
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergistic effect of coordinating interface and promoter for enhancing ammonia synthesis activity of Ru@N–C catalyst†
Dongwei
Wang
ab,
Zhanwei
Ma
 *a,
Farong
Gou
a and
Bin
Hu
*a
*a,
Farong
Gou
a and
Bin
Hu
*a
aState Key Laboratory for Oxo Synthesis and Selective Oxidation, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou 730000, China. E-mail: zhanweima@licp.cas.cn; hcom@licp.cas.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 2nd October 2023
Abstract
Triruthenium dodecacarbonyl (Ru3(CO)12) was applied to prepare the Ru-based ammonia synthesis catalysts. The catalyst obtained from this precursor exhibited higher activity than the other Ru salts owing to its unique atomic reorganization under mild temperatures. Herein, Ru3(CO)12 as a guest metal source incorporated into the pore of ZIF-8 formed the Ru@N–C catalysts. The results indicated that the Ru nanoparticle (1.7 nm) was dispersed in the confined N coordination environment, which can increase the electron density of the Ru nanoparticles to promote N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond cleavage. The promoters donate the basic sites for transferring the electrons to Ru nanoparticles, further enhancing ammonia synthesis activity. Ammonia synthesis investigations revealed that the obtained Ru@N–C catalysts exhibited obvious catalytic activity compared with the Ru/AC catalyst. After introducing the Ba promoter, the 2Ba–Ru@N–C(450) catalyst exhibited the highest ammonia synthesis activity among the catalysts. At 360 °C and 1 MPa, the activity of the 2Ba–Ru@N–C(450) is 16
N bond cleavage. The promoters donate the basic sites for transferring the electrons to Ru nanoparticles, further enhancing ammonia synthesis activity. Ammonia synthesis investigations revealed that the obtained Ru@N–C catalysts exhibited obvious catalytic activity compared with the Ru/AC catalyst. After introducing the Ba promoter, the 2Ba–Ru@N–C(450) catalyst exhibited the highest ammonia synthesis activity among the catalysts. At 360 °C and 1 MPa, the activity of the 2Ba–Ru@N–C(450) is 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 817.3 µmol h−1 gRu−1, which is 1.1, 1.6, and 2 times higher than those of 2Cs–Ru@N–C(450) (14925.4 µmol h−1 gRu−1), 2K–Ru@N–C(450) (10736.7 µmol h−1 gRu−1), and Ru@N–C(450) (8604.2 µmol h−1 gRu−1), respectively. A series of characterizations were carried out to explore the 2Ba–Ru@N–C(450) catalysts, such as H2-TPR, XPS, and NH3-TPD. These results suggest that the Ba promoter played the role of an electronic and structural promoter; moreover, it can promote the NH3 desorption from the Ru nanoparticles.
817.3 µmol h−1 gRu−1, which is 1.1, 1.6, and 2 times higher than those of 2Cs–Ru@N–C(450) (14925.4 µmol h−1 gRu−1), 2K–Ru@N–C(450) (10736.7 µmol h−1 gRu−1), and Ru@N–C(450) (8604.2 µmol h−1 gRu−1), respectively. A series of characterizations were carried out to explore the 2Ba–Ru@N–C(450) catalysts, such as H2-TPR, XPS, and NH3-TPD. These results suggest that the Ba promoter played the role of an electronic and structural promoter; moreover, it can promote the NH3 desorption from the Ru nanoparticles.
1. Introduction
Ammonia is a vitally important fertilizer feedstock, chemical precursor, and viable chemical energy carrier. Currently, world ammonia production has reached more than 140 million tons per year, which consumes 1–2% of the world's energy using the Haber–Bosch process with high operating temperature (400–500 °C) and pressure (20–30 MPa).1 As a result, much effort has been paid to Ru-based catalysts with much higher catalytic activity under mild conditions relative to those of the conventional Fe-based catalysts. To date, none but graphite carbon-supported Ru catalysts (Ru–Ba–Cs/graphite) have been used in the ammonia synthesis industry. To further enhance the catalytic activity, many studies have focused on the Ru morphology,2–4 support materials,5–7 and promoters.8–10 However, developing an efficient Ru-based catalyst is still the greatest challenge.Ammonia synthesis over Ru nanoparticles is a structurally sensitive reaction,11 and it is observed that there is a close relationship between Ru particle size and catalytic performance. Moreover, the injection of electrons into the antibonding π*-orbital of the N2 molecule can promote the NN bond cleavage.9 Thus, accurate control over the Ru nanostructure and interfacial electronic environment is crucial for enhancing ammonia synthesis activity. The metal support interaction effects can change the interfacial electronic environment for altering the catalytic activity, such as the strong metal support interaction12 and electronic metal support interaction.13,14 The results clearly demonstrated that tuning the nature of the interfacial boundary or interfacial bonding environment is an important strategy to enhance the catalytic ammonia synthesis performance.12,15 For example, the ammonia synthesis activity of N-doped carbon nanotubes loaded with Ru was 3–5 times that of un-doped carbon nanotubes under mild reaction conditions, which was attributed to the electron-donating nitrogen bonding with Ru and the graphitization.16 Shao et al.17 confirmed that carbon nanotube surface after N atoms doping intensified the electron donating effect against metal and promoted the dispersion of metal particles and catalytic performance. Li et al.18 reported that N in the carbon material can have a stronger interaction with Ru and thus increase the electron density of Ru. Ma et al.5 reported that the load of Ru on an electron-rich graphitic carbon nitride (g-C3N4) led to a Ru-dispersed layer with a mean diameter of 3.2 nm, which contributed to interfacial N bonding with Ru. In summary, tuning the interfacial coordinating environment at the atom level can tune the electronic property and control the Ru nanostructure. To achieve the modulation of the metal-support interface at the atomic scale, it has been proved that metal organic frameworks (MOF) could host the organometallic molecule and further produce small nano metal, which is attributed to the metal bonding with the metal nodes or N atoms in situ to form bimetallic clusters or metal-N species.19 Recently, Wu et al.20 reported that Fe(acac)3 as a guest metal source can be incorporated into the cavities of MOF during the in situ synthesis process, thus increasing access to the node-coordinated Cu ions for efficient Fe–Cu diatomic site generation. Using Ru3(CO)12 as a precursor, Li et al.21 reasonably designed the assembly of Ru and Co in the limited space of ZIF and carried out fine control on the atomic scale. Inspired by these studies, the direct thermolysis of organometallic molecules in a confined space is indeed a promising method for the controlled synthesis of Ru specific structures or electronic environments.
Herein, we employed a simple and efficient method to confine Ru nanoparticles to the pore of ZIF-8 through the well-defined Ru3(CO)12 as a guest metal source and then incorporated it into the pore of ZIF-8. Transmission electron microscopy (TEM) revealed the well-dispersed Ru particles with a mean size of 1.7 nm in Ru@N–C catalyst. Ammonia synthesis investigations revealed that the obtained Ru@N–C catalysts exhibited obvious catalytic activity compared with the Ru/AC catalyst. After introducing the Ba promoter, the 2Ba–Ru@N–C(450) catalyst exhibited the highest ammonia synthesis activity among the catalysts.
2. Experimental
2.1 Chemicals
All chemicals were purchased from the Macklin Industrial Corporation in China. They were directly used without any further purification. All gases used in the experiment were 99.999% pure. The deionized water resistivity in all reactions was 18.25 MΩ cm.2.2 Preparation of ZIF-8
2-Methylimidazole (3.0 g) was first dissolved in 20 ml methanol; then, 30 mL methanol of dissolved Zn (NO3)2·6H2O (1.0 g) was quickly added into the above solution. After that, the resulting mixture was stirred at room temperature for 3 hours to obtain a milky suspension, which was centrifuged at 10000 rpm for 1 minute, washed with methanol 3 times, and dried in an oven at 80 °C to obtain ZIF-8.
2.3 Preparation of catalysts
Ru3(CO)12 (56.6 mg) was dissolved in tetrahydrofuran solution and then added to the prepared ZIF-8 (1.0 g). After stirring at room temperature for 24 hours, the mixture was placed on a rotary evaporator to remove the solvent, and the obtained sample was denoted as Ru3(CO)12@ZIF-8. The prepared Ru3(CO)12@ZIF-8 was placed in an ark and calcined in a tubular furnace at 450 °C for one hour at a heating rate of 5 °C min−1 under an argon atmosphere. After the calcination, the sample was naturally cooled to room temperature, removed and recorded as Ru@N–C(450), loading 3% Ru. The samples were thermally treated at 400 °C, 500 °C, and 900 °C and donated as Ru@N–C(400), Ru@N–C(500), and Ru@N–C(900), respectively.The promoters were introduced using the wet impregnation method with an aqueous solution of nitrates (KNO3, CsNO3 or Ba(NO3)2). The prepared Ru@N–C was used as the support, impregnated at room temperature for 24 hours, and then dried in an oven at 110 °C for 12 hours to obtain 2Cs–Ru@N–C. (The molar ratio of Cs and Ru is 2:1) 2K–Ru@N–C and 2Ba–Ru@N–C were prepared using the same method.
2.4 Catalyst evaluation
Catalyst activity was measured in a fixed-bed reactor. In the ammonia synthesis reaction, the amount of catalyst in each experiment is 0.2 g. The catalyst was loaded into a stainless–steel reaction tube φ = 6 mm, and a mixture of nitrogen and hydrogen gas (1:3) was introduced. The pressure was stabilized at 1 MPa, and the total gas flow was 60 mL min−1. After the catalyst was stabilized at different temperatures, the concentration of ammonia at the outlet was measured. The ammonia synthesis rate was determined by chemical titration and Nessler's reagent spectrophotometry.
2.5 Catalyst characterization
The morphology and size of the samples were observed using a JEM-2010 transmission electron microscope (TEM) at a 200 KV accelerating voltage. The crystal structure of the samples was analyzed by X-ray powder diffraction (XRD) (X'pert, PANalytical, Dutch) using Cu Kα radiation (λ = 1.54050 Å). The surface elemental composition of the sample was detected using an X-ray photoelectron spectrometer (ESCALAB 250Xi), and the electron binding energy scale of all spectra was calibrated using C 1s at 284.8 eV. Using Tianjin first right company TP-5080 typed automatic multi-purpose adsorption instrument, NH3-TPD, CO2-TPD, and H2-TPR tests were conducted. For the TPD tests, a 100 mg sample was placed in a quartz tube, heated to 300 °C in a He gas flow at a flow rate of 36 mL min−1, and pretreated for 1 hour. After cooling to room temperature, the He purged to a stable baseline and automatically switched to 10% NH3 or CO2/He mix gas with a flow rate of 40 mL min−1. Programmed temperature was applied. The heating rate was 10 °C min, and the temperature increased to 900 °C. For the H2-TPR tests, a 50 mg sample was pretreated with a He flow (27 mL min−1) at 300 °C for 1 hour and then cooled to room temperature. The test was performed by heating the sample in a H2/He (H2, 10%) mixture flow (30 mL min−1) at a linear heating rate ranging from 10 °C min to 900 °C. Thermogravimetric (TG) measurement was conducted using a DTG-60H analyzer (Shimadza, Tokyo, Japan) with a heating rate of 10 °C min−1. The Fourier transform infrared (FT-IR) spectra of the samples were obtained using a NEXUS 670 FT-IR spectrometer with KBr pellets prepared by manual grinding.3. Results and discussion
Scheme 1 shows the fabrication process of Ru3(CO)12@ZIF-8. Briefly, Ru3(CO)12 first dissolved in the THF formed a homogeneous solution. Then, the prepared ZIF-8 supporter was mixed with the above solution for Ru3(CO)12 transporting in the confined space. After that, the Ru3(CO)12@ZIF-8 was achieved by vacuum distillation. Interestingly, the colour of the mixed solution turned from orange to white; this indicates that the Ru3(CO)12 molecule would be adsorbed within the porosities of ZIF-8 owing to the Ru3(CO)12 cluster with a molecular size of 8.4 Å × 6.1 Å and the ZIF-8 with 12.5 Å porosity.22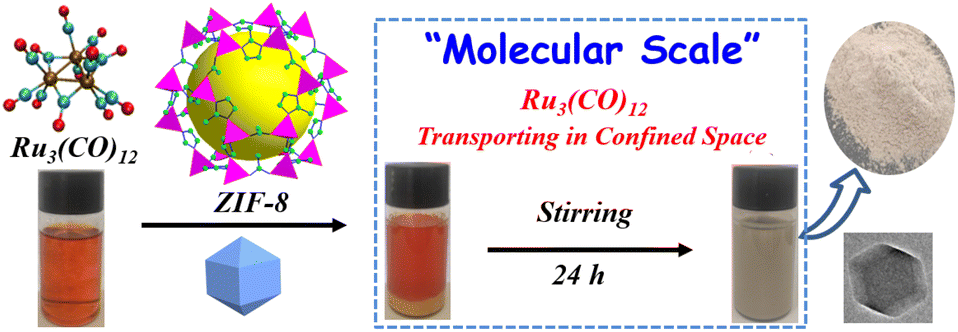 | ||
| Scheme 1 Synthesis of the Ru3(CO)12@ZIF-8. | ||
Fig. 1a and b shows the typical TEM images of the ZIF-8. It can be clearly observed that the regular dodecahedron structure was formed with a diameter of 100–120 nm. The specific surface area of the precursor ZIF-8 is 1107.7 m2 g−1 and the pore size is 6.3 Å, which is larger than the theoretical size of Ru3(CO)12, indicating that Ru3(CO)12 can enter the pore of ZIF-8. After calcination in inert Ru3(CO)12@ZIF-8 at 450 °C, the Ru@N–C(450) catalyst was obtained. The Ru@N–C(450) with surface area (1109.24 m2 g−1) retained the basic structure of ZIF-8, which is 239.68 m2 g−1 higher than ZIF-8(450) with surface area (869.56 m2 g−1). These results can be attributed to the CO molecule in the fabricated porous Ru3(CO)12 (Fig. S1 and Table S1†). Fig. S2† shows the X-ray diffraction (XRD) patterns, and the diffraction peaks of Ru@N–C(450) were well indexed to ZIF-8, which agrees with the TEM results. However, no diffraction peaks related to Ru species are observed in the XRD patterns of Ru@N–C(450), which implies that the Ru species are highly dispersed in the catalysts. Moreover, the small-dotted circles with yellow colour were the well-dispersed Ru particles depicted in Fig. 1c. A previous study23 has shown that the most active site for N2 dissociation and ammonia synthesis is called the B5-type site, which is preferentially formed on small particles ranging from 1.8–3.5 nm. Fig. 1d shows the well-dispersed Ru particles with a mean size of 1.7 nm. Energy-dispersive spectroscopy (EDS) elemental mapping shows that the C, N, O Zn and Ru elements are uniformly distributed in the whole detection region (Fig. S3†). These results suggest that the confined pore space and rich N coordination environment can stabilize the Ru nanoparticles.
 | ||
| Fig. 1 TEM images of (a, b) ZIF-8, (c) Ru@N–C(450), and (d) the histograms of Ru particle size distribution in Ru@N–C(450) catalyst. | ||
To illustrate the catalytic performance of the Ru@N–C catalysts, five catalysts Ru@N–C(400), Ru@N–C(450), Ru@N–C(500), Ru@N–C(900) and N–C(450) were prepared, and ammonia synthesis reaction was used as the model reaction. Fig. 2 shows the ammonia synthesis activity of the five catalysts under conditions of 1 MPa and 360 °C. It can be observed that the ammonia synthesis reaction rate of Ru@N–C(450) was 8604.2 µmol h−1 gRu−1, which is 7.4 and 10.5 times higher than those of Ru@N–C(400) (1157.5 µmol h−1 gRu−1) and Ru@N–C(500) (816.2 µmol h−1 gRu−1), respectively. However, no ammonia synthesis activity of the Ru@N–C(900) and N–C(450) catalysts was observed.
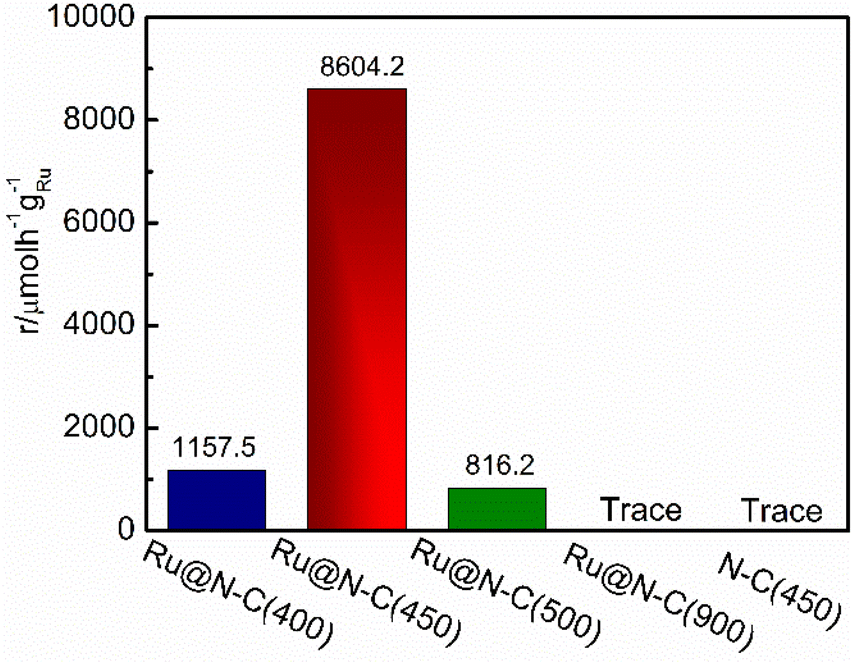 | ||
| Fig. 2 NH3 synthesis rate at 360 °C, 1 Mpa over Ru@N–C(400), Ru@N–C(450), Ru@N–C(500) and Ru@N–C(900) catalysts. | ||
Furthermore, the FTIR and TEM were used to detect the structure of four catalysts at various calcination temperatures. It can be noted that the characteristic IR spectrum is similar to that of the ZIF-8 (Fig. 3). Interestingly, the facet partial collapse of ZIF-8 was observed after calcination at 400–500 °C in the TEM graph (Fig. S4a–d†). With the increase in roasting temperature, the size of the Ru particles on the support increased (Fig. S4e–h†). At 900 °C, the dodecahedron structure was destroyed, which is in accordance with the TG and XRD results (Fig. S5 and S6,† respectively), and most MOFs are thermally stable only between 250 and 500 °C.24 Moreover, the XPS was employed to detect surface composition and electron property. The peaks of Zn 2p in Ru@N–C(400), Ru@N–C(450), and Ru@N–C(500) catalysts were shifted to the high bonding energy (Fig. S7a†) compared with Zn2+ at 1021.4 and 1044.3 eV. Moreover, the peaks of Ru 3d can be assigned to Run+ (n = 1–3) (Fig. S7b†). Thus, the Ru3(CO)12 molecule released Ru atoms with N in ZIF-8 to form RuN species or generated an interaction between the Ru nanoparticles and the support during calcination, which further caused the chemical shift of Zn 2p and Ru 3d.
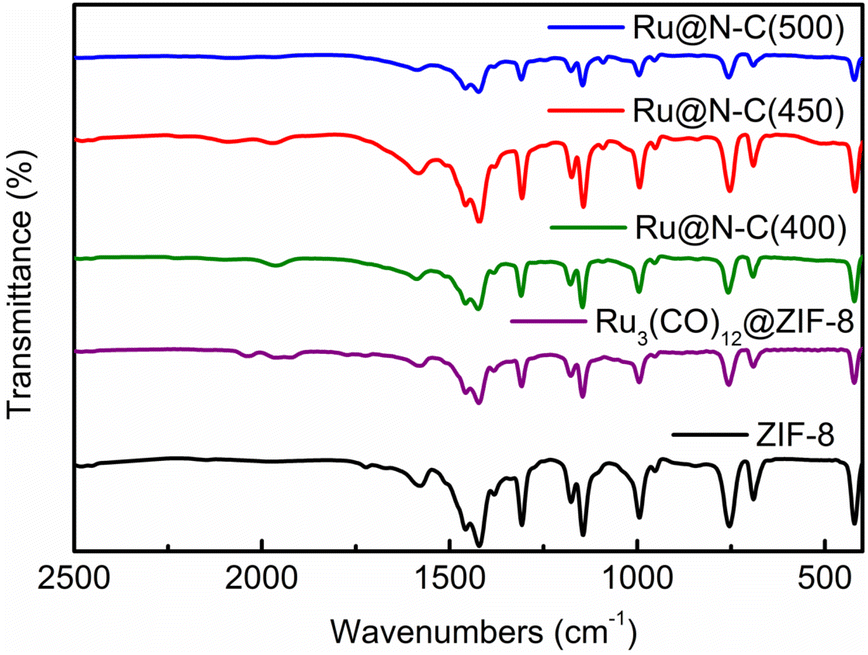 | ||
| Fig. 3 FTIR spectra of ZIF-8, Ru3(CO)12@ZIF-8, Ru@N–C(400), Ru@N–C(450), and Ru@N–C(500) catalysts. | ||
The alkali and alkaline earth metals (K, Cs, and Ba) are used as promoters for ammonia synthesis.25,26 Fig. S8† depicts the elemental mapping of 2K–Ru@N–C(450), 2Ba–Ru@N–C(450) and 2Cs–Ru@N–C(450). It can be observed that K, Ba and Cs are evenly distributed on the support. Fig. 4a shows the effect of K, Cs, and Ba promoters introduced into the Ru@N–C(450) catalysts on the ammonia synthesis reaction rate. In the temperature range of 300–360 °C, the catalytic activity increased with the introduction of Cs, K, and Ba promoters, and the 2Ba–Ru@N–C(450) catalyst activity was the highest. At 360 °C, the activity of 2Ba–Ru@N–C(450) is 16817.3 µmol h−1 gRu−1, which is 1.1, 1.6, and 2 times higher than those of 2Cs–Ru@N–C(450) (14925.4 µmol h−1 gRu−1), 2K–Ru@N–C(450) (10736.7 µmol h−1 gRu−1), and Ru@N–C(450) (8604.2 µmol h−1 gRu−1), respectively. The Ea was also evaluated using the four Ru@N–C catalysts (Fig. S9†). It can be observed that the Ea of Ru@N–C(450) is 87.5 kJ mol−1, which is lower than that of Ru/AC catalyst (92.4–134.4 kJ mol−1).27 After introducing the Ba promoter, the Ea of the 2Ba–Ru@N–C(450) catalyst is 49.9 kJ mol−1, which indicates that the Ba promoter significantly affects the ammonia synthesis activity. To further investigate the relationship between the amount of Ba promoter and the activities, three catalysts were employed to illustrate the catalytic performance (Fig. 4b). It is noteworthy that the molar ratio Ba/Ru = 2 exhibited the most catalytic activity among the three catalysts.
 | ||
| Fig. 4 (a) Temperature dependence of NH3 synthesis activity over the catalyst. (b) The effect of the Ba promoter amount on the ammonia synthesis activity. | ||
The traditional industrial ammonia synthesis catalyst used the AC act as the support. For comparison, the Ru/AC catalyst was used to illustrate the effect of the carbon support (Fig. 5), and other catalysts are summarized in Table S2.† However, the Ru/AC(450) catalyst without the promoter was completely inactive at 360 °C, indicating that the rich N coordination environment with strong electron-donating ability can obviously enhance N2 molecule dissociation and promote ammonia synthesis activity. This result is in accordance with a previous report.8 RuCl3 is the most common Ru salt. The catalyst RuCl3@N–C(450) is completely inactive at 360 °C. Even if it was washed using NaOH solution to remove the Cl− ion on the surface of the catalyst, the ammonia cannot be detected. Further, we used NaOH to etch away Zn from Ru@N–C(450) and tested the catalyst for ammonia synthesis at 360 °C. The activity of the catalyst was 1672.1 µmol h−1 gRu−1, which was much lower than that of Ru@N–C(450) (8604.2 µmol h−1 gRu−1). These results indicate that there is an interaction between the support and Ru nanoparticles, which is in accordance with the XPS analysis. More specifically, the Ru3(CO)12 released the Ru atoms to form RuN species with the N ligand or generated the interaction with Zn species at the atom level.
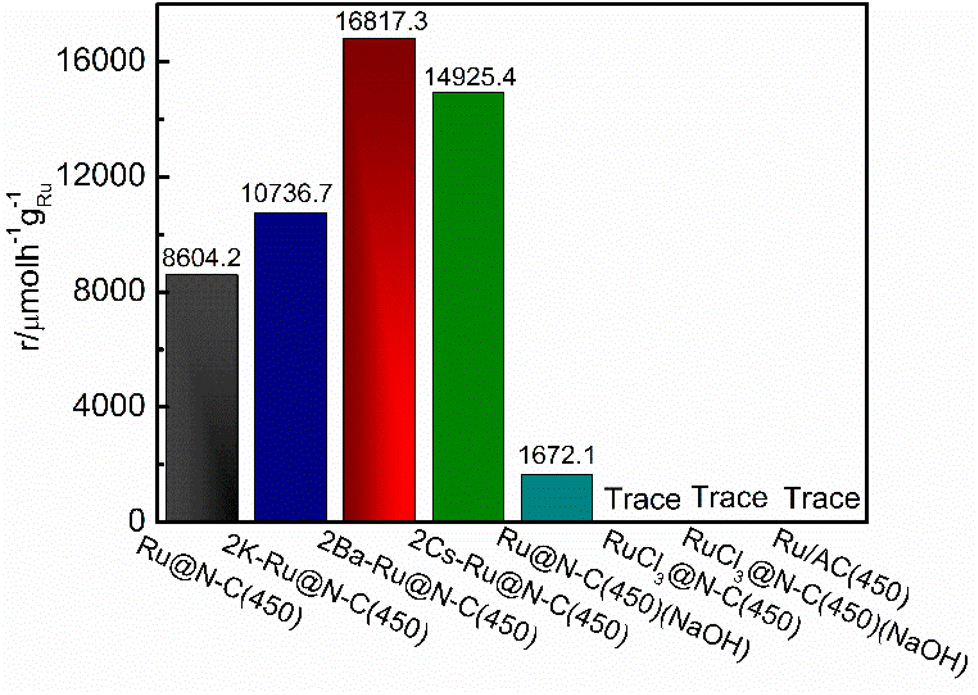 | ||
| Fig. 5 NH3 synthesis rate at 360 °C and 1 MPa for comparison. | ||
Previously, it was found that the promoters (K, Cs, and Ba) have a major effect on the Ru electron to promote ammonia synthesis activities.8,25,28,29 Based on these results, it can be rationally speculated that the K and Cs promoters slightly affect the Ru electron density in a rich N coordination environment. The Ba promoter may act as the electron and structure promoter so that it shows higher activities. XRD was used to detect the existing species of promoters. Fig. S10† shows the XRD patterns of 2K–Ru@N–C(450), 2Ba–Ru@N–C(450), and 2Cs–Ru@N–C(450) catalysts. It is noteworthy that the diffraction peaks are well indexed to ZIF-8, However, the peaks of the species of the promoters have not been evidently detected in the samples, which may have resulted from its low amounts as well as high dispersion.
To further investigate the effect of the promoters (K, Cs, and Ba), XPS was employed to detect the surface composition and electron property. The fitted N 1s peaks of Ru@N–C(450) centered at 399.0, 399.7, and 400.9 eV represented pyridinic, pyrrolic, and graphitic N, respectively (Fig. 6a).30 After introducing promoters (K, Cs, and Ba), the binding energy of N 1s shifted from the −0.4 to −0.5 eV range to the lower position, indicating that the promoters transfer electrons to the support. For the Ru 3d binding energy, it can be clearly observed that the binding energy at 281.7 eV of Ru@N–C catalysts shifted −0.4 eV to the lower position at 281.3 eV (Fig. 6b). Thus, the promoters (K, Cs, and Ba) affect the Ru electron properties, which is beneficial to weaken the NN bond and results in cleavage.
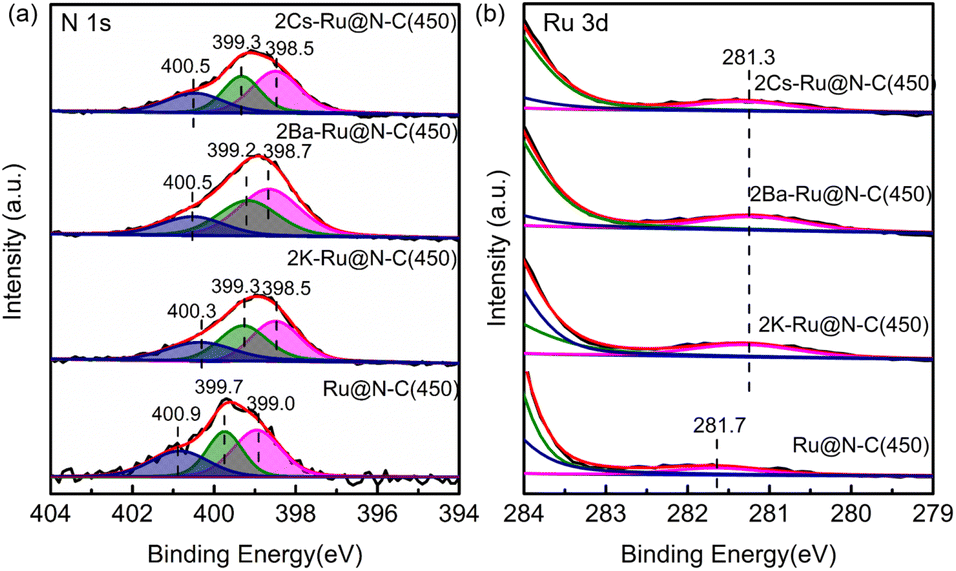 | ||
| Fig. 6 XPS spectra of (a) N 1s and (b) Ru 3d of 2Cs–Ru@N–C(450), 2Ba–Ru@N–C(450), 2K–Ru@NC(450), and Ru@N–C(450) catalysts. | ||
Fig. 7a shows the O 1s high-resolution XPS spectra. It can be clearly observed that a new band at the binding energy 531.2 eV occurred after introducing the promoters (K, Cs, and Ba), which can be attributed to the O bond with the promoters. Furthermore, the XPS spectra of K 2p, Cs 3d, and Ba 3d indicated that the promoters were in an oxidation state (Fig. S11†). Fig. 7b shows the H2-TPR of the catalysts for investigating reducibility, such as surface oxygen and the oxidation state of metal. The peaks in the high-temperature region (>500 °C) were similar to those in Ru@N–C(450) after introducing the promoters. Interestingly, the peak at 341.5 °C starting at 241.2 °C still existed in 2Ba–Ru@N–C(450) catalyst. Given that the samples were pretreated with Ar at 300 °C for 1 h, the peak at 341.5 °C can be attributed to the oxygen surrounding or binding with Ru nanoparticles owing to its exhibited Ba structural promoter property.
 | ||
| Fig. 7 (a) XPS spectra of O 1s of 2Cs–Ru@N–C(450), 2Ba–Ru@N–C(450), 2K–Ru@NC(450), and Ru@N–C(450) catalysts and (b) H2-TPR profiles of 2Cs–Ru@N–C(450), 2Ba–Ru@N–C(450), 2K–Ru@N–C(450), and Ru@N–C(450) catalysts. | ||
The ammonia synthesis rate was found to have a linear relationship with the electronegativity of the promoter or support.31 Thus, basic support is effective at promoting ammonia synthesis. Fig. 8a shows CO2-TPD profiles for the various catalysts containing promoters. The new peaks below 400 °C occurred after introducing promoters, indicating that the promoters offered the new basic sites. These catalysts with various degrees of enhancing activity are related to basic sites, especially Ba promoters. For 2Ba–Ru@N–C(450) catalyst, the CO2-desorbed temperatures were 356, 422, 461, and 480 °C, which are higher than those in 2Cs–Ru@N–C(450) and 2K–Ru@N–C(450) catalysts. The result indicates that the basicity of the 2Ba–Ru@N–C(450) is the highest among the catalysts. The Ba promoters can markedly weaken the NN bond of adsorbed N2, which is consistent with the activity test result. Generally, a lower N2 dissociation barrier implies stronger adsorption of N atoms with higher NHx desorption energy.32,33 Fig. 8b shows the NH3-TPD profiles for the catalysts. Compared with Ru@N–C(450) catalyst (>500 °C), the NH3-desorbed temperature of the catalyst with promoters (<450 °C) was much lower, indicating that the promoters can reduce the NHx desorption energy. Among the promoters (K, Cs, and Ba), the NH3-desorbed temperature of the Ba promoter was slightly higher than that of the K and Cs promoters. However, the 2Ba–Ru@N–C(450) catalyst exhibited the highest ammonia synthesis activity among the catalysts. Thus, it can be rationally attributed to the structural promoter effect of Ba promoter in contact with Ru particles and the high dispersion of Ru nanoparticles on the support, which is in accordance with Hara and Hosono et al. report that most Ru nanoparticles in Ru/BaO–CaH2 are immobilized onto the BaO phase.34 After the reaction, the C, N, O, Ba, Zn and Ru elements are uniformly distributed in the whole detection region (Fig. 9), and no significant decrease in reactivity was observed for more than 33 hours (Fig. S12†).
 | ||
| Fig. 8 (a) CO2-TPD profiles of 2Cs–Ru@N–C(450), 2Ba–Ru@N–C(450), 2K–Ru@N–C(450), Ru@N–C(450) and ZIF-8(450) catalysts and (b) NH3-TPD profiles of 2Cs–Ru@N–C(450), 2Ba–Ru@N–C(450), 2K–Ru@N–C(450), and Ru@N–C(450) catalysts. | ||
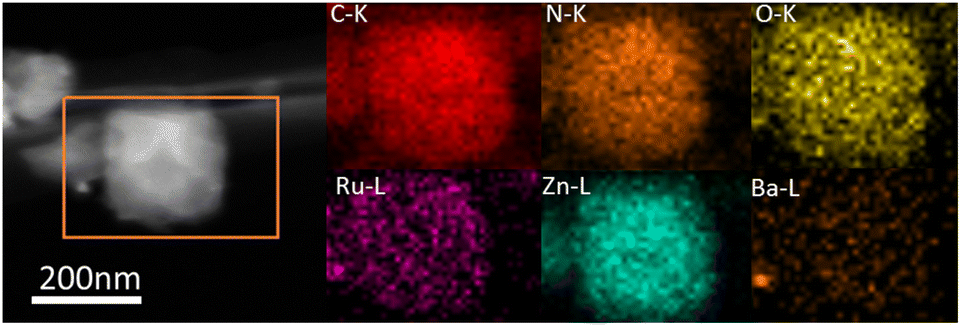 | ||
| Fig. 9 Elemental mapping of the used 2Ba–Ru@N–C(450) catalyst. | ||
Based on the above results, the matching size between Ru3(CO)12 volume and ZIF-8 pore diameter formed the confined coordinating interface, which provided the rich N coordination environment with a strong electron-donating ability that can obviously enhance N2 molecule dissociation and promote ammonia synthesis activity. The Ru3(CO)12 can release high-energy Ru atoms to form the RuN species. The promoters as electronic promoters donated the new basic sites for transferring the electron to Ru nanoparticles; moreover, Ba promoters were used as the electronic and structural promoters. The results indicate that the Ba promoter is located on the surface of the Ru nanoparticle and is the support for stabilizing the Ru nanoparticle. After introducing the Ba promoter, the desorption of adsorbed NH3 on Ru nanoparticle was much easier, which can contribute to improving the ammonia synthesis performance.
4. Conclusions
In summary, Ru3(CO)12 as a guest metal source incorporated into the pore of ZIF-8 can form the confined coordinating interfacial structure of Ru@N–C catalysts. The dispersed Ru nanoparticle (1.7 nm) in the confined N coordination environment obviously increased the electron density of the Ru nanoparticles and enhanced the ammonia synthesis activity compared with the Ru/AC catalyst. The promoters donated the basic sites for transferring the electrons to Ru nanoparticles. Furthermore, the Ba promoters are used as both electronic and structural promoters, which are located on the surface of Ru nanoparticles and support stabilizing the Ru nanoparticle and promoting the desorption of adsorbed NH3 on Ru nanoparticles. The 2Ba–Ru@N–C(450) catalyst exhibited the highest ammonia synthesis activity among the catalysts. This study provides an efficient strategy for constructing the Ru-base catalyst at a confined interface and at the Ru atom level to improve ammonia synthesis activity.Author contributions
Dongwei Wang: conceptualization, investigation, data curation, formal analysis, writing original draft. Zhanwei Ma: conceptualization, project administration, funding acquisition, writing review & editing. Farong Gou: funding acquisition, conceptualization, writing review & editing. Bin Hu: resources, project administration, writing review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the National Natural Science Foundation of China (22102194), the Science and Technology Plan of Gansu Province (20JR10RA044, 20YF3GA009), and the Youth Innovation Promotion Association of CAS (2022427).References
- S. L. Foster, S. Bakovic, R. D. Duda, S. Maheshwari and L. F. Greenlee, Catalysts for nitrogen reduction to ammonia, Nat. Catal., 2018, 1, 490–500 CrossRef.
- S. Dahl, A. Logadottir, R. C. Egeberg, J. H. Larsen, E. Törnqvist and J. K. Nørskov, Role of Steps in N2 Activation on Ru(0001), Phys. Rev. Lett., 1999, 83, 1814–1817 CrossRef.
- C. Jacobsen, S. Dahl, P. L. Hansen, E. Törnqvist, L. Jensen, H. Topsøe, D. V. Prip, P. B. Møenshaug and I. Chorkendorff, Structure sensitivity of supported ruthenium catalysts for ammonia synthesis, J. Mol. Catal. A: Chem., 2000, 163, 19–26 CrossRef CAS.
- J. He, K. Ding, X. Yang, X. Cheng, C. Huo and H. Liu, Effects of Stearic Acid Modification on Ru-Ba-MgO Interaction and the Underlying Mechanism, ChemistrySelect, 2022, 7, e202200924 CrossRef CAS.
- Z. Ma, S. Zhao, X. Xiong, B. Hu and C. Song, Effect of Graphitic Carbon Nitride on the Electronic and Catalytic Properties of Ru Nanoparticles for Ammonia Synthesis, Catal. Lett., 2016, 146, 1–6 CrossRef.
- M. Nishi, S. Y. Chen and H. Takagi, A Mesoporous Carbon-Supported and Cs-promoted Ru Catalyst with Enhanced Activity and Stability for Sustainable Ammonia Synthesis, ChemCatChem, 2018, 10, 3411–3414 CrossRef CAS.
- M. Nishi, S. Y. Chen and H. Takagi, X-ray absorption spectroscopy of Ba- and Cs-promoted Ru/mesoporous carbon catalysts for long-term ammonia synthesis under intermittent operation conditions, Sustain. Energy Fuels, 2020, 4, 832–842 RSC.
- H. S. Zeng, K. Inazu and K. I. Aika, The Working State of the Barium Promoter in Ammonia Synthesis over an Active-Carbon-Supported Ruthenium Catalyst Using Barium Nitrate as the Promoter Precursor, J. Catal., 2002, 211, 33–41 CrossRef CAS.
- M. Kitano, Y. Inoue, Y. Yamazaki, F. Hayashi, S. Kanbara, S. Matsuishi, T. Yokoyama, S. W. Kim, M. Hara and H. Hosono, Ammonia synthesis using a stable electride as an electron donor and reversible hydrogen store, Nat. Chem., 2012, 4, 934–940 CrossRef CAS PubMed.
- C. Jacobsen, Boron Nitride: A Novel Support for Ruthenium-Based Ammonia Synthesis Catalysts, J. Catal., 2001, 200, 1–3 CrossRef CAS.
- H. Kim, A. Jan, D. H. Kwon, H. I. Ji, K. J. Yoon, J. H. Lee, Y. Jun, J. W. Son and S. Yang, Exsolution of Ru Nanoparticles on BaCe0.9Y0.1O3-δ Modifying Geometry and Electronic Structure of Ru for Ammonia Synthesis Reaction Under Mild Conditions, Small, 2022, 19, 2205424 CrossRef PubMed.
- J. Ni, Z. Tan, Q. Sai, J. Zhu, X. Wang, B. Lin, J. Lin, C. Au and L. Jiang, Target-oriented confinement of Ru-Co nanoparticles inside N-doped carbon spheres via a benzoic acid guided process for high-efficient low-temperature ammonia synthesis, J. Energy Chem., 2021, 57, 140–146 CrossRef CAS.
- Z. Ma, X. Xiong, C. Song, B. Hu and Q. Zhang, Electronic metal–support interactions enhance the ammonia synthesis activity over ruthenium supported on Zr-modified CeO2 catalysts, RSC Adv., 2016, 6, 51106–51110 RSC.
- C. T. Campbell, Catalyst-support interactions: electronic perturbations, Nat. Chem., 2012, 4, 597–598 CrossRef CAS PubMed.
- A. Parastaev, V. Muravev, E. H. Osta, A. J. F. V. Hoof and E. J. M. Hensen, Boosting CO2 hydrogenation via size-dependent metal–support interactions in cobalt/ceria-based catalysts, Nat. Catal., 2020, 3, 526–533 CrossRef CAS.
- W. Gao, S. Guo, H. Zhang, X. Pan and X. Bao, Enhanced Ammonia Synthesis Activity of Ru Supported on Nitrogen-Doped Carbon Nanotubes, Chin. J. Catal., 2014, 32, 1418–1423 Search PubMed.
- Y. Shao, J. Sui, G. Yin and Y. Gao, Nitrogen-doped carbon nanostructures and their composites as catalytic materials for proton exchange membrane fuel cell, Appl. Catal. B Environ., 2008, 79, 89–99 CrossRef CAS.
- Y. Ma, G. Lan, X. Wang, G. Zhang, W. Han, H. Tang, H. Liu and Y. Li, Effect of nitrogen co-doping with ruthenium on the catalytic performance of Ba/Ru–N-MC catalysts for ammonia synthesis, RSC Adv., 2019, 9, 22045–22052 RSC.
- J. Mao, J. Li, J. Pei, Y. Liu, D. Wang and Y. Li, Structure regulation of noble-metal-based nanomaterials at an atomic level, Nano Today, 2019, 26, 164–175 CrossRef CAS.
- M. Feng, X. Wu, H. Cheng, Z. Fan, X. Li, F. Cui, S. Fan, Y. Dai, G. Lei and G. He, Well-defined Fe–Cu diatomic sites for efficient catalysis of CO2 electroreduction, J. Mater. Chem. A, 2021, 9, 23817–23827 RSC.
- J. Yang, D. He, W. Chen, W. Zhu, H. Zhang, S. Ren, X. Wang, Q. Yang, Y. Wu and Y. Li, Bimetallic Ru–Co Clusters Derived from a Confined Alloying Process within Zeolite–Imidazolate Frameworks for Efficient NH3 Decomposition and Synthesis, ACS Appl. Mater. Interfaces, 2017, 9, 39450–39455 CrossRef CAS PubMed.
- Y. Jian, F. Zhang, H. Lu, H. Xun and Y. Li, Hollow Zn/Co ZIF Particles Derived from Core–Shell ZIF-67@ZIF-8 as Selective Catalyst for the Semi-Hydrogenation of Acetylene, Angew. Chem., Int. Ed., 2015, 54, 10889–10893 CrossRef PubMed.
- C. Jacobsen, S. Dahl, P. L. Hansen, E. Törnqvist and I. Chorkendorff, Structure sensitivity of supported ruthenium catalysts for ammonia synthesis, J. Mol. Catal. Chem., 2000, 163, 19–26 CrossRef CAS.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, ChemInform Abstract: The Chemistry and Applications of Metal-Organic Frameworks, Science, 2013, 341, 1230444 CrossRef PubMed.
- Z. Ma, S. Zhao, X. Pei, X. Xiong and B. Hu, New insights into the support morphology-dependent ammonia synthesis activity of Ru/CeO2 catalysts, Catal. Sci. Technol., 2017, 7, 191–199 RSC.
- W. Al Maksoud, R. K. Rai, N. Morlanés, M. Harb, R. Ahmad, S. Ould-Chikh, D. Anjum, M. N. Hedhili, B. E. Al-Sabban and K. Albahily, et al., Active and stable Fe-based catalyst, mechanism, and key role of alkali promoters in ammonia synthesis, J. Catal., 2021, 394, 353–365 CrossRef CAS.
- K. I. Aika, H. Hori and A. Ozaki, Activation of nitrogen by alkali metal promoted transition metal I. Ammonia synthesis over ruthenium promoted by alkali metal, J. Catal., 1972, 27, 424–431 CrossRef CAS.
- B. Lin, K. Wei, X. Ma, J. Lin and J. Ni, Study of potassium promoter effect for Ru/AC catalysts for ammonia synthesis, Catal. Sci. Technol., 2013, 3, 1367 RSC.
- M. Osozawa, A. Hori, K. Fukai, T. Honma, K. Oshima and S. Satokawa, Improvement in ammonia synthesis activity on ruthenium catalyst using ceria support modified a large amount of cesium promoter, Int. J. Hydrogen Energy, 2022, 47, 2433–2441 CrossRef CAS.
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong and Z. Deng, et al., Single Cobalt Atoms with Precise N-Coordination as Superior Oxygen Reduction Reaction Catalysts, Angew. Chem., Int. Ed., 2016, 55, 10800–10805 CrossRef CAS PubMed.
- K. I. Aika, T. Takano and S. Murata, Preparation and characterization of chlorine-free ruthenium catalysts and the promoter effect in ammonia synthesis: 3. A magnesia-supported ruthenium catalyst, J. Catal., 1992, 136, 126–140 CrossRef CAS.
- F. Calle-Vallejo, D. Loffreda, M. T. M. Koper and P. Sautet, Introducing structural sensitivity into adsorption–energy scaling relations by means of coordination numbers, Nat. Chem., 2015, 7, 403–410 CrossRef CAS PubMed.
- M. M. Montemore and J. W. Medlin, Scaling relations between adsorption energies for computational screening and design of catalysts, Catal. Sci. Technol., 2014, 4, 3748–3761 RSC.
- M. Hattori, T. Mori, T. Arai, Y. Inoue, M. Sasase, T. Tada, M. Kitano, T. Yokoyama, M. Hara and H. Hosono, Enhanced Catalytic Ammonia Synthesis with Transformed BaO, ACS Catal., 2018, 8, 10977–10984 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra04824a |
| This journal is © The Royal Society of Chemistry 2023 |