
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of a cationic polyethyleneimine-poly(lactic-co-glycolic acid) nanoparticle system for enhanced intracellular delivery of biologics†
Shannon R. Traceya,
Peter Smytha,
Una M. Herrona,
James F. Burrows b,
Andrew J. Porterc,
Caroline J. Barellec and
Christopher J. Scott*a
b,
Andrew J. Porterc,
Caroline J. Barellec and
Christopher J. Scott*a
aQueen's University Belfast, The Patrick G Johnston Centre for Cancer Research, 97 Lisburn Road, Belfast, Northern Ireland BT9 7AE, UK
bQueen's University Belfast, School of Pharmacy, Belfast, BT9 7BL, UK
cElasmogen, Aberdeen, UK
First published on 17th November 2023
Abstract
Intracellular delivery of proteins, peptides and biologics is an emerging field which has the potential to provide novel opportunities to target intracellular proteins, previously deemed ‘undruggable’. However, the delivery of proteins intracellularly remains a challenge. Here, we present a cationic nanoparticle delivery system for enhanced cellular delivery of proteins through use of a polyethyleneimine and poly-(lactic-co-glycolic acid) polymer blend. Cationic nanoparticles were shown to provide increased cellular uptake compared to anionic and neutral nanoparticles, successfully delivering Variable New Antigen Receptors (vNARs), entrapped within the nanoparticle core, to the cell interior. vNARs were identified as ideal candidates for nanoparticle entrapment due to their remarkable stability. The optimised 10% PEI-PLGA nanoparticle formulation displayed low toxicity, was uniform in size and possessed appropriate cationic charge to limit cellular toxicity, whilst being capable of escaping the endo/lysosomal system and delivering their cargo to the cytosol. This work demonstrates the ability of cationic nanoparticles to facilitate intracellular delivery of vNARs, novel biologic agents with potential utility towards intracellular targets.
Introduction
Completion of the human genome project almost 20 years ago led to the identification of a number of novel prospective drug targets, particularly within the field of cancer.1,2 A vast majority of these targets are intracellular and although several have been identified as prevalent tumour drivers, many remain ‘undruggable’, largely due to a lack of a suitable binding pocket, as well as sub-optimal therapeutic delivery by conventional approaches. Clinically approved drugs can be divided into two broad classes; small molecules, and biologics (which include monoclonal antibodies, proteins, peptides, and vaccines).3 Whilst small molecules have been successfully employed as targeted cancer therapies, they have been shown to be ineffective against a range of intracellular proteins, mostly due to their inability to block the large surface area of the target protein and/or impede protein–protein interactions due to their hydrophobic nature. As a result, interest has grown in utilising protein-based therapeutics in this setting. Following advancements in the design process and manufacturability, protein therapeutics have becoming increasingly fruitful, comprising ∼50% of the top ten best selling drugs globally in 2020.4,5To date, a large portion of protein delivery approaches have been effectively directed towards extracellular targets, as exemplified by a range of monoclonal antibodies.6 However, intracellular delivery has proven challenging due to the intrinsic properties of proteins, such as their high molecular weight, as well as an intrinsic polarity which prevents them from being transported across the cell membrane. Various approaches have been investigated in efforts to deliver proteins intracellularly such as physical/mechanical methods (e.g., microinjection and electroporation) and covalent protein modification including the integration of cell penetrating peptides. Whilst showing promise, these methods are invasive and have been shown to elicit some level of inherent cellular toxicity.7–9 As an alternative approach, the use of nanocarriers has sparked significant interest for the intracellular delivery of therapeutic agents, such as small molecules, peptides, mRNA, DNA, and larger proteins. Nanocarriers represent a versatile formulation platform enabling development of nanoparticles with a breadth of size and surface functionalisation, in turn facilitating enhanced cellular uptake, prolonged circulation times as well as active cell targeting.10,11
Cationic polymeric nanoparticles exhibit attractive characteristics for the delivery of biologics intracellularly; namely enhanced cellular uptake as a result of electrostatic interactions between cationic nanoparticles and the anionic cell membrane proteins and an ability to facilitate endosomal escape whilst preserving entrapped protein integrity.12–15
In this study, we wish to examine the capabilities of a cationic nanoparticle formulation for the intracellular delivery of proteins, through the development of a PEI-PLGA based nanoparticle entrapping Variable New Antigen Receptors (vNARs) as a model protein for enhanced intracellular delivery. vNARs possess many unique properties attributed from their distinctive origin. Other than their small size and high affinity for their target, the inherent stability of vNARs make them ideal candidates for the harsh conditions within the cell.16,17 Furthermore, the presence of disulfide bridges, results in vNARs having a protruding structure, which in turn leaves the structure of vNARs predisposed to binding inside deep fissures, where they have the ability to bind into deep cryptic epitopes which would usually be concealed to conventional antibodies and other proteins.17–20 As a result of exposure to harsh conditions within in their natural environment, of the shark sera, vNARs have adapted to their environment resulting in a molecule which possesses considerable stability. vNARs have been shown to be stable over a range of external conditions, including changes in temperature, pH, and the presence of proteases.16,18–21 Following synthesis and determination of the physical characteristics of the vNAR PEI-PLGA nanoparticle system, the enhanced ability of the cationic nanoparticle to escape the endo–lysosomal system was assessed using in vitro cell based assays. Overall, this study identifies a potential platform through which the well-established limitations of biologics in targeting intracellular proteins may be overcome.
Materials and methods
All chemicals were purchased from Sigma-Aldrich and were >97.0% purity unless stated otherwise.Experimental
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :glycolide 50:50; Mw 7000–17000 Da) (PLGA) was dissolved DCM, where a 10–25% (w/v) of polyethyleneimine (PEI) was added to the organic phase resulting in a PEI-PLGA polymer blend, resulting in 10 mg of polymer in 2 mL DCM. For example, for the final 10% PEI-PLGA formulation, nanoparticles were prepared by dissolving 9 mg Resomer® RG502H PLGA in 1.8 mL of DCM, where 200 μL of 5 mg mL−1 PEI in DCM was added to the organic phase, resulting in a 10% w/v PEI-PLGA polymer blend. The organic phase was injected dropwise into the aqueous phase consisting of 0.5 mL of 1% PVA in 50 mM MES hydrate buffer (pH 5). While stirring at 1000 rpm (multichannel stirrer, model MS-53M, Lab Companion) on ice. Emulsification was achieved by sonication in pulse mode (3 seconds on, followed by two seconds off) for 36 seconds at an amplitude of 50% using a Model FB120 sonic dismembrator (Fisher Scientific). 10 mL of 1% PVA/MES buffer solution was then added and emulsified by sonication in pulse mode (3 seconds on, followed by two seconds off) for 54 seconds at an amplitude of 50%. Vials were left stirring at 1000 rpm overnight to ensure evaporation of the organic solvent. The formed nanoparticles were purified via centrifugation (17000×g, at 4 °C, 20 min) using PBS, by three wash–spin cycles. Nanoparticles were adjusted to a concentration of 10 mg polymer per mL in PBS and stored at 4 °C until further usage. Alternatively, non-cationic, control PLGA nanoparticles were formulated. Where necessary, vNAR entrapped nanoparticles were formed using the method described, with the addition of 10 μg of D8 FITC tagged vNAR (unless otherwise stated) added per mg polymer to the initial aqueous of the nanoparticle formulation.000 magnification.:50 dilution in blocking buffer) or goat anti-rabbit EEA1 antibody (cell signalling technology (c45b10); 1:50 dilution in blocking buffer). Following this, incubation with goat anti-mouse IgG secondary antibody Alexafluor568 (1:1000 dilution in blocking buffer, ThermoFisher Scientific (A-11031) for LAMP-1) or goat anti-rabbit IgG secondary antibody Alexafluor568 (1:1000 dilution in blocking buffer, ThermoFisher Scientific (A-1103) for EEA1). Cells were washed ×3 with ice cold 1× PBS. Coverslips were mounted onto the glass slides using Vectashield antifade mounting medium with DAPI (Vector Laboratories, H-1200) and sealed before imaging using confocal microscopy.
:glycolide 50:50; Mw 7000–17000 Da) (PLGA) was dissolved DCM, where a 10–25% (w/v) of polyethyleneimine (PEI) was added to the organic phase resulting in a PEI-PLGA polymer blend, resulting in 10 mg of polymer in 2 mL DCM. For example, for the final 10% PEI-PLGA formulation, nanoparticles were prepared by dissolving 9 mg Resomer® RG502H PLGA in 1.8 mL of DCM, where 200 μL of 5 mg mL−1 PEI in DCM was added to the organic phase, resulting in a 10% w/v PEI-PLGA polymer blend. The organic phase was injected dropwise into the aqueous phase consisting of 0.5 mL of 1% PVA in 50 mM MES hydrate buffer (pH 5). While stirring at 1000 rpm (multichannel stirrer, model MS-53M, Lab Companion) on ice. Emulsification was achieved by sonication in pulse mode (3 seconds on, followed by two seconds off) for 36 seconds at an amplitude of 50% using a Model FB120 sonic dismembrator (Fisher Scientific). 10 mL of 1% PVA/MES buffer solution was then added and emulsified by sonication in pulse mode (3 seconds on, followed by two seconds off) for 54 seconds at an amplitude of 50%. Vials were left stirring at 1000 rpm overnight to ensure evaporation of the organic solvent. The formed nanoparticles were purified via centrifugation (17000×g, at 4 °C, 20 min) using PBS, by three wash–spin cycles. Nanoparticles were adjusted to a concentration of 10 mg polymer per mL in PBS and stored at 4 °C until further usage. Alternatively, non-cationic, control PLGA nanoparticles were formulated. Where necessary, vNAR entrapped nanoparticles were formed using the method described, with the addition of 10 μg of D8 FITC tagged vNAR (unless otherwise stated) added per mg polymer to the initial aqueous of the nanoparticle formulation.000 magnification.:50 dilution in blocking buffer) or goat anti-rabbit EEA1 antibody (cell signalling technology (c45b10); 1:50 dilution in blocking buffer). Following this, incubation with goat anti-mouse IgG secondary antibody Alexafluor568 (1:1000 dilution in blocking buffer, ThermoFisher Scientific (A-11031) for LAMP-1) or goat anti-rabbit IgG secondary antibody Alexafluor568 (1:1000 dilution in blocking buffer, ThermoFisher Scientific (A-1103) for EEA1). Cells were washed ×3 with ice cold 1× PBS. Coverslips were mounted onto the glass slides using Vectashield antifade mounting medium with DAPI (Vector Laboratories, H-1200) and sealed before imaging using confocal microscopy.Results
Preparation and characterisation of PEI cationic nanoparticles
To facilitate formulation of a cationic nanoparticle, a polymer blend of PLGA and PEI was chosen. PLGA was selected as the bulk polymer due to its slow release, as well as its FDA ‘generally regarded as safe’ status.24,25 Polyethyleneimine (PEI) is a branched polycationic polymer which has been extensively used for delivery of proteins and nucleic acids into living cells.26,27 This is as a result of its high positive charge density and chain flexibility when compared to other polycations.27–29 PEI-PLGA nanoparticles were prepared using various percentage polymer blends (10–25%), with PEI content represented as a percentage of the total polymer content. Increasing the percentage of PEI polymer added in the organic phase resulted in nanoparticles which displayed a greater cationic charge (+6.01 mV ± 3.86 – +14.44 mV ± 7.79), when compared to PLGA nanoparticles (−11.07 mV ± 0.98) (Table 1). Not only was there a change to nanoparticle charge when the percentage of PEI was increased, an increase in nanoparticle size was also observed, with blank PLGA nanoparticle diameter of 232.98 nm ± 9.07 increasing to a diameter of 250–260 nm for PEI nanoparticle formulations. This was associated with a marginal increase in PDI (Table 1).| Nanoparticle | Particle diameter (nm) | PDI | Zeta potential (mV) |
|---|---|---|---|
| PLGA NP | 232.98 ± 9.07 | 0.103 ± 0.015 | −11.07 ± 0.98 |
| 10% PEI-PLGA NP | 250.29 ± 1.45 | 0.114 ± 0.001 | +6.01 ± 3.86 |
| 15% PEI-PLGA NP | 260.28 ± 16.80 | 0.142 ± 0.005 | +8.04 ± 4.08 |
| 20% PEI-PLGA NP | 261.28 ± 11.07 | 0.142 ± 0.003 | +11.65 ± 1.40 |
| 25% PEI-PLGA NP | 259.02 ± 13.55 | 0.149 ± 0.020 | +14.44 ± 7.79 |
In order to evaluate nanoparticle toxicity, each formulation was tested in vitro. For the purposes of this study, HeLa cells were selected as a relevant cell model for this proof-of-concept study due to their ease of use in ability to analyse cellular toxicity and applicability for confocal microscopy with nanoparticle uptake/localisation studies. HeLa cells were treated with varied concentrations of the formulations (0.012–0.4 mg per mL polymer) for up to 72 h and cell viability assessed. At 24 h post-treatment, cells treated with 20% and 25% PEI-PLGA nanoparticles showed a small but significant decrease in cell viability at nanoparticle concentrations >0.1 mg mL−1 (Fig. 1A). At both 48- and 72 h timepoints, a decrease in cell viability was observed with 15–25% PEI-PLGA nanoformulations as polymer concentration increased (Fig. 1B and C), whereas 10% PEI-PLGA nanoparticles showed little to no toxicity at the highest assessed concentration; comparable to the PLGA nanoparticle control. Therefore, the 10% PEI-PLGA nanoformulation was selected for all further studies.
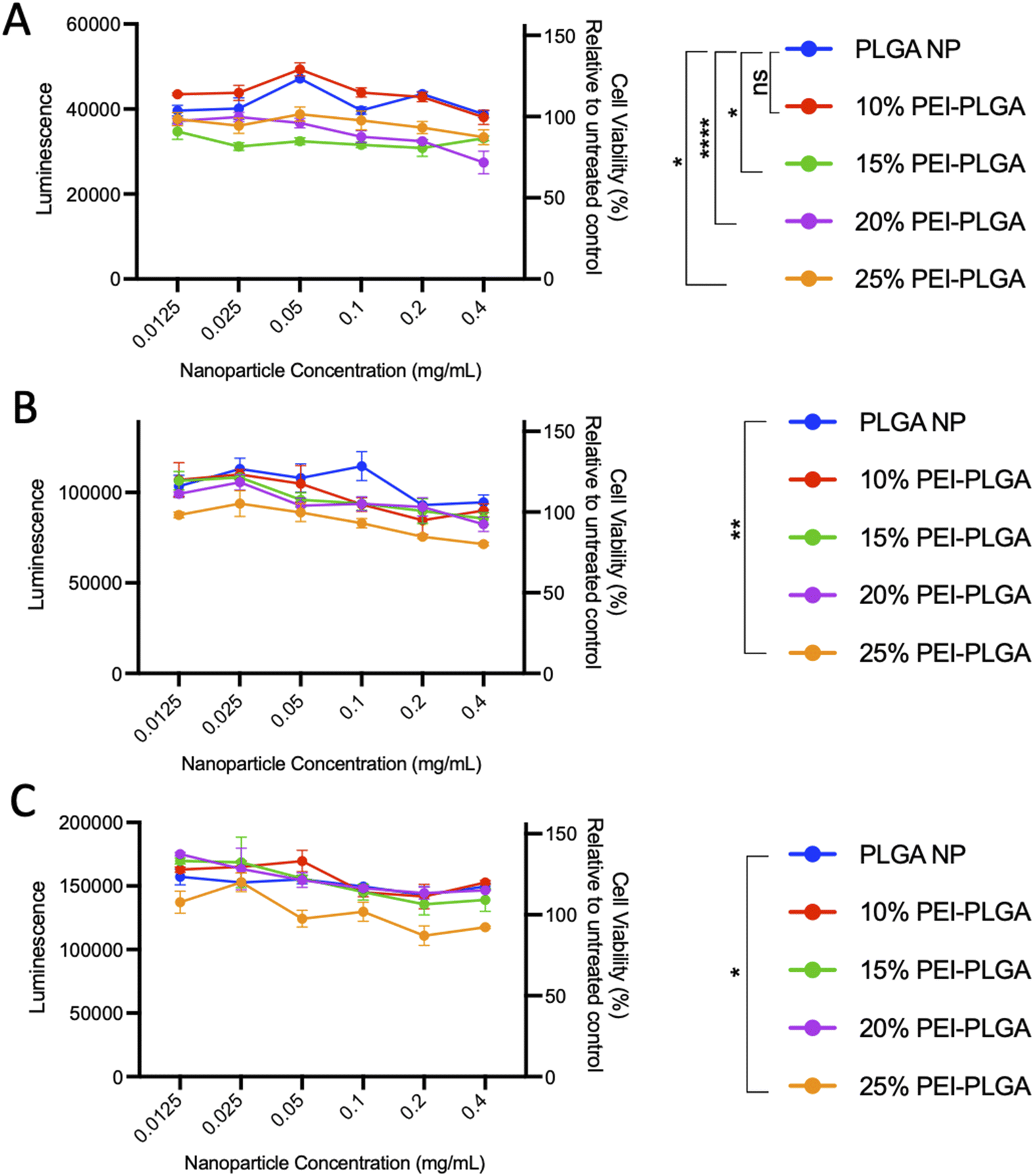 | ||
| Fig. 1 Evaluation of PEI-PLGA nanoparticle formulation. To determine cell viability, HeLa cells were treated with 0.012–0.4 mg per mL of PLGA or 10–25% PEI-PLGA nanoformulations. Cell viability was assessed (A) 24, (B) 48 and (C) 72 h post-treatment using CTG assay (Promega). Assay preformed in triplicate; data expressed as mean ± SEM. Statistical significance determined using a two-way ANOVA and Dunnett's multiple comparison's test (ns or no line denotes no significance, *p < 0.1, **p < 0.01, ***p < 0.001, ****p < 0.0001) where significance is denoted in legend. | ||
Cytotoxicity of the final 10% PEI-PLGA nanoparticle formulation was further assessed via Cell Titer- Glo® (CTG), by measuring cell viability in HeLa cells after 72 h treatment. The incubation of positively charged 10% PEI-PLGA nanoparticles lead to a significant reduction cell viability in HeLa cells, particularly after 48- and 72 h treatment at the highest concentration (0.6 mg per mL polymer), compared to the PLGA nanoparticle control (Fig. 2). However, at polymer concentrations <0.5 mg mL−1 nanoparticles show a negligible decrease in cell viability. Next, the stability of each nanoformulation was assessed, where both PLGA and 10% PEI-PLGA nanoparticles were subjected to a range of storage conditions (+4 °C, −20 °C and RT). For a period of up to 21 days, the physical characteristics of each nanoparticle formulation (diameter, PDI and zeta potential) was assessed. The PLGA and 10% PEI-PLGA nanoparticles were both found to have a diameter of ∼250 nm, where no significant change was observed in the baseline characteristics of both PLGA and 10% PEI-PLGA nanoparticles, whilst PDI remained <0.2, demonstrating a monodisperse nanoparticle suspension and the zeta potential for each formulation remained relatively consistent over the time course of the study at all three storage conditions (ESI 2†).
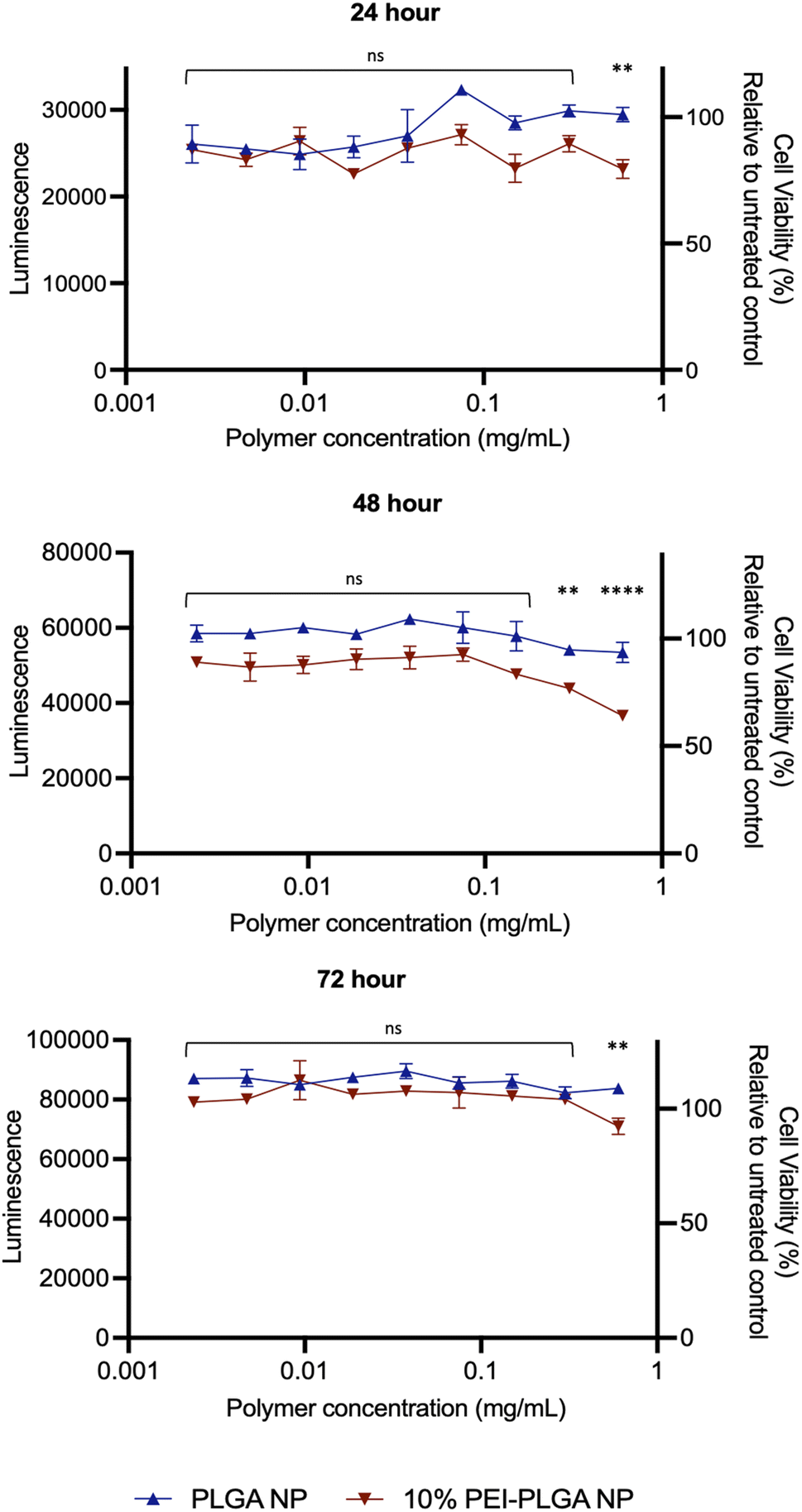 | ||
| Fig. 2 Cationic nanoparticles show minimal decrease in cell viability compared to anionic control. To determine cell viability, HeLa cells were treated with 0.003–0.6 mg per mL of PLGA or 10% PEI-PLGA nanoparticles (NP). Cell viability was assessed 24, 48 and 72 h post-treatment using CTG assay (Promega). Assay performed in triplicate; data expressed as mean ± SEM. Representative of three independent experiments. Statistical significance determined using a two-way ANOVA (ns denotes no significance, **p < 0.01, ****p < 0.0001) where significance is denoted in legend. | ||
Assessment of endo–lysosomal disruption
The behaviour of the nanoparticles once internalised and ability to escape the endo–lysosomal system was next studied. Firstly, calcein tracking confocal microscopy and acridine orange staining via flow cytometry was employed to assess the ability of the cationic nanoparticle formulation to enhance nanoparticle escape form the endo/lysosomal pathway. Calcein is a cell impermeable, fluorescent dye, which has previously been utilised to assess endosomal membrane stability.30,31 As calcein is cell impermeable, internalisation results in staining of the endosomal lumen following endocytosis. Cells incubated with calcein alone (untreated), showed no staining indicating there was no internalisation of the dye alone, without nanoparticle uptake (Fig. 3). Whereas those cells incubated with the anionic PLGA nanoparticle (charge −11.07 mV ± 0.98) in combination with calcein displayed punctate staining, which is indicative of the presence of the dye within the endocytic vesicles, showing that the calcein has been up taken alongside the PLGA nanoparticles via endocytosis resulting in staining of the endosomal lumen. The staining pattern observed is very clear punctate staining, highlighting vesicle integrity has been preserved, and nanoparticles are within the endosome (Fig. 3). Upon destabilisation and disruption of the endo–lysosomal membrane, this results in membrane leakage, thus resulting in leaching of calcein from the endosome. As a result, the calcein dye is released into the wider cytosolic region, appearing as a dispersed cytosolic staining pattern. This is observed following treatment with the cationic 10% PEI-PLGA nanoparticle formulation (charge +6.01 mV ± 3.86), where staining is observed as being more diffuse throughout the cytosolic space, in stark contrast to the aforementioned punctate staining seen with control PLGA nanoparticles (Fig. 3). This signifies that the 10% PEI-PLGA nanoparticles are capable of disrupting the endosomal membrane sufficiently to escape the endo–lysosomal pathway.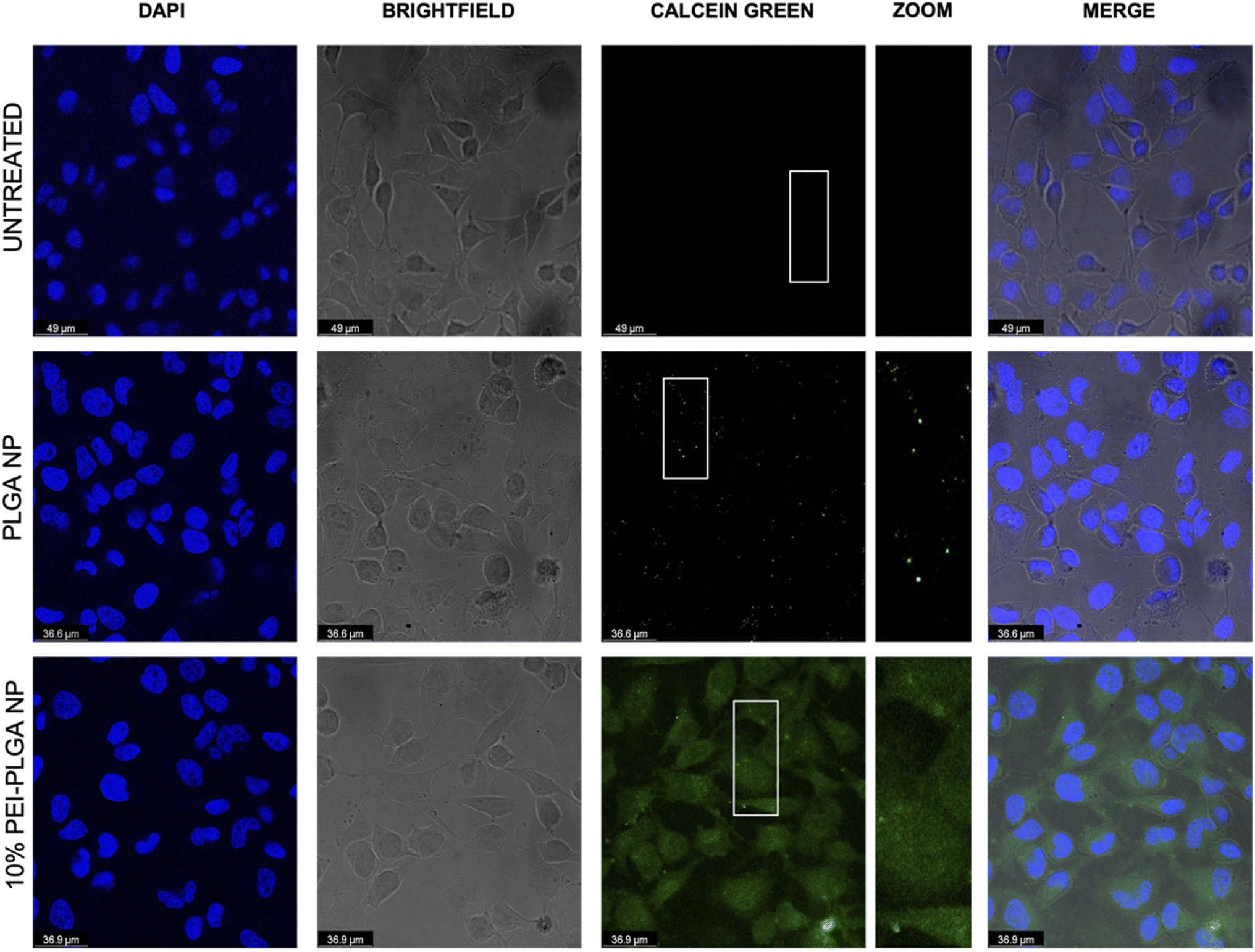 | ||
| Fig. 3 Assessment of endo/lysosomal disruption following nanoparticle treatment using calcein. HeLa cells were treated simultaneously with calcein (2 mg mL−1) and PLGA nanoparticles, 10% PEI-PLGA nanoparticles (0.2 mg mL−1) or PBS (untreated control). Following 3 h of treatment cells were fixed, permeabilised and stained with DAPI for visualisation of nuclear regions. Slides were imaged using confocal microscopy with DAPI (blue), calcein (green) and merged images shown. Images representative of three independent experiments. | ||
Endo–lysosomal escape was further confirmed via an acridine orange assay. HeLa cells were treated with 0.02–0.5 mg mL−1 of both PLGA and 10% PEI-PLGA nanoparticles for 24 and 48 h. Following treatment, cells were collected and stained by incubating with acridine orange. Acridine orange is a fluorescent dye which accumulates within acidic regions, such as the lysosome. Following lysosomal disruption, the fluorescent signal of acridine orange diminishes and becomes more diffuse, suggesting the dye is being released within the more neutral environment of the cytosol, essentially escaping the lysosome.32
This diminished staining profile is observed by a loss of fluorescence (a shift to the left, (ESI 3†)) when compared to the untreated control. Following treatment of HeLa cells with 10% PEI-PLGA nanoparticles, the acridine orange signal is significantly reduced in comparison to PLGA nanoparticles and untreated cells. Indeed, fluorescent signal was seen to be concentration dependant, with respect to the 10% PEI-PLGA nanoparticles at both 24 and 48 h time points (Fig. 4). This highlights again the ability of the cationic nanoparticle formulation to escape the endo–lysosomal system and subsequently delivery their cargo to the cytosol.
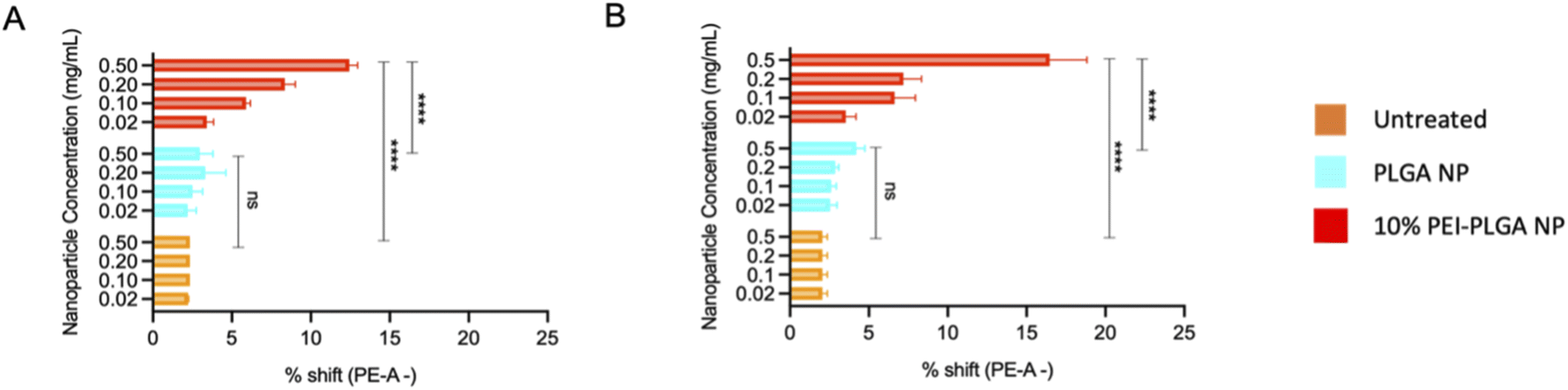 | ||
| Fig. 4 Cationic nanoparticles are capable of causing endo/lysosome disruption. The ability of cationic nanoparticles to escape the lysosome was assessed via acridine orange staining. HeLa cells were treated with 0.02–0.5 mg mL−1 of PLGA or 10% PEI-PLGA nanoparticles. Following treatment cells were stained with acridine orange and cell fluorescence detected via flow cytometry. Data shown as % shift of PE-cells (shift to the left), where disruption in observed in a concentration dependant manner following (A) 24 h and (B) 48 h relative to untreated control. Data expressed as mean ± SEM (n = 3). Statistical significance was established by one-way ANOVA and Tukey's post hoc test (ns or no line denotes no significance and ****p < 0.0001). | ||
Entrapment of a hydrophilic vNAR within nanoparticle core
For the purpose of these studies, a model vNAR was selected, containing a FITC tag to accommodate quantification of vNAR entrapment, as well as downstream localisation studies. In the first instance, the ability to entrap the vNAR within the nanoparticle core was assessed using the PLGA nanoparticle control, where varied quantities of vNAR (5–20 μg per mg polymer) were added to the aqueous phase during nanoparticle synthesis. After washing, vNAR loaded nanoparticles (subsequently referred to as vNAR nanoparticles) were assessed to determine physiochemical properties and vNAR entrapment. vNAR nanoparticles displayed diameters (∼200 nm) comparable to the PLGA nanoparticle control (Fig. 5A), whilst all formulations demonstrated a PDI <0.2, indicating a monodisperse nanoparticle suspension. vNAR entrapment was assessed following addition of between 5-20 μg vNAR per mg polymer. Interestingly, it was observed that there was no clear correlation of vNAR concentration in the formulation input and resultant entrapment, with entrapment efficiency demonstrating a reducing trend with increased vNAR concentration (Fig. 5C and D). Optimal vNAR entrapment was observed with 5 μg vNAR added to the formulation mix (1.70 ± 0.98 μg per mg polymer), with the optimal entrapment efficacy (Fig. 5D). Given that increasing amounts of vNAR input did not lead to enhanced entrapment (Fig. 5C), this formulation was selected for further studies.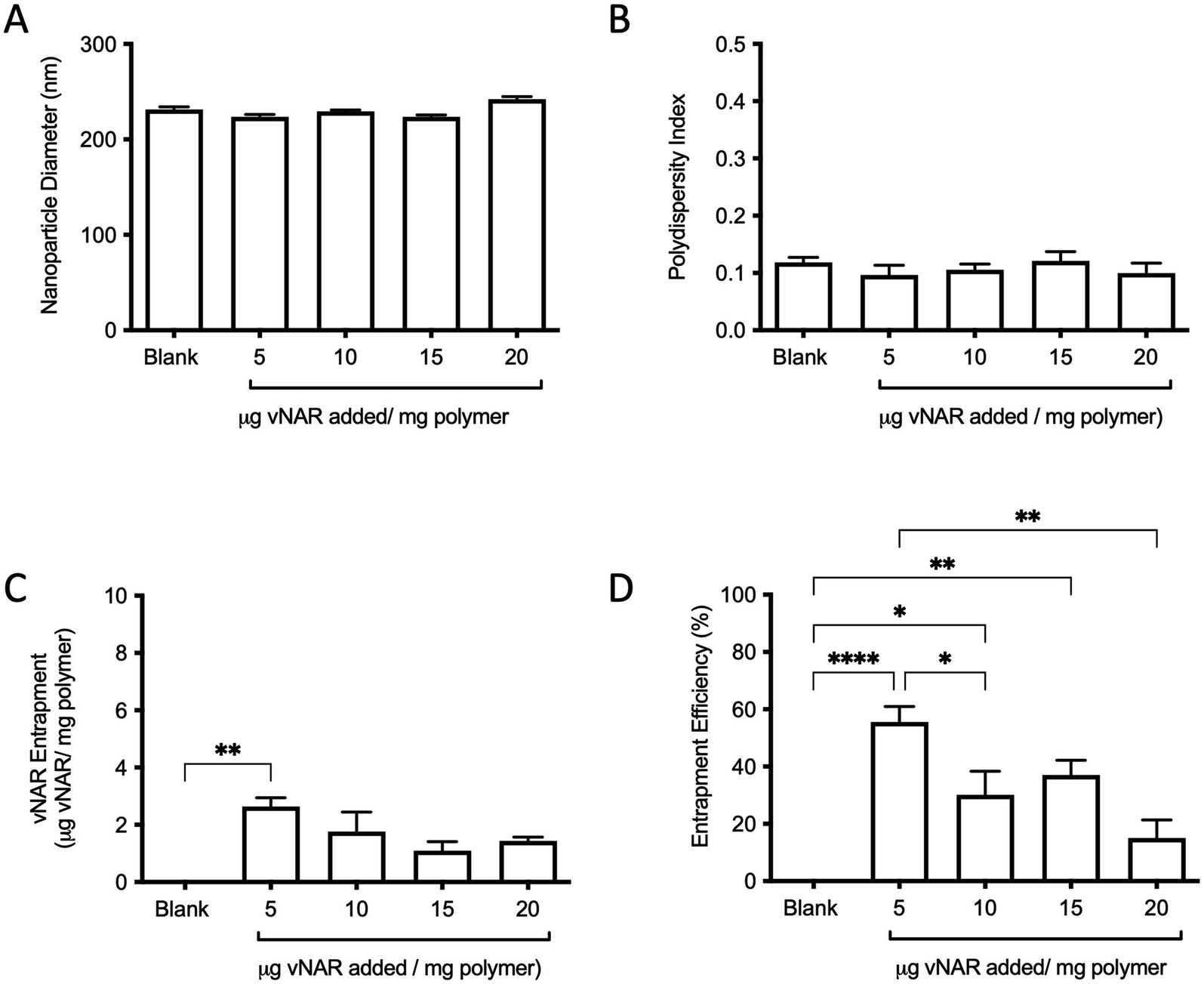 | ||
| Fig. 5 Characterisation of vNAR entrapment within nanoparticle core. Various amounts of fluorescent D8-FITC vNAR (5–20 μg per mg polymer) were added during nanoparticle synthesis, with physicochemical properties of blank PLGA nanoparticles and vNAR nanoparticles subsequently assessed by DLS in terms of (A) particle diameter, and (B) polydispersity index. (C) vNAR entrapment and entrapment efficiency (D) were assessed through measurement of fluorescence and extrapolated from a standard curve. Measurements were conducted in triplicate and presented as mean ± SEM. Statistical significance determined using one-way ANOVA and Tukey's post hoc test (ns or no line denotes no significance, *p < 0.1, **p < 0.01, ***p < 0.001, ****p < 0.0001). | ||
Following formulation of both 10% PEI-PLGA cationic and vNAR entrapped nanoformulations, both formulation parameters were combined to obtain a cationic nanoparticle delivery system for enhanced delivery of entrapped cargo, in this instance, vNARs. vNAR loaded 10% PEI-PLGA nanoparticles were synthesised as above, and physiochemical characteristics assessed via DLS (Fig. 6), and SEM (ESI 4A–D†). Nanoparticle diameter was in anticipated range (∼200–250 nm), with a negligible increase in nanoparticle diameter observed for vNAR loaded nanoparticles (Fig. 6). Importantly, vNAR 10% PEI-PLGA nanoparticles displayed similar vNAR entrapment to that of the vNAR PLGA nanoparticles (Table 2) where an increase in cationic nanoparticle charge observed with the PEI nanoformulation. Nanoparticle size and dispersity were further assessed, with nanoparticles resuspended in dH2O (0.1 mg mL−1) and analysed via NTA. NTA measurements were equivalent to trends obtained by DLS and further emphasised the monodispersity of these systems (ESI 4E†).
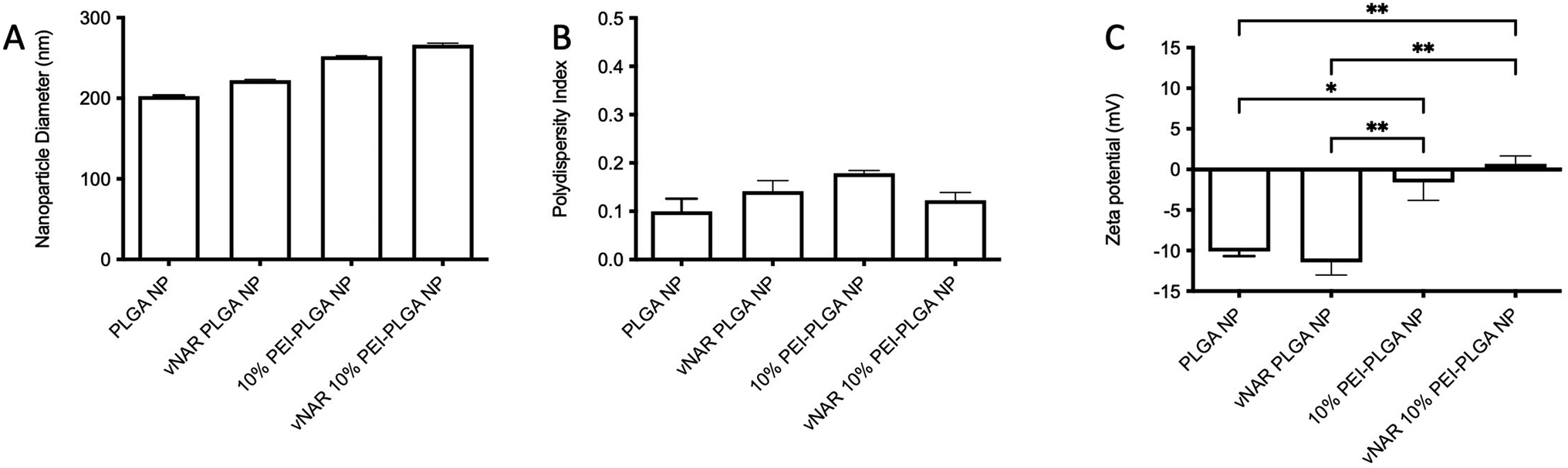 | ||
| Fig. 6 Characterisation of 10% PEI-PLGA NP formulation. Physicochemical properties of PLGA nanoparticles and 10% PEI-PLGA vNAR entrapped nanoparticles were assessed via DLS in terms of (A) particle diameter and (B) polydispersity index. (C) Zeta potential was measured via phase analysis light scattering (PALS). Nanoparticles were resuspended at 100 μg mL−1 in 0.05% PBS (v/v) in dH2O for analysis. Measurements conducted in triplicate and presented as mean ± SEM (n = 3). Statistical significance determined the using one-way ANOVA and Tukey's multiple comparisons test (ns or no line denotes no significance, *p < 0.1, **p < 0.01). | ||
| Average diameter (nm) | vNAR entrapment (μg per mg polymer) | Entrapment efficiency (%) | |
|---|---|---|---|
| vNAR PLGA NP | 252.50 ± 1.05 | 2.63 ± 0.30 | 52.66 ± 6.16 |
| vNAR 10% PEI-PLGA NP | 249.26 ± 2.13 | 2.80 ± 0.23 | 56.00 ± 4.62 |
Nanoparticle stability is a critical aspect of formulation development, as it can affect nanoparticle biodistribution resulting in unwarranted systemic toxicity.33 Therefore, stability of the nanoparticle systems was assessed. Nanoparticle physical characteristics, such as size, PDI, zeta potential and vNAR entrapment were assessed over a period of up to 10 days (ESI 5†). Throughout the course of this study, all nanoparticles synthesised were of ∼250 nm in size, with a slight size increase observed for those which were loaded with vNAR. There were no significant changes observed in the baseline characteristics of all nanoformulations assessed throughout the duration of study, demonstrating the stability of both cationic nanoparticle charge and vNAR entrapment in formulations.
vNAR retains binding affinity for target following nanoparticle formulation
Although it has been found that vNARs exhibit remarkable stability, such as temperature range and fluctuations in pH,16 their ability to withstand the harsh sonication conditions and solvents of nanoparticle synthesis is unknown. Therefore, following confirmation of successful vNAR entrapment, it was important to evaluate vNAR activity following the nanoparticle formulation process. vNAR loaded 10% PLGA-PEI nanoparticles were physically disrupted and supernatant collected for assessment via SDS-PAGE. The bands observed from the supernatant released from the nanoparticle appear to all be of the same size (∼11 kDa), indicating that the vNAR remains intact following release, where as expected, bands appear weaker than that of the free vNAR band (equivalent to 100% entrapment) (Fig. 7A). The ability of vNARs to refold and retain binding activity in a range of harsh biological conditions is a key attribute to their use for nanoformulation and intracellular delivery, therefore in addition to confirmation of vNAR release from nanoparticles, it is important to assess whether the vNAR can retain its functional activity after escaping the nanoparticle and is not somehow impaired within the nanoparticle synthesis process. In order to assess the vNAR binding, nanoparticles were disrupted, and supernatants collected as above. An ELISA was then performed in order to assess binding of disrupted vNAR nanoparticle supernatants to its cognate antigen (not disclosed) using anti-his HRP detection of the vNAR C-terminal his-tag. An increase in relative binding response was observed for the vNAR disrupted nanoparticle, in comparison to blank PLGA nanoparticle control binding (Fig. 7B). This suggests that despite the solvents and sonication techniques used within nanoparticle synthesis, the vNAR retains its binding activity. Despite there being a significant increase in binding observed between free vNAR and the disrupted vNAR nanoparticle, this is likely due to the case of residual vNAR still retained within the particle due to the mild nanoparticle disruption approach employed. Moreover, a significant increase in binding is observed with the vNAR disrupted nanoparticle in comparison to PBS and blank nanoparticle control, indicating that any vNAR released from the particle is still functional and retains its binding activity for its cognitive antigen, despite the reduction in binding observed compared to the free vNAR, which is likely due to not all vNAR being released from the nanoparticle.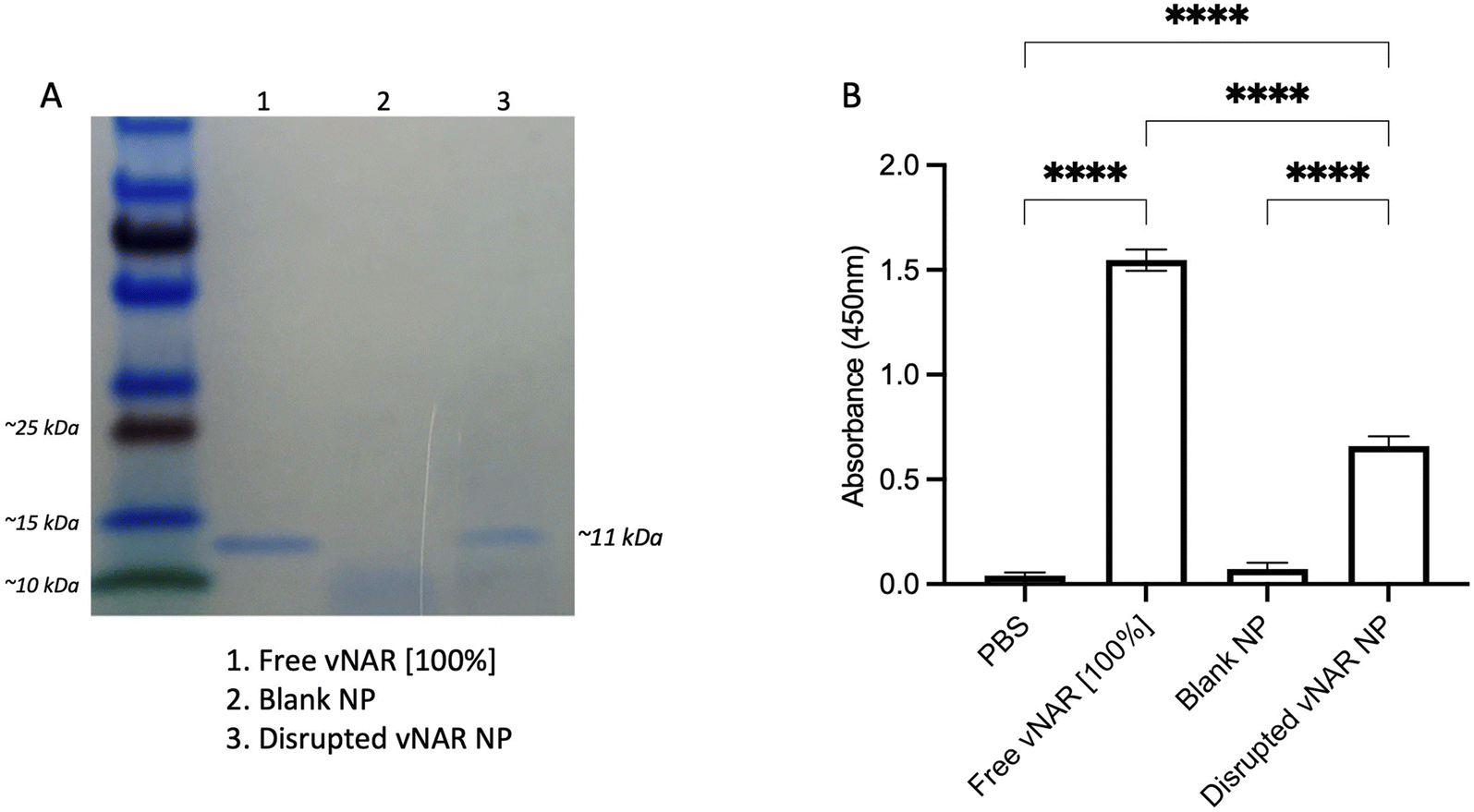 | ||
| Fig. 7 vNAR retains binding affinity for its target antigen following release from nanoparticle. 10% PEI-PLGA nanoparticles were physically disrupted via incubation at 95 °C for 2 h followed by intense sonication in an attempt to release entrapped vNAR. The supernatant was collected, and polymer pellet discarded. (A) Supernatant were prepared for analysis by SDS-PAGE. Lane (1) free vNAR (indicative of 100% nanoparticle entrapment) (2) blank nanoparticle supernatant (3) disrupted vNAR nanoparticle supernatant. The bands observed from the supernatant released from the disrupted vNAR nanoparticle appear to be of the same size of the free vNAR (∼11 kDa). (B) Binding of vNAR to its cognate antigen was assessed via ELISA, where free vNAR (indicative of 100% nanoparticle entrapment) was used as a control compared to supernatants collected from disrupted nanoparticles. Data presented as mean ± SEM (n = 3). Statistical significance determined using one-way ANOVA and Tukey's post hoc test (ns or no line denotes no significance, ****p < 0.0001). | ||
Cationic nanoparticles exhibit enhanced cellular uptake
Cationic nanoparticles are thought to exhibit a greater internalisation rate in comparison to neutral or anionic nanoparticles, owing to the overall net negative charge of the plasma membrane resulting in the electrostatic attraction of cationic nanoparticles.34,35 Thus, the effect of cationic nanoparticles on cellular uptake was evaluated following treatment of HeLa cells with vNAR loaded PLGA and 10% PEI-PLGA nanoformulations. Cells were treated with 200 μg per mL of vNAR 10% PEI-PLGA nanoparticles and vNAR PLGA control nanoparticles, in order to assess nanoparticle uptake in cells. Cells were imaged using confocal microscopy (Fig. 8), where nanoparticle uptake was quantified by assessing number of fluorescent points per cell, representative of FITC tagged vNAR localisation within the cell (Fig. 9).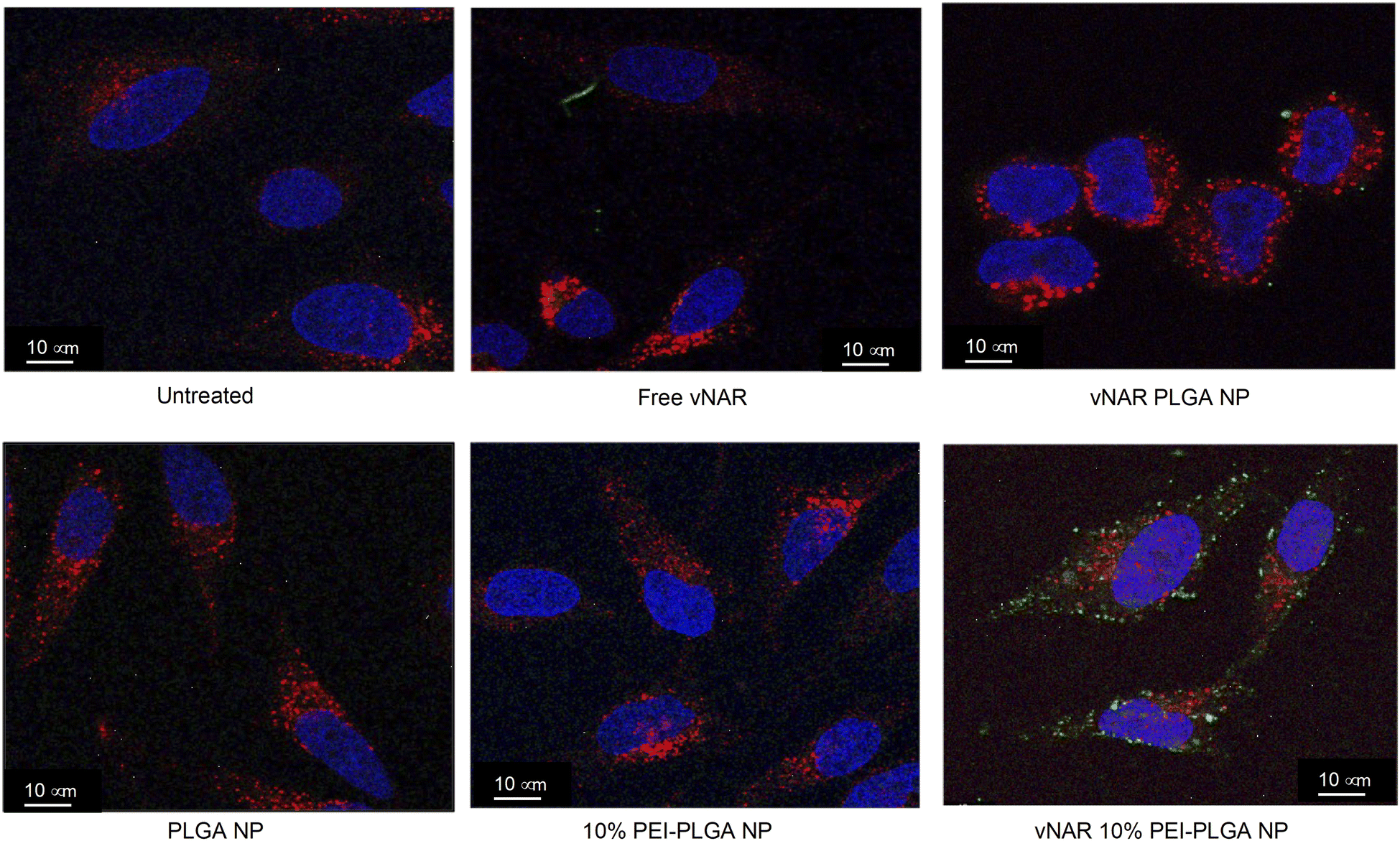 | ||
| Fig. 8 10% PEI-PLGA nanoparticles exhibit enhanced cellular uptake in comparison to PLGA nanoparticles. Nanoparticle internalisation was assessed in HeLa cells by confocal microscopy. Cells were seed at 50000 cells per well in an 8-well chamber slide and left overnight to adhere. Cells were treated with 0.2 mg mL−1 of various nanoparticles (PLGA, 10% PEI-PLGA, vNAR PLGA and vNAR 10% PEI-PLGA) and relevant controls for 1 h. Post treatment, cells were washed with both PBS and an acid strip wash before fixing cells for permeabilization and staining. Lysosomal regions were stained using anti-LAMP1 antibody (red) and nuclear regions stained (blue) using DAPI. FITC conjugated vNAR entrapped within the nanoparticle core can be visualised in green. Images representative of three independent experiments. Scale bars = 10 μm. | ||
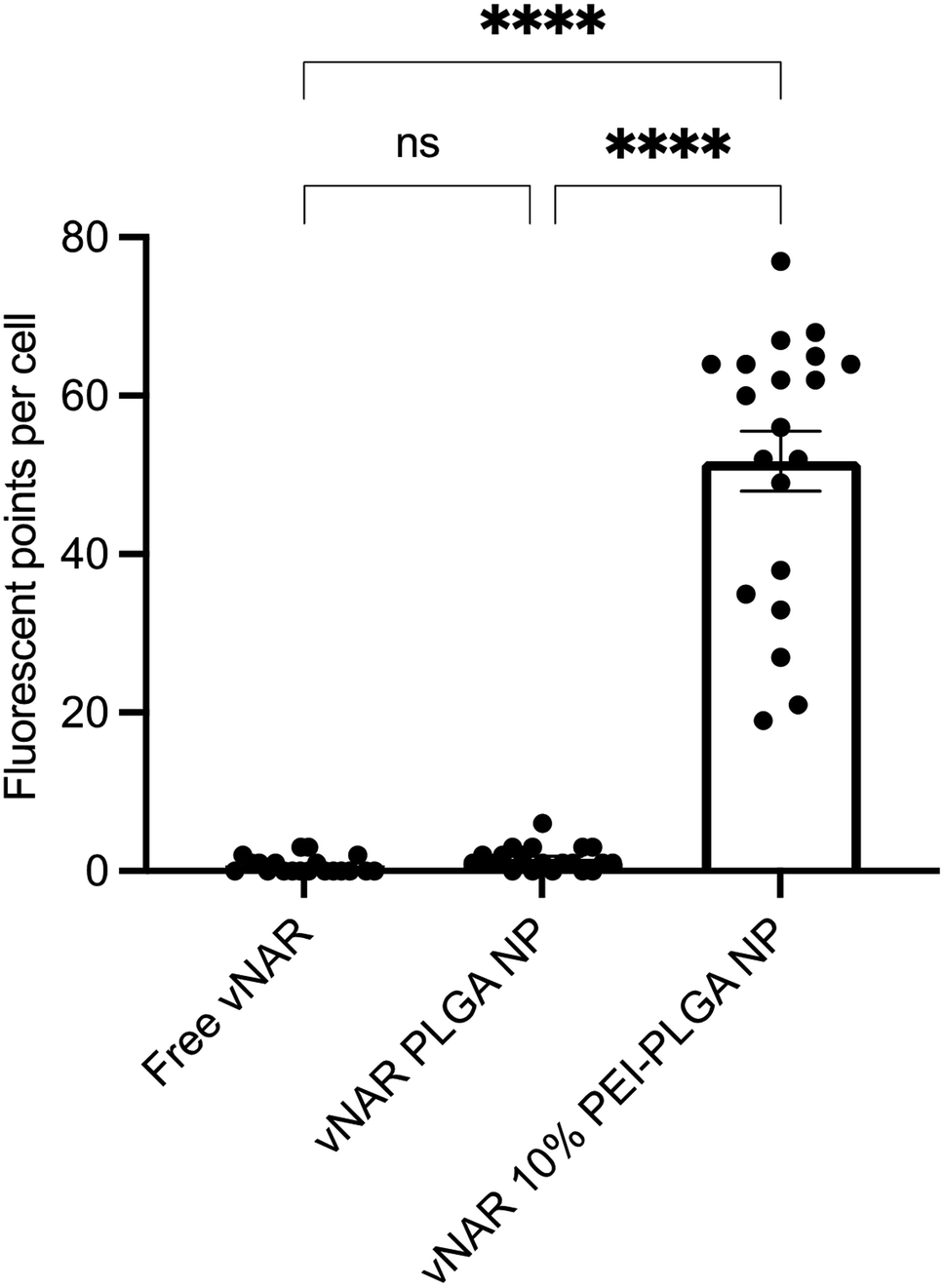 | ||
| Fig. 9 Quantification of nanoparticle uptake in cells. Nanoparticle uptake between treatment groups (free vNAR, vNAR PLGA nanoparticles and vNAR 10% PEI-PLGA nanoparticles) was quantified by determining the number of fluorescent points per cell per individual experiment. Each treatment was conducted in duplicate (2 wells), where 20 cells were selected at random for counting per treatment group. Experiment performed in triplicate, with data presented as mean ± SEM (n = 3). Statistical significance was established by one-way ANOVA and Tukey's post hoc test (ns or no line denotes no significance and ****p < 0.0001). | ||
Cellular localisation studies with nanoparticle formulations
To determine the subcellular localisation of both nanoformulations after internalisation, cells were treated with each nanoformulation as above, and subsequently stained with organelle-specific fluorescent markers, including DAPI, EEA1 and LAMP1 in order to visualise nuclei, early endosomes and lysosomes respectively. Merged images detail that not only is there a significant increase in cationic vNAR 10% PEI-PLGA nanoparticle uptake as previously observed, but after 1 h of treatment, vNAR 10% PEI-PLGA nanoparticles are present within the endosome, denoted by the colocalisation of staining with EEA1 (Fig. 10). Little to no colocalization of nanoparticles was observed with LAMP1 within the lysosome (after timepoints of 30 min–4 h (images shown for 1 h timepoint).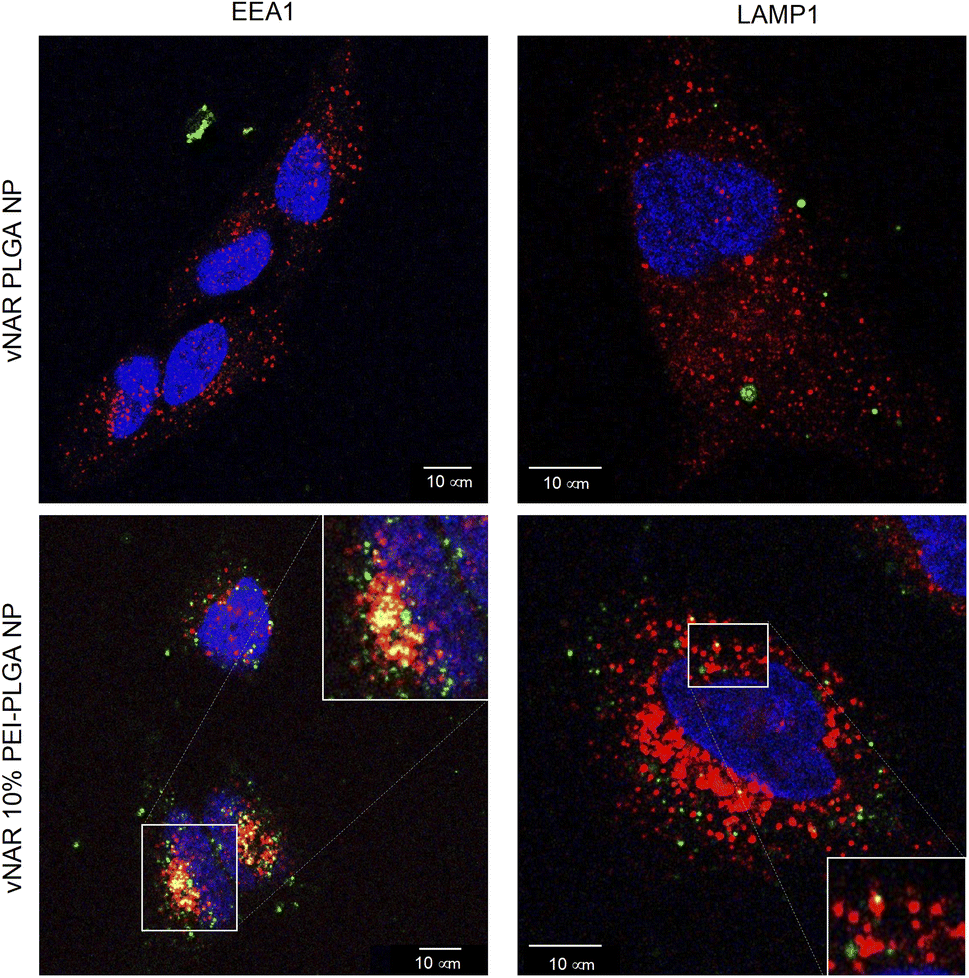 | ||
| Fig. 10 Assessment of nanoparticle localisation within the cell. Localisation of nanoparticles following treatment was assessed in HeLa cells using confocal microscopy. Cells were seeded at 50000 cells per well in an 8-well chamber slide and left overnight to adhere. Cells were treated with 0.2 mg mL−1 of either vNAR PLGA or vNAR 10% PEI-PLGA nanoparticles for 1 h. Post treatment, cells were washed with both PBS and an acid strip wash before fixing cells for permeabilization and staining. Lysosomal and endosomal regions were stained using anti-LAMP1/anti-EEA1 antibody respectively, visualised in red. Nuclear regions were stained using DAPI, visualised in blue. FITC conjugated vNAR entrapped within the nanoparticle core can be visualised in green. Images representative of three independent experiments. Scale bars = 10 μm. | ||
Moreover, there was no apparent localisation of vNAR PLGA nanoparticles within any endo–lysosomal compartment. The degree of colocalisation was quantified via Pearson's correlation coefficient (R) using ImageJ colocalisation analysis, where R +1 indicated perfect association and R < 0 indicated no correlation. Pearson's correlation values indicated there was moderate correlation in colocalisation of staining with vNAR 10% PEI-PLGA nanoparticles and EEA1 (R = 0.2), whereas there was no association with LAMP1 (R = −0.42). After demonstrating enhanced uptake, next we assessed whether we could detect the nanoparticle cargo within the cell after treatment. HeLa cells were treated for 3 h and 6 h timepoints with PLGA and 10% PEI-PLGA nanoparticles containing vNAR which contains a C-terminal his tag. Following treatment cells were collected and upon lysis, each sample was incubated with Ni-NTA IMAC resin to allow capture of the HIS-tagged vNAR before assessment by western blotting. Upon blotting against the x6-his tag, a distinctive band is observed (∼11 kDa, the approximate weight of the x6-his tagged vNAR) in lane 5, indicative of those cells treated with the cationic 10% PEI-PLGA nanoparticle, where a larger band is observed after 3 h treatment compared to 6 h (Fig. 11A and B).
 | ||
| Fig. 11 Delivery of vNAR into cells using cationic nanoparticle delivery system. HeLa cells were seeded and left overnight to adhere. Cells were treated with either vNAR PLGA or vNAR 10% PEI nanoparticles along with relevant controls for both (A) 3 and (B) 6 h timepoints. (1) Untreated, (2), PLGA NP, (3) 10% PEI NP, (4) vNAR PLGA NP, (5) vNAR 10% PEI NP. Post treatment, cells were washed with both PBS and an acid strip wash to remove any nanoparticles yet to be internalised before collecting cells for assessment via western blot. Lysates were collected, with cells incubated with his-NTA beads to select those cells containing the x6-his tag present on the C terminal of the vNAR. Samples were centrifuged to pull down any intact nanoparticles or polymer, ensuring the isolated supernatant contained only the soluble fraction representing what is inside the cell. Soluble fraction of supernatant was separated via SDS-PAGE and western blotting. Protein was detected using an anti-x6-his antibody, where band at ∼11 kDa is the approximate weight of vNARs. β-Actin was used as a protein loading control where a band is visible at ∼42 kDa. Data representative of three independent experiments. | ||
This is suggestive that the vNAR remains intact once delivered intracellularly, where more protein is observed 3 h post-treatment indicating that this is the optimal timepoint for detection, where after 6 h, it is possible that the his tag may be subject to proteolytic cleavage. These findings correlate with previous data presented, where the vNAR 10% PEI-PLGA nanoparticle demonstrated the ability for enhanced uptake into the cell.
Discussion
Despite the identification of promising intracellular targets within a range of disease states, the progression of associated therapeutics to the clinic has been underwhelming. Attempts to date have been limited by the inefficiency of small molecules in binding targets of interest, whilst more promising biologics have been restricted by stability issues, and sub-optimal delivery mechanisms. Here we report a PEI nanoparticle delivery system which is not only capable of endo–lysosomal escape to achieve intracellular delivery of a vNAR cargo, but also has limited cellular toxicity. Nanoparticles were characterised by assessment of nanoparticle diameter, monodispersity and zeta potential. Nanoparticle size and PDI can affect particle biodistribution, as any fluctuation in particle diameter can alter both biodistribution and bioavailability, with the ideal size of nanoparticles for endocytosis and tumour filtration has been said to range between 100–300 nm.33,36–40Furthermore, cationic nanoparticles have been shown to accelerate the rate of nanoparticle uptake as well as facilitate endo/lysosomal escape facilitating delivery of nanoparticle cargo to the cytosol.30,41,42 Physical characterisation of particles indicated that the 10% PEI-PLGA nanoformulation possessed the optimal characteristics for delivery, whilst also remaining stable for a period of up to 3 weeks. Whilst this nanoparticle formulation has been shown to be stable for storage at a range of conditions for this time period, stability is a key component for drug formulation, thus once a final formulation has been established where a vNAR has been identified against a specific intracellular target/disease state, stability will need to be further assessed. Therefore, future work should include assessment of vNAR binding following nanoparticle release in a biological setting, where vNAR functionality can be assessed in vitro after an extended period of time.
In this work, a vNAR was chosen as the nanoparticle cargo. As mentioned above, vNARs provide many attractive properties which are desirable when considering intracellular targets. vNARs are both the oldest (420 million years) and smallest (∼11 kDa) single chain domains acknowledged within vertebrates17,43 and exhibit many distinctive properties attributed to their unique origin within the adaptive immune system of sharks, such as significant stability and high affinity for their target, desirable features for intracellular delivery.17,44 Within their structure, the presence of a fourth binding loop and protruding paratopes facilitated by non-canonical disulfide bonds enables a distinctive ‘canyon binder’ targeting approach. The unique structure of vNARs also facilitates their propensity to bind unique and cryptic epitopes, inaccessible to more conventional biologics.16 Aside from their remarkable specificity and affinity for their target antigen, vNARs are subject to exposure to harsh conditions within their natural shark sera environment, which is characterized by having high levels of salt and urea. This has resulted in a molecule which possesses considerable stability, making them ideal for the harsh conditions which are characteristic of the intracellular environment, such as extreme pH, as well as exposure to proteases, all whilst retaining their binding affinity.16,17,45,46 The remarkable stability associated with vNARs make them ideal candidates for nanoformulation, where it has been demonstrated that vNARs remain physically intact as well as retaining all binding activity after enduring the harsh sonication techniques and presence of solvents during the process of nanoparticle synthesis, however further work should consider the relationship with pH and charge on encapsulation and subsequent release.
Upon cellular internalisation, nanoparticles preferentially accumulate within the endo/lysosomal pathway owing to their size profile.47,48 Nanoparticles are internalised via endocytosis, where they fuse into the early endosome. This vesicle then matures into the late endosome and subsequently lysosome.31 Endosomal escape is essential for intracellular delivery of therapeutics, as failure of nanoparticles to escape the endosome leads to accumulation within the lysosome, where degradation of the entrapped therapeutic is probable.30 In this study we have demonstrated that our 10% PEI-PLGA nanoparticle delivery system not only enhances nanoparticle uptake, demonstrated by an 8-fold increase in nanoparticle uptake, but shown that nanoparticles are visibly present within the endo/lysosomal pathway via colocalisation studies. Furthermore, it is shown that 10% PEI-PLGA nanoparticles are able to effectively escape the endo–lysosomal pathway as a result of endo–lysosomal disruption, clearly demonstrated by calcein and acridine orange staining profiles. However, the precise mechanism of action has not been determined.
Numerous theories as to how nanoparticles can escape the endosome have been proposed, one being the ‘proton sponge’ effect.28,49 This phenomenon states that once incorporated within the acidic pH of the endosomal lumen, the amine groups present on the PEI branched chains become protonated, resulting in buffering of the endosomal pH. In turn, this creates an influx of chlorine ions and water resulting in rupture of the lysosomal membrane as a consequence of osmotic swelling. In addition, the repulsive forces between similarly charged amine groups leading to swelling of the cationic nanoparticle may also contribute to loss of membrane integrity of the endosome. As a result, the ‘proton sponge’ theory itself may account for this cellular toxicity.49 However, validity of the proton sponge effect as a mechanism of endosomal disruption has been questioned, where reports have shown no observational change in lysosomal pH after treatment with cationic polymers, therefore other potential mechanisms of endo/lysosomal escape have been proposed.49 Such methods include endosomal release as a consequence of direct interaction of the cationic nanoparticle with the endosomal membrane itself, resulting in membrane disruption, as well as the effect of nanoparticle polymer degradation resulting in increased osmotic pressure. In addition, mechanisms reported also include, membrane fusion, nanoparticle swelling and membrane destabilisation.30,50 Furthermore, the nanoparticle formulation could be further modified through the addition of pH sensitive polymers, to further tailor the nanoformulation to react to fluctuations in pH within the tumour microenvironment, where pH sensitivity can be further investigated.51–55
Whilst promising, utilisation of cationic particles has proven challenging. One of the key concerns regarding the potential clinical translation of such cationic nanoparticles is their increased cellular cytotoxicity, as demonstrated by studies which have shown that cationic nanoparticles at high concentrations may induce caspase 3/7 activation and PARP cleavage ultimately leading to cell death.15 Furthermore, internalisation of cationic nanoparticle formulations can often induce cell death due to their effect on various intracellular mechanisms such as lysosomal disruption and damage to the mitochondria.35 Cationic nanoparticles must have a sufficient positive charge to increase cellular uptake and subsequently escape the endosome; however, this must be balanced by the need to maintain biocompatibility and not induce significant cell death. Although cationic nanoparticles facilitate a greater rate of internalisation, it is essential that the nanoparticles must be well tolerated with negligeable risk to healthy tissues.32,56–58 Hence, in addition to stability, toxicity screening of cationic nanoparticles is key. Encouragingly, our preliminary studies indicate the cationic 10% PEI-PLGA nanoparticles show little to no toxicity as observed in a panel of cancer cell lines, with only minimal toxicity observed even after 72 h at the top polymer concentration.
Conclusion
In summary, the application of a double emulsion and evaporation nanoparticle formulation technique successfully resulted in the formation of a cationic nanoparticle (∼250 nm) which can be successfully loaded with a protein cargo, in this case a vNAR. The vNAR was not only successfully entrapped within the nanoparticle core but was also shown to retain binding capacity following nanoparticle formulation and subsequent release. The incorporation of the cationic polymer (PEI) within the nanoformulation significantly improved nanoparticle uptake into cancer cells in vitro, as well as facilitating endosomal escape after nanoparticle endocytosis, all whilst causing minimal cytotoxicity. Future work should include assessment of vNAR functionality post-delivery against their target antigen as well as, examination into the exact mechanism of vNAR escape. In addition, further investigation of nanoparticle toxicity, biodistribution and pharmacokinetics should be assessed in vivo. Taken together, this study provides rationale and evidence for utilising PLGA-PEI nanoparticles for the delivery of biologics towards intracellular targets, whilst also highlighting the enormous untapped potential for using vNARs therapeutically within this space.Data availability
All data generated in this study are included within the main article and/or ESI† accompanying this paper.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was funded a grant from the BBSRC (BB/R009112/1) and a studentship funded by the Northern Ireland Department for Economy (DFE).References
- J. Wang, S. Li and H. Li, Drug Design of “Undruggable” Targets, Chin J Chem, 2019, 37(5), 501–512 CrossRef CAS.
- D. A. Wheeler and L. Wang, From human genome to cancer genome: The first decade, Genome Res., 2013, 23(7), 1954 CrossRef PubMed . Available from: http://genome.cshlp.org/content/23/7/1054.abstract.
- J. S. Lazo and E. R. Sharlow, Drugging Undruggable Molecular Cancer Targets, Annu. Rev. Pharmacol. Toxicol., 2016, 56(1), 23–40 CrossRef CAS PubMed.
- P. L. Bedard, D. M. Hyman, M. S. Davids and L. L. Siu, Small molecules, big impact: 20 years of targeted therapy in oncology, Lancet, 2020, 395(10229), 1078–1088 CrossRef CAS PubMed.
- L. Zhong, Y. Li, L. Xiong, W. Wang, M. Wu and T. Yuan, et al., Small molecules in targeted cancer therapy: advances, challenges, and future perspectives, Signal Transduction Targeted Ther., 2021, 6(1), 201 CrossRef PubMed.
- K. Strebhardt and A. Ullrich, Paul Ehrlich ’ s magic bullet concept : 100 years of progress, Nat. Rev. Cancer, 2008, 8(june), 473–480 CrossRef CAS PubMed.
- K. A. Jinturkar, M. N. Rathi and A. Misra, Gene Delivery Using Physical Methods, in Challenges in Delivery of Therapeutic Genomics and Proteomics, ed. A. Misra, Elsevier, London, 2011, pp. 83–126, available from: http://www.sciencedirect.com/science/article/pii/B9780123849649000037 Search PubMed.
- A. Bolhassani, A. Khavari and Z. Orafa, Electroporation – Advantages and Drawbacks for Delivery of Drug, Gene and Vaccine, Application of Nanotechnology in Drug Delivery [Internet], 2014. pp. 369–98, available from: https://www.intechopen.com/books/advanced-biometric-technologies/liveness-detection-in-biometrics Search PubMed.
- Y. Zhang and L. C. Yu, Microinjection as a tool of mechanical delivery, Curr. Opin. Biotechnol., 2008, 19(5), 506–510 CrossRef CAS PubMed . Available from: http://www.sciencedirect.com/science/article/pii/S0958166908000864.
- A. Albanese, P. S. Tang and W. C. W. Chan, The effect of nanoparticle size, shape, and surface chemistry on biological systems, Annu. Rev. Biomed. Eng., 2012, 14, 1–16 CrossRef CAS PubMed.
- A. Kumari, S. K. Yadav and S. C. Yadav, Biodegradable polymeric nanoparticles based drug delivery systems, Colloids Surf., B, 2010, 75(1), 1–18 CrossRef CAS PubMed.
- G. Chen, A. A. Abdeen, Y. Wang, P. K. Shahi, S. Robertson and R. Xie, et al., A biodegradable nanocapsule delivers a Cas9 ribonucleoprotein complex for in vivo genome editing, Nat. Nanotechnol., 2019, 14(10), 974–980 CrossRef CAS PubMed.
- M. R. Villegas, A. Baeza and M. Vallet-Regí, Nanotechnological strategies for protein delivery, Molecules, 2018, 23(5), 1–21 CrossRef PubMed.
- R. R. Arvizo, O. R. Miranda, M. A. Thompson, C. M. Pabelick, R. Bhattacharya and J. David Robertson, et al., Effect of nanoparticle surface charge at the plasma membrane and beyond, Nano Lett., 2010, 10(7), 2543–2548 CrossRef CAS PubMed.
- M. G. Bexiga, J. A. Varela, F. Wang, F. Fenaroli, A. Salvati and I. Lynch, et al., Cationic nanoparticles induce caspase 3-, 7-and 9-mediated cytotoxicity in a human astrocytoma cell line, Nanotoxicology, 2011, 5(4), 557–567 CrossRef CAS PubMed.
- K. Griffiths, O. Dolezal, K. Parisi, J. Angerosa, C. Dogovski and M. Barraclough, et al., Shark Variable New Antigen Receptor (VNAR) Single Domain Antibody Fragments: Stability and Diagnostic Applications, Antibodies, 2013, 2(4), 66–81 CrossRef CAS.
- C. Barelle and A. Porter, VNARs: An Ancient and Unique Repertoire of Molecules That Deliver Small, Soluble, Stable and High Affinity Binders of Proteins, Antibodies, 2015, 4(3), 240–258 CrossRef CAS . Available from: http://www.mdpi.com/2073-4468/4/3/240/.
- M. Feng, H. Bian, X. Wu, T. Fu, Y. Fu and J. Hong, et al., Construction and next-generation sequencing analysis of a large phage-displayed VNARsingle-domain antibody library from six naïve nurse sharks, Antibiot. Ther., 2018, 1(1), 1–11 Search PubMed.
- J. Steven, M. R. Müller, M. F. Carvalho, O. C. Ubah, M. Kovaleva and G. Donohoe, et al., In vitro maturation of a humanized shark VNAR domain to improve its biophysical properties to facilitate clinical development, Front. Immunol., 2017, 8, 1361 CrossRef PubMed.
- O. Cabanillas-Bernal, S. Dueñas, M. Ayala-Avila, A. Rucavado, T. Escalante and A. F. Licea-Navarro, Synthetic libraries of shark vNAR domains with different cysteine numbers within the CDR3, PLoS One, 2019, 14(6), 1–24 CrossRef PubMed.
- O. C. Ubah, M. J. Buschhaus, L. Ferguson, M. Kovaleva, J. Steven and A. J. Porter, et al., Next-generation flexible formats of VNAR domains expand the drug platform's utility and developability, Biochem. Soc. Trans., 2018, 46(6), 1559–1565 CrossRef CAS PubMed.
- M. R. Müller, K. Saunders, C. Grace, M. Jin, N. Piche-Nicholas and J. Steven, et al., Improving the pharmacokinetic properties of biologics by fusion to an anti-HSA shark VNAR domain, mAbs, 2012, 4(6), 673–685 CrossRef PubMed.
- O. C. Ubah, E. W. Lake, G. S. Gunaratne, J. P. Gallant, M. Fernie and A. J. Robertson, et al., Mechanisms of SARS-CoV-2 neutralization by shark variable new antigen receptors elucidated through X-ray crystallography, Nat. Commun., 2021, 12(1), 1–12 CrossRef PubMed.
- D. Bobo, K. J. Robinson, J. Islam, K. J. Thurecht and S. R. Corrie, Nanoparticle-Based Medicines: A Review of FDA-Approved Materials and Clinical Trials to Date, Pharm. Res., 2016, 33(10), 2373–2387, DOI:10.1007/s11095-016-1958-5.
- F. Cogo, M. Poreba, W. Rut, K. Groborz, P. Smyth and M. C. Johnston, et al., Development of an advanced nanoformulation for the intracellular delivery of a caspase-3 selective activity-based probe, Nanoscale, 2019, 11(2), 742–751 RSC.
- A. Zakeri, M. A. J. Kouhbanani, N. Beheshtkhoo, V. Beigi, S. M. Mousavi and S. A. R. Hashemi, et al., Polyethylenimine-based nanocarriers in co-delivery of drug and gene: a developing horizon, Nano Rev. Exp., 2018, 9(1), 1488497, DOI:10.1080/20022727.2018.1488497.
- C. N. Lungu, M. V. Diudea, M. V. Putz and I. P. Grudziński, Linear and branched PEIs (polyethylenimines) and their property space, Int. J. Mol. Sci., 2016, 17(4), 555 CrossRef PubMed.
- A. Akinc, M. Thomas, A. M. Klibanov and R. Langer, Exploring polyethylenimine-mediated DNA transfection and the proton sponge hypothesis, J. Gene Med., 2005, 7(5), 657–663 CrossRef CAS PubMed.
- Q. D. Hu, H. Fan, Y. Ping, W. Q. Liang, G. P. Tang and J. Li, Cationic supramolecular nanoparticles for co-delivery of gene and anticancer drug, Chem. Commun., 2011, 47(19), 5572–5574 RSC.
- S. A. Smith, L. I. Selby, A. P. R. Johnston and G. K. Such, The Endosomal Escape of Nanoparticles: Toward More Efficient Cellular Delivery, Bioconjug. Chem., 2019, 30(2), 263–272 CrossRef CAS PubMed.
- T. G. Iversen, T. Skotland and K. Sandvig, Endocytosis and intracellular transport of nanoparticles: present knowledge and need for future studies, Nano Today, 2011, 6(2), 176–185, DOI:10.1016/j.nantod.2011.02.003.
- W. Song, L. Popp, J. Yang, A. Kumar, V. S. Gangoli and L. Segatori, The autophagic response to polystyrene nanoparticles is mediated by transcription factor EB and depends on surface charge, J. Nanobiotechnology, 2015, 13(87) CAS.
- T. L. Moore, L. Rodriguez-Lorenzo, V. Hirsch, S. Balog, D. Urban and C. Jud, et al., Nanoparticle colloidal stability in cell culture media and impact on cellular interactions, Chem. Soc. Rev., 2015, 44, 6287–6305 RSC.
- H. Li, T. Y. Tsui and W. Ma, Intracellular delivery of molecular cargo using cell-penetrating peptides and the combination strategies, Int. J. Mol. Sci., 2015, 16(8), 19518–19536 CrossRef CAS PubMed.
- X. Wang, D. Niu, C. Hu and P. Li, Polyethyleneimine-Based Nanocarriers for Gene Delivery, Curr. Pharm. Des., 2015, 21(42), 6140–6156 CrossRef CAS PubMed.
- S. K. Hobbs, W. L. Monsky, F. Yuan, W. G. Roberts, L. Griffith and V. P. Torchilin, et al., Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment, Proc. Natl. Acad. Sci. USA, 1998, 95(8), 4607–4612 CrossRef CAS PubMed.
- V. Saxena, M. Sadoqi and J. Shao, Polymeric nanoparticulate delivery system for Indocyanine green: biodistribution in healthy mice, Int. J. Pharm., 2006, 308(1–2), 200–204 CrossRef CAS PubMed.
- M. K. Greene, D. A. Richards, J. C. F. Nogueira, K. Campbell, P. Smyth and M. Fernández, et al., Forming next-generation antibody-nanoparticle conjugates through the oriented installation of non-engineered antibody fragments, Chem. Sci., 2017, 9(1), 79–87 RSC.
- L. Zhang, Y. Hu, X. Jiang, C. Yang, W. Lu and Y. H. Yang, Camptothecin derivative-loaded poly(caprolactone-co-lactide)-b-PEG-b- poly(caprolactone-co-lactide) nanoparticles and their biodistribution in mice, J. Contr. Release, 2004, 96(1), 135–148 CrossRef CAS PubMed.
- A. Saez, M. Guzmán, J. Molpeceres and M. R. Aberturas, Freeze-drying of polycaprolactone and poly(D,L-lactic-glycolic) nanoparticles induce minor particle size changes affecting the oral pharmacokinetics of loaded drugs, Eur. J. Pharm. Biopharm., 2000, 50(3), 379–387 CrossRef CAS PubMed.
- D. Pei and M. Buyanova, Overcoming Endosomal Entrapment in Drug Delivery, Bioconjug. Chem., 2019, 30(2), 273–283 CrossRef CAS PubMed.
- L. Huang and S. Guo, Nanoparticles escaping RES and endosome: Challenges for siRNA delivery for cancer therapy, J. Nanomater., 2011, 11, 1–12 Search PubMed.
- O. C. Ubah, M. J. Buschhaus, L. Ferguson, M. Kovaleva, J. Steven and A. J. Porter, et al., Next-generation flexible formats of VNAR domains expand the drug platform's utility and developability, Biochem. Soc. Trans., 2018, 46(6), 1559–1565 CrossRef CAS PubMed.
- M. Kovaleva, L. Ferguson, J. Steven, A. Porter and C. Barelle, Shark variable new antigen receptor biologics – a novel technology platform for therapeutic drug development, Expert Opin. Biol. Ther., 2014, 14(10), 1527–1539 CrossRef CAS PubMed.
- M. F. Flajnik, N. Deschacht and S. Muyldermans, A case of convergence: Why did a simple alternative to canonical antibodies arise in Sharks and Camels?, PLoS Biol., 2011, 9(8), e1001120 CrossRef CAS PubMed.
- H. Dooley and M. F. Flajnik, Shark immunity bites back: Affinity maturation and memory response in the nurse shark, Ginglymostoma cirratum, Eur. J. Immunol., 2005, 35(3), 936–945 CrossRef CAS PubMed.
- I. Canton and G. Battaglia, Endocytosis at the nanoscale, Chem. Soc. Rev., 2012, 41, 2718–2739 RSC.
- N. D. Donahue, H. Acar and S. Wilhelm, Concepts of nanoparticle cellular uptake, intracellular trafficking, and kinetics in nanomedicine, Adv. Drug Deliv. Rev., 2019, 143, 68–96, DOI:10.1016/j.addr.2019.04.008.
- R. V. Benjaminsen, M. A. Mattebjerg, J. R. Henriksen, S. M. Moghimi and T. L. Andresen, The possible “proton sponge ” effect of polyethylenimine (PEI) does not include change in lysosomal pH, Mol. Ther., 2013, 21(1), 149–157 CrossRef CAS PubMed.
- E. Bilensoy, Cationic nanoparticles for cancer therapy, Expert Opin. Drug. Deliv., 2010, 7(7), 795–809, DOI:10.1517/17425247.2010.485983.
- S. R. Tracey, P. Smyth, C. J. Barelle and C. J. Scott, Development of next generation nanomedicine-based approaches for the treatment of cancer: we’ve barely scratched the surface, Biochem. Soc. Trans, 2021, 49(5), 2253–2269 CrossRef CAS PubMed.
- N. Deirram, C. Zhang, S. S. Kermaniyan, A. P. R. Johnston and G. K. Such, pH-Responsive Polymer Nanoparticles for Drug Delivery, Macromol. Rapid. Commun., 2019, 40(10), 1–23 CrossRef PubMed.
- Y. Bae, S. Fukushima, A. Harada and K. Kataoka, Design of environment-sensitive supramolecular assemblies for intracellular drug delivery: Polymeric micelles that are responsive to intracellular pH change, Angew. Chem., Int. Ed., 2003, 42(38), 4640–4643 CrossRef CAS PubMed.
- T. J. Gibson, P. Smyth, W. J. Mcdaid, D. Lavery, J. Thom and G. Cotton, et al., Single-Domain Antibody-Functionalized pH-Responsive Amphiphilic Block Copolymer Nanoparticles for Epidermal Growth Factor Receptor Targeted Cancer Therapy, ACS Macro Lett., 2018, 7(8), 1010–1015 CrossRef CAS PubMed.
- P. Smyth, T. J. Gibson, G. Irvine, G. Black, D. Lavery and M. Semsarilar, et al., pH-Responsive benzaldehyde-functionalized PEG-based polymeric nanoparticles for drug delivery: effect of preparation method on morphology, dye encapsulation and attachment, Eur. Polym. J., 2020, 124, 109471 CrossRef CAS.
- T. P. Thomas, I. Majoros, A. Kotlyar, D. Mullen, M. M. Banaszak Holl and J. R. Baker, Cationic poly(amidoamine) dendrimer induces lysosomal apoptotic pathway at therapeutically relevant concentrations, Biomacromolecules, 2009, 10(12), 3207–3214 CrossRef CAS PubMed.
- W. Yan, W. Chen and L. Huang, Reactive oxygen species play a central role in the activity of cationic liposome based cancer vaccine, J. Contr. Release, 2008, 130(1), 22–28 CrossRef CAS PubMed.
- C. M. Goodman, C. D. McCusker, T. Yilmaz and V. M. Rotello, Toxicity of gold nanoparticles functionalized with cationic and anionic side chains, Bioconjug. Chem., 2004, 15(4), 897–900 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra06050k |
| This journal is © The Royal Society of Chemistry 2023 |