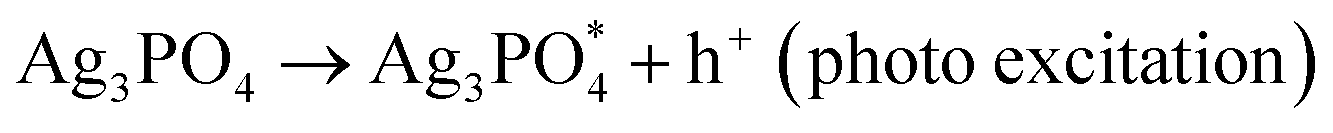DOI:
10.1039/D3RA05023H
(Paper)
RSC Adv., 2023,
13, 30643-30648
Thin silica shell on Ag3PO4 nanoparticles augments stability and photocatalytic reusability†
Received
25th July 2023
, Accepted 16th September 2023
First published on 24th October 2023
Abstract
Semiconductor photocatalysts are promising cost-effective materials for degrading hazardous organic contaminants in water. Ag3PO4 is an efficient visible-light photocatalyst for the oxidation of water and dye degradation. The excited Ag3PO4 photocatalyst uses a hole to oxidise water or organic contaminants except the electron, which reduces Ag+ to Ag0. In the present study, the inherited disadvantage was overcome by a thin silica shell overcoating on Ag3PO4 nanoparticles. The silica-coated Ag3PO4 nanoparticles retain the photocatalytic activity even after five cycles of photodegradation, while the bare Ag3PO4 nanoparticles show a photocatalytic activity declined to half. The study demonstrates that the thin silica shell enhances the photostability, keeping the photocatalytic activity unaffected, even after several cycles of photodegradation of dyes. XPS analysis showed that the Ag0 formation on the surface of bare Ag3PO4 is greater than that on silica-coated Ag3PO4, which declines the photocatalytic activity of Ag3PO4 after five cycles of photodegradation. Electrochemical studies identified that the intermediates, such as OH˙ and O2−, formed during water oxidation play a crucial role in the photodegradation of dyes. This study can provide insights into the design of core–shell semiconductor nanostructures for reusable photocatalytic applications.
Introduction
Freshwater aquatics are contaminated by the disposal of various untreated molecules of interest from industries, such as textiles and pesticides.1–4 The cost-effective decontamination of organic contaminants in water is urgently needed for the present scenario. Semiconductor photocatalysts have the ability to degrade organic contaminants present in water using solar energy.5–12 Semiconductors with a wide band gap, such as TiO2 and ZnO, are widely utilised for the photodegradation of organic contaminants. However, their application is limited by their UV wavelength absorption.13,14 However, visible light photocatalysts are promising candidates for the photodegradation of organic contaminants in applications outside the lab scenario.15–17 Among them, a Ag3PO4 photocatalyst attracts the attention of the scientific community with its capability to produce highly efficient holes for the oxidation of water or organic contaminants under visible light.18–20 Thus far, the Ag3PO4 photocatalyst has emerged as a versatile material for the ease of synthesis, usability and its high ability of water oxidation. Besides, capabilities, such as recyclability, photostability, and the ability to degrade all kinds of organic contaminants, are essential for an ideal photocatalyst. However, one of its non-negotiable disadvantages is its self-reduction of Ag+ to Ag0 during the photocatalysis.21,22 Such unwanted side reactions decline the efficiency in recycles of photocatalysis and may vary with the kind of molecules. There are various methods to overcome such drawbacks: (i) increasing the surface area by changing the crystallinity, reducing the size and making it porous,23 (ii) adding ingredients like H2O2 to prevent the side reaction or recycling the catalyst,24 and (iii) incorporating other materials to extract electrons and protect the catalyst from self-degradation.25–27
Apart from this, some of the recent research studies focus on the coating of an appropriate material onto a semiconductor photocatalyst, which will protect the surface of the semiconductor and subsequently facilitate the adsorption of molecules near the photocatalyst, leading to negotiable loss of photocatalytic efficiency.28,29 The present work focuses on designing and synthesising thin silica-coated Ag3PO4 nanostructures and studies their reusability for the photocatalytic degradation of methylene blue and rhodamine B dyes in water. The mechanism of photodegradation has been done electrochemically by cyclic voltammetric studies.
Experimental section
Materials
Experiments were all carried out at room temperature. All chemicals were used as received. Tetrahydrofuran (C4H8O 99.5% Isochem), acetonitrile (CH3CN 99.8% Merck), di-sodium hydrogen phosphate (Na2HPO4 > 98% Merck), silver nitrate (AgNO3 99.8% Nice Chemicals), triethoxy n-octyl silane (C4H32O3Si > 97% TCI), tetrabutylammonium hexafluorophosphate (C16H36F6NP > 98% TCI), and acetone ((CH3)2CO 99% Isochem) were used for washing purposes. Methylene blue (Nice Chemicals) and rhodamine B (Sigma Aldrich >95%) were used as organic pollutants.
Synthesis of Ag3PO4@SiO2 photocatalysts
Silane-capped Ag3PO4 was synthesised by the in situ overcoating of silica during the precipitation of Ag3PO4. Then, 300 μL of triethoxy n-octyl silane was added to an RB flask containing 0.44 mmol of AgNO3 in 25 mL of tetrahydrofuran, and 2.5 mL of 0.1 M Na2HPO4 was added dropwise into the solution. The resulting solution was stirred for 1 hour to ensure complete precipitation. The formation of Ag3PO4 was confirmed by the colour change of the solution from colourless to yellow. The synthesised Ag3PO4@SiO2 photocatalyst was purified by repeated centrifugation and kept in a hot oven at 100 °C for 1 hour. Bare Ag3PO4 was also prepared by the same method without triethoxy n-octyl silane and purified by repeated centrifugation.
Photocatalytic studies
The photocatalytic activities of Ag3PO4@SiO2 and Ag3PO4 were evaluated by the degradation of methylene blue and rhodamine B dyes. First, 0.86 mg of synthesised material is used for 1.6 × 10−5 M methylene blue (MB) and rhodamine B (RhB). Photocatalytic degradation was monitored every 10 minutes under 365 nm light of 125 W, and a xenon lamp of 300 W was used for 450 nm light. The bandpass filter was used to transmit a wavelength of 450 nm (from 440 nm to 480 nm) from the xenon lamp. The solution was stirred in the darkness for 30 min to reach the adsorption–desorption equilibrium between the organic molecules and the photocatalyst surface. At illumination intervals, 3 mL reacted solutions were taken out and then analysed using a T90+ UV-Vis spectrophotometer. The degradability of the above pollutants was represented by C/C0, where C0 and C denoted the main absorption peak intensities of the above-mentioned pollutants (rhodamine B at 553 nm and MB at 667 nm) before and after photocatalytic reactions. More than 90% of the dyes were degraded within 30 minutes of irradiation, the remaining dye solution was washed off, and a fresh solution was taken for other recycles. Further TOC analysis of the photodegraded dye solution (first cycle) was carried out to ensure more than 90% conversion, and the results show 92% degradation (ESI Table 1†). The used Ag3PO4@SiO2 and Ag3PO4 nanoparticles were washed with water after each recycling of the material was carried out up to 6 cycles of photodegradation.
Cyclic voltammetric studies
Cyclic voltammetry is deployed for electrochemical studies. Cyclic voltammetry is a three-electrode setup. The glassy carbon working electrode is replaced with FTO, sandwiched by a Ag3PO4@SiO2-coated glass plate, a Pt counter electrode and an Ag/AgCl reference electrode. Ag3PO4@SiO2 was spin-coated onto a glass plate of dimension 2 cm length and 1 cm breadth. Then, 1 M tetrabutyl ammonium hexafluorophosphate (TBAHFP) in an acetonitrile–10% water mixture was used as a supporting electrolyte. The electrolyte is degassed using N2 gas before all the experiments, and uniform stirring for 5 minutes has been done before each cyclic voltammetric measurement.
Results and discussion
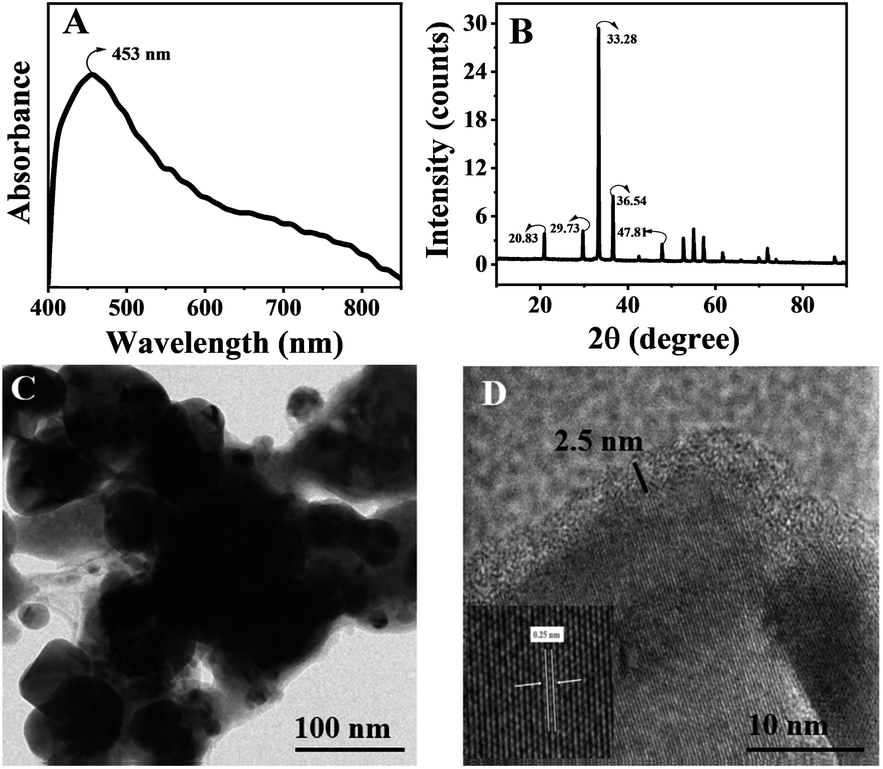
We synthesised thin silica-coated Ag3PO4-based semiconductor nanostructures by a chemical method.30 The lack of solubility of Ag3PO4 in polar solvents limits various synthetic possibilities such as incorporation of other materials (other semiconductor or metal nanomaterials) and surface modification like core–shell structures.31–34 In this case, direct silica overcoating is inappropriate due to the lack of silane loving groups on the surface of Ag3PO4 nanostructures. Thus, triethoxy n-octyl silane was chosen as the silane coating agent, which overcoats the in situ formed Ag3PO4 nanoparticles. Octyl silane facilitated the anchoring of octyl silane groups on the surface of Ag3PO4, where the triethoxy silane group condensed to form a thin silica layer. A basic pH of ∼10 of Na2HPO4 enables the alkaline hydrolysis of octyl silane, resulting in thin silica coating onto the surface of Ag3PO4 nanostructures. The formed silica-coated Ag3PO4 (Ag3PO4@SiO2) is yellow; the unreacted reactants were removed by repeated centrifugation in water. The thin silica shell overcoating enhances the versatility of usage by dispersing in various solvents such as chloroform, tetrahydrofuran and water. The synthesised semiconductor photocatalyst was characterised by the UV-Vis absorption spectrum, which is taken by coating Ag3PO4@SiO2 onto a glass surface, and BaSO4 coated onto glass was used as the baseline. The absorption spectra displayed in Fig. 1A show an absorption peak at 453 nm, equal to the bandgap (2.36 eV) of Ag3PO4. Further, the characteristic peaks at 20°, 29°, 33° and 36° in the PXRD pattern shown in Fig. 1B confirm the crystallinity of Ag3PO4@SiO2 and is found to be similar to the previously reported values.10 The HR-TEM images of Ag3PO4@SiO2 were recorded by drop casting Ag3PO4@SiO2 onto a coated Cu grid, as presented in Fig. 1C. The morphological analysis of Fig. 1D reveals the amorphous 2.5 nm silica coated on crystalline Ag3PO4 with an average size of 20 ± 5 nm (ESI Fig. S1A†). Further, the EDX spectra of Ag3PO4@SiO2 confirm the presence of elements Si, Ag, P, and O (ESI Fig. S1B†). Purified Ag3PO4@SiO2 is dispersed in distilled water for photocatalytic studies. Methylene blue and rhodamine B were chosen as two different kinds of organic contaminants. Time-dependent absorption spectra were recorded and used to monitor the photocatalytic degradation of dyes.
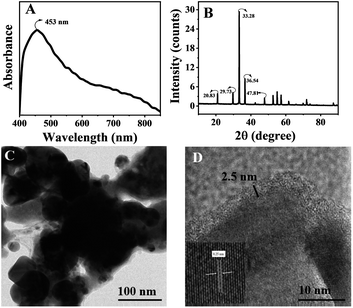 |
| | Fig. 1 (A) Absorption spectra of Ag3PO4@SiO2 nanoparticles coated onto a glass. (B) XRD patterns of Ag3PO4@SiO2 coated onto a glass surface. (C and D) High-resolution transmission electron microscopic (HRTEM) images of Ag3PO4@SiO2. Magnified image of 2.5 nm silica-coated crystalline Ag3PO4 nanoparticles (D). | |
Photocatalytic activity of Ag3PO4@SiO2
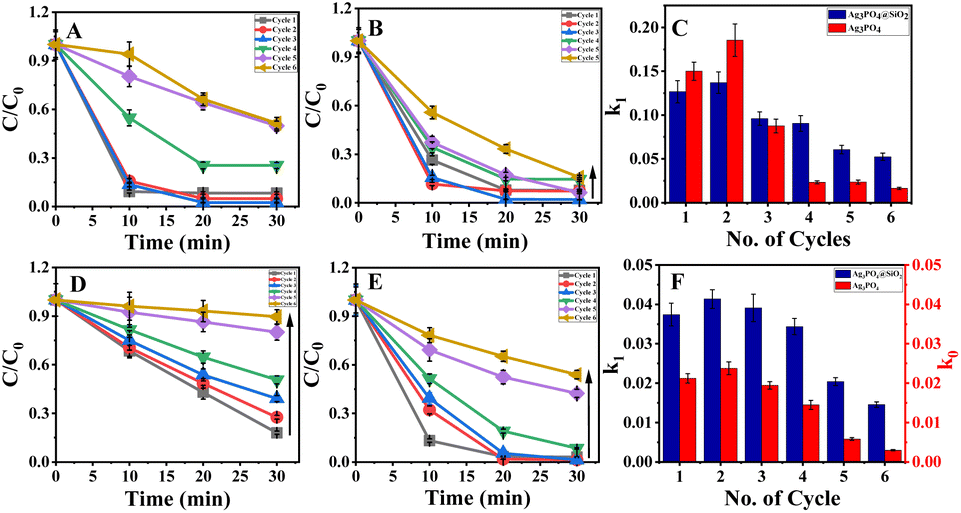
The photocatalytic efficiency of Ag3PO4@SiO2 was initially evaluated by the photodegradation of MB (methylene blue) incubated for 30 min to achieve equilibrium of adsorption of dyes on Ag3PO4@SiO2. Then, it was irradiated with 365 nm light with a power of 125 W, and Ag3PO4@SiO2 showed a remarkable degradation rate of 90% within 20 minutes, which is similar to bare Ag3PO4 presented in ESI Fig. S4.† Five more cycles of photodegradation were carried out to ensure the recyclability of photocatalysts under 365 nm light having the same power and experimental conditions. For each cycle, 90% completion of photodegradation of MB was ensured by monitoring the UV-vis spectra and correlated with TOC analysis. Unreacted dyes and degradation products were removed by repeated centrifugation. Then, purified Ag3PO4@SiO2 nanoparticles were dispersed in 1.6 × 10−5 M of MB solution, and photodegradation was carried out again. The MB concentration was estimated and presented in ESI Fig. S5.† It should be noted that both silica-coated and bare Ag3PO4 showed significant photocatalytic degradation of MB even after 6 cycles. The results indicate the photodegradation efficiency of Ag3PO4@SiO2 in parity with bare Ag3PO4. Further, the photocatalytic efficiency of Ag3PO4 and Ag3PO4@SiO2 was studied under visible light, where a Xe lamp was used as a light source with a bandpass filter (λ ∼ 450 nm) having a power of 300 W. The difference in the photocatalytic efficiency of Ag3PO4 and Ag3PO4@SiO2 was more pronounced in the photodegradation of MB under visible light irradiation, which is presented in Fig. 2A and B respectively. In contrast to the previous experiment, methylene blue showed 2 times enhanced degradation after 4 cycles of photodegradation in Ag3PO4@SiO2 compared to bare Ag3PO4. The rate constant was calculated and plotted as a function of number of cycles of photocatalysis by following the Langmuir–Hinshelwood first-order kinetic model, presented in Fig. 2C and ESI Table 3.† Initially, the rate constant of Ag3PO4 is comparable to Ag3PO4@SiO2. However, the photocatalytic activity of Ag3PO4 deteriorates in subsequent cycles, where Ag3PO4@SiO2 showed four times enhancement in photodegradation after the 5th cycle (ESI Table 2†). TOC analysis was carried out to ensure more than 90% photodegradation (ESI Table 1†).
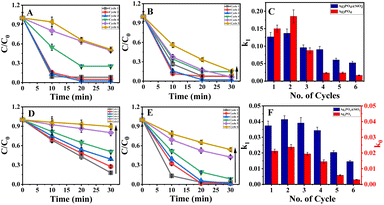 |
| | Fig. 2 Photocatalytic degradation of methylene blue (MB) in the presence of (A) Ag3PO4 and (B) Ag3PO4@SiO2 under 450 nm light. (C) Comparison of the rate constant of Ag3PO4 (red) and Ag3PO4@SiO2 (blue) for methylene blue. Photocatalytic degradation of rhodamine (RhB) in the presence of (D) Ag3PO4 and (E) Ag3PO4@SiO2 under 450 nm light. (F) The rate constant comparison of Ag3PO4 (red) and Ag3PO4@SiO2 (blue) for rhodamine B. | |
The photodegradation studies were extended into other dyes as well, carrying a negative charge, such as rhodamine B. Contrary to methylene blue, the photocatalytic degradation of rhodamine B by Ag3PO4 showed a pseudo-first-order kinetics with the increase in cycles, as shown in Fig. 2D and E. Nevertheless, the photocatalytic efficiency of Ag3PO4@SiO2 remains unaffected by changing the molecule from MB to rhodamine B, and is presented in Fig. 2D–F. Besides, the photocatalytic reusability enhanced when rhodamine B was used as a dye in the presence of Ag3PO4@SiO2 compared to Ag3PO4. The plot C/C0 vs. time of Ag3PO4 showed a linear plot indicating the reaction that proceeds through pseudo zero order, whereas Ag3PO4@SiO2 showed the same kinetics as followed in the MB degradation (ESI Table 4†).
The thin silica prevents direct contact between Ag3PO4 and methylene blue, although the amorphous silica layer traps the dye molecules and keeps them in the vicinity of Ag3PO4. Thus, the reactive intermediates produced due to the oxidation of water molecules by Ag3PO4 react effectively and oxidise methylene blue. The porous nature of thin silica allows the easy penetration of water molecules and hinders the interaction between methylene blue and Ag3PO4, which makes the Ag3PO4 surface less contaminated.33–36 It should be noted that various other studies showed that the solvent medium35 water plays a significant role in the photodegradation of organic contaminants. It is well documented that in the presence of light, Ag3PO4 will react with water molecules to form O2, O2−˙, OH˙, all of which oxidise organic molecules into CO2 and other inorganic products. Here, excited Ag3PO4 follows the same mechanism for the photodegradation of organic molecules. It is expected that SiO2 prevents direct contact with the molecules. Therefore, the reaction proceeds via the solvent-mediated mechanism. However, the direct contact between photocatalysts and organic contaminants will leave a footprint on the surface of Ag3PO4. This may decline the efficiency of photocatalysts in further recycling. Even though Ag3PO4 has high photocatalytic efficiency, it has an unavoidable drawback of self-reduction of Ag+ on the surface of Ag3PO4. The deposition of photodegraded organic contaminants on the surface of Ag3PO4 leads to catalytic poisoning. The self-reduction of Ag+ to Ag0 on the surface of Ag3PO4 is initially enhancing, but the photocatalytic processes decrease at later stages.
X-ray photoelectron spectroscopic studies were carried out to analyse the surface contamination of Ag3PO4 and Ag3PO4@SiO2 nanostructures. ESI Fig. S7† shows the deconvoluted spectra of Ag 3d5/2 and Ag 3d3/2 peaks of bare Ag3PO4 and Ag3PO4@SiO2 before photocatalysis to be 368.21 eV (Ag+ 3d5/2), 374.10 eV (Ag+ 3d3/2) and 369.10 eV (Ag0 3d5/2), 374.81 eV (Ag0 3d3/2) respectively. After 6 cycles of photocatalysis, the peak corresponding to Ag0 is more pronounced in bare Ag3PO4 than in Ag3PO4@SiO2, and the ratio of peak area of Ag0 to Ag+ showed a two-time increase in Ag0 content in bare Ag3PO4 than in Ag3PO4@SiO2. This explains why bare Ag3PO4 declined its efficiency of photodegradation after recycles while Ag3PO4@SiO2 retained.37 The XRD pattern of Ag3PO4 and Ag3PO4@SiO2 before and after photodegradation agrees with the conclusions (ESI Fig. S8†). The HR-TEM images of Ag3PO4 and Ag3PO4@SiO2 presented in ESI Fig. S2† clearly distinguish the Ag0 accumulation on the surface of Ag3PO4 rather than Ag3PO4@SiO2.
To evaluate the photocatalytic performance of bare Ag3PO4 and Ag3PO4@SiO2, we calculated the photocatalytic degradation efficiency and compared with other works reported in the literature.34–43 The maximum photocatalytic degradation efficiency of bare Ag3PO4 obtained after adsorption–desorption of methylene blue shows 1.29 × 10−2 mg min−1 mg−1 and Ag3PO4@SiO2 showed a maximum efficiency of 1.45 × 10−2 mg min−1 mg−1. For rhodamine B, the maximum photocatalytic degradation efficiency of bare Ag3PO4 showed 1.62 × 10−2 mg min−1 mg−1 and Ag3PO4@SiO2 showed an enhanced photocatalytic activity of 2.18 × 10−2 mg min−1 mg−1 efficiency. The above reported values of degradation efficiency for Ag3PO4@SiO2 are better than those for pure Ag3PO4 and the other reported values of methylene blue and rhodamine B.38–43 Further reusability of Ag3PO4@SiO2 is well proved in recyclability experiments while retaining the photocatalytic activity even after six cycles of photodegradation of methylene blue and rhodamine B. In the case of Ag3PO4@SiO2, the dye molecule gets adsorbed onto the surface of the SiO2 shell irrespective of charge and it exposes the molecule to reactive intermediates formed in the vicinity of Ag3PO4 (ESI Table 2†). Considering all these factors, SiO2-coated Ag3PO4 showed better performance, unaffected by the dyes' charges and reusability. Thus, SiO2 coating will be a better method for making the photocatalyst a versatile and reusable material for the decontamination of water.
Elucidating the mechanism via cyclic voltammetry
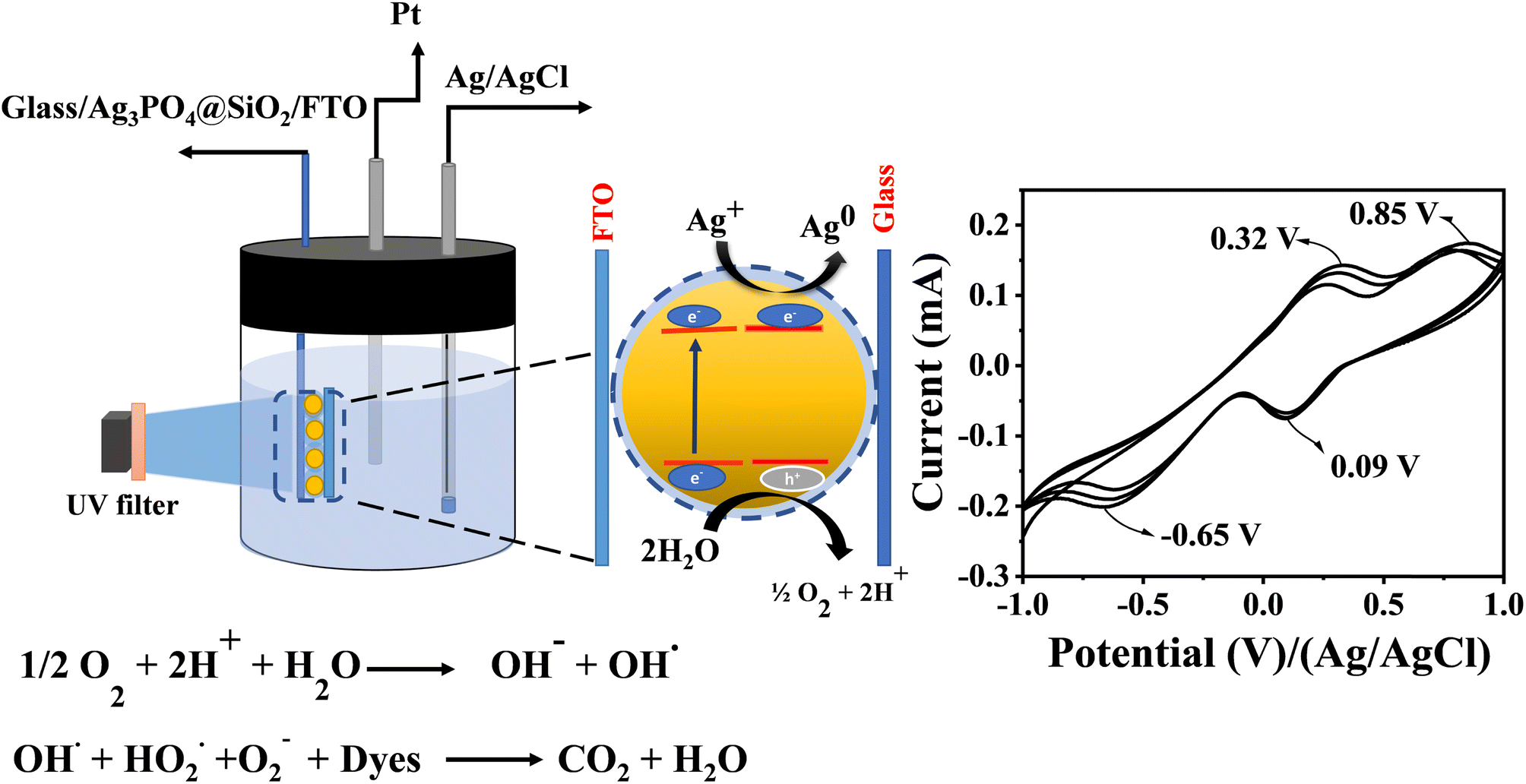
Cyclic voltammetric analysis was carried out to elucidate the mechanism and identify the intermediates formed during the experiment. This research gives clear-cut evidence for the mechanism of oxidation of dyes via O2−, OH˙ and OOH− intermediate formation. A three-electrode setup was used for the study; a glass plate spin coated with Ag3PO4@SiO2 sandwiched with the FTO plate was used as the working electrode, Ag/AgCl as the reference electrode and platinum as the counter electrode. The photocatalytic activity of Ag3PO4@SiO2 and its intermediate formation were monitored electrochemically via the fabricated working electrode. In order to get proper solvation, the working electrode was immersed into electrolytes before sandwiching them (Fig. 3).
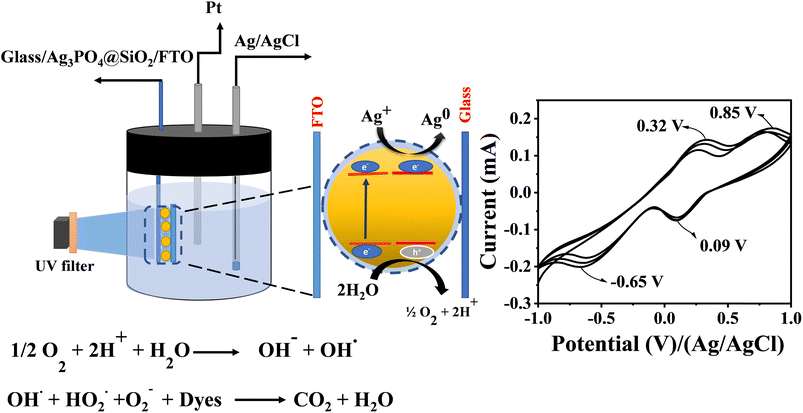 |
| | Fig. 3 Schematic of Ag3PO4@SiO2 sandwiched between the FTO and glass and the mechanism of photodegradation. The set up shows the monitoring of intermediates ((O2−˙), peroxide (HOO−) and OH˙) formed during the photocatalytic oxidation of water by cyclic voltammetry. Cyclic voltammogram of Ag3PO4@SiO2 sandwiched between FTO and the glass plate during the photocatalytic oxidation of water, light irradiated at 450 nm. | |
The stability of Ag3PO4@SiO2 was confirmed by cycling the modified electrode from a potential range of 1 V to −1 V vs. Ag/AgCl at different scan rates (ESI Fig. S9†). Even after several cycles, the current remains stable, indicating the stability of Ag3PO4@SiO2. Cyclic voltammetric studies were conducted in the presence of light and water under deaerated conditions. In the presence of light, Ag3PO4@SiO2 produces electron–hole pairs, which further react with water to form superoxides (O2−˙), peroxide (HOO−) and OH˙. Fig. 3 shows the cyclic voltammetric response of Ag3PO4@SiO2 from a potential range of −1 V to +1 V vs. Ag/AgCl at a scan rate of 0.1 V s−1; it shows two oxidation peaks at 0.32 V and 0.85 V and two reduction peaks at 0.09 V and −0.65 V vs. Ag/AgCl. The oxidation peak at 0.32 V corresponds to the oxidation of super oxides (O2−˙) produced as a result of photochemical reactions. On prolonged exposure of light, current response goes on increasing, which indicates the continuous generation of O2− in the system.44,45
| |
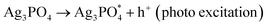 | (1) |
| | |
H2O + h+ + O2−˙ → OOH− + OH˙ (photochemical reaction)
| (2) |
| | |
O2 → O2− + e− (0.32 V, Ag/AgCl)
| (3) |
The oxidation peaks at 0.85 V are due to the formation of Ag+ from metallic silver (Ag0), which is confirmed by conducting a blank experiment with bare Ag3PO4 under the same experimental conditions in the presence and absence of light. During photoexcitation, the recombination of electron–hole results in the decomposition of Ag+ to Ag0 and weakens the photocatalytic activity. From the CV analysis, the oxidation peak at 0.85 V appears only in the presence of light, confirming the Ag-to-Ag+ oxidation; the peak current increases with time, confirming the photocatalytic generation of Ag. There is no characteristic oxidation peak of Ag+ observed for the experiments conducted in the absence of light. In the case of Ag3PO4@SiO2, the oxidation peak of Ag0 emerges only after 30 minutes because the SiO2 shell inhibits the direct contact of the Ag3PO4 surface to the reaction medium (ESI Fig. S11†).
| | |
4Ag3PO4 + 6H2O + 12h+ + 12e− → 12Ag + 4H3PO4 + 3O2 (photochemical reaction)
| (4) |
| | |
Ag0 → Ag+ + e− (0.85 V, Ag/AgCl)
| (5) |
In the reverse scan, the reduction peak at 0.09 V is quasi reversible with the oxidation peak at 0.32 V by a potential difference of 0.23 V. According to the reported work by G. Crompton et al., this peak is due to the reduction of oxygen to super oxide. This super oxide formation is followed by its fast reaction with water producing OH˙ and OOH−; this is observed as the reduction peak at −0.65 V. OOH− further reacts with water producing hydrogen peroxide and OH˙ followed by the disproportionation reaction of H2O2, which leads to water formation as follows:46,47
| | |
O2−˙ + H2O + e− → HOO− + OH˙
| (7) |
| | |
OOH− + H2O → H2O2 + OH−
| (8) |
The effect of the sacrificial electron acceptor on the photocatalytic effect was evaluated by experimenting in the presence of AgNO3 (ESI Fig. S13†). In the presence of AgNO3, the electrons are exported from the conduction band, and Ag+ will be reduced to Ag0, which is observed as the rise in peak current from 0.4 mA (in the absence of AgNO3) to 0.8 mA at 0.75 V. The reduction peak current increases with time in the presence of light. No peaks corresponding to the superoxide and peroxide are observed in the absence of light. The above cyclic voltammetric study reveals that light and water are necessary for the photocatalytic generation of intermediates such as OH˙, O2−˙ and OOH− and are responsible for the photodegradation of dyes. SiO2 coating on Ag3PO4 gives stability to the photocatalyst, which can be recycled and reused.
Conclusions
Thin silica-coated Ag3PO4 was synthesized by in situ addition and subsequent condensation of octyl silane during the precipitation of Ag3PO4. The photocatalytic activity of silica-coated Ag3PO4 and bare Ag3PO4 was studied using methylene blue and rhodamine B as organic contaminants. Thin silica improves the photostability of Ag3PO4 by retaining the photocatalytic efficiency even after six cycles of photodegradation. The photocatalytic efficiency of bare Ag3PO4 declined in both dyes after three cycles, and the percentage of photodegradation became half after five cycles of photodegradation. After photodegradation, XPS and HR-TEM analysis of Ag3PO4 and Ag3PO4@SiO2 revealed that the self-reduction of Ag+ to Ag0 was more predominant in Ag3PO4 than in Ag3PO4@SiO2. Photocatalytic generations of such intermediates were monitored electrochemically by a cyclic voltammetric technique. The intermediate formation was confirmed by conducting experiments in the presence and absence of light. It is evident from the study that water and light are indispensable parts for producing intermediate species O2−˙, OH− and OOH−, which further results in the photodegradation of dyes. SiO2-coated Ag3PO4 degrades various organic contaminants irrespective of their charge. Thus, SiO2 coating will be a better method for making the Ag3PO4 photocatalyst a versatile and reusable material for the decontamination of water.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
We are very thankful to Prof. K. George Thomas IISER-TVM, for providing instrumentation facilities for characterisation. We thank Ms Nimisha Krishnan, Research Dept. of Physics, Govt. Victoria College Palakkad for giving support for photodegradation studies. We thank Dr J. P. Vivek for valuable suggestions in electrochemical studies. We thank Dr Raj Sankar C, Assistant Professor, Dept. of Chemistry, Kerala Varma College Thrissur for providing instrumentation facilities for XRD characterisation. Ms P. Kavya thank SC/ST Development Department, Govt. of Kerala for providing Post-metric Scholarship.
Notes and references
- L. Tan, X. Wang, S. Wang, X. Qin, L. Xiao, C. Li, S. Sun and S. Hu, New J. Chem., 2023, 47, 11723–11735 RSC.
- J. Zhao, R. Song, H. Li, Q. Zheng, S. Li, L. Liu, X. Li, L. Bai and K. Liu, ACS Omega, 2022, 7, 29027–29037 CrossRef CAS PubMed.
- Z. Zhu, F. Guo, Z. Xu, X. Di and Q. Zhang, RSC Adv., 2020, 10, 11929–11938 RSC.
- J. T. DuBose and P. V. Kamat, J. Am. Chem. Soc., 2023, 145, 4601–4612 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- L. Zhang, P. Ma, L. Dai, S. Li, W. Yu and J. Guan, Catal. Sci. Technol., 2021, 11, 3834–3844 RSC.
- J. Zhang, Y. Li, X. Zhao, H. Zhang, L. Wang, H. Chen, S. Wang, X. Xu, L. Shi, L.-C. Zhang, J.-P. Veder, S. Zhao, G. Nealon, M. Wu, S. Wang and H. Sun, ACS Nano, 2020, 14, 17505–17514 CrossRef CAS PubMed.
- G. Zheng, S. de Marchi, V. López-Puente, K. Sentosun, L. Polavarapu, I. Pérez-Juste, E. H. Hill, S. Bals, L. M. Liz-Marzán and I. Pastoriza-Santos, Small, 2016, 12, 3935–3943 CrossRef CAS PubMed.
- S. Jatav, M. Herber, H. Xiang and E. H. Hill, ACS Appl. Mater. Interfaces, 2022, 14, 22790–22798 CrossRef CAS PubMed.
- C. Hanske, E. H. Hill, D. Vila-Liarte, G. González-Rubio, C. Matricardi, A. Mihi and L. M. Liz-Marzán, ACS Appl. Mater. Interfaces, 2019, 11, 11763–11771 CrossRef CAS PubMed.
- C. Chen, W. Ma and J. Zhao, Chem. Soc. Rev., 2010, 39, 4206–4219 RSC.
- G. Xi and J. Ye, Chem. Commun., 2010, 46, 1893–1895 RSC.
- J. T. DuBose and P. V. Kamat, J. Am. Chem. Soc., 2020, 142, 5362–5370 CrossRef CAS PubMed.
- S. J. Moniz, J. Zhu and J. Tang, Adv. Energy Mater., 2014, 4, 1301590 CrossRef.
- H. Yan, J. Yang, G. Ma, G. Wu, X. Zong, Z. Lei, J. Shi and C. Li, J. Catal., 2009, 266, 165–168 CrossRef CAS.
- D. Chen and J. Ye, Adv. Funct. Mater., 2008, 18, 1922–1928 CrossRef CAS.
- K. Sandeep and K. T. Hamida, Phys. Status Solidi A, 2021, 218, 2100101 CrossRef CAS.
- K. Sandeep, ChemistrySelect, 2020, 5, 4034–4039 CrossRef CAS.
- Z. Yi, J. Ye, N. Kikugawa, T. Kako, S. Ouyang, H. Stuart-Williams, H. Yang, J. Cao, W. Luo and Z. Li, Nat. Mater., 2010, 9, 559–564 CrossRef CAS PubMed.
- Y. Meng, J. Huang, J. Li, Y. Jian, S. Yang and H. Li, Green Chem., 2023, 25, 4453–4462 RSC.
- H. Wang, Y. Bai, J. Yang, X. Lang, J. Li and L. Guo, Chem.–Eur. J., 2012, 18, 5524–5529 CrossRef CAS PubMed.
- A. Houas, H. Lachheb, M. Ksibi, E. Elaloui, C. Guillard and J.-M. Herrmann, Appl. Catal., B, 2001, 31, 145–157 CrossRef CAS.
- M. Shanthil, K. Sandeep and P. Sajith, Phys. Chem. Chem. Phys., 2020, 22, 4788–4792 RSC.
- W. Teng, X. Li, Q. Zhao and G. Chen, J. Mater. Chem. A, 2013, 1, 9060–9068 RSC.
- H. Zhang, H. Huang, H. Ming, H. Li, L. Zhang, Y. Liu and Z. Kang, J. Mater. Chem., 2012, 22, 10501–10506 RSC.
- A. N. Nair, M. F. Sanad, V. S. N. Chava and S. T. Sreenivasan, Chem. Commun., 2022, 58, 10368–10371 RSC.
- B.-A. Chen, S. Ptasinska and P. V. Kamat, J. Phys. Chem. C, 2022, 126, 11907–11914 CrossRef CAS.
- M. Sharma, K. Ojha, A. Ganguly and A. K. Ganguli, New J. Chem., 2015, 39, 9242–9248 RSC.
- M. Sharma, K. Ojha, A. Ganguly and A. K. Ganguli, New J. Chem., 2015, 39, 9242–9248 RSC.
- P. Mulvaney, M. Giersig, T. Ung and L. M. Liz-Marzán, Adv. Mater., 1997, 9, 570–575 CrossRef.
- V. R. Nair, M. Shanthil, K. Sandeep, K. U. Savitha, A. Archana, V. Deepamol, C. Swetha and P. V. Vaishag, ACS Omega, 2023, 8, 29468–29474 CrossRef CAS PubMed.
- M. Shanthil, H. Fathima and K. George Thomas, ACS Appl. Mater. Interfaces, 2017, 9, 19470–19477 CrossRef CAS PubMed.
- K. Iwasaki, T. Torimoto, T. Shibayama, H. Takahashi and B. Ohtani, J. Phys. Chem. B, 2004, 108, 11946–11952 CrossRef CAS.
- N. Tavker, U. K. Gaur and M. Sharma, Nanoscale Adv., 2020, 2, 2870–2884 RSC.
- T. Yan, W. Guan, W. Li and J. You, RSC Adv., 2014, 4, 37095–37099 RSC.
- E. Nyankson, R. Amedalor, G. Chandrabose, M. Coto, S. Krishnamurthy and R. V. Kumar, ACS Omega, 2020, 5, 13641–13655 CrossRef CAS PubMed.
- Y. Liu, D. Yang, R. Yu, J. Qu, Y. Shi, H. Li and Z.-Z. Yu, J. Phys. Chem. C, 2017, 121, 25172–25179 CrossRef CAS.
- Y. Naciri, A. Hsini, A. Bouziani, R. Djellabi, Z. Ajmal, M. Laabd, J. A. Navío, A. Mills, C. L. Bianchi, H. Li, B. Bakiz and A. Albourine, Crit. Rev. Environ. Sci. Technol., 2022, 52, 2339–2382 CrossRef CAS.
- G. He, W. Yang, W. Zheng, L. Gong, X. Wang, Y. An and M. Tian, RSC Adv., 2019, 9, 18222–18231 RSC.
- S. Zhang, T. Yu, H. Wen, R. Guo, J. Xu, R. Zhong, X. Li and J. You, RSC Adv., 2020, 10, 16892–16903 RSC.
- E. Nyankson, J. K. Efavi, B. Agyei-Tuffour and G. Manu, RSC Adv., 2021, 11, 17032–17045 RSC.
- X. Yang, H. Cui, Y. Li, J. Qin, R. Zhang and H. Tang, ACS Catal., 2013, 3, 363–369 CrossRef CAS.
- A. N. Nair, V. S. N. Chava, S. Bose, T. Zheng, S. Pilla and S. T. Sreenivasan, ACS Sustainable Chem. Eng., 2020, 8, 16565–16576 CrossRef CAS.
- M. Kardeş, H. C. Yatmaz and K. Öztürk, ACS Appl. Nano Mater., 2023, 6, 6605–6613 CrossRef.
- P. Amornpitoksuk, K. Intarasuwan, S. Suwanboon and J. Baltrusaitis, Ind. Eng. Chem. Res., 2013, 52, 17369–17375 CrossRef CAS.
- P. M. Wood, Biochem. J., 1988, 253, 287–289 CrossRef CAS PubMed.
- Y. Guo, M. Yang, R.-C. Xie and R. G. Compton, Chem. Sci., 2021, 12, 397–406 RSC.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Karisseri P. Saranya
a,
Karisseri P. Saranya