
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceC(sp3)–H cyanation by a formal copper(III) cyanide complex†
Jamey K.
Bower‡
,
Maxwell S.
Reese‡
,
Ilia M.
Mazin
 ,
Lina M.
Zarnitsa
,
Andrew D.
Cypcar
,
Curtis E.
Moore
,
Alexander Yu.
Sokolov
* and
Shiyu
Zhang
*
,
Lina M.
Zarnitsa
,
Andrew D.
Cypcar
,
Curtis E.
Moore
,
Alexander Yu.
Sokolov
* and
Shiyu
Zhang
*
Department of Chemistry and Biochemistry, The Ohio State University, 100 W. 18th Ave, Columbus, OH 43210, USA. E-mail: zhang.8941@osu.edu
First published on 16th January 2023
Abstract
High-valent metal oxo complexes are prototypical intermediates for the activation and hydroxylation of alkyl C–H bonds. Substituting the oxo ligand with other functional groups offers the opportunity for additional C–H functionalization beyond C–O bond formation. However, few species aside from metal oxo complexes have been reported to both activate and functionalize alkyl C–H bonds. We herein report the first example of an isolated copper(III) cyanide complex (LCuIIICN) and its C–H cyanation reactivity. We found that the redox potential (Eox) of substrates, instead of C–H bond dissociation energy, is a key determinant of the rate of PCET, suggesting an oxidative asynchronous CPET or ETPT mechanism. Among substrates with the same BDEs, those with low redox potentials transfer H atoms up to a million-fold faster. Capitalizing on this mechanistic insight, we found that LCuIIICN is highly selective for cyanation of amines, which is predisposed to oxidative asynchronous or stepwise transfer of H+/e−. Our study demonstrates that the asynchronous effect of PCET is an appealing tool for controlling the selectivity of C–H functionalization.
Introduction
Nature has evolved metalloenzymes that rapidly and selectively hydroxylate aliphatic C–H bonds under ambient conditions with earth-abundant metals.1 The rate-limiting step of C–H hydroxylation – hydrogen atom abstraction (HAA) – is often mediated by high-valent metal-oxo intermediates (Scheme 1A).2–4 The remarkable HAA ability of metal-oxo intermediates has motivated intense efforts to understand the factors that underpin the reactivity. A large body of experimental and theoretical studies were dedicated to investigating the movement of protons and electrons from C–H substrates to metal oxo complexes.5,6 These studies have important implications in understanding the activation energies of HAA reactions, which can predominately trend with the driving force of proton transfer (PT,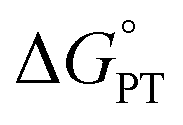 ), electron transfer (ET,
), electron transfer (ET,  ), or proton-coupled electron transfer
), or proton-coupled electron transfer 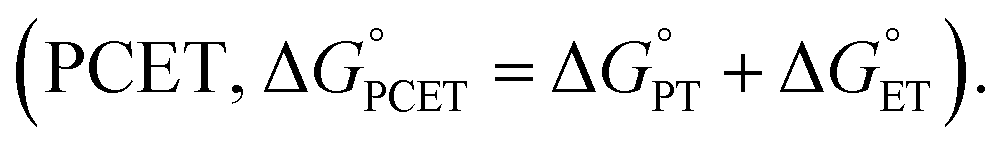 7,8 While C–H bond cleavage is often considered as a synchronous PCET that is governed by bond dissociation free energy, recent studies by Anderson,9 Borovik,10,11 Tolman, Cramer,12 Kojima,13,14 and Fukuzumi15 show that the basicity (pKa) or redox potential (E°) of the C–H-activating metal complexes can also be the determining factor for C–H activation.
7,8 While C–H bond cleavage is often considered as a synchronous PCET that is governed by bond dissociation free energy, recent studies by Anderson,9 Borovik,10,11 Tolman, Cramer,12 Kojima,13,14 and Fukuzumi15 show that the basicity (pKa) or redox potential (E°) of the C–H-activating metal complexes can also be the determining factor for C–H activation.
 | ||
| Scheme 1 | ||
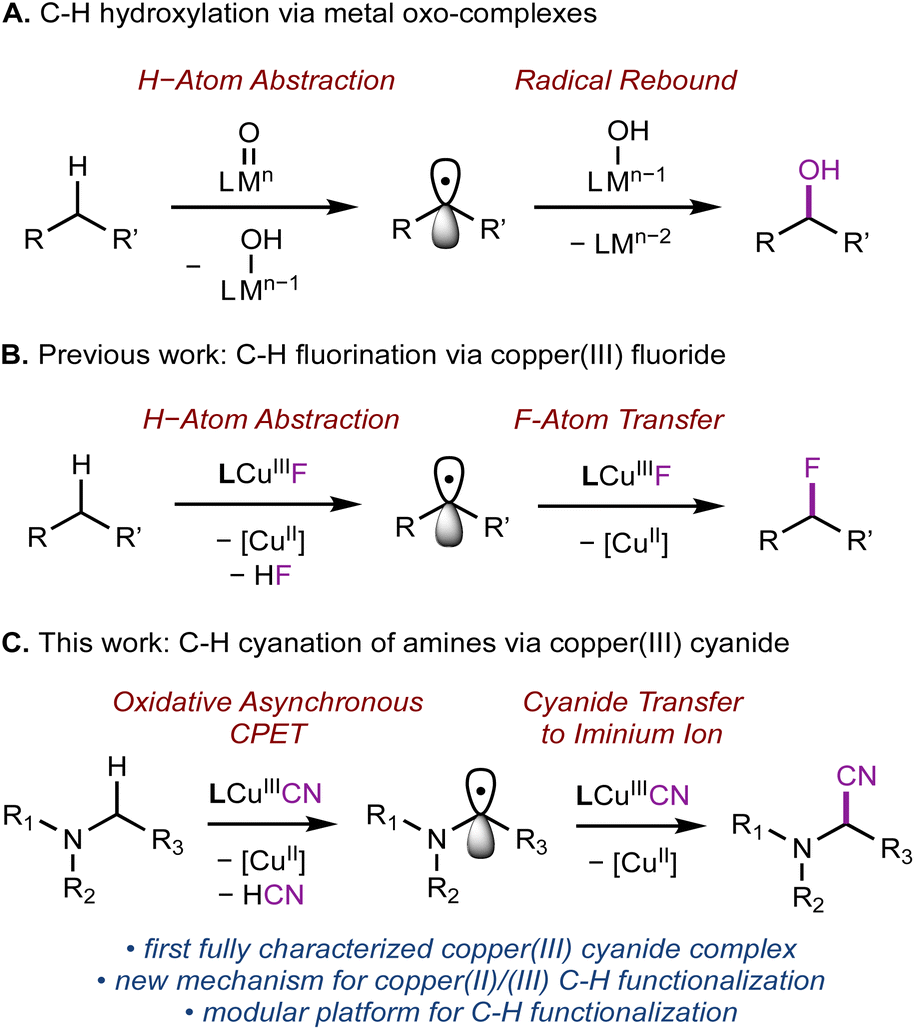
In addition to the successful demonstration of metal-oxo species for C–H activation and functionalization,4,16–18 a variety of synthetic high-valent metal complexes carrying other functional groups M–FG, e.g., OH, superoxo, carboxylate, halides, nitrite, and nitrate, have been shown to activate C–H bonds. The studies by Tolman,12,19–23 McDonald,24–28 us,29 and others30–32 have demonstrated that C–H activation can be achieved in the absence of the oxo ligand through mechanistically similar HAA/PCET processes with M–FG. However, in contrast to metal oxo species, few M–FG complexes have been shown to subsequently functionalize the substrate following C–H activation.23,29 Seminal research by Tolman12,19–23 and McDonald24–28 showed that pyridinedicarboxamide (L) Cu(III) and Ni(III) complexes can activate strong C–H bonds. Employing the same ligand L, we established that a copper(III) fluoride complex (LCuIIIF) can perform sp3 C–H fluorination by sequential HAA and fluorine atom transfer, similar to the mechanism of metal-oxo-mediated C–H functionalization (Scheme 1B).29 The modularity of L is particularly attractive, as one could envision substituting fluorine with other desirable functional groups in C–H functionalization sequences.
Toward this end, we were motivated to investigate the reactivity of copper(III) cyanide complexes for C–H cyanation. Nitriles are particularly valuable synthetic motifs since the –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N group provides a handle to develop molecular complexity through further functional group manipulation.33 For example, α-aminonitriles are a common motif in anti-tumor therapeutics and important synthetic precursors to 1,2-diamines and β-amino acids.33 Recently, many copper-catalyzed cyanation reactions have been developed.25–36 High-valent copper(III) cyanide complexes are often proposed as reactive intermediates in cyanation reactions, e.g., cross-coupling,34–36 radical-relay C–H cyanation,37–42 or photoinduced cyanation.43–45 The key C–CN bond formation steps are often proposed to involve reductive elimination from the organocopper(III) cyanide species. However, copper(III) cyanide intermediates are often too short-lived for isolation, preventing rigorous characterization and interrogation of their participation in key mechanistic steps.
N group provides a handle to develop molecular complexity through further functional group manipulation.33 For example, α-aminonitriles are a common motif in anti-tumor therapeutics and important synthetic precursors to 1,2-diamines and β-amino acids.33 Recently, many copper-catalyzed cyanation reactions have been developed.25–36 High-valent copper(III) cyanide complexes are often proposed as reactive intermediates in cyanation reactions, e.g., cross-coupling,34–36 radical-relay C–H cyanation,37–42 or photoinduced cyanation.43–45 The key C–CN bond formation steps are often proposed to involve reductive elimination from the organocopper(III) cyanide species. However, copper(III) cyanide intermediates are often too short-lived for isolation, preventing rigorous characterization and interrogation of their participation in key mechanistic steps.
Herein, we report the synthesis, characterization, and C–H cyanation reactivity of the first fully characterized formal copper(III) cyanide complex, LCuIIICN (Scheme 1C). Interestingly, while benzylic and allylic substrates (BDE ∼90 kcal mol−1) are not amenable for C–H cyanation by LCuIIICN, trialkylamines with essentially the same C–H BDE (∼89–93 kcal mol−1)46 readily undergo C–H cyanation in up to 90% yields. The contrasting reactivity of benzylic and α-amino C–H bonds with LCuIIICN was attributed to the large thermodynamic bias between ET and PT.7
Results and discussion
Synthesis, characterization, and electronic structure of a formal copper(III) cyanide complex
We first synthesized the copper(II) cyanide complex using a procedure analogous to previously reported copper(II) hydroxide and halide complexes.19,29 Treatment of the copper(II) acetonitrile complex, LCuIIMeCN, with tetrabutylammonium cyanide (TBACN) affords the anionic copper(II) cyanide complex, [TBA]LCuIICN in 94% yield. X-ray diffraction analysis of single crystals obtained from a THF/diethyl ether solution shows a square planar copper(II) center with a linear Cu–CN unit (Fig. 2A). The cyclic voltammogram (CV) of [TBA]LCuIICN in CH2Cl2 exhibits a quasi-reversible redox couple at E1/2 = 0.585 V (vs. Ag/AgNO3) (Fig. 1B), which is the highest among the LCuIII/II halide/pseudohalide series (F 0.395 V, Cl 0.458 V, Br 0.463 V in CH2Cl2).29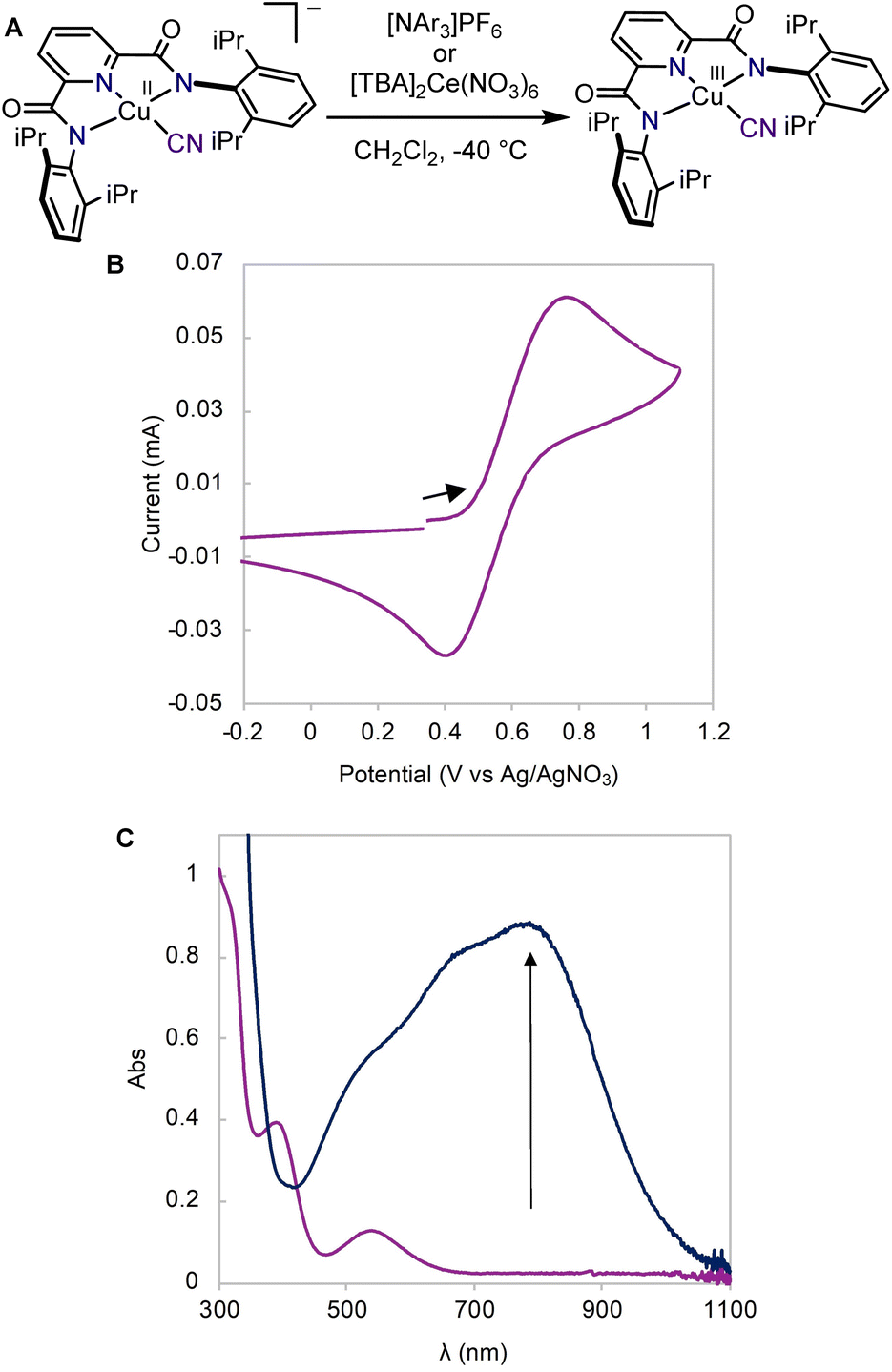 | ||
| Fig. 1 (A) Synthesis of LCuIIICN. (B) Cyclic voltammogram of [TBA]LCuIICN (3 mM in CH2Cl2 with 0.1 M [TBA]ClO4 at 23°. (C) UV-vis spectra showing the formation of LCuIIICN (dark blue trace) by chemical oxidation of [TBA]LCuIICN (purple trace) in CH2Cl2 at −40 °C. | ||
Having demonstrated the chemical reversibility of the formal copper(II)/(III) cyanide redox couple, we investigated the chemical oxidation of [TBA]LCuIICN with variable temperature UV-vis spectroscopy. Treatment of [TBA]LCuIICN with the triarylaminium radical cation, [NAr3]PF6 (Ar = 4-bromophenyl, E1/2 = 0.70)47 at −40 °C in CH2Cl2 reveals the rapid formation of a new species with intense absorption at 790 nm (ε = 8800 M−1 cm−1) (Fig. 1C). The observed optical features are very similar to other formal copper(III) species in the same ligand framework and are thus attributed to the LCuIIICN complex. Despite its highly oxidizing nature, the formal LCuIIICN complex is metastable in solution even at room temperature in CH2Cl2. LCuIIICN can also be generated by oxidation with [TBA]2Ce(NO3)6 at −40 °C. When prepared from the CeIV oxidant, LCuIIICN exhibits lower thermal stability (slow decay at 0 °C), perhaps due to the extraction of the cyanide anion by the Lewis acidic CeIII ion. Nonetheless, the CeIV route is better for large-scale preparation due to the ease of separation of LCuIIICN from the CeIII byproduct. Extraction of LCuIIICN into diethyl ether led to its isolation as an analytically pure microcrystalline solid in 95% yield. The isolated solid LCuIIICN can be stored at −35 °C indefinitely.
Spectroscopic and structural characterization further confirms the formation of a formal copper(III) cyanide complex. The 1H NMR spectrum of LCuIIICN in CD2Cl2 at room temperature exhibits sharp resonances within the 0–9 ppm range that account for all ligand protons (Fig. S4†). Furthermore, LCuIIICN is EPR silent at cryogenic and room temperatures (Fig. S6†). Collectively, the NMR and EPR studies suggest a diamagnetic ground state for LCuIIICN, similar to the analogous halide, hydroxo, and alkoxo complexes.29,48 Infrared spectroscopy was used to gain further insight into the bonding between CN and the Cu center. Coordination of [TBA]CN to LCuIIMeCN shows a shift in the CN stretching frequency from 2055 cm−1 to 2141 cm−1, indicating that binding to the Lewis acidic copper(II) strengthens the CN bond. Upon oxidation of [TBA]LCuIICN to LCuIIICN, the CN stretching frequency shifts to 2164 cm−1 (Fig. S7†). We attribute the higher CN stretching frequency in LCuIIICN to the decreased π backbonding resulting from the increase in Cu effective nuclear charge. Single-crystals of LCuIIICN were grown from vapor diffusion of pentane into a THF solution of LCuIIICN at −35 °C (Fig. 2B). The structure of LCuIIICN displays retention of the square planar geometry with contraction of the copper–ligand bond distances by 0.07–0.1 Å compared to the copper(II) complex, in agreement with the higher effective nuclear charge of LCuIIICN. The CN bond length remains essentially the same, consistent with the minimal change in the CN stretching frequency upon oxidation. LCuIIICN is the first isolated and crystallographically characterized formal copper(III) complex bearing a cyanide and sp hybridized C donor ligand.
 | ||
| Fig. 2 Single-crystal X-ray diffraction structures and selected structural parameters of (A) [TBA]LCuIICN and (B) LCuIIICN with thermal ellipsoids shown at 50% level of probability. Counterions, co-crystallized solvents, and hydrogen atoms are omitted for clarity. | ||
Multireference calculations with complete active space self-consistent field (CASSCF) and N-electron valence second-order perturbation theory (NEVPT2) were performed to compare the electronic structure of LCuIIICN and LCuIII halides. Similar to the halide analogs, LCuIIICN has a singlet ground state and multiconfigurational electronic structure. The adiabatic singlet–triplet energy gap computed by NEVPT2 with 8 electrons in 8 active orbitals is 36.6 kcal mol−1 for LCuIIICN and 22.6 kcal mol−1 for LCuIIIF. The most significant difference between the electronic structure of LCuIIICN and LCuIII halides is in the composition and occupation of the lowest unoccupied natural orbital (LUNO), which is the key orbital for both HAA and radical capture (RC). For both LCuIIIF and LCuIIICN, the LUNO is comprised of the σ antibonding interactions between the Cu dx2–y2 and ligand π orbitals (Fig. 3). However, while the LUNO of LCuIIIF has orbital density equally distributed on either side of the fluorine atom, the LUNO of LCuIIICN is centered mainly on the C atom of CN, partly due to the π interactions between CN and Cu. In addition, the LCuIIICN LUNO has a lower occupation (0.14 e−) than the LCuIIIF LUNO (0.20 e−), in agreement with the larger singlet–triplet gap of LCuIIICN.
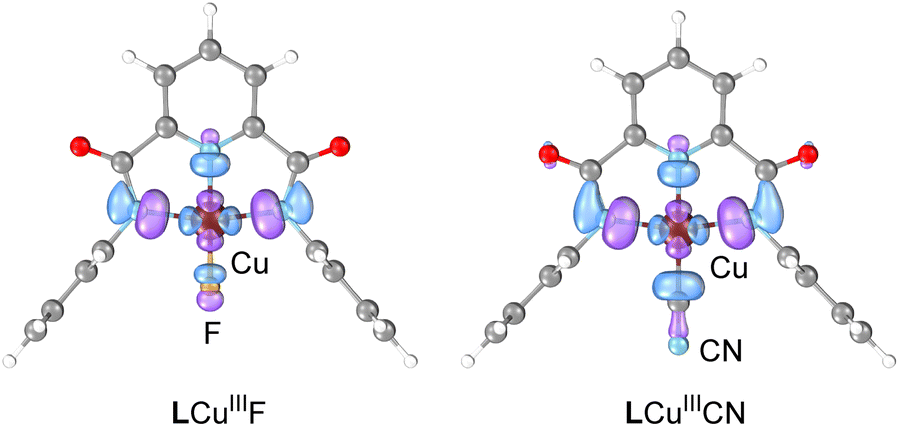 | ||
| Fig. 3 Lowest unoccupied natural orbitals of LCuIIIF and LCuIIICN from CASSCF calculation with 8 electrons in 8 active orbitals. Orbitals are shown at an isovalue of 0.05. | ||
Hydrogen atom abstraction reactivity of LCuIIICN
The spectroscopic and structural resemblance of LCuIIICN to LCuIIIF motivated us to expand the scope of CuIII/CuII-promoted C–H functionalization. We began by studying the reaction between LCuIIICN and the hydrogen atom donor, 9,10-dihydroanthracene (DHA, C–H BDE 76.3 kcal mol−1). The second-order rate constant of HAA with LCuIIICN is approximately three orders of magnitude lower than that of LCuIIIF and five orders of magnitude lower than that of LCuIIIOH (Fig. S12†). Monitoring the reaction between LCuIIICN and DHA at varying temperatures from 0 to 30 °C gave activation parameters ΔH‡ = 9.4(1.6) kcal mol−1 and ΔS‡ = −37(5) eu (Fig. S13†). The self-decay of the LCuIIICN complex at 30 °C is negligible (Fig. S14†). Notably, the enthalpy of activation is significantly disfavored for LCuIIICN compared to LCuIIIF and LCuIIIOH (Table 1), while the entropy of activation is comparable.21,29The coupled movement of electron and proton during HAA has important implications on the kinetic barrier. The extent of proton/electron coupling in PCET reactions ranges from concerted proton electron transfer (CPET), in which the proton and electron are transferred in a single kinetic step, to stepwise electron transfer–proton transfer (ET–PT) or proton transfer–electron transfer (PT–ET, Scheme 2).11,49,50 For example, it has been demonstrated that LCuIIIOH activates C–H bonds via a synchronous CPET reaction.19–22 The mechanistic space between CPET and stepwise reactions is occupied by asynchronous CPET reactions, in which the proton and electron are transferred together, but with one leading the other (Scheme 2). Mid- to late-transition metal-oxo complexes are often highly basic and undergo C–H activation via basic asynchronous PCET, where the transition state exhibits predominant PT characters.7,9,12,51,52 Tolman and Cramer et al. provide computational evidence that LCuIIIO2CAr complexes activate C–H bonds through a rare oxidative asynchronous process with a transition state bearing ET character (Scheme 2).12 The different dependence of kPCET on thermodynamic factors of PCET 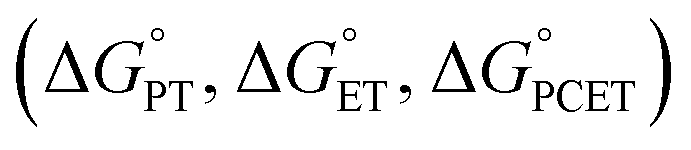 suggests that C–H activation selectivity can be tuned by the identity of the functional group in high-valent M–FG complexes.
suggests that C–H activation selectivity can be tuned by the identity of the functional group in high-valent M–FG complexes.
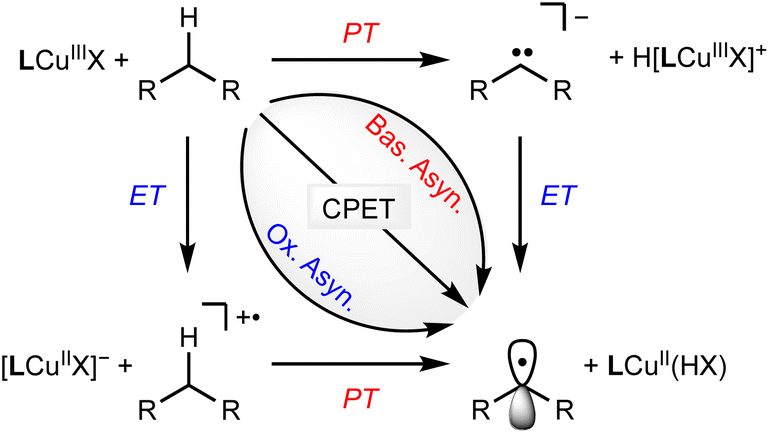 | ||
| Scheme 2 Possible mechanisms of PCET by LCuIIIFG complexes. | ||
To futher understand HAA by LCuIIICN, we examined the reactivity of LCuIIICN with a series of H-atom donors. All reactions were monitored by UV-vis spectroscopy at 20 °C in CH2Cl2. Gas chromatography-mass spectrometry analysis of the reaction mixtures revealed the corresponding oxidized hydrocarbons, but no C–H cyanation products (see the ESI†). The pseudo-first-order rate constants, kobs, were plotted versus the BDE, pKa, and E° of the C–H substrates (Fig. 4). The resulting plots show a linear correlation between HAA rates and C–H BDEs with an R2 value of 0.91, supporting a concerted, homolytic C–H bond cleavage reaction (Fig. 5A). The Brønsted α value (Fig. 4A) obtained from this plot is 0.048, which is quite low for concerted CPET process,53 indicating a high degree of transition state asymmetry.54 The correlations of kobs with E° and pKa of the C–H substrates are poor with R2 values of 0.39 and 0.09, respectively (Fig. 4Band C). We also attempted to correlate the rates of C–H activation with the driving forces of ΔGPCET, ΔGPT, and ΔGET using semiempirical models based on the work of Anderson and Borovik (see the ESI, Fig. S75 and S76†).52,55 The results show a predominant contribution from ΔGPCET with minor influence from ΔGET, further supporting concerted C–H activation with benzylic and allylic C–H substrates.
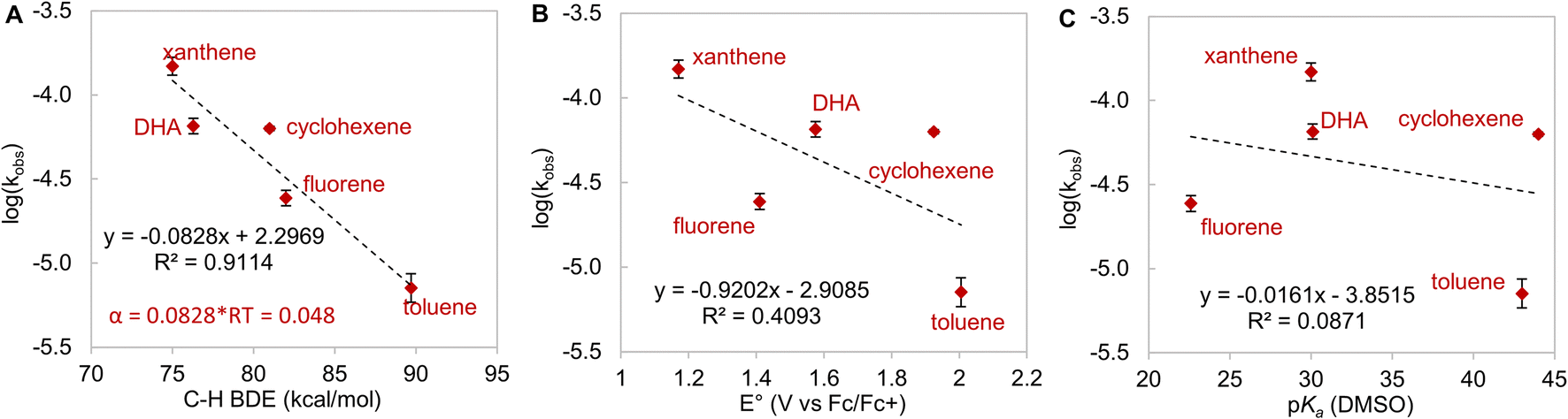 | ||
| Fig. 4 Plot of log(kobs) versus (A) C–H bond dissociation energy (R2 = 0.91), (B) pKa (R2 = 0.41) and (C) Eox (R2 = 0.09) of the substrates for the reaction of LCuIIICN with 100 eq. of hydrocarbon C–H substrates. | ||
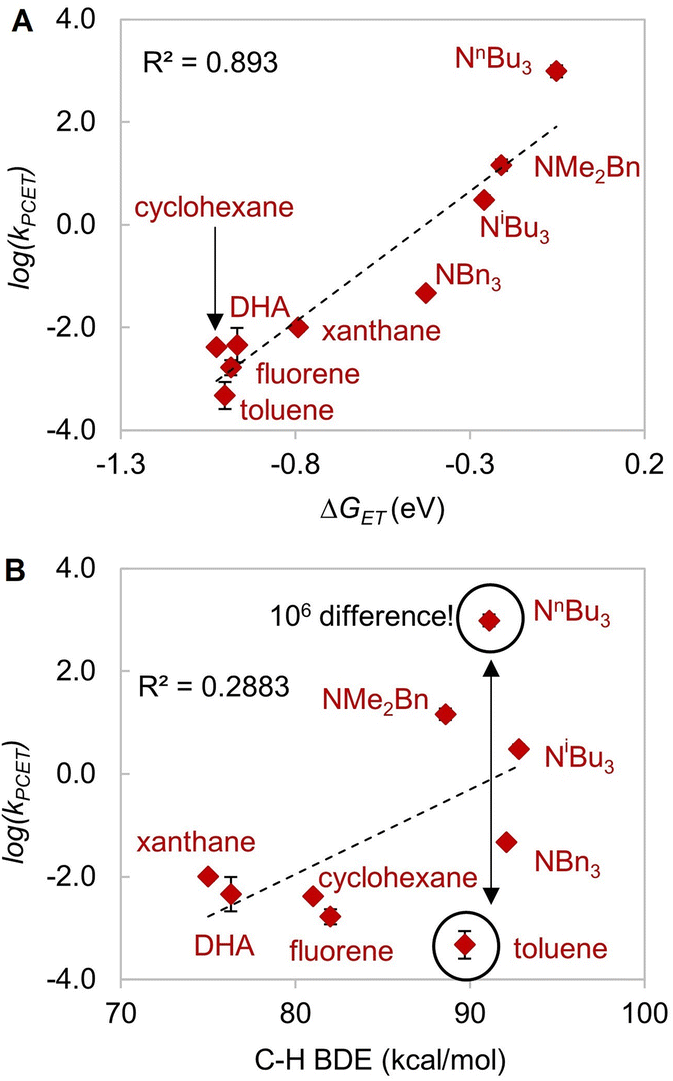 | ||
| Fig. 5 Plot of log(kobs) versus (A) driving force of electron transfer −ΔGET (R2 = 0.893), and (B) BDE (R2 = 0.2883). | ||
Reactivity of LCuIIICN with amines
The lack of C–H cyanation reactivity toward benzylic and allylic substrates by LCuIIICN prompted us to consider other strategies to reduce the barrier of HAA/PCET. A recent study by Srnec et al. suggests that more asynchronous PCET processes, as characterized by a larger |η| value, give rise to a lower activation free energy and faster PCET.7 The η value can be calculated by considering the thermodynamics of ET and PT:52| η = 2−1/2 × (ΔE° − RT/F × ln(10) × ΔpKa,ox). |
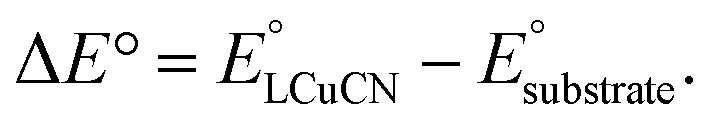
| ΔpKa,ox = pKLCuCN,oxa − pKsubstrate,oxa. |
The LCuIIICN complex is unstable toward protonation, preventing the experimental determination of pKa. Nonetheless, based on the reactivity of LCuIIICN with various acids, we estimated the pKa of LCuIIICN/[LCuIIICN]H+ to be less than 4.4 (see the ESI†). As shown in the equations above, the absolute pKa value of LCuIIICN/[LCuIIICN]H+ (pKLCuCN,oxa) does not change the difference in η value between substrates, since pKLCuCN,oxa is a common term for all the η calculation. Combining the estimated pKa with the experimentally determined E1/2 of LCuIIICN in DMSO (0.295 V vs. Fc/Fc+), we have found that the η values of C–H substrates are generally positive, indicating oxidative asynchronous CPET or ET–PT (Table 2).
| Substrate | C–H BDE | η (V) | −ΔGET (eV) | k PCET or kET (M−1 s−1) |
|---|---|---|---|---|
| Fluorene | 82.0 | 0.076 | −0.985 | 1.7(6) × 10−3 |
| DHA | 76.3 | 0.146 | −0.967 | 5(5) × 10−3 |
| Toluene | 89.7 | 0.441 | −1.00 | 5(4) × 10−4 |
| Xanthene | 75.0 | 0.470 | −0.792 | 1.0(2) × 10−2 |
| Cyclohexene | 81 | — | −1.03 | 4.1(6) × 10−3 |
| NBn3 | 92.1 | 1.22 | −0.427 | 4.7(4) × 10−2 |
| NiBu3 | 92.8 | 1.34 | −0.259 | 3.1(6) |
| NMe2Bn | 88.6 | 1.34 | −0.210 | 15(4) |
| NnBu3 | 91.1 | 1.41 | −0.052 | 10(3) × 102 |
Since C–H activation by LCuIIICN is biased for oxidative asynchronous processes, substrates that exhibit even more positive η values (more oxidative asynchronicity) than benzylic/allylic C–H substrates for HAA by LCuIIICN should have a lower kinetic barrier of HAA/PCET. Therefore, we consider amines as possible substrates due to their mild oxidation potentials (E° = ∼0.4–1 V vs. Fc/Fc+ in MeCN) and low acidities (pKa ≥ 40).56,57 Indeed, the η values of amines (1.22–1.41 V) are substantially higher than those of benzylic and allylic substrates (0.076–0.470 V). The comparison between trialkylamines and toluene is noteworthy as they have similar bond dissociation enthalpies (BDEs, 89–93 kcal mol−1 (ref. 46) and 89.7 kcal mol−1) but very different η values (ηtributylamine = 1.41 V; ηtoluene = 0.441 V).
According to Srnec's theory, LCuIIICN is expected to activate trialkylamines at a much faster rate because CPET with tributylamine is more asynchronous (larger |η|). Indeed, we found that LCuIIICN activates trialkylamines faster than toluene by 103 to 106-fold (Table 2)! While the plot of log(kPCET) versus BDEs with C–H substrates shows good correlation with benzylic and allylic substrates (Fig. 4A), the R2 value drops down to 0.2883 if amine substrates are included in the plot (Fig. 5A). In contrast, the plot of log(kPCET) versus the driving force of electron transfer (−ΔGET) now shows a good correlation with R2 = 0.893 (Fig. 5B), suggesting that electron transfer is the determining factor for the rate of PCET. The PCET rate of NnBu3 appears to be an outlier (Fig. 5B), potentially due to a mechanism crossover from oxidative asynchronous PCET to stepwise ET–PT. It is possible that the amine substrates lie on a different line due to stepwise ET–PT. Similar phenomena have been observed by Kojima13,14 and Fukuzumi.15
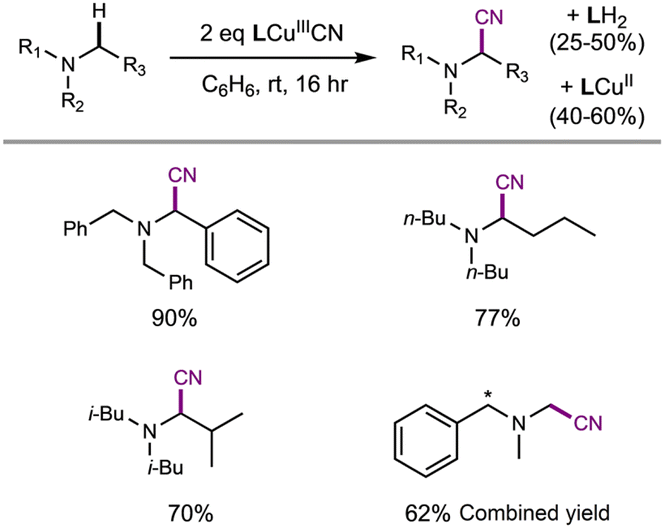
Additionally, LCuIIICN not only activates α-amino C–H bonds efficiently but also furnishes the amine C–H cyanation products. Treatment of trialkylamines with two equivalents of LCuIIICN in benzene at room temperature affords the corresponding α-aminonitrile products in 62–90% yield (Table 3). The reaction of LCuIIICN with N,N-dimethyl-1-phenylmethanamine furnishes a mixture of nitrile products at the aliphatic and benzylic positions in a combined 62% yield. To evaluate the role of Cu, control experiments were performed by treating amine substrates with two equivalents of the one-electron oxidant [NArBr3]+ in the presence of TBACN. The cyanation products were also observed in 10–55% yield (Fig. S54–S57†), indicating that an ET-initiated cyanation is possible.58 The higher yields by LCuIIICN can be attributed to the fact that LCuIIICN can serve as both the oxidant and the source of cyanide.
|
Following the stoichiometric C–H cyanation reactions, we observed varying amounts of protonated ligand LH2 (25–50%) by 1H NMR, indicating that the dianionic ligand L2− serves as the terminal proton acceptor. Furthermore, the copper(II) byproduct can be recovered as LCuIIMeCN in 40–60% spectroscopic yields according to UV-vis and EPR analysis (see the ESI†). This dual C–H activation/radical “rebound” type of reactivity has only been observed with LCuIIIF (ref. 29) and LCuIIINO2.23 However, the narrow substrate scope, as well as the use of stoichiometric amount of LCuIIICN, limits the use of this method compared to state-of-the-art C–H cyanation protocols.58–60
Conclusion
In summary, we report the synthesis and characterization of the first formal copper(III) cyanide complex. Despite the similar spectroscopic properties of LCuIIIF and LCuIIICN, their reactivities stand in stark contrast. While LCuIIIF is quite reactive towards benzylic and allylic C–H bonds, HAA by LCuIIICN is approximately 103 times slower. A very striking result of our study is that the rate of C–H activation with LCuIIICN can be increased by 103–107-fold by polarity matching, as characterized by the asynchronicity factor η. This observation is consistent with the prediction by Srnec that asynchronous processes have smaller activation barriers than synchronous processes with the same driving force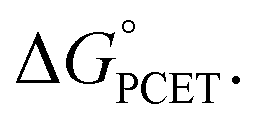 We further demonstrate that LCuIIICN is capable of the C–H cyanation of tertiary amines. The reaction proceeds through 2
We further demonstrate that LCuIIICN is capable of the C–H cyanation of tertiary amines. The reaction proceeds through 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry with LCuIIICN performing both HAA/PCET and CN group transfer.
:1 stoichiometry with LCuIIICN performing both HAA/PCET and CN group transfer.
Polarity matching strategies have been demonstrated in sp3 C–H activation with organic radicals to facilitate selectivity beyond the conventional bond dissociation energy model.61–63 Herein, we show that a similar strategy can be applied to metal-based oxidants. Pairing the oxidative ability of LCuIIICN with the oxidizable nature of amines allows the development of HAA/PCET processes not dictated by the thermodynamic driving force of the reaction itself. While it is still early to conclude whether high asynchronicity factors |η| generally lead to faster HAA rates, the success of this model in our case is part of a growing number of studies that suggest the (im)balance between 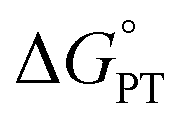 and
and 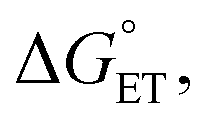 considered in the form of asynchronicity factor η (the difference between
considered in the form of asynchronicity factor η (the difference between 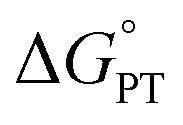 and
and 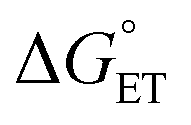 ) or empirical models, plays a critical role in understanding the rates of HAA.9,52,53,55 The comparison of LCuIIICN and LCuIII halides shows that changing the functional group bound to the copper(III) center opens new avenues for accessing distinct mechanistic pathways. Studies to further generalize the copper(II)/(III)-mediated C–H functionalization with other functional groups are ongoing in our laboratory.
) or empirical models, plays a critical role in understanding the rates of HAA.9,52,53,55 The comparison of LCuIIICN and LCuIII halides shows that changing the functional group bound to the copper(III) center opens new avenues for accessing distinct mechanistic pathways. Studies to further generalize the copper(II)/(III)-mediated C–H functionalization with other functional groups are ongoing in our laboratory.
Data availability
All other data supporting the findings of this study are available in the ESI.†Author contributions
Conceptualization: JKB, AYS, SZ; methodology: JKB, MR, IMM; investigation: JKB, MR, LMZ, IMM, ADC, CEM; writing – original draft: JKB, AYS, SZ; writing – review & editing: MR, AYS, SZ; funding acquisition: SZ, AYS.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
The authors thank Prof. Christine Thomas for access to an IR spectrometer and Dr Wenjie Tao for assistance with low-temperature NMR data collection. This material is based on work supported by the U.S. National Institute of Health (NIH) under award number R01-GM145746 (SZ) and the U.S. National Science Foundation CHE-2044648 (AYS). JKB was supported by a Presidential Fellowship from the Ohio State University Graduate School. The authors thank the Ohio State University Department of Chemistry and Biochemistry for additional financial support.References
- J. Rittle and M. T. Green, Science, 2010, 330, 933–937 CrossRef CAS PubMed.
- X. Huang and J. T. Groves, J. Biol. Inorg Chem., 2017, 22, 185–207 CrossRef CAS PubMed.
- A. R. McDonald and L. Que, Coord. Chem. Rev., 2013, 257, 414–428 CrossRef CAS.
- A. S. Borovik, Chem. Soc. Rev., 2011, 40, 1870–1874 RSC.
- M. H. V Huynh and T. J. Meyer, Chem. Rev., 2007, 107, 5004–5064 CrossRef PubMed.
- J. J. Warren, T. A. Tronic and J. M. Mayer, Chem. Rev., 2010, 110, 6961–7001 CrossRef CAS PubMed.
- D. Bím, M. Maldonado-Domínguez, L. Rulísek and M. Srnec, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E10287–E10294 CrossRef PubMed.
- R. G. Agarwal, S. C. Coste, B. D. Groff, A. M. Heuer, H. Noh, G. A. Parada, C. F. Wise, E. M. Nichols, J. J. Warren and J. M. Mayer, Chem. Rev., 2022, 122, 1–49 CrossRef CAS PubMed.
- M. K. Goetz and J. S. Anderson, J. Am. Chem. Soc., 2019, 141, 4051–4062 CrossRef CAS PubMed.
- B. S. K., Y. Meng-Yin, P. T. H., G. M. T. and B. A. S., Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2108648118 CrossRef PubMed.
- T. H. Parsell, M. Y. Yang and A. S. Borovik, J. Am. Chem. Soc., 2009, 131, 2762–2763 CrossRef CAS PubMed.
- M. Mandal, C. E. Elwell, C. J. Bouchey, T. J. Zerk, W. B. Tolman and C. J. Cramer, J. Am. Chem. Soc., 2019, 141, 17236–17244 CrossRef CAS PubMed.
- S. Miyazaki, T. Kojima, J. M. Mayer and S. Fukuzumi, J. Am. Chem. Soc., 2009, 131, 11615–11624 CrossRef CAS PubMed.
- H. Kotani, S. Kaida, T. Ishizuka, M. Sakaguchi, T. Ogura, Y. Shiota, K. Yoshizawa and T. Kojima, Chem. Sci., 2015, 6, 945–955 RSC.
- Y. Morimoto, J. Park, T. Suenobu, Y.-M. Lee, W. Nam and S. Fukuzumi, Inorg. Chem., 2012, 51, 10025–10036 CrossRef CAS PubMed.
- A. Gunay and K. H. Theopold, Chem. Rev., 2010, 110, 1060–1081 CrossRef CAS PubMed.
- W. Liu and J. T. Groves, Acc. Chem. Res., 2015, 48, 1727–1735 CrossRef CAS PubMed.
- M. Puri, A. N. Biswas, R. Fan, Y. Guo and L. Que, J. Am. Chem. Soc., 2016, 138, 2484–2487 CrossRef CAS PubMed.
- P. J. Donoghue, J. Tehranchi, C. J. Cramer, R. Sarangi, E. I. Solomon and W. B. Tolman, J. Am. Chem. Soc., 2011, 133, 17602–17605 CrossRef CAS PubMed.
- D. Dhar and W. B. Tolman, J. Am. Chem. Soc., 2015, 137, 1322–1329 CrossRef CAS PubMed.
- D. Dhar, G. M. Yee, A. D. Spaeth, D. W. Boyce, H. Zhang, B. Dereli, C. J. Cramer and W. B. Tolman, J. Am. Chem. Soc., 2016, 138, 356–368 CrossRef CAS PubMed.
- C. E. Elwell, M. Mandal, C. J. Bouchey, L. Que, C. J. Cramer and W. B. Tolman, Inorg. Chem., 2019, 58, 15872–15879 CrossRef CAS PubMed.
- C. J. Bouchey and W. B. Tolman, Inorg. Chem., 2022, 61, 2662–2668 CrossRef CAS PubMed.
- P. Pirovano, E. R. Farquhar, M. Swart and A. R. McDonald, J. Am. Chem. Soc., 2016, 138, 14362–14370 CrossRef CAS PubMed.
- P. Mondal, P. Pirovano, A. Das, E. R. Farquhar and A. R. McDonald, J. Am. Chem. Soc., 2018, 140, 1834–1841 CrossRef CAS PubMed.
- D. Unjaroen, R. Gericke, M. Lovisari, D. Nelis, P. Mondal, P. Pirovano, B. Twamley, E. R. Farquhar and A. R. Mcdonald, Inorg. Chem., 2019, 58, 16838–16848 CrossRef CAS PubMed.
- P. Mondal, M. Lovisari, B. Twamley and A. R. McDonald, Angew. Chem., Int. Ed., 2020, 59, 13044–13050 CrossRef CAS PubMed.
- C. Panda, L. M. Doyle, R. Gericke and A. R. McDonald, Angew. Chem., Int. Ed., 2021, 60, 26281–26286 CrossRef CAS PubMed.
- J. K. Bower, A. D. Cypcar, B. Henriquez, S. C. E. Stieber and S. Zhang, J. Am. Chem. Soc., 2020, 142, 8514–8521 CrossRef CAS PubMed.
- T. Corona, A. Draksharapu, S. K. Padamati, I. Gamba, V. Martin-Diaconescu, F. Acuna-Parés, W. R. Browne and A. Company, J. Am. Chem. Soc., 2016, 138, 12987–12996 CrossRef CAS PubMed.
- K. J. Fisher, M. L. Feuer, H. M. C. Lant, B. Q. Mercado, R. H. Crabtree and G. W. Brudvig, Chem. Sci., 2020, 11, 1683–1690 RSC.
- Y. M. Kwon, Y. Lee, G. E. Evenson, T. A. Jackson and D. Wang, J. Am. Chem. Soc., 2020, 142, 13435–13441 CrossRef CAS PubMed.
- D. Enders and J. P. Shilvock, Chem. Soc. Rev., 2000, 29, 359–373 RSC.
- T. S. Ratani, S. Bachman, G. C. Fu and J. C. Peters, J. Am. Chem. Soc., 2015, 137, 13902–13907 CrossRef CAS PubMed.
- N. Miwa, C. Tanaka, S. Ishida, G. Hirata, J. Song, T. Torigoe, Y. Kuninobu and T. Nishikata, J. Am. Chem. Soc., 2020, 142, 1692–1697 CrossRef CAS PubMed.
- K. Kim and S. H. Hong, Adv. Synth. Catal., 2017, 359, 2345–2351 CrossRef CAS.
- W. Zhang, F. Wang, S. D. McCann, D. Wang, P. Chen, S. S. Stahl and G. Liu, Science, 2016, 353, 1014–1018 CrossRef CAS PubMed.
- Z. Zhang, X. Zhang and D. A. Nagib, Chem, 2019, 5, 3127–3134 CAS.
- C. Y. Wang, Z. Y. Qin, Y. L. Huang, R. X. Jin, Q. Lan and X. S. Wang, iScience, 2019, 21, 490–498 CrossRef CAS PubMed.
- R. Lu, T. Yang, X. Chen, W. Fan, P. Chen, Z. Lin and G. Liu, J. Am. Chem. Soc., 2021, 143, 14451–14457 CrossRef CAS PubMed.
- P. F. Zhu, Y. X. Si and S. L. Zhang, Org. Biomol. Chem., 2020, 18, 9216–9220 RSC.
- J. Li, Z. Zhang, L. Wu, W. Zhang, P. Chen, Z. Lin and G. Liu, Nature, 2019, 574, 516–521 CrossRef CAS PubMed.
- H. Chen, W. Jin and S. Yu, Org. Lett., 2020, 22, 5910–5914 CrossRef CAS PubMed.
- D. Wang, N. Zhu, P. Chen, Z. Lin and G. Liu, J. Am. Chem. Soc., 2017, 139, 15632–15635 CrossRef CAS PubMed.
- F. D. Lu, D. Liu, L. Zhu, L. Q. Lu, Q. Yang, Q. Q. Zhou, Y. Wei, Y. Lan and W. J. Xiao, J. Am. Chem. Soc., 2019, 141, 6167–6172 CrossRef CAS PubMed.
- J. Lalevée, X. Allonas and J. P. Fouassier, J. Am. Chem. Soc., 2002, 124, 9613–9621 CrossRef PubMed.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- V. M. Krishnan, D. Y. Shopov, C. J. Bouchey, W. D. Bailey, R. Parveen, B. Vlaisavljevich and W. B. Tolman, J. Am. Chem. Soc., 2021, 143, 3295–3299 CrossRef CAS PubMed.
- M. Asaka and H. Fujii, J. Am. Chem. Soc., 2016, 138, 8048–8051 CrossRef CAS PubMed.
- M. J. Zdilla, J. L. Dexheimer and M. M. Abu-Omar, J. Am. Chem. Soc., 2007, 129, 11505–11511 CrossRef CAS PubMed.
- D. Usharani, D. C. Lacy, A. S. Borovik and S. Shaik, J. Am. Chem. Soc., 2013, 135, 17090–17104 CrossRef CAS PubMed.
- S. K. Barman, M. Y. Yang, T. H. Parsell, M. T. Green and A. S. Borovik, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2108648118 CrossRef CAS PubMed.
- J. W. Darcy, S. S. Kolmar and J. M. Mayer, J. Am. Chem. Soc., 2019, 141, 10777–10787 CrossRef CAS PubMed.
- G. Qiu and R. R. Knowles, J. Am. Chem. Soc., 2019, 141, 2721–2730 CrossRef CAS PubMed.
- J. E. Schneider, M. K. Goetz and J. S. Anderson, Chem. Sci., 2021, 12, 4173–4183 RSC.
- Y. L. Chow, S. F. Nelsen and D. H. Rosenblatt, Chem. Rev., 1978, 78, 243–274 CrossRef CAS.
- S. V. Kessar and P. Singh, Chem. Rev., 1997, 97, 721–737 CrossRef CAS PubMed.
- A. J. J. Lennox, S. L. Goes, M. P. Webster, H. F. Koolman, S. W. Djuric and S. S. Stahl, J. Am. Chem. Soc., 2018, 140, 11227–11231 CrossRef CAS PubMed.
- D. H. R. Barton, A. Billion and J. Boivin, Tetrahedron Lett., 1985, 26, 1229–1232 CrossRef CAS.
- D. B. Ushakov, K. Gilmore, D. Kopetzki, D. T. McQuade and P. H. Seeberger, Angew. Chem., Int. Ed., 2014, 53, 557–561 CrossRef CAS PubMed.
- J. M. Tedder, Angew. Chem., Int. Ed. Engl., 1982, 21, 401–410 CrossRef.
- B. P. Roberts, Chem. Soc. Rev., 1999, 28, 25–35 RSC.
- C. Le, Y. Liang, R. W. Evans, X. Li and D. W. C. MacMillan, Nature, 2017, 547, 79–83 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2154012 and 2154013. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc06573h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |