
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExperimental and computational insights into the mechanism of FLP mediated selective C–F bond activation†
Richa
Gupta‡
a,
Dániel
Csókás‡
a,
Kenneth
Lye
a and
Rowan D.
Young
 *ab
*ab
aDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, 117543, Singapore. E-mail: rowan.young@nus.edu.sg
bSchool of Chemistry and Molecular Biosciences, The University of Queensland, St Lucia, 4072, Queensland, Australia
First published on 13th January 2023
Abstract
Frustrated Lewis pairs (FLP) comprising of B(C6F5)3 (BCF) and 2,4,6-triphenylpyridine (TPPy), P(o-Tol)3 or tetrahydrothiophene (THT) have been shown to mediate selective C–F activation in both geminal and chemically equivalent distal C–F sites. In comparison to other reported attempts of C–F activation using BCF, these reactions appear surprisingly facile. We investigate this reaction through a combination of experimental and computational chemistry to understand the mechanism of the initial C–F activation event and the origin of the selectivity that prevents subsequent C–F activation in the monoactivated salts. We find that C–F activation likely occurs via a Lewis acid assisted SN1 type pathway as opposed to a concerted FLP pathway (although the use of an FLP is important to elevate the ground state energy), where BCF is sufficiently Lewis acidic to overcome the kinetic barrier for C–F activation in benzotrifluorides. The resultant intermediate salts of the form [ArCF2(LB)][BF(C6F5)3] (LB = Lewis base) are relatively thermodynamically unstable, and an equilibrium operates between the fluorocarbon/FLP and their activation products. As such, the use of a fluoride sequestering reagent such as Me3SiNTf2 is key to the realisation of the forward C–F activation reaction in benzotrifluorides. Selectivity in this reaction can be attributed to both the installation of bulky Lewis bases geminal to residual C–F sites and from electronic re-ordering of kinetic barriers (of C–F sites in products and starting materials) arising from the electron withdrawing nature of the pyridinium, phosphonium and sulfonium groups.
Introduction
Selective functionalization of a single C–F bond in difluoro and trifluoromethyl groups is a simple strategy to access a vast array of fluorocarbon products. However, this approach is complicated by the fact that the resulting singly functionalized products contain weaker C–F bonds than their starting materials and a mixture of ‘over-reaction’ products is commonly obtained.1A number of solutions have been offered to address this problem, however, most are limited in fluorocarbon substrate and functionalization scope. For example, electron poor benzotrifluorides and trifluoromethylamides can be functionalised via SET reactions,2 CF3 and CF2H groups vicinal to carbonyl or vinyl groups are able to generate fluoroalkene products with thermodynamically stable sp2 C–F bonds,3 and monoselective nucleophilic substitution of aromatic CF3 and CF2H groups ortho to hydrosilyl groups is possible.4
Recently, frustrated Lewis pairs (FLPs) have been used to selectively activate aliphatic polyfluorocarbons.5 Notably, we have been able to employ a combination of B(C6F5)5 (BCF) with triarylphosphines, 2,4,6-triphenylpyridine (TPPy) and dialkylsulfides to enable selective activation of CF3 and CF2H groups (Fig. 1).5a–d This reaction can be run with catalytic Lewis acid in the presence of Me3SiNTf2. Indeed, the reaction performs much better under catalytic conditions.
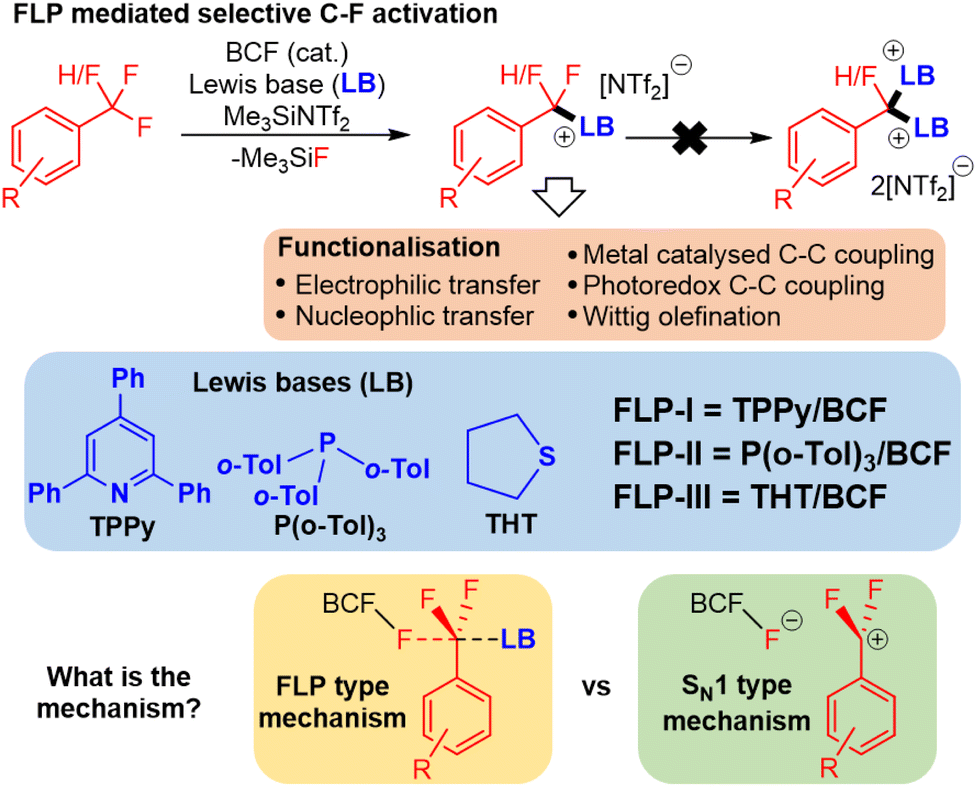 | ||
| Fig. 1 FLP mediated C–F bond activation allows single fluoride functionalisation in difluoromethyl and trifluoromethyl groups. TPPy = 2,4,6-triphenylpyridine, THT = tetrahydrothiophene, BCF = tris(pentafluorophenyl)borane. | ||
The observed activity of this FLP mediated C–F bond activation reaction is surprising given past reports that have failed to activate benzotrifluorides using BCF alone.6 Indeed, other reports of FLP mediated C–F bond activation have employed stoichiometric silylium and dicationic phosphonium, both of which are very strong Lewis acids.5e,f Computational evidence that BCF and TPPy exhibit FLP type cooperative activity was recently reported by Fernández (and later Chattaraj) (Fig. 1), whose calculations suggested that the TPPy base plays an important role in lowering the activation barrier for the Lewis acid mediated C–F scission.7 Despite the insight that Fernández's model offered, it relied upon stabilizing π–π stacking interactions between arenes on the benzotrifluoride substrate and TPPy base that would not be present in other Lewis bases that are competent in FLP C–F activation. In addition, computational examination of the initial C–F activation event does not explain the high selectivity that these FLPs have for controlled single fluoride abstraction in polyfluoroalkanes, nor the enhanced reactivity observed under catalytic conditions (as opposed to stoichiometric reactions).
In an effort to illuminate unexplored mechanistic aspects of the selective C–F activation FLP reaction, we have undertaken a combined computational and experimental study. In addition to the BCF/TPPy FLP system studied by Fernández,7a we have expanded the study to include sulfide and phosphine FLP systems, which have also been reported to facilitate selective C–F activation.5a,c Importantly, we are able to benchmark our computational results to experimentally obtained kinetic and thermodynamic data, which corroborate each other in suggesting that the reaction mirrors a Lewis acid assisted SN1 reaction (independent of Lewis base) more closely than an FLP cooperative reaction (as suggested by Fernández).
Results and discussion
The initial FLP mediated C–F activation event
We chose to examine three FLPs that have previously demonstrated selective C–F activation; namely BCF/TPPy (FLP-I), BCF/P(o-Tol)3 (FLP-II) and BCF/THT (THT = tetrahydrothiophene) (FLP-III) (Fig. 1).5a–c Based upon Fernández's report,7a we expected to observe a reaction rate dependence directly proportional to the concentration of substrate (fluorocarbon), Lewis acid (BCF) and Lewis base used, consistent with a Lewis acid assisted SN2 (FLP type) mechanism. A first-order rate dependence was established for both substrate and BCF, however, we found that the reaction rate was independent of the concentration of Lewis base used. For example, the activation of 4-Me-C6H4CF3 (1a) with FLP-I or FLP-II showed no increase in rate with increasing base concentration. Indeed, if anything a slight inhibition was observed at very high TPPy and P(o-Tol)3 concentrations.We reasoned that an FLP type mechanism was still viable if the apparent base independence (or inhibition) resulted from a competing equilibrium of FLP and Lewis adduct forms of FLP-I and FLP-II, where higher concentrations of Lewis base favoured the inactive Lewis adduct form. As such, we undertook a Hammett plot analysis8 for the activation of a series of benzotrifluorides with FLP-I and FLP-II, the results of which would not be affected by any FLP/Lewis adduct equilibrium (Table 1).
|
|
||||
|---|---|---|---|---|
| Entry | FLP system | Substrate | ρ-Value | ρ +-Value |
| a N.R. = No reaction observed with any of ArCF3 substrates. | ||||
| 1 | FLP-I | ArCF3 | −3.0 ± 0.3 | −2.6 ± 0.5 |
| 2 | FLP-II | ArCF3 | −4.8 ± 0.7 | −4.2 ± 0.4 |
| 3 | FLP-III | ArCF3 | N.R. | N.R. |
| 4 | FLP-I | ArCF2H | −3.1 ± 0.4 | −2.8 ± 0.2 |
| 5 | FLP-II | ArCF2H | −7.0 ± 1.2 | −6.6 ± 0.9 |
| 6 | FLP-III | ArCF2H | −3.5 ± 0.7 | −3.2 ± 0.5 |
We observed a ρ-value of −3.0 ± 0.3 for FLP-I, indicative of a significant positive charge accumulation at the benzylic position (Table 1, entry 1). Such a value is common for SN1 transition states, but could also be due to an open or early transition state SN2 process.9 Repeating the Hammett plot analysis with FLP-II gave an even lower ρ-value of −4.8 ± 0.7, clearly indicative of an SN1 type process (Table 1, entry 2). A Hammett plot analysis of FLP-III with benzotrifluorides was not possible due to no observed reaction in this system (Table 1, entry 3).
Hammett plot analyses were also carried out for FLP-I, FLP-II and FLP-III for difluoromethylarene substrates, where the ρ-values were indicative of SN1 processes (Table 1, entries 4–6). In theory, for an SN1 process the observed ρ-values for each FLP system should be identical for either of the substrates sets (i.e. ArCF3 or ArCF2H). In practice we would expect error in these measurements, but given the wide spread of ρ-values, we suspect that secondary intermolecular interactions in the transition states – such as H-bonding or π–π stacking – may have subtle influences on the ρ-value for each FLP system (see ESI, Fig. S47†).
We also modelled these data using σ+ values (Table 1). The corresponding ρ+-values cannot be directly compared to ρ-values, however, a better Hammett plot fit of rate constants to σ+ values is indicative of an SN1 type process. In all cases, the calculated standard error for the σ+ plots was lower than their σ plot counterparts, except in the case of FLP-I with benzotrifluorides (Table 1).
The intermediary ρ-value of −3.0 ± 0.3 for FLP-I taken with its poorer fit to σ+ values does not allow conclusive assignment of FLP-I to an SN1 mechanism or an FLP type mechanism as proposed by Fernández. To clarify the situation, we decided to conduct our own theoretical analysis at DFT level. Geometry optimisations and transition state locations were performed at the level of PCM(DCM)-B3LYP-D3/Def2SVP. The reported Gibbs free energies in our study were obtained by combining PCM(DCM)-B3LYP-D3/def2TZVPP electronic energies with the thermal and entropic contributions computed at the PCM(DCM)-B3LYP-D3/Def2SVP level followed by quasi-harmonic correction (for more details on testing different DFT functionals, solvation models and dispersion corrections see Computational studies section in ESI†). Considering the large conformational space that a Lewis base may occupy, a thorough conformational study was performed to obtain the lowest energy transition state. Additionally, various conformers were assessed for the FLP adducts and different ion pairs.
The FLP systems (FLP-I, FLP-II, FLP-III) were computationally interrogated with two model substrates; PhCF3 (1b) and PhCF2H (2b) (Fig. 2). We investigated two possible pathways for the C–F activation reaction. Namely, a stepwise SN1 type reaction where BCF abstracts fluoride from the fluorocarbon to form a carbocation intermediate (SN1 pathway) and a concerted FLP type activation involving both Lewis acid and base in the transition state, similar to that reported by Fernández (FLP pathway). Fernández calculated that an SN2 pathway in the absence of Lewis acid has a barrier of over 80 kcal mol−1,7a and therefore this mechanism was not considered in our studies.
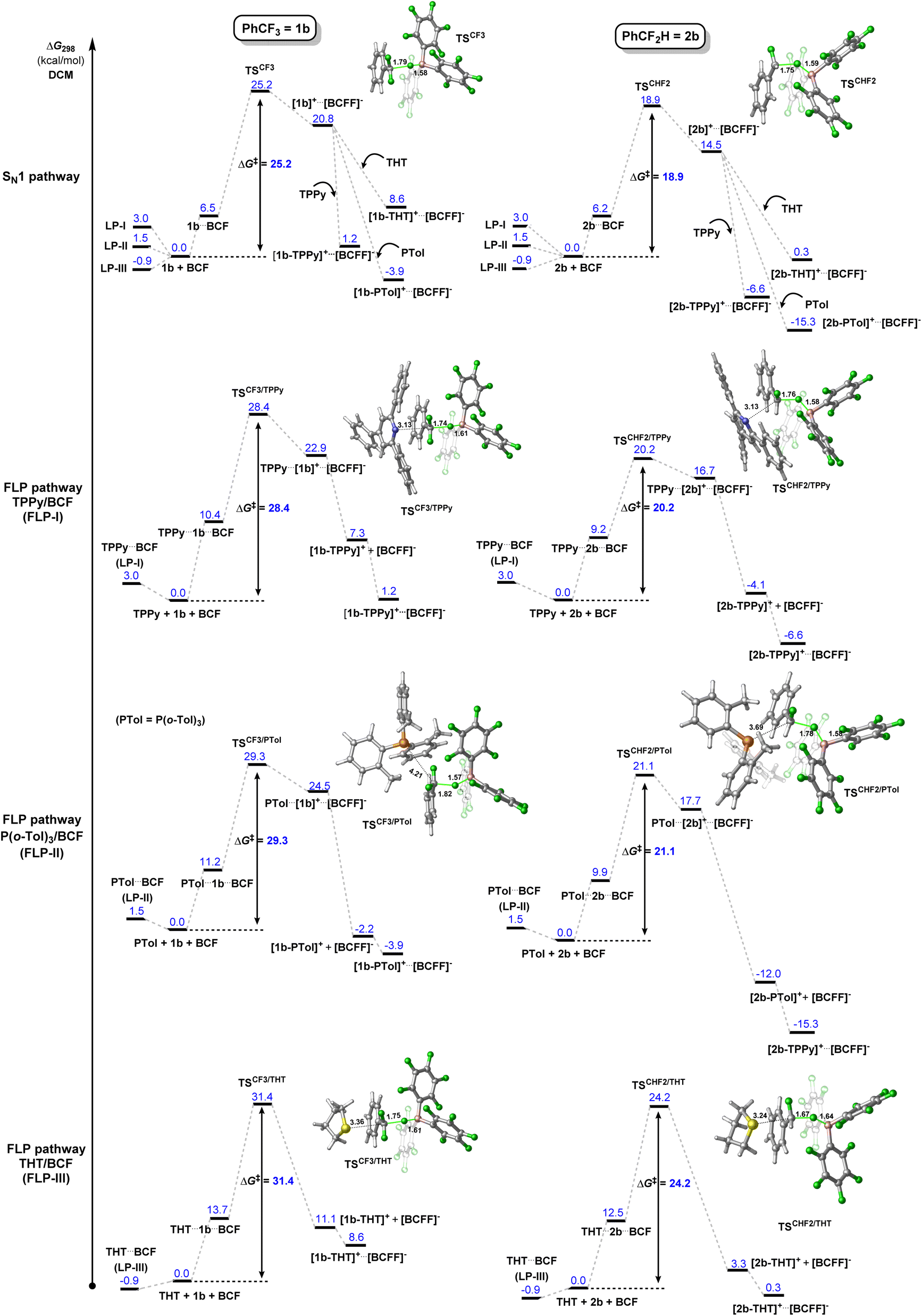 | ||
| Fig. 2 Calculated SN1 and FLP reaction pathways for the reactions of FLP-I, FLP-II and FLP-III with PhCF3 (1b, left hand side) and PhCF2H (2b, right hand side). Note: PTol = P(o-Tol)3, LP = Lewis pair/adduct. Level of theory: PCM(DCM)-B3LYP-D3/Def2TZVPP//PCM(DCM)-DB3LYP-D3/Def2SVP (quasi-harmonic entropic correction). Displayed calculated bond distances given in Å. | ||
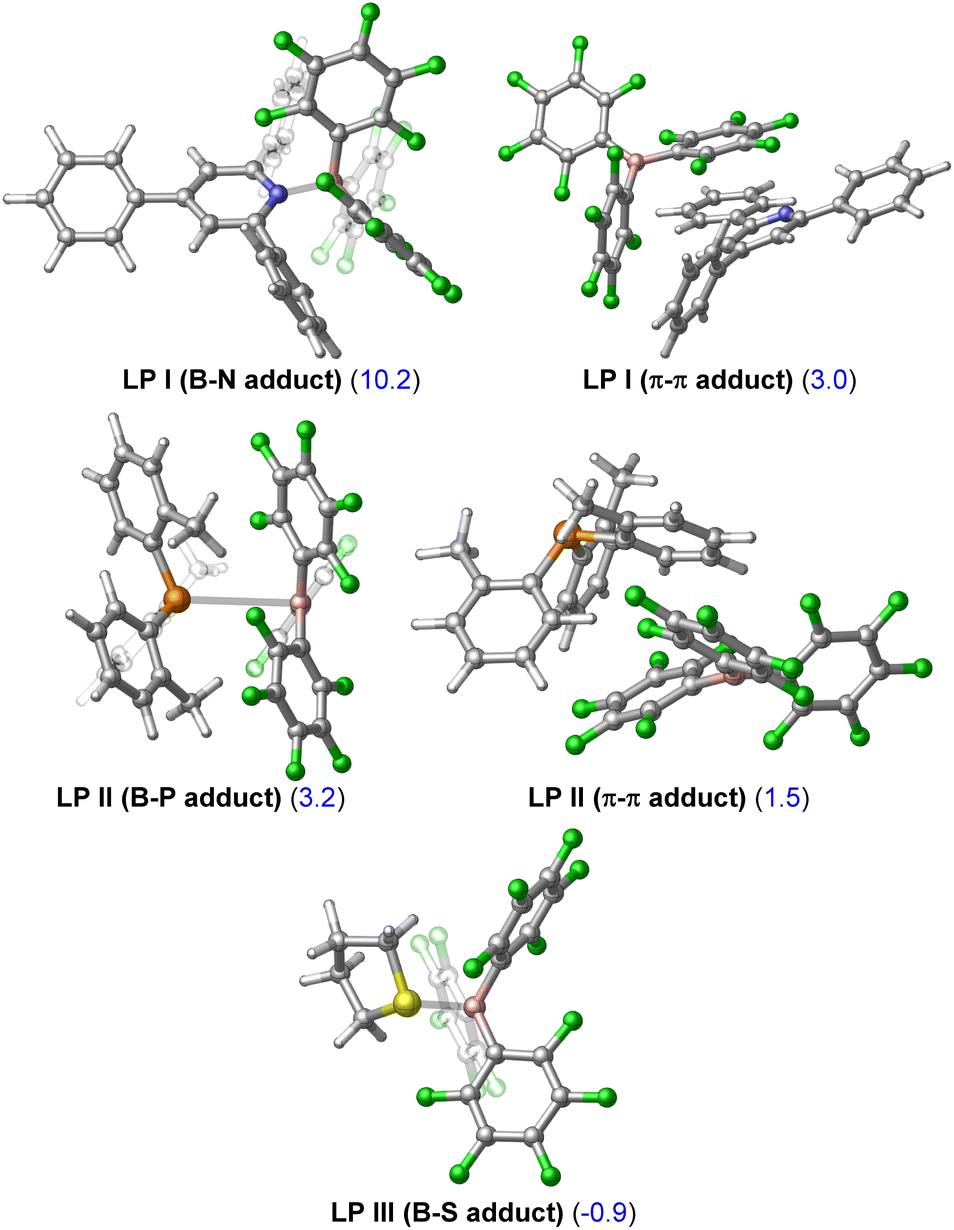
The first computational result of note is that calculated equilibria between Lewis pair/adduct (LP) and FLP forms greatly favour the FLP forms in FLP-I and FLP-II (Table 2). The classical B–N adduct form of FLP-I is thermodynamically disfavoured by 10.2 kcal mol−1 in comparison to the FLP form and the lowest energy adduct conformer (an arene π–π stacking adduct) is disfavoured by 3.0 kcal mol−1. Similarly, the lowest energy B–P adduct form of FLP-II located is disfavoured by 3.2 kcal mol−1 as compared to the FLP form, however, once again the lowest energy adduct conformer found corresponded to an arene π–π stacking adduct, which was disfavoured by 1.5 kcal mol−1. In the case of FLP-III, the classical B–S adduct represented the lowest energy isomer, and (in contrast to FLP-I and FLP-II) was slightly thermodynamically preferred by 0.9 kcal mol−1.10 The consequences of these calculated equilibria are that (i) if there is no base dependence arising in the rate determining step, then higher concentrations of base that perturb the FLP/LP equilibria will result in an inverse observed rate, and that (ii) the ground state of FLP-III is likely the Lewis adduct form (i.e.LP-III), and the additional energy required to break this adduct will result in higher observed kinetic barriers for reactions involving FLP-III.
|
|
|||
|---|---|---|---|
| Entry | FLP system | Conformer | Calc. ΔG0298 (kcal mol−1) |
| 1 | FLP-I/LP-I | B–N adduct | +10.2 |
| 2 | FLP-I/LP-I | π–π adduct | +3.0 |
| 3 | FLP-II/LP-II | B–P adduct | +3.2 |
| 4 | FLP-II/LP-II | π–π adduct | +1.5 |
| 5 | FLP-III/LP-III | B–S adduct | −0.9 |
|
|||
Calculated differences in free energies of FLP and adduct forms of FLP-I, FLP-II and FLP-III could not be verified experimentally due to fast dynamic exchange between the two forms that could not be resolved through 19F NMR spectroscopy upon cooling. However, in support of the adduct form of FLP-III being thermodynamically viable, the solid state molecular structure for this adduct has been reported.11
Turning our attention to the initial C–F activation event, in the case of FLP-I, calculated barriers for C–F activation in 1b show that an SN1 pathway (25.2 kcal mol−1) is slightly favourable to an FLP pathway (28.4 kcal mol−1). The ordering of these energy barriers is the reverse of that which Fernández reported and they are more aligned to a slow ambient temperature reaction. We found that the previously reported relative Gibbs free energy profile by Fernández lacks single-point refinements with a larger basis set (the indicated relative free energies were computed at the PCM(DCM)-B3LYP-D3/def2SVP level). Besides, thorough conformational analysis for transition states was not performed.
Even higher kinetic barriers are calculated for the activation of 1b with FLP-II and FLP-IIIvia FLP pathways, corroborating with experimental data that suggest a preferred SN1 pathway for FLP-II, while no product is observed experimentally in the case of FLP-III.
For C–F activation in benzodifluoride 2b, FLP-I was calculated to have similar kinetic barriers for SN1 and FLP reaction pathways, with an SN1 pathway slightly preferred by 1.3 kcal mol−1. The SN1 pathway was clearly preferred over FLP pathways for FLP-II and FLP-III. The barrier for C–F activation in CF2H groups was found to be lowered by as much as ca. 2 kcal mol−1 through a hydrogen-bonding interaction between the CF2H hydrogen and an ortho-fluoroaryl group present on the BCF catalyst. These computational data support an SN1 mechanism for C–F activation reactions of 1b and 2b in all FLP systems studied.
A series of kinetic experiments under catalytic conditions were undertaken to obtain experimental ΔG‡ values for the C–F activation event in difluoromethyl and trifluoromethyl arenes (listed in Table 3). Using the substrate 4-Me-C6H4-CF3 (1a), ΔG‡ was determined to be 22.4 ± 2.3 kcal mol−1 for FLP-I and 22.9 ± 1.7 kcal mol−1 for FLP-II, with the calculated barrier for SN1 activation of 1a found to be 24.4 kcal mol−1 (see ESI, Fig. S54†). Substrates 2a and 2b were found to react too quickly under our standard conditions, so 2-Br-C6H4-CF2H (2c) was employed to obtain experimental kinetic data. With 2c, ΔG‡ was determined to be 20.7 ± 1.3 kcal mol−1 for FLP-I, 21.1 ± 0.7 kcal mol−1 for FLP-II and 22.2 ± 0.6 kcal mol−1 for FLP-III, against calculated values of 19.3 kcal mol−1 for FLP-I and FLP-II, and 20.2 kcal mol−1 for FLP-III (Fig. 4 and S61†).
| Entry | FLP system | Substrate | ΔG‡ (kcal mol−1) |
|---|---|---|---|
| 1 | FLP-I | 1a | 22.4 ± 2.3 (exp.) |
| 2 | FLP-II | 1a | 22.9 ± 1.7 (exp.) |
| 3 | FLP-I, FLP-II | 1a | 24.4 (calc.) |
| 4 | FLP-III | 1a | 25.3 (calc.) |
| 5 | FLP-I, FLP-II | 1b | 25.2 (calc.) |
| 6 | FLP-III | 1b | 26.1 (calc.) |
| 7 | FLP-I, FLP-II | 2b | 18.9 (calc.) |
| 8 | FLP-III | 2b | 19.8 (calc.) |
| 9 | FLP-II | [2b-P(o-Tol)3][NTf2] | 27.1 (calc.) |
| 10 | FLP-I | 2c | 20.7 ± 1.3 (exp.) |
| 11 | FLP-II | 2c | 21.1 ± 0.7 (exp.) |
| 12 | FLP-III | 2c | 22.2 ± 0.6 (exp.) |
| 13 | FLP-I, FLP-II | 2c | 19.3 (calc.) |
| 14 | FLP-III | 2c | 20.2 (calc.) |
| 15 | FLP-I | 3b | 19.3 (calc.) |
| 16 | FLP-I | [3b-TPPy][NTf2] | 20.3 (calc.) |
| 17 | FLP-III | 3b | 20.2 (calc.) |
| 18 | FLP-III | [3b-THT][NTf2] | 24.0 (calc.) |
The similar activation barrier values for FLP-I and FLP-II for each substrate strongly support an SN1 process with a common transition state, and verify the calculated activation barriers in the SN1 pathway. Additionally, the higher experimental activation barrier for FLP-III with 2c (as compared to FLP-I and FLP-II) agrees with the adduct form of FLP-III being thermodynamically preferred. Indeed, a control experiment where 1b was reacted with FLP-I under standard catalytic conditions but with the addition of 1.5 equivalents of THT resulted in no activation occurring after 48 hours, confirming that the presence of THT inhibits activation of benzotrifluorides through the formation of a thermodynamically preferred Lewis adduct.
The role of Me3SiNTf2
As our catalytic protocol includes the use of Me3SiNTf2, we attempted to calculate pathways involving Me3SiNTf2 in the rate determining C–F activation step. Such attempts resulted in higher kinetic barriers for both 1b and 2b as compared to C–F activation by BCF alone with the lowest energy transition states located for Me3SiNTf2 mediated C–F bond cleavage of 1b and 2b 5.4 kcal mol−1 and 6.4 kcal mol−1 (respectively) higher in energy than those mediated by BCF (Fig. 3A). Interestingly, the lowest energy transition states for Me3SiNTf2 mediated C–F activation involved oxygen bridged Me3SiNTf2 (see ESI, Fig. S55†), similar to tin bistriflimide isomers reported by Pápai and Ashley.12 The non-active role of Me3SiNTf2 in the C–F activation step was confirmed through kinetic experiments that varied the concentration of Me3SiNTf2 in reactions of FLP-I with 1a and 2c, and FLP-II with 2c, all of which showed no rate dependence on Me3SiNTf2 concentration (see ESI, Fig. S7 and S8†).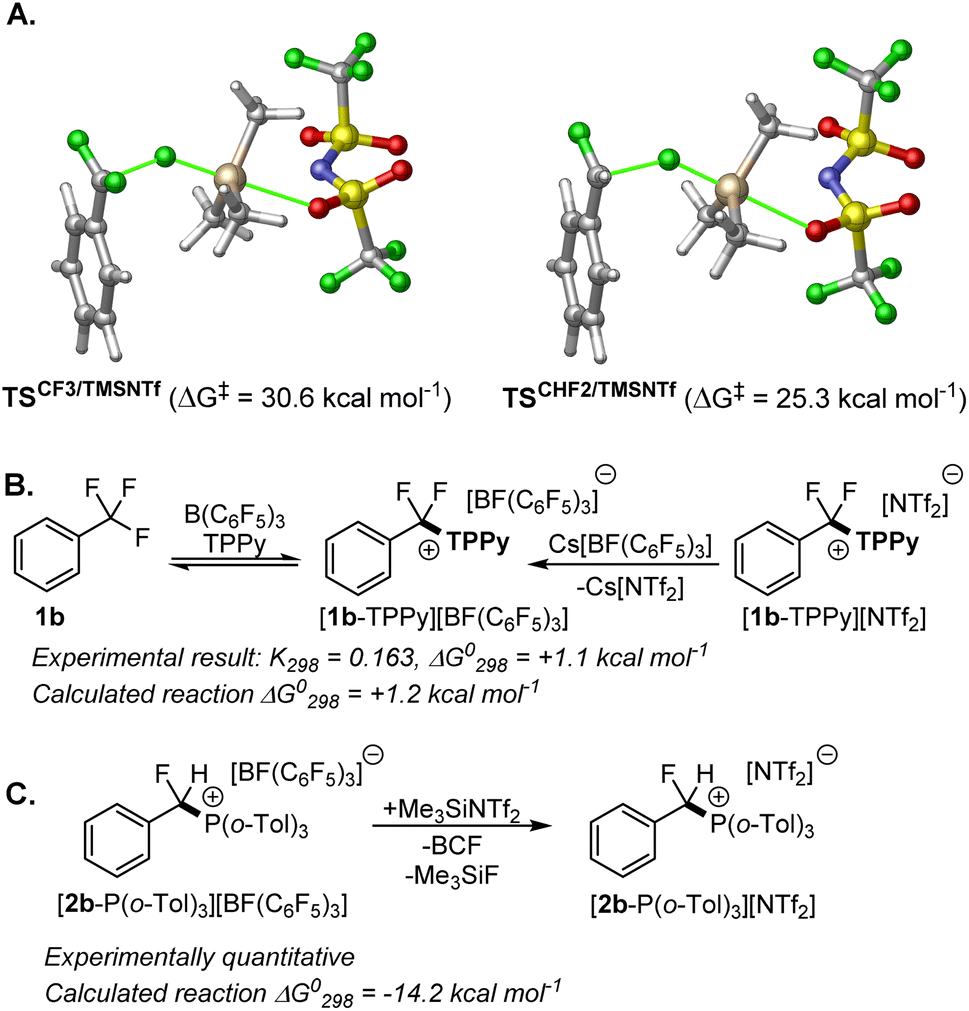 | ||
| Fig. 3 (A) Lowest energy transition states for C–F activation of 1b and 2b by Me3SiNTf2. (B) Experimental and computational analysis for the equilibrium between FLP-I/1b and their activation product. (C) Experimental and computational evidence supporting the role of Me3SiNTf2 in abstracting fluoride from [BF(C6F5)3]− and providing thermodynamic preference to the reaction products. | ||
In the absence of Me3SiNTf2, calculated free energies of the starting materials and the FLP activation products revealed that very little thermodynamic gradient existed in FLP-I with 1b, with the product, [1b-TPPy][BF(C6F5)3], a mere 1.2 kcal mol−1 less stable than the starting materials (Fig. 3B). These comparable free energies, combined with the low kinetic barrier, result in an equilibrium between FLP-I and 1b with their activation products. The equilibrium constant for this reaction was determined from both reaction directions through stoichiometric reaction of FLP-I with 1b and then with stoichiometric reaction of Cs[BF(C6F5)3] with [PhCF2(TPPy)][NTf2], that provided an equilibrium constant of K298 = 0.163, corresponding to an experimentally derived free energy difference of ΔG298 = +1.1 kcal mol−1 and agreeing with computed energies remarkably well (Fig. 3B).
In the case of FLP-II, the equilibrium lies well in the direction of the products and the kinetic barrier for the reverse reaction is consequently higher. However, the possibility of the reverse reaction using FLP-II with certain benzotrifluoride substrates was confirmed through reaction of [4-OMe-C6H4CF2(P(o-Tol)3)][NTf2] with Cs[BF(C6F5)3] to generate 4-OMe-C6H4CF3. It is proposed in the presence of Me3SiNTf2 that the anion [BF(C6F5)3]− is quickly converted to BCF and Me3SiF, preventing the reverse reaction and improving reaction rate and yield.
In support of this assertion, a control reaction between [2b-P(o-Tol)3][BF(C6F5)3] and Me3SiNTf2 resulted in the fast generation of BCF, Me3SiF and [2b-P(o-Tol)3][NTf2] (Fig. 3C). The calculated free energy change for the reaction of Me3SiNTf2 with [2b-P(o-Tol)3][BF(C6F5)3] shows a significantly exergonic reaction (ΔG298 = −14.2 kcal mol−1) (see ESI, Fig. S53†). Providing further evidence that the main role of Me3SiNTf2 is to sequester fluoride from the reaction, thermodynamically driving it in a productive direction whilst regenerating the BCF catalyst.
Indication that Me3SiNTf2 can play a more direct role in the activation reaction was observed in the activation of 2c with FLP systems FLP-I and FLP-II. In these reactions, the formation of an intermediate was observed, assigned as [2-Br-C6H4-CFH(NTf2)] (2c-NTf2), that formed quickly and was then consumed to generate the final product [2c-LB][NTf2] (LB = TPPy, P(o-Tol)3). In the case of FLP-III, this intermediate was not observed. The intermediate was easily identifiable via a distinct shift in the bistriflimide 19F NMR signal from δF −80.0 to −71.5 that is characteristic for N-alkyl bistriflimide adducts.13 Additionally, a signal for the benzylic fluoride was observed at δF −143.8 and the intermediate's composition was confirmed via ESI-MS. Repeating the activation reaction in the absence of Lewis base provided 2c-NTf2 as the sole product in 60% NMR yield. However, the instability of this compound prevented its isolation.
The rate of reaction of 2c-NTf2 with Lewis bases followed the order TPPy > P(o-Tol)3, and was concentration dependent, implying an SN2 type mechanism for NTf2 substitution (see ESI, Fig. S22 and S23†). DFT calculations supported this observation with SN2 pathways at least 1.2 kcal mol−1 lower in energy than the calculated SN1 pathway and following the ordering of TPPy < THT < P(o-Tol)3 in the transition barrier height (Fig. 4).
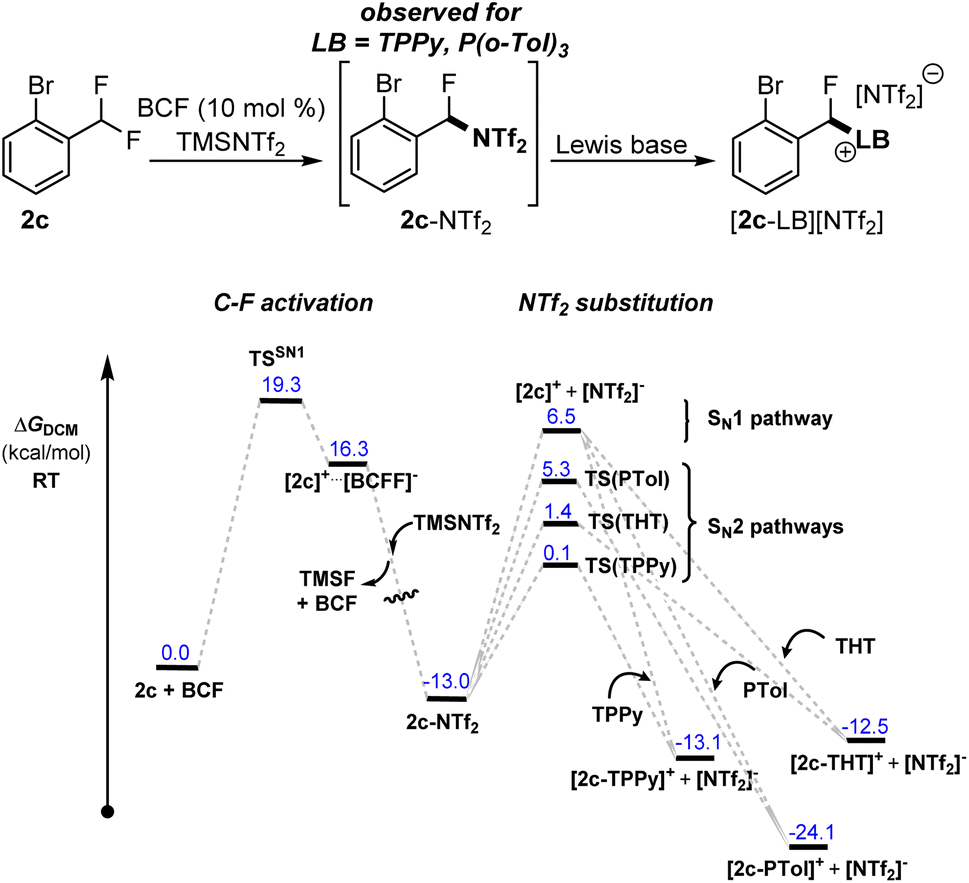 | ||
| Fig. 4 Calculated reaction profiles for formation and nucleophilic substitution of 2-NTf2 with TPPy, P(o-Tol)3 and THT. | ||
The transient detection of 2c-NTf2 under standard reaction conditions implies that (i) Me3SiNTf2 (or [NTf2]−) reacts with the carbocation [2c]+ faster than TPPy and P(o-Tol)3 and (ii) the barrier for the substitution of [NTf2]− with TPPy/P(o-Tol)3 is similar to the barrier for C–F activation (and formation of 2c-NTf2).14 Our calculations support the similar barriers between C–F activation and substitution steps, with substitution with P(o-Tol)3 only 0.4 kcal mol−1 lower in energy than C–F activation and substitution with TPPy 5.4 kcal mol−1 lower in energy.
In the case of FLP-III, intermediate 2c-NTf2 was not observed. This could be due to either THT reacting with the carbocation intermediate [2c]+ faster than Me3SiNTf2/[NTf2]− or due to a relatively higher C–F activation barrier as compared to the substitution barrier arising from the need to dissociate the Lewis adduct (LP-III) prior to C–F activation. Similarly, in benzotrifluoride and benzodifluoride substrates 1 and 2 (other than 2c), 1-NTf2 and 2-NTf2 are not observed under standard reactions, however, this does not discount their formation if the C–F activation step is significantly higher in energy than NTf2 substitution. The veracity of bistriflimide adducts as a universal intermediate in the C–F activation reaction is dependent on the kinetic barriers for combination of intermediate carbocations [1]+ and [2]+ with Lewis bases (TPPy, P(o-Tol)3, THT) as compared to with [NTf2]−.
Attempts to computationally model nucleophilic attack of the free carbocations [1]+ and [2]+ by NTf2, TPPy, THT and P(o-Tol)3 failed to provide a definitive ordering of relative kinetic barriers. However, the calculations did suggest that carbocation combination with THT was a barrierless processes, while nucleophilic attack with TPPy and P(o-Tol)3 had barriers similar to that of the SN1 C–F activation event (based upon endergonic ion separation) (see ESI, Fig. S57–S60†). Observations that 2c-NTf2 forms preferentially to [2c-TPPy]+ and [2c-P(o-Tol)3]+ but is not observed with FLP-III support our calculations, and imply that the barrier to nucleophilic attack by Me3SiNTf2 lies between those of THT and TPPy/P(o-Tol)3. However, our failure to locate convergent transition states for the addition of both TPPy and P(o-Tol)3 to the free carbocations points towards a more complicated nucleophilic substitution process that is beyond the scope of this study. Unfortunately, the facile substitution reactions of bistriflimide, THT and TPPy adducts to form thermodynamic products also precludes any experimentally determined relative distribution of initial kinetic products formed immediately after C–F activation.
Although the formulation of bistriflimide adducts as intermediates in cannot be discounted, C–F activation of benzodifluorides using FLP-I, FLP-II and FLP-III has been reported in the absence of Me2SiNTf2 (i.e. under stoichiometric conditions). Thus, the formation of 2-NTf2 is not necessary for C–F activation and might only occur in cases where the barrier for Lewis base combination with the carbocation fragment [2]+ is greater than that for [NTf2]−.
The observed difference in reaction rates of bases with the carbocation fragment arising from the SN1-type C–F activation of 2c may help explain other anecdotal observations made in related systems which did not observe C–F activation using BCF. For example, Ozerov reported that BCF does not catalyse the hydrodefluorination reaction between benzotrifluorides and Et3SiH, and Stephan reported BCF does not catalyse the Friedel–Crafts arylation of benzotrifluorides.6a,b Our studies suggest that BCF is capable of SN1-type C–F activation in benzotrifluorides and that it is the subsequent kinetic barrier arising from reaction with poorer nucleophilic reagents15 (e.g. hydrosilane, arene) that prevented any observable reaction occurring in these cases. That is, C–F activation is kinetically accessible with BCF but the thermodynamic instability of the initial product (i.e. [ArCF2][BF(C6F5)3]) may hinder the ability of weak nucleophiles to overcome the barrier for combination with the carbocation fragment.
Subsequent C–F activation (from where selectivity arises)
Pertinent to the development of FLP mediated selective C–F activation is obtaining an understanding of why the reaction is monoselective. Most substitutions of fluoride in CF2R groups (R = H or F) result in dramatic decreases in remaining C–F bond activation energies in the products. For example, the BCF mediated C–F activation energy barrier in 2b is calculated to be 18.9 kcal mol−1 (Fig. 2), while in BnF (the initial product of hydrodefluorination for 2b) it is calculated to be only 13.5 kcal mol−1 (see ESI, Fig. S56†).In the cases of secondary C–F activation at geminal positions in substrates with installed bulky TPPy and P(o-Tol)3 groups, it might be expected that the approach of BCF would be sterically hindered resulting in an increase in the kinetic barrier, however, THT is an unassuming base, and we have shown that chemically equivalent distal C–F sites may also undergo selective activation using our approach. Thus, we suspected that the substitution of a fluoride in polyfluorocarbons with a neutral Lewis base might re-order the kinetic barriers for C–F activation in the starting materials and products through electronic factors.
To investigate the second C–F activation event (i.e. C–F activation of the monoactivated products) we utilised 2b and 1,4-(CF2H)2C6H4 (3b) as model substrates for (de)activation at geminal and distal sites respectively. The first activation of 2b is described above (Fig. 2) and we have previously reported on the selective activation of 3b to generate [1-CF2H-4-CFH(LB)C6H4][NTf2] {[3b-LB][NTf2]} (LB = TPPy, THT).5c Of note in previous reports, no diactivation side products were observed in the case of 2b, whereas we have observed small amounts of diactivation products when employing distal polyfluorides 3a and 3b (Fig. 5).
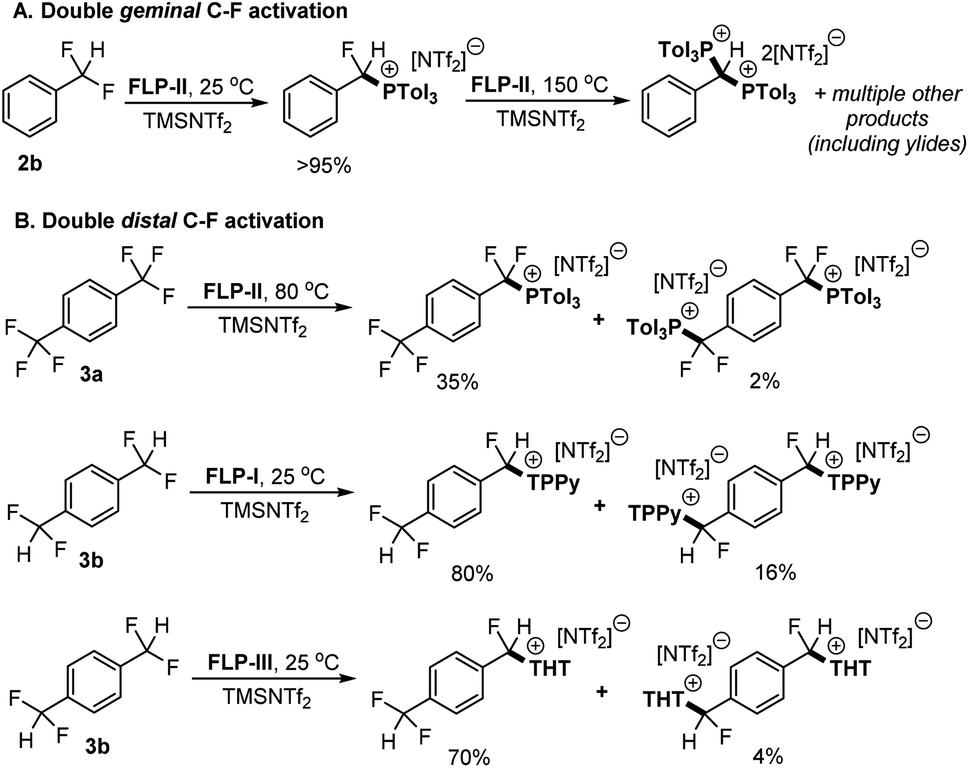 | ||
| Fig. 5 (A) Sequential activation of 2b by FLP-II. The initial activation product was isolated before the second activation reaction was undertaken. (B) Examples of diactivation occurring in distal (and chemically equivalent) C–F positions in substrates 3a and 3b. The mono and diactivation products were observed in the same reaction (in the yields indicated).5a,c, Yields determined by 19F and/or 31P NMR spectroscopy. | ||
Although we do not observe double C–F activation of 2b under ambient temperatures, subjecting the isolated salt [2-P(o-Tol)3][NTf2] to more forcing conditions (130–150 °C) resulted in further C–F activation and formation of bisphosphonium salt [2-(P(o-Tol)3)2][NTf2]2 (Fig. 5A). 31P NMR spectroscopy revealed that multiple products are present once [2-P(o-Tol)3][NTf2] is consumed. ESI-MS confirmed the presence of [2-(P(o-Tol)3)2][NTf2]2 in solution, however, a number of isomers of [2-(P(o-Tol)3)2][NTf2]2 are possible, with the incoming P(o-Tol)3 foreseeably attacking the α-carbon position to generate [α-2-(P(o-Tol)3)2][NTf2]2 or the aryl ring at the ortho or para positions to generate [ortho-2-(P(o-Tol)3)2][NTf2]2 or [para-2-(P(o-Tol)3)2][NTf2]2 (Fig. 6). In addition to the dication, a major signal is observed at 697.97 m/z with a monocationic isotopic distribution pattern and attributed to ylide formation from deprotonation of [2-(P(o-Tol)3)2][NTf2]2. As such, [2-(P(o-Tol)3)2][NTf2]2 could not be isolated, but it is shown to be kinetically accessible (albeit with difficulty).
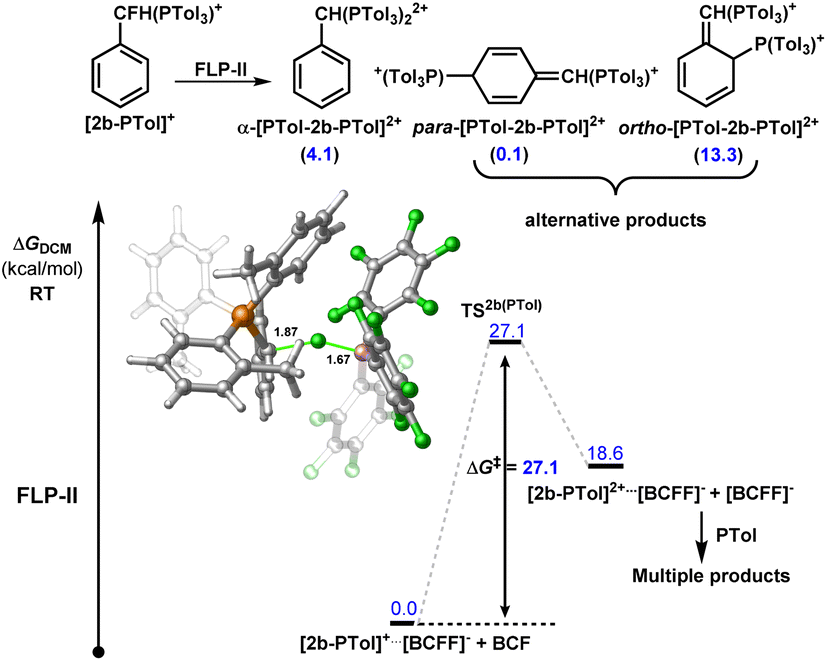 | ||
| Fig. 6 Calculated energy profile for the reaction of [PhCHFP(o-Tol)3][BF(C6F5)3] {[2b-PTol][BCFF]} with FLP-II. Note: P(o-Tol)3 is represented by PTol in figure. The relative stabilities of possible products are shown in parentheses in kcal mol−1. Displayed calculated bond distances given in Å. | ||
To understand the origin of the higher kinetic barrier for the second C–F activation event in 2b, we modelled the second activation event computationally for FLP-II. A transition state corresponding to an SN1 C–F activation was located 27.1 kcal mol−1 higher in energy than the ground state {i.e. [2-P(o-Tol)3]+⋯[BF(C6F5)3]−} (Fig. 6), representing an increase in activation barrier of 8.2 kcal mol−1 as compared to the initial C–F activation event in 2b, and 13.6 kcal mol−1 greater than in benzyl fluoride (as a comparison to another monofluoride substrate).
Much of this increase in barrier may be attributed to the steric profile of P(o-Tol)3, hindered access of BCF to the remaining fluoride position and dissociation of the ion pair. In contrast, distal C–F positions are equally accessible in starting materials and monoactivation products. As stated above, we do see diactivation products in the case of distal fluorides, however, these are minor products as compared to the desired monoactivated salts, suggesting that the initial activation products are less reactive than the starting materials.
Modelling the double C–F bond activation of 3b with FLP-I, we found that the initial C–F activation is calculated to have a kinetic barrier of 19.3 kcal mol−1 (Fig. 7, for a detailed energy profile see Fig. S63†). This value is slightly higher than in 2b, as would be expected with the inclusion of an additional electron withdrawing para-CF2H group. The second barrier for C–F bond activation (i.e. C–F activation of [3b-TPPy][NTf2]) was calculated to be 20.3 kcal mol−1. Although the increase in activation barrier from 3b to [3b-TPPy][NTf2] is small, under controlled reaction conditions, a high yield of the monoactivation product can be obtained when using stoichiometric amounts of TPPy.
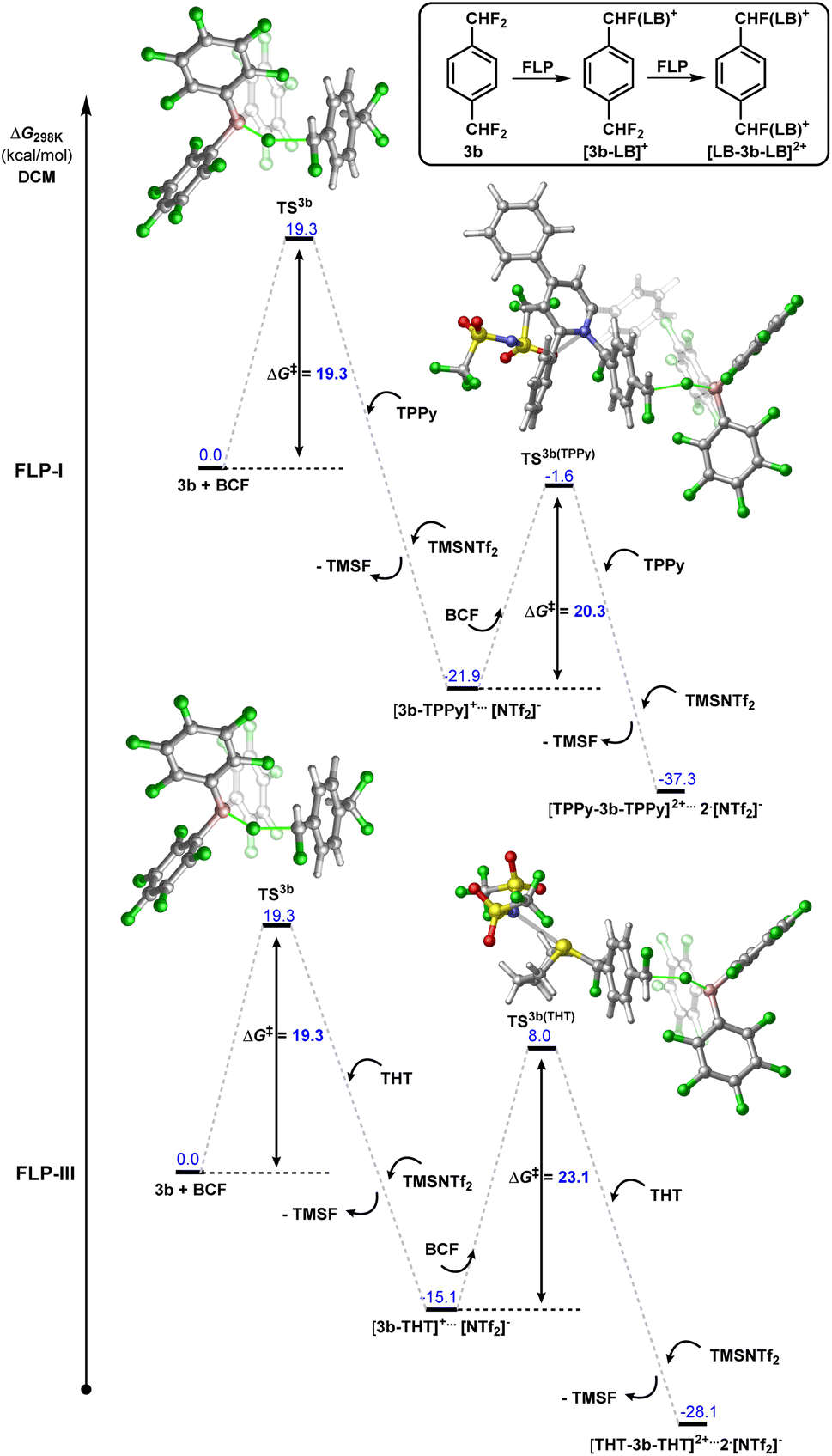 | ||
| Fig. 7 Calculated energy profile for the 1st and 2nd C–F activation events between 1,4-(CF2H)2-C6H4 (3b) and FLP-I or FLP-III. | ||
In contrast, we found that the barrier for the second C–F activation using FLP-III (i.e. C–F activation of [3b-THT][NTf2]) was 23.1 kcal mol−1, representing a barrier increase of 3.8 kcal mol−1 as compared to 3b (Fig. 8). The result of this is corroborated with experimental observations, where we generally see much higher selectivity of the monoactivation product over the diactivation product (Fig. 5B). In both cases, the effect of installing a neutral Lewis base is similar to the conversion of CF2H to a slightly more electron withdrawing group, providing an electronic deterrent for further C–F activation in the product.
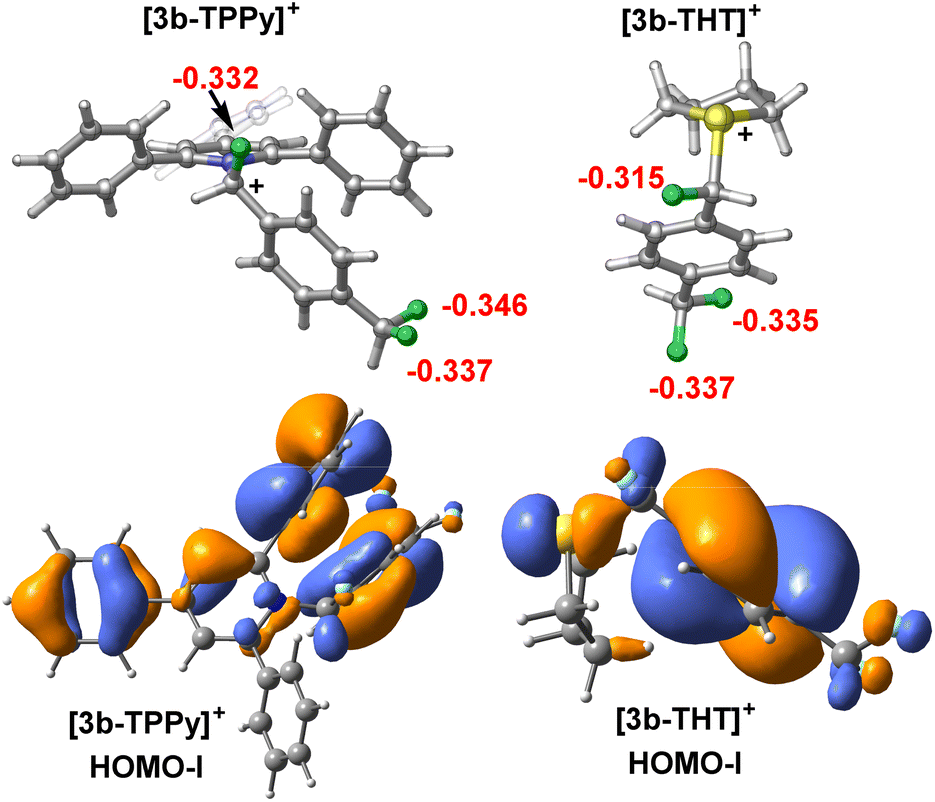 | ||
| Fig. 8 Calculated atom NBO charges for fluorine atoms and HOMO for [3b-TPPy]+ and [3b-THT]+. | ||
To understand from where the difference in 1st and 2nd C–F activation barrier heights between FLP-I and FLP-III arises, we generated frontier orbitals and charge assignments for [3b-LB]+ (LB = TPPy, THT) (Fig. 8 and S67†). The charge assignment for the geminal and distal fluorides in [3b-THT]+ are more positive than in [3b-TPPy]+ meaning that there is potentially less electrostatic interaction between the BCF catalyst and fluoride positions in [3b-THT]+. Examining the HOMO-I (and LUMO-I) of [3b-TPPy]+ and [3b-THT]+, it is clear that the frontier orbitals are significantly delocalised throughout the TPPy motif, whereas the frontier orbitals have little occupancy on the THT backbone. This can be interpreted as a delocalisation of positive charge throughout the TPPy fragment in [3b-TPPy]+, whereas in [3b-THT]+ the positive charge is maintained on the sulfonium position and throughout the fluorocarbon fragment. So the cationic charge that inhibits further C–F activation is more prevalent in the ‘3b+’ fragment in [3b-THT]+, resulting in a higher activation barrier for subsequent fluoride abstraction as compared to [3b-TPPy]+.
It is also apparent from the frontier orbitals of [3b-TPPy]+ that significant π–π stacking occurs between one of the TPPy ortho phenyl substituents and the ‘3b’ aromatic fragment. By modelling a single phenyl substituent on TPPy (i.e. monophenylpyridine) in the 2, 4 and 6 positions, inductive and through space stabilization provided by the phenyl substituents could be estimated (for details see Fig. S68†). It was found that π–π stacking of the phenyl group stabilized the diactivation product [3b′-TPPy]2+ by up to 2.7 kcal mol−1, and as such, it might be expected to also stabilise the transition state for the second C–F activation event in [3b-TPPy]+. This effect would not be active in [3b-THT]+, resulting in a higher transition state barrier.
Conclusions
The results of this study suggest that C–F activation in benzotrifluorides (1) and benzodifluorides (2) occurs via an SN1 type mechanism as opposed to a concerted FLP mechanism. Although this is not a classical FLP activation mechanism involving both the FLP components in the activation transition state, the use of an FLP system is an important aspect of the reaction given that thermodynamically preferred Lewis adducts (as in the case of FLP-III) can increase the kinetic activation barriers and suppress C–F activation. Indeed, given that BCF has become a mainstay of main group catalysis,16 a number of facets of this system are important to consider in other FLP and C–F activation systems. In addition to ‘reversible’ FLPs17 suffering from an energy penalty to dissociate the adduct form, this study suggests that steps that occur after the C–F activation event can be reaction limiting. This finding will be of interest to main group chemists who employ BCF in C–F and other bond activation chemistry.We have also shown that the inclusion of Me3SiNTf2 is important for the thermodynamic stabilization of C–F activation products. Indeed, the reverse reaction (attack of the activation product by [BF(C6F5)3]− anion) is thermodynamically preferred for FLP-I with 1a and 1b in the absence of Me3SiNTf2. However, despite Me3SiNTf2 being a competent Lewis acid, we have found that it does not affect the kinetic barrier for C–F activation.
Lastly, we have provided mechanistic evidence of where the monoselective C–F activation reaction arises. We have shown that subsequent C–F activation events incur a higher kinetic barrier (as compared to the initial C–F activation step), and that this is true for geminal and distal fluorides (i.e. selectivity has an electronic component). Moreover, we have shown that the Lewis bases incorporated into the activation products affect the kinetic barriers for subsequent C–F activation, and that this should be a consideration when choosing FLP for monoselective C–F activation.
Data availability
Experimental and computational data are available in the accompanying ESI.†Author contributions
RG and KL performed all experimental procedures including the collection and processing of kinetic data. DC performed all of the computations. RDY wrote the manuscript and devised the project. RG, DC, KL and RDY planned the experiments.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank the Singapore Agency for Science, Technology and Research for funding (A*STAR grant no. A1983c0033 and M21K2c0111) and Dr Jason B. Harper for helpful discussions.Notes and references
- (a) F. Jaroschik, Chem.–Eur. J., 2018, 24, 14572 CrossRef CAS PubMed; (b) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308 RSC.
- (a) K. Chen, N. Berg, R. Gschwind and B. Konig, J. Am. Chem. Soc., 2017, 139, 18444 CrossRef CAS PubMed; (b) H. Wang and N. T. Jui, J. Am. Chem. Soc., 2018, 140, 163 CrossRef CAS PubMed; (c) D. B. Vogt, C. P. Seath, H. Wang and N. T. Jui, J. Am. Chem. Soc., 2019, 141, 13203 CrossRef CAS PubMed; (d) C. Luo and J. S. Bandar, J. Am. Chem. Soc., 2019, 141, 14120 CrossRef CAS PubMed; (e) Y.-C. Luo, F.-F. Tong, Y. Zhang, C.-Y. He and X. Zhang, J. Am. Chem. Soc., 2021, 143, 13971 CrossRef CAS PubMed; (f) Y.-J. Yu, F.-L. Zhang, T.-Y. Peng, C.-L. Wang, J. Cheng, C. Chen, K. N. Houk and Y.-F. Wang, Science, 2021, 371, 1232 CrossRef CAS PubMed; (g) J.-H. Ye, P. Bellotti, C. Heusel and F. Glorius, Angew. Chem., Int. Ed., 2021, 61, e202115456 Search PubMed.
- (a) G. Meißner, K. Kretschmar, T. Braun and E. Kemnitz, Angew. Chem., Int. Ed., 2017, 56, 16338 CrossRef PubMed; (b) M. Bergeron, T. Johnson and J.-F. Paquin, Angew. Chem., Int. Ed., 2011, 50, 11112 CrossRef CAS PubMed; (c) L. Tang, Z.-Y. Liu, W. She and C. Feng, Chem. Sci., 2019, 10, 8701 RSC; (d) T. Xiao, L. Li and L. Zhou, J. Org. Chem., 2016, 81, 7908 CrossRef CAS PubMed; (e) S. B. Lang, R. J. Wiles, C. B. Kelly and G. A. Molander, Angew. Chem., Int. Ed., 2017, 56, 15073 CrossRef CAS PubMed; (f) T. Ichitsuka, T. Fujita and J. Ichikawa, ACS Catal., 2015, 5, 5947 CrossRef CAS; (g) S.-S. Yan, D.-S. Wu, J.-H. Ye, L. Gong, X. Zeng, C.-K. Ran, Y.-Y. Gui, J. Li and D.-G. Yu, ACS Catal., 2019, 9, 6987 CrossRef CAS; (h) X. Lu, X.-X. Wang, T.-J. Gong, J.-J. Pi, S.-J. He and Y. Fu, Chem. Sci., 2019, 10, 809 RSC; (i) Y. Lan, F. Yang and C. Wang, ACS Catal., 2018, 8, 9245 CrossRef CAS; (j) Z. Lin, Y. Lan and C. Wang, ACS Catal., 2019, 9, 775 CrossRef CAS; (k) Z. Lin, Y. Lan and C. Wang, Org. Lett., 2019, 21, 8316 CrossRef CAS PubMed; (l) Z. Liu, X.-S. Tu, L.-T. Guo and X.-C. Wang, Chem. Sci., 2020, 11, 11548 RSC; (m) J.-P. Bégué, D. Bonnet-Delpon and M. H. Rock, Synlett, 1995, 659 CrossRef; (n) J. P. Bégué, D. Bonnet-Delpon and M. H. Rock, Tetrahedron Lett., 1995, 36, 5003 Search PubMed; (o) J. Ichikawa, H. Fukui and Y. Ishibashi, J. Org. Chem., 2003, 68, 7800 CrossRef CAS PubMed; (p) J. Yang, A. Mao, Z. Yue, W. Zhu, X. Luo, C. Zhu, Y. Xiao and J. Zhang, Chem. Commun., 2015, 51, 8326 RSC; (q) J. Ichikawa, J. Synth. Org. Chem., Jpn., 2010, 68, 1175 CrossRef CAS; (r) K. Fuchibe, M. Takahashi and J. Ichikawa, Angew. Chem., Int. Ed., 2012, 51, 12059 CrossRef CAS PubMed; (s) T. Fujita, M. Takazawa, K. Sugiyama, N. Suzuki and J. Ichikawa, Org. Lett., 2017, 19, 588 CrossRef CAS PubMed; (t) K. Fuchibe, H. Hatta, K. Oh, R. Oki and J. Ichikawa, Angew. Chem., Int. Ed., 2017, 56, 5890 CrossRef CAS PubMed; (u) M. Bergeron, D. Guyader and J.-F. Paquin, Org. Lett., 2012, 14, 5888 CrossRef CAS PubMed.
- (a) S. Yoshida, K. Shimomori, Y. Kim and T. Hosoya, Angew. Chem., Int. Ed., 2016, 55, 10406 CrossRef CAS PubMed; (b) Y. Kim, K. Kanemoto, K. Shimomori, T. Hosoya and S. Yoshida, Chem.–Eur. J., 2020, 26, 6136 CrossRef CAS PubMed; (c) R. Idogawa, Y. Kim, K. Shimomori, T. Hosoya and S. Yoshida, Org. Lett., 2020, 22, 9292 CrossRef CAS PubMed.
- (a) D. Mandal, R. Gupta and R. D. Young, J. Am. Chem. Soc., 2018, 140, 10682 CrossRef CAS PubMed; (b) D. Mandal, R. Gupta, A. K. Jaiswal and R. D. Young, J. Am. Chem. Soc., 2020, 142, 2572 CrossRef CAS PubMed; (c) R. Gupta, D. Mandal, A. K. Jaiswal and R. D. Young, Org. Lett., 2021, 23, 1915 CrossRef CAS PubMed; (d) R. Gupta, A. K. Jaiswal, D. Mandal and R. D. Young, Synlett, 2020, 31, 933 CrossRef CAS; (e) S. Khanapur, K. Lye, D. Mandal, X. J. Wee, E. G. Robins and R. D. Young, Angew. Chem., Int. Ed., 2021, 61, e202210917 Search PubMed; (f) I. Mallov, A. J. Ruddy, H. Zhu, S. Grimme and D. W. Stephan, Chem.–Eur. J., 2017, 23, 17692 CrossRef CAS PubMed; (g) P. Mehlmann, T. Witteler, L. F. B. Wilm and F. Dielmann, Nat. Chem., 2019, 11, 1139 CrossRef CAS PubMed.
- (a) V. J. Scott, R. Celenligil-Cetin and O. V. Ozerov, J. Am. Chem. Soc., 2005, 127, 2852 CrossRef CAS PubMed; (b) J. Zhu, M. Perez, C. B. Caputo and D. W. Stephan, Angew. Chem., Int. Ed., 2016, 55, 1417 CrossRef CAS PubMed; (c) H.-J. Ai, X. Ma, Q. Song and X.-F. Wu, Sci. China: Chem., 2021, 64, 1630 CrossRef CAS.
- (a) J. J. Cabrera-Trujillo and I. Fernández, Chem.–Eur. J., 2021, 27, 3823 CrossRef CAS PubMed; (b) M. Ghara, S. Giri, P. Das and P. K. Chattaraj, J. Chem. Sci., 2022, 134, 14 CrossRef CAS.
- H. M. Yau, A. K. Croft and J. B. Harper, Chem. Commun., 2012, 48, 8937 RSC.
- H. Tinnermann, S. Sung, D. Csókás, Z. H. Toh, C. Fraser and R. D. Young, J. Am. Chem. Soc., 2021, 143, 10700 CrossRef CAS PubMed.
- The ground state for FLP-III is −0.9 kcal. Kinetic barriers should be calculated from this point, however, to compare with FLP-I and FLP-II, we have taken the FLP state (i.e. fully dissociated) as the ground state.
- F. Schaper and H.-H. Brintzinger, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2002, 58, o77 CrossRef CAS.
- J. S. Sapsford, D. Csókás, R. C. Turnell-Ritson, L. A. Parkin, A. D. Crawford, I. Pápai and A. E. Ashley, ACS Catal., 2021, 11, 9143 CrossRef CAS.
- (a) A. K. Jaiswal, P. K. Kishore and R. D. Young, Chem.–Eur. J., 2019, 25, 6290 CrossRef CAS PubMed; (b) R. Gupta and R. D. Young, Synthesis, 2022, 54, 1671 CrossRef CAS.
- If the barrier for [NTf2]− substitution was much less than C–F activation then the rate of formation of 2c-NTf2 would be less than its consumption at all observable concentrations. If the barrier for [NTf2]− substitution was much greater than C–F activation then 2c would be consumed before formation of [2c-LB][NTf2] was observed. If the barrier for [NTf2]− substitution was similar to (or slightly less than) C–F activation, then the rate of formation of 2c-NTf2 would exceed its consumption assuming that the consumption rate of 2c-NTf2 is dependent on [2c-NTf2] and that [2c-NTf2] ≪ [2c].
- Mayr nucleophilic parameters: pyridine: N = 12.90, sN = 0.67; P(o-Tol)3: N = 8.56, sN = 0.70; THT: N = 13.10, sN = 0.72; Et3SiH: N = 3.58, sN = 0.70; toluene: N = −4.36, sN = 1.77. See: (a) F. Brotzel, B. Kempf, T. Singer, H. Zipse and H. Mayr, Chem.–Eur. J., 2006, 13, 336 CrossRef PubMed; (b) J. Ammer, C. Nolte and H. Mayr, J. Am. Chem. Soc., 2012, 134, 13902 CrossRef CAS PubMed; (c) H. Mayr and A. R. Ofial, J. Phys. Org. Chem., 2008, 21, 584 CrossRef CAS.
- G. Kumar, S. Roy and I. Chatterjee, Org. Biomol. Chem., 2021, 19, 1230 RSC.
- T. C. Johnstone, G. N. J. H. Wee and D. W. Stephan, Angew. Chem., Int. Ed., 2018, 57, 5881 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc05632a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |