24–7.15 (m, 2H), 7.11–6.92 (m, 4H), 6.88 (dd, J = 8.2, 1.5 Hz, 1H), 3.37 (s, 2H). 13C NMR (101 MHz, DMSO, δ = ppm): 169.96, 141.04, 136.98, 136.54, 134.29, 131.79, 130.55, 129.88, 129.71, 128.02, 127.84, 127.65, 125.10, 124.68, 124.46, 122.40, 121.46, 119.08, 118.85, 111.79, 108.47, 33.08. Anal. calcd. for C26H21N3O4S: C, 66.23; H, 4.49; N, 8.91; S, 6.80%. Found: C, 66.16; H, 4.57; N, 8.79; S, 6.76%.:4), colour: white to off-white solid, mp: 169–172 °C, FT-IR (, cm−1): 3282 (N–H, stretching), 2947 (C–H, stretching), 1613 (CO, stretching), 1097 (SO, stretching); 1H NMR (400 MHz, DMSO, δ = ppm): 10.98 (s, 1H), 9.42 (s, 1H), 7.83 (d, J = 8.1 Hz, 2H), 7.76 (d, J = 8.0 Hz, 1H), 7.57 (d, J = 7.8 Hz, 1H), 7.37 (dd, J = 8.1, 3.4 Hz, 3H), 7.29–7.20 (m, 3H), 7.11 (dt, J = 15.3, 7.8 Hz, 2H), 7.00 (t, J = 7.4 Hz, 1H), 3.59 (s, 2H). 13C NMR (101 MHz, DMSO, δ = ppm): 169.89, 140.14, 138.94, 136.56, 134.21, 131.43, 129.91, 128.09, 127.72, 125.22, 124.65, 124.51, 123.22, 121.54, 119.24, 118.95, 111.86, 108.60, 104.88, 33.41. Anal. calcd. for C22H17IN2O4S: C, 49.64; H, 3.22; N, 5.26; S, 6.02%. Found: C, 49.76; H, 3.31; N, 5.29; S, 6.11%.:4), colour: off-white solid, mp: 114–116 °C, FT-IR (, cm−1): 3407 (N–H, stretching), 2922 (C–H, stretching), 1618 (CO, stretching), 1066 (SO, stretching); 1H NMR (400 MHz, DMSO, δ = ppm): 10.93 (s, 1H), 10.13 (s, 1H), 7.65–7.54 (m, 2H), 7.53–7.39 (m, 2H), 7.38–7.29 (m, 2H), 7.26 (d, J = 2.4 Hz, 1H), 7.07 (m, 1H), 6.98 (m, 1H), 3.77 (s, 2H). 13C NMR (101 MHz, DMSO, δ = ppm): 170.38, 143.06, 136.55, 131.08, 129.49, 127.75, 127.60, 127.10, 124.53, 122.52, 121.44, 119.04, 118.83, 111.78, 108.32, 32.99. Anal. calcd. for C17H13F3N2O4S: C, 51.26; H, 3.29; N, 7.03; S, 8.05%. Found: C, 51.22; H, 3.35; N, 7.08; S, 8.14%.:4), colour: off-white solid, mp: 139–141 °C, FT-IR (, cm−1): 3412 (N–H, stretching), 2958 (C–H, stretching), 1605 (CO, stretching), 1076 (SO, stretching); 1H NMR (400 MHz, DMSO, δ = ppm): 11.01 (s, 1H), 9.29 (s, 1H), 7.88–7.81 (m, 1H), 7.58 (d, J = 7.9 Hz, 1H), 7.37 (d, J = 8.1 Hz, 1H), 7.34–7.25 (m, 3H), 7.22–7.14 (m, 1H), 7.13–7.04 (m, 1H), 6.99 (t, J = 7.4 Hz, 1H), 3.80 (s, 2H), 1.21 (d, J = 6.8 Hz, 6H). 13C NMR (101 MHz, DMSO, δ = ppm): 170.28, 140.52, 136.63, 131.26, 127.58, 127.51, 125.68, 125.23, 124.92, 123.02, 121.60, 119.05, 119.02, 111.88, 108.25, 53.26, 33.81, 16.56. Anal. calcd. for C19H20N2O4S: C, 61.27; H, 5.41; N, 7.52; S, 8.61%. Found: C, 61.19; H, 5.36; N, 7.43; S, 8.73%.:4), colour: off-white solid, mp: 186–188 °C, FT-IR (, cm−1): 3376 (N–H, stretching), 2948 (C–H, stretching), 1598 (CO, stretching), 1071 (SO, stretching); 1H NMR (400 MHz, DMSO, δ = ppm): 10.98 (s, 1H), 9.44 (s, 1H), 7.75 (d, J = 8.1 Hz, 1H), 7.64 (d, J = 8.3 Hz, 2H), 7.56 (d, J = 8.0 Hz, 1H), 7.50 (d, J = 8.3 Hz, 2H), 7.37 (d, J = 8.1 Hz, 1H), 7.25 (q, J = 8.0 Hz, 3H), 7.11 (m, 2H), 6.99 (t, J = 7.5 Hz, 1H), 3.60 (s, 2H). 13C NMR (101 MHz, DMSO, δ = ppm): 169.91, 140.63, 140.15, 136.55, 133.39, 131.43, 130.50, 130.16, 128.14, 127.72, 125.26, 124.70, 124.53, 123.25, 121.54, 119.21, 118.92, 111.86, 108.56, 33.37. Anal. calcd. for C22H17ClN2O4S: C, 59.93; H, 3.89; N, 6.35; S, 7.27%. Found: C, 61.01; H, 3.86; N, 6.39; S, 7.25%.:4), colour: off-white solid, mp: 178–180 °C, FT-IR (, cm−1): 3428 (N–H, stretching), 2922 (C–H, stretching), 1613 (CO, stretching), 1073 (SO, stretching); 1H NMR (400 MHz, DMSO, δ = ppm): 10.93 (s, 1H), 9.53 (s, 1H), 9.19 (dd, J = 4.3, 1.7 Hz, 1H), 8.65 (dd, J = 8.4, 1.8 Hz, 1H), 8.49 (m, 2H), 8.05–7.98 (m, 1H), 7.84 (t, J = 7.8 Hz, 1H), 7.77 (dd, J = 8.3, 4.3 Hz, 1H), 7.48 (d, J = 7.9 Hz, 1H), 7.33 (dt, J = 8.1, 0.8 Hz, 1H), 7.25 (m, 2H), 7.14 (dd, J = 8.2, 1.6 Hz, 1H), 7.06 (m, 2H), 6.93 (m, 1H), 3.67 (s, 2H). 13C NMR (101 MHz, DMSO, δ = ppm): 170.16, 153.03, 143.46, 139.53, 138.03, 137.06, 136.55, 134.34, 132.25, 131.93, 129.42, 127.88, 127.63, 126.30, 124.84, 124.58, 123.64 (d, J = 6.2 Hz), 123.09, 121.49, 118.98, 118.89, 111.81, 108.27, 33.77. Anal. calcd. for C25H19N3O4S: C, 65.63; H, 4.19; N, 9.18; S, 7.01%. Found: C, 65.57; H, 4.24; N, 9.39; S, 7.25%.:4), colour: off-white solid, mp: 180–183 °C, FT-IR (, cm−1): 3431 (N–H, stretching), 2925 (C–H, stretching), 1649 (CO, stretching), 1083 (SO, stretching); 1H NMR 1H NMR (400 MHz, DMSO, δ = ppm): 10.96 (s, 1H), 9.42 (s, 1H), 8.06 (d, J = 5.0 Hz, 1H), 7.83 (d, J = 8.1 Hz, 1H), 7.56 (d, J = 8.1 Hz, 2H), 7.36 (d, J = 8.1 Hz, 1H), 7.29–7.18 (m, 3H), 7.17–7.03 (m, 3H), 6.99 (t, J = 7.6 Hz, 1H), 3.66 (s, 2H). 13C NMR (101 MHz, DMSO, δ = ppm): 170.01, 140.20, 137.76, 136.75, 136.57, 133.40, 131.72, 128.80, 128.15, 127.75, 125.05, 124.61, 124.37, 123.13, 121.51, 119.25, 118.90, 111.83, 108.61, 33.50. Anal. calcd. for C20H16N2O4S2: C, 58.24; H, 3.91; N, 6.79; S, 15.55%. Found: C, 58.31; H, 3.84; N, 6.70; S, 15.58%.:4), colour: white solid, mp: 160–162 °C, FT-IR (, cm−1): 3404 (N–H, stretching), 2955 (C–H, stretching), 1519 (CO, stretching), 1373 (SO, stretching); 1H NMR (400 MHz, DMSO, δ = ppm): 10.95 (s, 1H), 9.47 (s, 1H), 7.77 (m, 2H), 7.53 (dd = 8.06, 1.71 Hz, 2H), 7.42 (d = 8.11 Hz, 1H), 7.36 (d = 8.07 Hz, 1H), 7.26 (m, 3H), 7.15 (td = 7.79, 1.69 Hz, 1H), 7.08 (m, 1H), 6.98 (m, 1H), 3.58 (s, 2H), 2.35 (s, 3H).·13C NMR (101 MHz, DMSO, δ = ppm): 169.91, 140.63, 140.15, 136.55, 133.39, 131.43, 130.50, 130.16, 128.14, 127.72, 125.26, 124.70, 124.53, 123.25, 121.54, 119.21, 118.92, 111.86, 108.56, 33.37, 20.36. Anal. calcd. for C23H20N2O4S: C, 65.70; H, 4.79; N, 6.66; S, 7.62%. Found: C, 65.53; H, 4.84; N, 6.72; S, 7.55%.Enzyme inhibition assays
Enzyme kinetics studies
The enzyme kinetic research was carried out utilizing a series of substrate concentrations of 0, 1.25, 2.5, 5.0, 7.5, and 10 mM. For most potent inhibitors 5c, 5e, 5g, 5i and 5j concentrations utilized were based on the IC50 values from in vitro assays with two concentrations above and two below the determined IC50 value concentrations. The test plate was made by adding the enzyme per well (in vitro assay concentration of respective enzyme assays afore mentioned) to the assay buffer. A microplate reader (BIOTEK ELX800™, Instruments, Inc., USA) was used to record the reading after the assay mixture had been incubated at 37 °C for 25 minutes. Lineweaver–Burk graphs with PRISM 5.0 (Graph Pad, San Diego, California, USA) was used to analyze the data.Molecular docking
The Molecular Operating Environment software (MOE) 2019.0102 (ref. 82) was employed to perform molecular docking studies on the synthesized derivatives (5a–5o). The docking studies was carried out by using the Protein data Bank https://www.rcsb.org/ under PDB ID 6WEW and 4H2G for human ectonucleotide pyrophosphatase/phosphodiesterase 1 (h-ENPP1) and h-e5′NT while homology models were used for r-e5′NT, h-ENPP3 and h-TNAP. Standard inhibitors were docked along with synthesized ligands to validate the docking results. N-(4-[(7-methoxyquinolin-4-yl)oxy]phenyl)sulfuric diamide (TZV) a co-crystal was used as standard for both human h-ENPP1 and h-ENPP3. Suramin was taken as standard inhibitor for human and rat ecto-5′-nucleotidase and in case of h-TNAP, levamisole was considered as standard inhibitor.Chemical structures were sketched using chemDraw Ultra 12.0 and energy was minimized using MMFF-94 force field implemented in MOE.83 The addition of hydrogen atoms was carried out to the model structure and then ionization states of metals ions were prepared to be fixed. Docking was performed by using the refinement features where induced fit receptor and triangular matcher was selected. The best pose was selected which exhibited top-ranked minimum binding energy and RMSD value less than 2.84 The re-docking of the identified derivatives was carried out for the validation of the docking results.
Author contributions
Muhammad Siraj Khan Jadoon: conceptualization, synthesis, characterization, methodology, formal analysis, validation investigation (bioactivity), docking studies, writing – original draft preparation, software. Julie Pelletier, Jean Sévigny: expression of recombinant enzymes. Jamshed Iqbal: supervision, resources (synthesis), data curation, writing – original draft reviewing and editing, supervision, resources (bioactivity and docking), funding acquisition.Conflicts of interest
The authors declare that they have no significant conflict of interest.Acknowledgements
The authors gratefully acknowledge the financial support for this research provided by the Higher Education Commission of Pakistan (HEC) via NRPU project no. 20-15846/NRPU/R&D/HEC/2021-2020, German–Pakistani Research Collaboration Program and Equipment Grant funded by DAAD, Germany. J. S. received support from the Natural Sciences and Engineering Research Council of Canada (RGPIN-2023-05498).References
- J. Candido and T. Hagemann, Cancer-related inflammation, J. Clin. Immunol., 2013, 33, 79–84 CrossRef CAS PubMed.
- A. Kalbasi and A. Ribas, Tumour-intrinsic resistance to immune checkpoint blockade, Nat. Rev. Immunol., 2020, 20(1), 25–39 CrossRef CAS PubMed.
- D. Bedognetti, et al., Toward a comprehensive view of cancer immune responsiveness: a synopsis from the SITC workshop, J. Immunother. Cancer, 2019, 7(1), 1–23 CrossRef PubMed.
- P. Sharma and J. P. Allison, Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential, Cell, 2015, 161(2), 205–214 CrossRef CAS PubMed.
- J. Stagg and M. Smyth, Extracellular adenosine triphosphate and adenosine in cancer, Oncogene, 2010, 29(39), 5346–5358 CrossRef CAS PubMed.
- L.-l. Feng, et al., The yin and yang functions of extracellular ATP and adenosine in tumor immunity, Cancer Cell Int., 2020, 20(1), 1–11 CrossRef PubMed.
- H. Zimmermann, 5'-Nucleotidase: molecular structure and functional aspects, Biochem. J., 1992, 285(Pt 2), 345 CrossRef CAS PubMed.
- L. Antonioli, et al., CD39 and CD73 in immunity and inflammation, Trends Mol. Med., 2013, 19(6), 355–367 CrossRef CAS PubMed.
- N. Sträter, Ecto-5′-nucleotidase: Structure function relationships, Purinergic Signal., 2006, 2(2), 343 CrossRef PubMed.
- D. Allard, et al., Targeting the CD73-adenosine axis in immuno-oncology, Immunol. Lett., 2019, 205, 31–39 CrossRef CAS PubMed.
- J. L. Millán, Alkaline phosphatases: structure, substrate specificity and functional relatedness to other members of a large superfamily of enzymes, Purinergic Signal., 2006, 2, 335–341 CrossRef PubMed.
- M. Picher, et al., Ecto 5′-nucleotidase and nonspecific alkaline phosphatase: two AMP-hydrolyzing ectoenzymes with distinct roles in human airways, J. Biol. Chem., 2003, 278(15), 13468–13479 CrossRef CAS PubMed.
- M. al-Rashida and J. Iqbal, Therapeutic potentials of ecto-nucleoside triphosphate diphosphohydrolase, ecto-nucleotide pyrophosphatase/phosphodiesterase, ecto-5′-nucleotidase, and alkaline phosphatase inhibitors, Med. Res. Rev., 2014, 34(4), 703–743 CrossRef CAS PubMed.
- D. Vijayan, et al., Targeting immunosuppressive adenosine in cancer, Nat. Rev. Cancer, 2017, 17(12), 709–724 CrossRef CAS PubMed.
- S. C. Robson, J. Sévigny and H. Zimmermann, The E-NTPDase family of ectonucleotidases: structure function relationships and pathophysiological significance, Purinergic Signal., 2006, 2, 409–430 CrossRef CAS PubMed.
- J. B. Volmer, L. F. Thompson and M. R. Blackburn, Ecto-5′-nucleotidase (CD73)-mediated adenosine production is tissue protective in a model of bleomycin-induced lung injury, J. Immun., 2006, 176(7), 4449–4458 CrossRef CAS PubMed.
- M. R. C. Schetinger, et al., NTPDase and 5′-nucleotidase activities in physiological and disease conditions: New perspectives for human health, Biofactors, 2007, 31(2), 77–98 CrossRef CAS PubMed.
- A. El-Tayeb, et al., Nucleoside-5′-monophosphates as prodrugs of adenosine A2A receptor agonists activated by ecto-5′-nucleotidase, J. Med. Chem., 2009, 52(23), 7669–7677 CrossRef CAS PubMed.
- B. B. Fredholm, et al., International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors—an update, Pharmacol. Rev., 2011, 63(1), 1–34 CrossRef CAS PubMed.
- S. Ryzhov, et al., Adenosinergic regulation of the expansion and immunosuppressive activity of CD11b+ Gr1+ cells, J. Immun., 2011, 187(11), 6120–6129 CrossRef CAS PubMed.
- A. Buffon, et al., Differential expression of nucleotide pyrophosphatase/phosphodiesterases by Walker 256 mammary cancer cells in solid tumors and malignant ascites, Life Sci., 2010, 86(11–12), 435–440 CrossRef CAS PubMed.
- N. A. Sowa, M. K. Voss and M. J. Zylka, Recombinant ecto-5′-nucleotidase (CD73) has long lasting antinociceptive effects that are dependent on adenosine A1 receptor activation, Mol. Pain, 2010, 6, 20 CrossRef PubMed.
- I. D. Goldfine, et al., The role of membrane glycoprotein plasma cell antigen 1/ectonucleotide pyrophosphatase phosphodiesterase 1 in the pathogenesis of insulin resistance and related abnormalities, Endocr. Rev., 2008, 29(1), 62–75 CrossRef CAS PubMed.
- L. Bavaresco, et al., The role of ecto-5′-nucleotidase/CD73 in glioma cell line proliferation, Mol. Cell. Biochem., 2008, 319, 61–68 CrossRef CAS PubMed.
- J. Stella, et al., Differential ectonucleotidase expression in human bladder cancer cell lines, in Urologic Oncology: Seminars and Original Investigations, Elsevier, 2010 Search PubMed.
- J. Spychala, et al., Role of estrogen receptor in the regulation of ecto-5′-nucleotidase and adenosine in breast cancer, Clin. Cancer Res., 2004, 10(2), 708–717 CrossRef CAS PubMed.
- L. Antonioli, et al., Switching off CD73: a way to boost the activity of conventional and targeted antineoplastic therapies, Drug Discovery Today, 2017, 22(11), 1686–1696 CrossRef CAS PubMed.
- I. Perrot, et al., Blocking antibodies targeting the CD39/CD73 immunosuppressive pathway unleash immune responses in combination cancer therapies, Cell Rep., 2019, 27(8), 2411–2425 CrossRef CAS PubMed.
- N. Sträter, Ecto-5′-nucleotidase: Structure function relationships, Purinergic Signal., 2006, 2, 343–350 CrossRef PubMed.
- R. Sadej, J. Spychala and A. C. Skladanowski, Expression of ecto-5′-nucleotidase (eN, CD73) in cell lines from various stages of human melanoma, Melanoma Res., 2006, 16(3), 213–222 CrossRef CAS PubMed.
- R. Resta, Y. Yamashita and L. F. Thompson, Ecto-enzyme and signaling functions of lymphocyte CD 7 3, Immunol. Rev., 1998, 161(1), 95–109 CrossRef CAS PubMed.
- J. Spychala, Tumor-promoting functions of adenosine, Pharmacol. Ther., 2000, 87(2–3), 161–173 CrossRef CAS PubMed.
- F. Gendron, et al., Purine signaling and potential new therapeutic approach: possible outcomes of NTPDase inhibition, Curr. Drug Targets, 2002, 3(3), 229–245 CAS.
- J. Iqbal, et al., A highly sensitive CE-UV method with dynamic coating of silica-fused capillaries for monitoring of nucleotide pyrophosphatase/phosphodiesterase reactions, Electrophoresis, 2008, 29(17), 3685–3693 CrossRef CAS PubMed.
- A. Tokumura, et al., Identification of human plasma lysophospholipase D, a lysophosphatidic acid-producing enzyme, as autotaxin, a multifunctional phosphodiesterase, J. Biol. Chem., 2002, 277(42), 39436–39442 CrossRef CAS PubMed.
- H.-J. Bühring, A. Streble and P. Valent, The basophil-specific ectoenzyme E-NPP3 (CD203c) as a marker for cell activation and allergy diagnosis, Int. Arch. Allergy Appl. Immunol., 2004, 133(4), 317–329 CrossRef PubMed.
- C. Stefan, S. Jansen and M. Bollen, Modulation of purinergic signaling by NPP-type ectophosphodiesterases, Purinergic Signal., 2006, 2, 361–370 CrossRef CAS PubMed.
- B. Grobben, P. De Deyn and H. Slegers, Rat C6 glioma as experimental model system for the study of glioblastoma growth and invasion, Cell Tissue Res., 2002, 310, 257–270 CrossRef CAS PubMed.
- I. Aerts, et al., The expression of ecto-nucleotide pyrophosphatase/phosphodiesterase 1 (E-NPP1) is correlated with astrocytic tumor grade, Clin. Neurol. Neurosurg., 2011, 113(3), 224–229 CrossRef PubMed.
- N. Ruf, et al., The mutational spectrum of ENPP1 as arising after the analysis of 23 unrelated patients with generalized arterial calcification of infancy (GACI), Hum. Mutat., 2005, 25(1), 98 CrossRef PubMed.
- M.-L. He, et al., Release and extracellular metabolism of ATP by ecto-nucleotidase eNTPDase 1–2 in hypothalamic and pituitary cells, Purinergic Signalling, 2005, 1, 135–144 CrossRef CAS PubMed.
- U. Sharma, D. Pal and R. Prasad, A novel role of alkaline phosphatase in the ERK1/2 dephosphorylation in renal cell carcinoma cell lines: a new plausible therapeutic target, Biochimie, 2014, 107, 406–409 CrossRef CAS PubMed.
- A. Kozlenkov, et al., Residues determining the binding specificity of uncompetitive inhibitors to tissue-nonspecific alkaline phosphatase, J. Bone Miner. Res., 2004, 19(11), 1862–1872 CrossRef CAS PubMed.
- J. M. Kim, et al., The effect of alkaline phosphatase and intrahepatic metastases in large hepatocellular carcinoma, World J. Surg. Oncol., 2013, 11, 1–8 CrossRef PubMed.
- I. Ali, et al., Heterocyclic scaffolds: centrality in anticancer drug development, Curr. Drug Targets, 2015, 16(7), 711–734 CrossRef CAS PubMed.
- S. S. Gholap, Pyrrole: An emerging scaffold for construction of valuable therapeutic agents, Eur. J. Med. Chem., 2016, 110, 13–31 CrossRef CAS PubMed.
- S. Ahmad, et al., Pyrrole: An insight into recent pharmacological advances with structure activity relationship, Eur. J. Med. Chem., 2018, 157, 527–561 CrossRef CAS PubMed.
- M. A. Tantawy, et al., Auspicious role of the steroidal heterocyclic derivatives as a platform for anti-cancer drugs, Bioorg. Chem., 2017, 73, 128–146 CrossRef CAS PubMed.
- G. M. das Neves, et al., Targeting ecto-5′-nucleotidase: A comprehensive review into small molecule inhibitors and expression modulators, Eur. J. Med. Chem., 2023, 247, 115052 CrossRef CAS PubMed.
- J. L. Jeffrey, K. V. Lawson and J. P. Powers, Targeting metabolism of extracellular nucleotides via inhibition of ectonucleotidases CD73 and CD39, J. Med. Chem., 2020, 63(22), 13444–13465 CrossRef CAS PubMed.
- L. Schäkel, et al., Nucleotide analog ARL67156 as a lead structure for the development of CD39 and dual CD39/CD73 ectonucleotidase inhibitors, Front. Pharmacol, 2020, 11, 1294 CrossRef PubMed.
- K. V. Lawson, et al., Discovery of AB680: a potent and selective inhibitor of CD73, J. Med. Chem., 2020, 63(20), 11448–11468 CrossRef CAS PubMed.
- M. I. Choudhary, et al., New biscoumarin derivatives-cytotoxicity and enzyme inhibitory activities, Bioorg. Med. Chem., 2006, 14(23), 8066–8072 CrossRef CAS PubMed.
- Y. Baqi, et al., Development of potent and selective inhibitors of ecto-5′-nucleotidase based on an anthraquinone scaffold, J. Med. Chem., 2010, 53(5), 2076–2086 CrossRef CAS PubMed.
- P. Frasson Corbelini, et al., Insights into Ecto-5’-nucleotidase as a new target for cancer therapy: a medicinal chemistry study, Curr. Med. Chem., 2015, 22(15), 1776–1792 CrossRef PubMed.
- R. B. Giordani, et al., Trichomonas vaginalis nucleoside triphosphate diphosphohydrolase and ecto-5′-nucleotidase activities are inhibited by lycorine and candimine, Parasitol. Int., 2010, 59(2), 226–231 CrossRef CAS PubMed.
- A. Fiene, et al., Inhibitors for the bacterial ectonucleotidase Lp1NTPDase from Legionella pneumophila, Bioorg. Med. Chem., 2016, 24(18), 4363–4371 CrossRef CAS PubMed.
- S. Ullah, et al., Synthesis and Biological Evaluation of Arylamide Sulphonate Derivatives as Ectonucleotide Pyrophosphatase/Phosphodiesterase-1 and-3 Inhibitors, ACS Omega, 2022, 7(30), 26905–26918 CrossRef CAS PubMed.
- H. Ahmad, et al., Synthesis of biphenyl oxazole derivatives via Suzuki coupling and biological evaluations as nucleotide pyrophosphatase/phosphodiesterase-1 and-3 inhibitors, Eur. J. Med. Chem., 2020, 208, 112759 CrossRef CAS PubMed.
- M. I. El-Gamal, et al., Synthesis, biological evaluation, and docking studies of new raloxifene sulfonate or sulfamate derivatives as inhibitors of nucleotide pyrophosphatase/phosphodiesterase, Eur. J. Med. Chem., 2019, 181, 111560 CrossRef PubMed.
- A. I. Shahin, et al., Design and synthesis of new adamantyl derivatives as promising antiproliferative agents, Eur. J. Med. Chem., 2023, 246, 114958 CrossRef CAS PubMed.
- M. Lind, Principles of systemic anticancer therapy, Medicine, 2020, 48(2), 90–96 CrossRef.
- L. Cyr, et al., Antiproliferative effects of a series of novel synthetic sulfonate esters on human breast cancer cell line MCF-7, Anticancer Res., 2007, 27(3B), 1437–1448 CAS.
- S. Ullah, et al., Synthesis, biological evaluation, and docking studies of new pyrazole-based thiourea and sulfonamide derivatives as inhibitors of nucleotide pyrophosphatase/phosphodiesterase, Bioorg. Chem., 2020, 99, 103783 CrossRef CAS PubMed.
- E. Mahmoud, et al., Recent progress in biologically active indole hybrids: A mini review, Pharmacol. Rep., 2022, 74(4), 570–582 CrossRef CAS PubMed.
- D. Kumar, et al., Synthesis and identification of α-cyano bis (indolyl) chalcones as novel anticancer agents, Bioorganic Med. Chem. Lett., 2014, 24(22), 5170–5174 CrossRef CAS PubMed.
- A. J. Yee and N. S. Raje, Panobinostat and multiple myeloma in 2018, Oncologist, 2018, 23(5), 516–517 CrossRef PubMed.
- S. Peters, et al., Alectinib versus crizotinib in untreated ALK-positive non–small-cell lung cancer, N. Engl. J. Med., 2017, 377(9), 829–838 CrossRef CAS PubMed.
- R. J. Motzer, et al., Sunitinib in patients with metastatic renal cell carcinoma, JAMA, 2006, 295(21), 2516–2524 CrossRef CAS PubMed.
- J.-C. Soria, et al., Osimertinib in untreated EGFR-mutated advanced non–small-cell lung cancer, N. Engl. J. Med., 2018, 378(2), 113–125 CrossRef CAS PubMed.
- G. J. Roth, et al., Nintedanib: from discovery to the clinic, J. Med. Chem., 2015, 58(3), 1053–1063 CrossRef CAS PubMed.
- S. I. Belli and J. W. Goding, Biochemical characterization of human PC-1, an enzyme possessing alkaline phosphodiesterase I and nucleotide pyrophosphatase activities, Eur. J. Biochem., 1994, 226(2), 433–443 CrossRef CAS PubMed.
- P. Jin-Hua, et al., Molecular cloning and chromosomal localization of PD-Iβ (PDNP3), a new member of the human phosphodiesterase I genes, Genomics, 1997, 45(2), 412–415 CrossRef CAS PubMed.
- J. Lecka, M. S. Rana and J. Sévigny, Inhibition of vascular ectonucleotidase activities by the pro-drugs ticlopidine and clopidogrel favours platelet aggregation, Br. J. Pharmacol., 2010, 161(5), 1150–1160 CrossRef CAS PubMed.
- Y. Misumi, et al., Primary structure of rat liver 5'-nucleotidase deduced from the cDNA. Presence of the COOH-terminal hydrophobic domain for possible post-translational modification by glycophospholipid, J. Biol. Chem., 1990, 265(4), 2178–2183 CrossRef CAS PubMed.
- T. Kiffer-Moreira, et al., Catalytic signature of a heat-stable, chimeric human alkaline phosphatase with therapeutic potential, PLoS One, 2014, 9(2), e89374 CrossRef PubMed.
- S. Lévesque, et al., Specificity of the ecto-ATPase inhibitor ARL 67156 on human and mouse ectonucleotidases, Br. J. Pharmacol., 2007, 152(1), 141–150 CrossRef PubMed.
- M. M. Bradford, A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding, Anal. Biochem., 1976, 72(1–2), 248–254 CrossRef CAS PubMed.
- S. Ullah, et al., Synthesis, biological evaluation, and docking studies of novel pyrrolo [2, 3-b] pyridine derivatives as both ectonucleotide pyrophosphatase/phosphodiesterase inhibitors and antiproliferative agents, Eur. J. Med. Chem., 2021, 217, 113339 CrossRef CAS PubMed.
- M. Adzic and N. Nedeljkovic, Unveiling the role of ecto-5′-nucleotidase/CD73 in astrocyte migration by using pharmacological tools, Front. Pharmacol., 2018, 9, 153 CrossRef CAS PubMed.
- I. Khan, et al., Influence of the diversified structural variations at the imine functionality of 4-bromophenylacetic acid derived hydrazones on alkaline phosphatase inhibition: synthesis and molecular modelling studies, RSC Adv., 2015, 5(110), 90806–90818 RSC.
- Molecular Operating Environment (MOE), Chemical Computing Group ULC, 1010 Sherbrooke St. West, Suite #910, Montreal, AC, Canada, H3A 2R7, 2020 Search PubMed.
- L. Heinzerling, R. Klein and M. Rarey, Fast force field-based optimization of protein–ligand complexes with graphics processor, J. Comput. Chem., 2012, 33(32), 2554–2565 CrossRef CAS PubMed.
- Z. Wang, et al., Comprehensive evaluation of ten docking programs on a diverse set of protein–ligand complexes: the prediction accuracy of sampling power and scoring power, Phys. Chem. Chem. Phys., 2016, 18(18), 12964–12975 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra04266a |
| This journal is © The Royal Society of Chemistry 2023 |