
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRadical ring-opening polymerization of sustainably-derived thionoisochromanone†
Emily A.
Prebihalo‡
 a,
Anna M.
Luke‡
a,
Yernaidu
Reddi
a,
Christopher J.
LaSalle
a,
Vijay M.
Shah
a,
Christopher J.
Cramer
b and
Theresa M.
Reineke
*a
a,
Anna M.
Luke‡
a,
Yernaidu
Reddi
a,
Christopher J.
LaSalle
a,
Vijay M.
Shah
a,
Christopher J.
Cramer
b and
Theresa M.
Reineke
*a
aDepartment of Chemistry, University of Minnesota, 207 Pleasant St. SE, Minneapolis, MN 55455, USA. E-mail: treineke@umn.edu
bUnderwriters Laboratories Inc., 333 Pfingsten Rd., Northbrook, Illinois 60620, USA
First published on 9th May 2023
Abstract
We present the synthesis, characterization and radical ring-opening polymerization (rROP) capabilities of thionoisochromanone (TIC), a fungi-derivable thionolactone. TIC is the first reported six-membered thionolactone to readily homopolymerize under free radical conditions without the presence of a dormant comonomer or repeated initiation. Even more, the resulting polymer is fully degradable under mild, basic conditions. Computations providing molecular-level insights into the mechanistic and energetic details of polymerization identified a unique S,S,O-orthoester intermediate that leads to a sustained chain-end. This sustained chain-end allowed for the synthesis of a block copolymer of TIC and styrene under entirely free radical conditions without explicit radical control methods such as reversible addition–fragmentation chain transfer polymerization (RAFT). We also report the statistical copolymerization of ring-retained TIC and styrene, confirmed by elemental analysis and energy-dispersive X-ray spectroscopy (EDX). Computations into the energetic details of copolymerization indicate kinetic drivers for ring-retaining behavior. This work provides the first example of a sustainable feedstock for rROP and provides the field with the first six-membered monomer susceptible to rROP, expanding the monomer scope to aid our fundamental understanding of thionolactone rROP behavior.
Introduction
Plastics are an integral and necessary part of nearly every industry in modern society. However, the vast majority of these materials are petroleum-derived1 and non-degradable, even by abiotic degradation.2 Problematically, their lifetimes in landfills can be indefinite.2 A major goal of the sustainable polymer community is thus to generate new and useful feedstocks and polymeric materials from renewable resources that can be degraded and/or chemically recycled easily. However, many commodity polymers are sourced from vinyl monomers and produced through free radical polymerization or using methods such as reversible addition–fragmentation chain transfer polymerization (RAFT).3–5 While these methods can impart control, radical methods often produce all-carbon backbones, limiting degradation efforts. Radical ring-opening polymerization (rROP), in which radical chemistry is exploited to promote ring-opening of cyclic monomers, is an especially useful technique for introducing degradable linkages along radical-derived polymer backbones.6,7 When paired with radical polymerization of common olefinic comonomers, rROP allows for incorporation of degradable linkages within otherwise nondegradable carbon–carbon backbones.8–11 rROP copolymerizations have been largely limited to cyclic ketene acetals (CKAs)12–15 and variations of an allylic sulfide.16–20Thionolactones, in particular, remain understudied in the sustainable polymer field despite their susceptibility to rROP.21 Due to their structural resemblance to chain-transfer agents, thionolactones can accept a radical and isomerize via ring-opening, resulting in a thioester moiety in the backbone and propagating carbon radical21 (Fig. 1A). Introducing thioester functionality has been explored via non-radical methods due to the utility of thiol–thioester exchange22,23 and optical properties.24 However, to date, few thionolactone monomers have been studied thoroughly as candidates for rROP and the relationship between their structure and their polymerization efficacy is largely unknown. For example, thionocaprolactone (thCL, Fig. 1B) copolymerizes with vinyl acetate and cyclic ketene acetals but remains undisturbed when subjected to acrylates or acrylamides.25,26 Alternatively, relatively well-studied dibenzo[c,e]oxepane-5-thione (DOT, Fig. 2B) has copolymerized successfully with acrylates,21,27–29 acrylamides,30 N-functional maleimides,31 and styrenics32,33 but remains inert in copolymerizations with vinyl acetate.21,31 The styrene-co-DOT systems published to date are susceptible to degradation under specific conditions, yet traditional degradation methods, such as aminolysis, are unable to degrade p(S-co-DOT).33 Recently published work modified the DOT structure by adding various substituents to the aromatic rings, resulting in predictable copolymerization with styrene to varying molar masses, followed by degradation and successful reworking back to established molar masses.34 However, homopolymerization of thionolactones has proven difficult and typically requires a dormant comonomer/initiator combination or copious and repeated additions of radical initiator AIBN.21,25,31 Most recently, a monomer similar in structure to DOT, 3,3-dimethyl-2,3-dihydro-5H-benzo[e][1,4]dioxepine-5-thione (DBT, Fig. 1B), has reported limited homopolymerization via RAFT conditions (and free-radical conditions, but to significantly lower conversions) and copolymerization with styrene and methacrylates.35
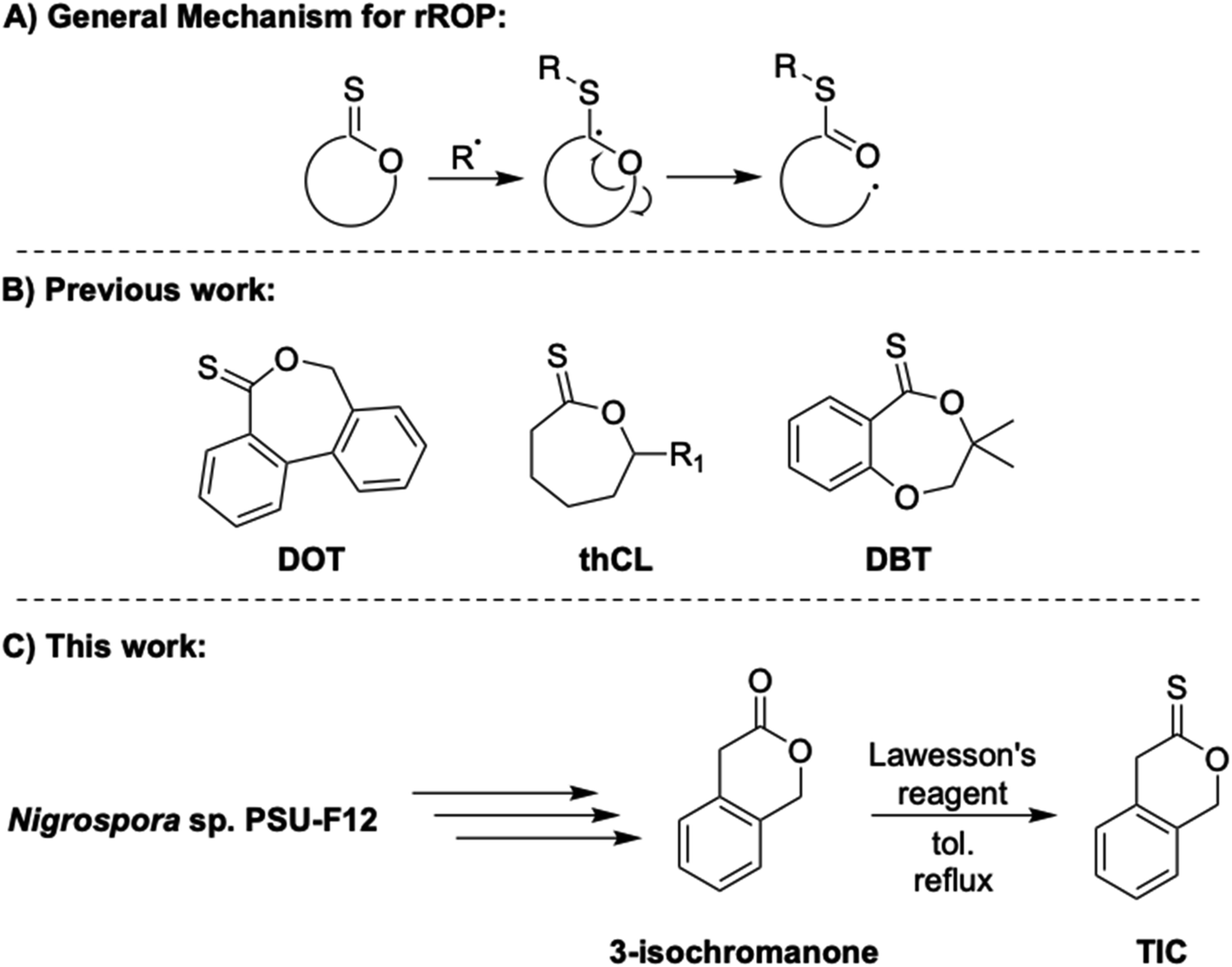 | ||
| Fig. 1 (A) General rROP mechanism via a generic thionolactone, (B) structures of DOT, thCL (R1 = H, C4H9) and DBT, and (C) TIC synthesis. | ||
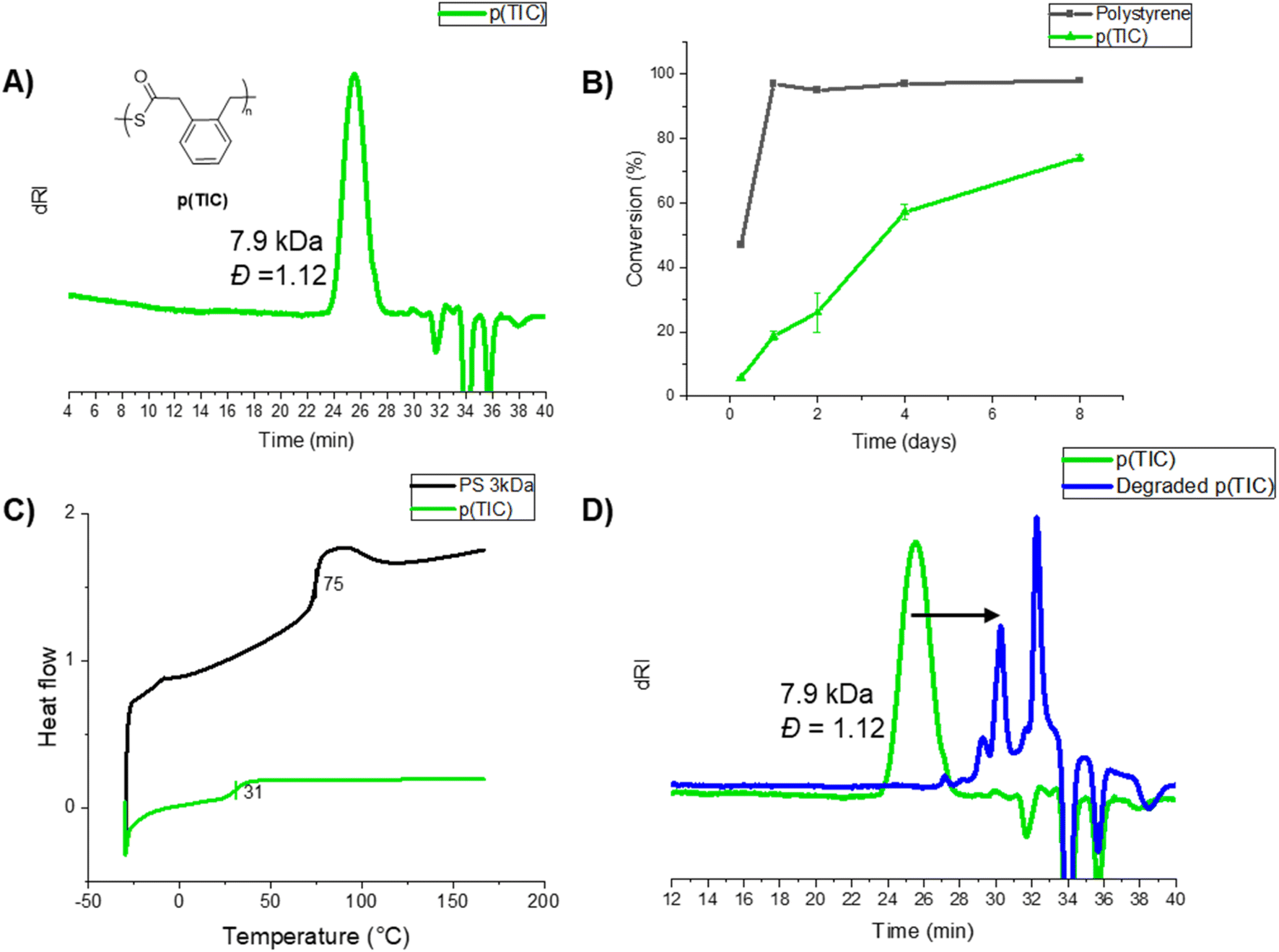 | ||
| Fig. 2 (A) SEC-MALS trace of p(TIC) using AIBN at 70 °C in DMF, (B) monomer conversion vs. time of TIC polymerization (green) and a styrene polymerization control (black), via free-radical conditions under N2 at 70 °C, (C) DSC thermogram of p(TIC) (green) and polystyrene of the same molar mass (black), and (D) SEC-MALS trace of p(TIC) before (green) and after degradation (blue) via sodium thiomethoxide-mediated thiolysis. | ||
Herein, we present the synthesis and characterization of a novel 6-membered thionolactone, thionoisochromanone (TIC, Fig. 1C), as the first six-membered thionolactone monomer to successfully homopolymerize without the need for a comonomer or repeated initiation, thus producing an entirely degradable polythioester. We also provide detailed computational analysis to support mechanistic understanding of radical ring-opening versus ring-retaining pathways. Synthesized from 3-isochromanone, a metabolite derivative from the fungi Nigrospora sp. PSU-F12,36 TIC is a renewably sourced monomer, making it an attractive alternative to the aforementioned thionolactones. Moreover, we present successful block copolymerization of TIC with styrene through entirely free-radical methods, illustrating a sustained chain-end capable of continued propagation past traditional radical lifetimes. Computational findings highlight the unique S,S,O-orthoester intermediate responsible for this interesting behavior. Statistical copolymerization of TIC with styrene yields incorporation of ring-retained TIC into polystyrene, an unexpected result due to thionolactones' general lack of ring-retaining tendencies.21 We present holistic computational findings that explore the energetics of polymerization and identify this ring-retaining polymerization to be a kinetic phenomenon. Overall, this work provides a fundamental look at six-membered thionolactone rROP behavior and introduces an exciting sustained chain-end under free radical conditions. These computational results add to the fundamental understanding of thionolactone rROP behavior.
At the time of publication of this manuscript as a preprint, this work was the first example of a six-membered thionolactone monomer for rROP. Since then, two further examples have been published of rROP of a thionolactone lactide derivative.37,38
Results and discussion
TIC was synthesized from 3-isochromanone through reflux with Lawesson's reagent39 and was produced as a bright orange powder in moderate yields (44%) and high purity following column chromatography. Yield of thionation (44%) is competitive and slightly higher with previous reports of DOT thionation via Lawesson's reagent (14–41%).27,34 Indications of successful transformation to TIC included significant shifts of the methylene peaks in the 1H NMR spectrum from that of 3-isochromanone (from 3.7 to 5.3 ppm and from 4.15 to 5.4 ppm, Fig. S1†). Infrared (IR) spectroscopy further confirmed the synthesis of TIC via a disappearance of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O peak of 3-isochromanone at 1732 cm−1 and a new presence of a CS peak at 1673 cm−1 (Fig. S2†). The purified TIC monomer proved to be stable in air at room temperature for several days.
O peak of 3-isochromanone at 1732 cm−1 and a new presence of a CS peak at 1673 cm−1 (Fig. S2†). The purified TIC monomer proved to be stable in air at room temperature for several days.
Homopolymerization of TIC under free-radical conditions proceeded slowly, reaching 75% conversion after eight days at 70 °C in DMF (Table S2†). Successful polymerization was indicated via1H NMR spectroscopy analysis by significantly shifted and broadened aromatic proton resonances, as well as the growth of broad methylene peaks at ∼4 ppm (Fig. S3†). IR analysis showed a strong thioester carbonyl stretch at 1671 cm−1 and a C–S stretching vibration at 907 cm−1 (Fig. S7†), matching previous reports of thioester carbonyl stretch (1678 cm−1 and 907 cm−1).27 SEC-MALS analysis of various homopolymer samples revealed moderate molecular weights in the range of 2–8 kDa (example SEC trace shown in Fig. 2B).
Several side reactions competed with polymerization under radical conditions. While TIC is air-stable, any adventitious water or oxygen present during radical polymerization conditions induced oxidation of TIC back to 3-isochromanone, as well as isomerization to its thioester analog (∼30%) when subjected to high temperatures (>100 °C). These side reactions were partially mitigated by performing polymerizations under rigorously air-free conditions (i.e., freeze/pump/thaw techniques via a Schlenk line), resulting in a maximum 18% of the monomer isomerizing or oxidizing during polymerization.
To further understand the homopolymerization system, a range of radical initiators and solvents were studied. TIC showed very little initiation with initiator V-70 at 30 °C, indicating that sufficient heat is needed to overcome the thermodynamic barriers of polymerization. Initiator benzoyl peroxide at 70 °C did initiate polymerization, although only 36% conversion to polymer was observed after four days. Increasing temperature while maintaining constant initiator AIBN concentration only increased undesirable isomerization to the corresponding thiolactone, not polymerization. Interestingly, decreasing initiator relative to monomer showed a decrease in Mn, speculated to be due to small reaction volumes (Table 1A). Blank experiments run without any radical initiator present showed auto-initiation of TIC when heated to 70 °C. Due to this interesting result, control experiments were run and quenched with acetic anhydride to investigate if the blank experiment was moving through a different, ionic mechanism than the typical homopolymerizations. After quenching with acetic anhydride, the blank experiment interestingly showed a complete shift of the acetic anhydride by NMR, indicating auto-initiation occurring via a nucleophilic pathway. However, when tested with the typical homopolymerization with AIBN, no reactivity of acetic anhydride was observed, indicating a radical pathway (Fig. S6†). Further work into this interesting nucleophilic pathway is underway.
| A. Polymerization data for TIC | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Initiator | TIC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Initiator :Initiator |
Time | Temp. (°C) | TIC conv.a | M n (kDa) | Đ |
| a Determined by 1H NMR spectroscopy. The X denotes not measured due to low conversion. b Determined by SEC using light scattering (MALS) detection with THF or DMF eluent (for SEC traces, see Fig. S11–S29, eluent specified for each trace). *Sample was bimodal with a first peak of large light scattering size but small concentration, Mn value shown is for second peak of significant concentration. c Sample was bimodal but with no clearly defined second peak, Mn value shown for first peak. | |||||||
| 1 | AIBN | 20:1 |
4 d | 70 | 55% | 7.90 | 1.12 |
| 2 | AIBN | 40:1 |
4 d | 70 | 70% | 4.14 | 1.28 |
| 3 | AIBN | 80:1 |
4 d | 70 | 56% | 3.40 | 1.26 |
| 4 | AIBN | 160:1 |
4 d | 70 | 25% | 2.18 | 1.18 |
| 5 | AIBN | 20:1 |
2 d | 100 | 36% | X | X |
| 6 | BPO | 20:1 |
4 d | 70 | 36% | X | X |
| 7 | V-70 | 20:1 |
11 d | 30 | 5% | X | X |
| C. Polymerization data for TIC-co-styrene | ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Styrene:TIC |
Concentration in DMF | Time | Temp. (°C) | Styrene conv.a | TIC conv.a | M n*b (kDa) | Đ |
| 1 | 1:1 |
N/A | 4 d | 70 | 40% | 3% | 5.01* | 1.34 |
| 2 | 5:1 |
N/A | 4 d | 70 | 23% | 55% | 64.3c | 1.80 |
| 3 | 19:1 |
N/A | 4 d | 70 | 19% | 79% | 2.89* | 1.31 |
| 4 | 50:1 |
N/A | 4 d | 70 | 12% | 57% | 3.29* | 1.45 |
| 5 | 100:1 |
N/A | 4 d | 70 | 28% | 75% | 8.91* | 1.75 |
Kinetic analysis of the TIC homopolymerization (Fig. 2B, green trace) showed a steady increase of monomer conversion throughout the entirety of polymerization. While TIC polymerizes more slowly than compared to styrene homopolymerization under the same conditions (Fig. 2B, black trace), TIC's steady conversion past four days indicates a relatively stable radical, atypical in free radical polymerization systems. This unusual result was probed via computational analysis, the results of which are discussed in the following sections. The thermal properties of p(TIC) were compared to polystyrene as a commercial vinyl analog via differential scanning calorimetry (DSC). p(TIC) yielded a Tg of 30 °C at a Mn of ∼3 kDa, a significantly lower value than the Tg of ∼3 kDa polystyrene (75 °C) (Fig. 2C).
p(TIC) was subjected to sodium thiomethoxide treatment overnight for degradation analysis. Fig. 2D compares the homopolymer SEC trace (green) and that of the recovered organic product from the degradation studies (blue). The shift in molecular weight indicates that the polymer is fully degraded via thiolysis into small molecule analogs. The identity of the degraded product was investigated via GC-MS, revealing that the major degradation product shared the same molecular weight as one repeat unit of the polymer, indicating complete degradation followed by cyclization to the isomerized thiolactone (proposed route shown in Fig. S32†).
Homopolymerization theory
Density functional theory (DFT) was used to develop insight at the atomic level for the mechanistic details of homopolymerization of TIC (relevant analogous details for homopolymerization of DOT were also computed and can be found in the ESI†). Importantly, ΔG of isodesmic ring-opening for TIC was −4.1 kcal mol−1, very similar to conventional rROP thionolactone DOT (−4.2 kcal mol−1), shown to ring-open in previous rROP reports (see Fig. S33†).21,27,28,30–33 Next, full reaction coordinates with Gibbs free energy profiles were investigated (Fig. 3). To begin, the isobutyronitrile radical generated from AIBN adds to the monomer to create a reactive radical intermediate Int1TIC, which is reached after passing through transition-state (TS) structure TS1TIC. The free energy of activation for TIC is 21.4 kcal mol−1 relative to separated reactants. Int1TIC can follow two possible paths, either C–O bond scission to open the ring or instead radical addition to another monomer. The former path, through corresponding TS structure TS2TIC, involves activation free energies of 36.1 kcal mol−1 while the latter path, through corresponding TS structure TS5TIC, has activation free energies of 35.3 kcal mol−1. The near degeneracy in the energies of the competing TS structures suggests that Int1TIC should partition about equally along both pathways. However, the Int5TIC radical derived from immediate addition to another monomer is itself considerably less prone to C–O bond scission, with the relevant TS6TIC free energies being computed to be roughly 50 kcal mol−1. Thus, any populations of Int5TIC are expected to revert to Int1TIC and subsequently react productively along the path involving ring-opened Int2TIC.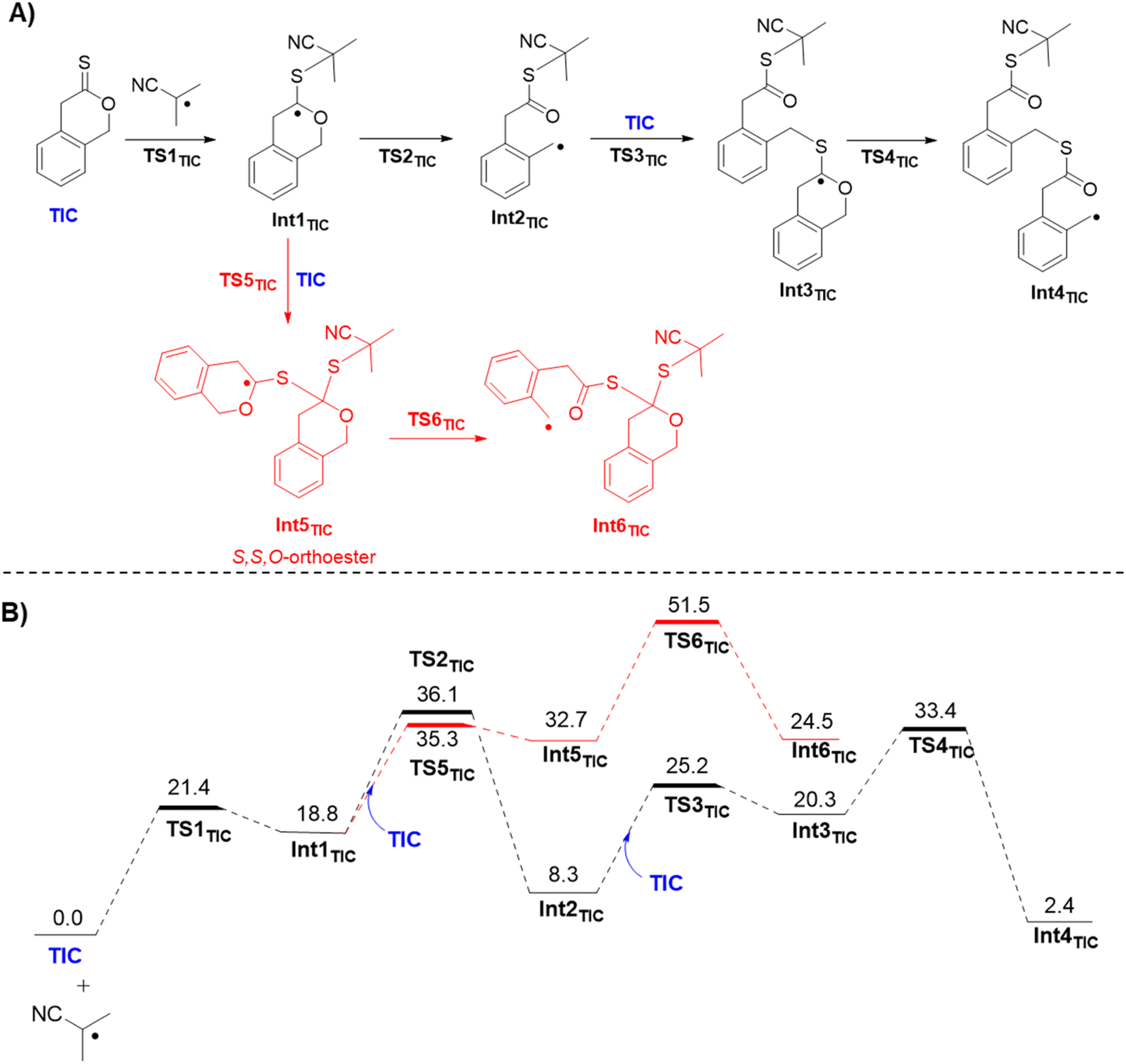 | ||
| Fig. 3 (A) Key mechanistic steps for the homopolymerization of TIC and (B) Gibbs free energy (kcal mol−1; 343.15 K; SMD(DMF)/PBE0-D3/6-311++G(d,p)//M06-2X/6-31+G(d,p) level of theory) profile on the potential energy surface for homopolymerization of TIC. | ||
That reactivity involves addition of Int2TIC to another monomer to generate Int3TIC with computed free energy of activation through TS3TIC of 25.2 kcal mol−1. Subsequently, Int3TIC can undergo ring-opening C–O bond cleavage to generate Int4TIC, which is the polymeric analog to Int2TIC. Computed activation free energy for ring-opening viaTS4TIC is 33.4 kcal mol−1. Continued homopolymerization would follow the energetics associated with repeatedly moving from Int2TIC to Int4TIC, which involves an activation free energy of 25.1 kcal mol−1 and an exergonicity of 5.9 kcal mol−1. We note that electronic structure theory calculations tend to underestimate polymerization exergonicities because they are not well suited to capturing the full entropy of the growing polymer chain.40,41 Examination of the structure of Int5TIC shows a S,S,O-orthoester structure, and will be referred to as thus in further work. This orthoester is proposed to stabilize the propagating radical past normal radical lifetimes through the equilibrium of Int1 and Int5. While this does not lead to full control of the system, the sustained chain-end could explain the slow and continuous monomer conversion observed during kinetic experiments.
This sustained chain-end is reminiscent of thioketone-mediated polymerization (TKMP), in which a thioketone acts as a reversible capping agent similar to a chain-transfer agent in controlled radical polymerization, but keeps chains living by creating a stabilized radical.42 TKMP leads to living conditions, as evidenced by linear increase of polymer molecular weight with conversion and modulation of molecular weight by changing initiator ratio.43,44 While the S,S,O-orthoester similarly provides a stabilized radical to the chain end of p(TIC), notably TIC reacts with itself to homopolymerize, behavior not seen in TKMP. Additionally, Table 1A highlights changes in the initiator:monomer ratio that do not result in a predictable change in polymer molecular weight, indicating this sustained chain-end does not give fully living conditions.
Intrigued by the utility of this S,S,O-orthoester intermediate, a chain extension of p(TIC) with styrene was demonstrated to yield a TIC-b-styrene block polymer (Fig. 4A). A significant and monomodal increase in molecular weight from the p(TIC) block to that of the copolymer indicated that chain extension was successful (Fig. 4B). Diffusion-ordered spectroscopy (DOSY) also showed identical diffusion coefficients for all polymer peaks, confirming the block nature of the polymer. Completed under free-radical conditions, these results suggest the S,S,O-orthoester leaves a sustained chain end radical that can be further polymerized by styrene. While this does not give fully living conditions, it does allow for block extension without the use of a chain transfer agent or other method of control. To the best of our knowledge there have been no other reports of block copolymer synthesis under free-radical conditions.
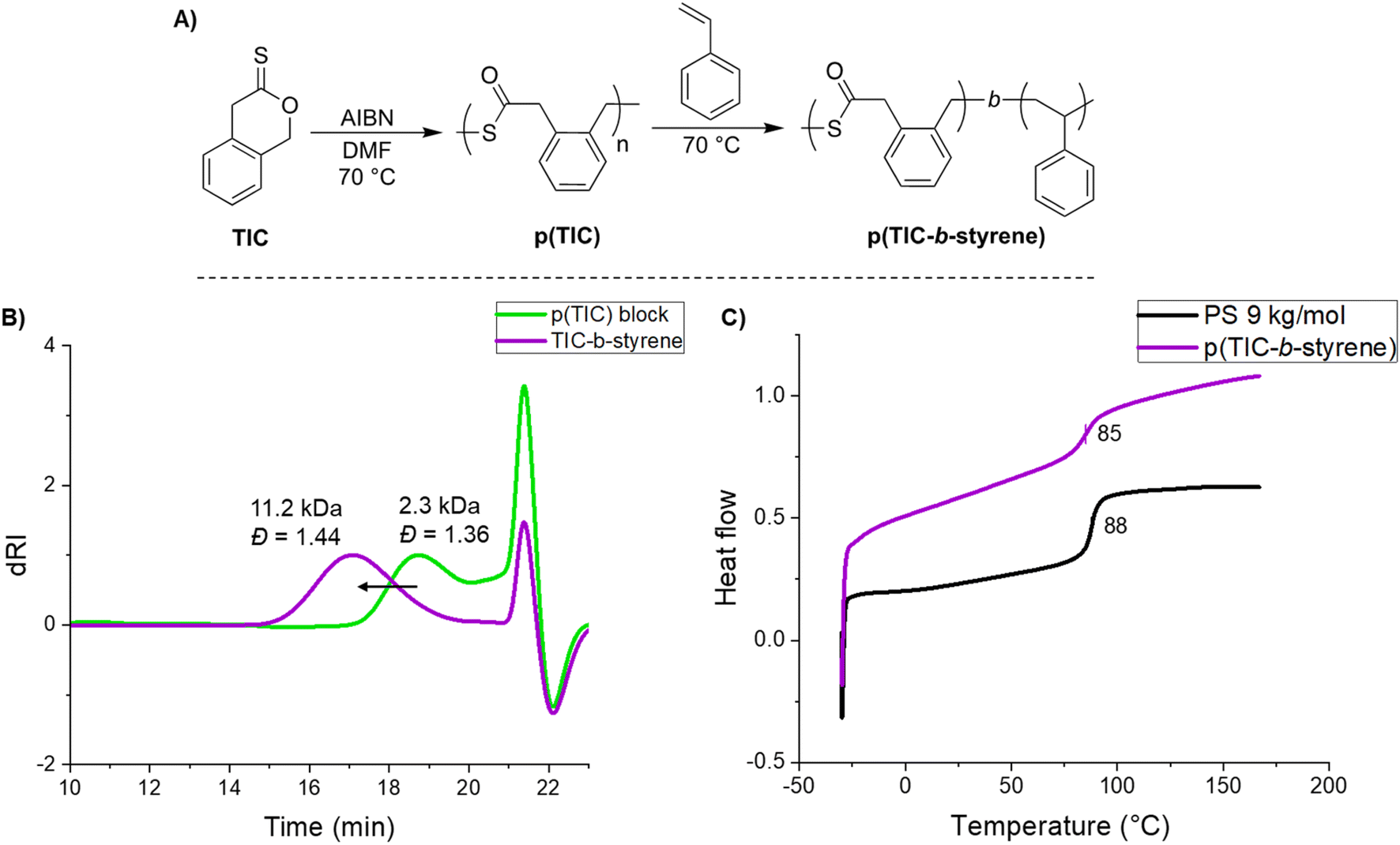 | ||
| Fig. 4 (A) Synthesis of p(TIC-b-styrene), (B) SEC-MALS traces of p(TIC) and the block copolymer resulting from chain-extension, and (C) DSC thermograms of p(TIC-b-styrene) (purple trace) and polystyrene of similar molar mass (black trace). | ||
The thermal properties of this block copolymer were investigated via DSC. p(TIC-b-styrene) at Mn ∼11 kDa yielded a Tg of 85 °C, compared to 88 °C of polystyrene at comparable Mn (Fig. 4C). We expected the similarity in Tg values based on the composition of the block copolymer of a ∼2 kDa p(TIC) block with a ∼9 kDa polystyrene block. Given that the majority of the copolymer is polystyrene, we expect the p(TIC) block to have little effect on the Tg.
Statistical copolymerization of TIC with vinyl monomers was also investigated. TIC showed no polymerization with acrylates, methacrylates, acrylamides, or acrylonitrile, but readily copolymerized with styrene in bulk at 70 °C, reaching 80% TIC conversion and 20% styrene conversion after four days. Various styrene:TIC ratios were investigated, showing a general trend of increasing TIC conversion as styrene:TIC ratio increased (Table 1B). In ratios greater than 1:1, TIC shows higher conversion than styrene, a trend also reported for copolymerization of DOT with vinyl monomers in work by Smith et al.3 The ability of TIC to copolymerize with styrene precludes the S,S,O-orthoester from yielding living conditions through TKMP, as for those conditions the thioketone must be inert to free radical polymerization with the monomer.
Kinetic analysis of styrene/TIC copolymerizations demonstrates that monomer to polymer conversion steadily increases with time, similar to that observed in the TIC homopolymerization (Table S3†). This result points to a similar chain end S,S,O-orthoester intermediate forming. Interestingly, no degradation of the copolymer was observed with addition of acid, base, amines, or methoxides. However, elemental analysis confirmed incorporation of TIC into the copolymer with 19:1 feed ratio of styrene:TIC via presence of 1.34 wt% sulfur in the purified polymer, denoting a 21:1 styrene:TIC incorporation ratio (see ESI†). EDX analysis further corroborated the presence of sulfur in the p(TIC-co-styrene) (Table S1†). Interestingly, IR analysis of purified copolymer samples did not contain a carbonyl stretch, which is representative of the ring-opened thionolactone (Fig. S8†). Together, these results pointed to a lack of thioester moieties within the copolymer, and suggested that the thionolactone was incorporated via a ring-retaining mechanism rather than a ring-opening mechanism (Fig. 5A).
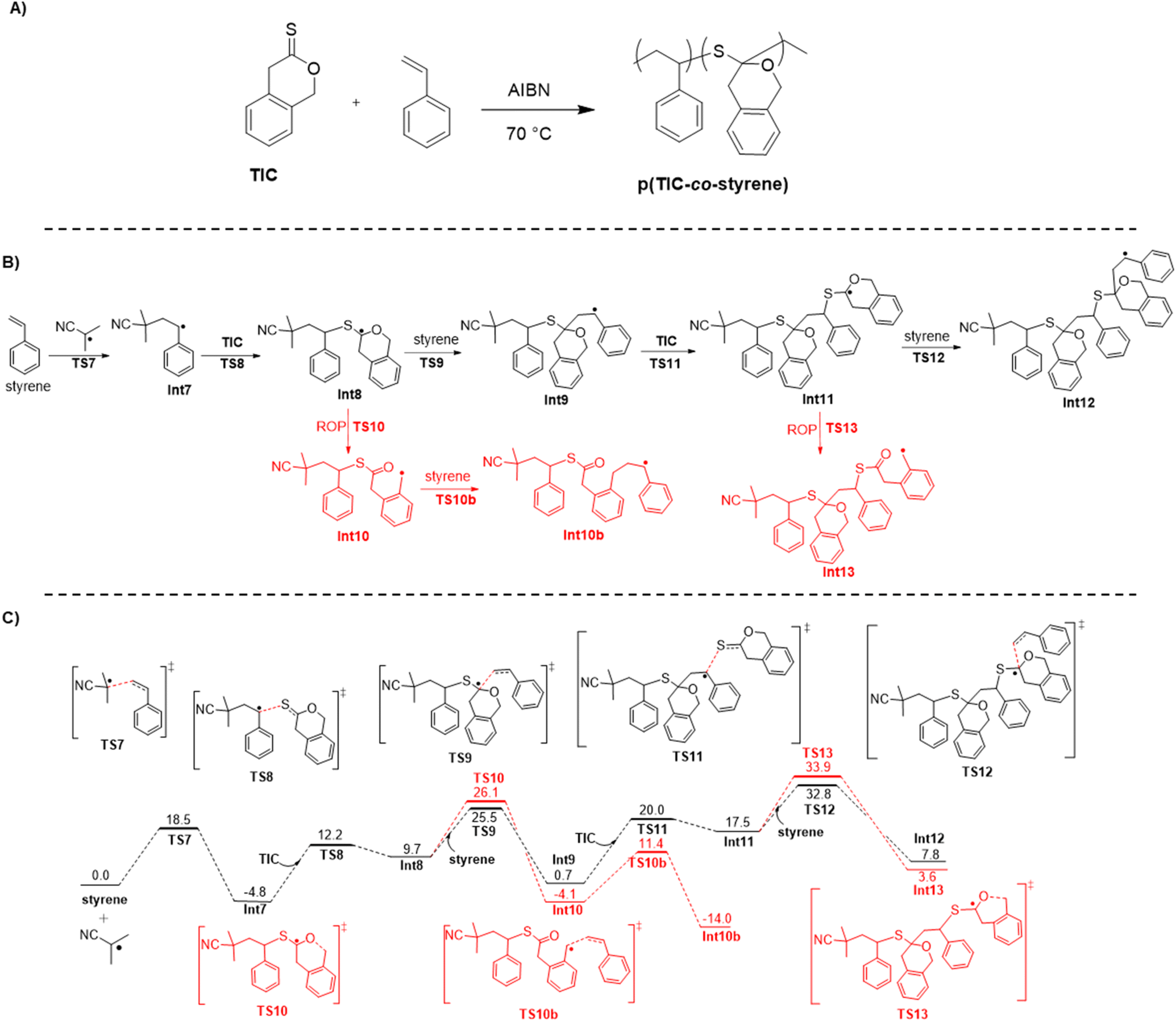 | ||
| Fig. 5 (A) Synthesis of p(TIC-co-styrene), (B) key mechanistic steps for the copolymerization of TIC and styrene, and (C) Gibbs free energy (kcal mol−1; 343.15 K; SMD(DMF)/PBE0-D3/6-311++G(d,p)//M06-2X/6-31+G(d,p) level of theory) profile on the potential energy surface for copolymerization of TIC and styrene. | ||
Ring-retaining polymerization has been frequently observed in rROP of cyclic ketene acetals (CKAs)45 as well as several thionolactones, including thionocaprolactone,26 and lactide-derived monomers.37,38 As demonstrated in work from the Satoh group, lactide-derived thionolactones show partial ring-retaining monomer units when copolymerized with styrene, with Mn values reaching 28 kDa.37 Similar work from the Destarac group shows copolymerization of thionolactide with various comonomers, including styrene, that results in ring-retained thioacetal units. In these polymerizations, the presence of thionolactide slowed the rate of polymerization and Mn values were around 5–7 kDa.38 Particularly in CKA systems it has been shown that ring-retaining behavior can be disfavored by decreasing the concentration of reaction solution.10 Therefore, TIC-co-styrene polymerizations (normally performed in bulk), were diluted via addition of DMF. The resulting copolymers yielded predictably lower monomer conversions (Table S3†), but also showed no difference in molar mass upon subjection to degradation conditions, indicating ring-opening was still not achieved. Future work to further investigate conditions to induce ring opening is underway.
The thermal properties of the TIC and styrene copolymers were again analyzed and compared to polystyrene at similar molecular weights as a commercial standard. p(TIC-co-styrene) at Mn ∼3 kDa revealed a Tg of 62 °C, lower than the ∼3 kDa polystyrene Tg of 75 °C (Fig. S30 and S31†). The significant deviation from the Tg of polystyrene standards in conjunction with the elemental analysis and EDX data supports that TIC has been incorporated into the copolymer. The DSC results also indicate that the presence of TIC lowers the Tg of the polystyrene system, despite the aromatic nature of the monomer.
DFT was used to survey key mechanistic details in the copolymerization of styrene and TIC (Fig. 5B and C). Of key importance is the partitioning of ring-retained vs. ring-opening polymerization. Although ring-opening is preferred from a thermodynamic standpoint, the addition of ring-closed intermediate to styrene is predicted to be kinetically favorable (recall that addition of the ring-closed radical to another TIC monomer is possible, but has no productive forward path, cf. Fig. 3). We note that, again, our electronic structure calculations fail to fully account for the entropy of the growing copolymer chain, as theory indicates polymerization to be endergonic, even though it is predicted to be exothermic if we consider only the enthalpy (Fig. S35†). Since some of that missing entropy may also be present in the propagation transition-state structures, it is possible that we are underestimating the differential free energy associated with propagation vs. ring-opening. Additionally, due to the difference of unimolecular (beta-scission) and bimolecular (radical addition) reactions, thermodynamics alone cannot fully represent the reaction and explain the difference between ring-opening and ring-retaining behavior. Irrespective of the quantitative details, theory leads us to conclude that the absence of thioester functionality in the copolymer is a kinetic phenomenon. As thionolactones are known to primarily show ring-opening behavior with little ring-retaining side product, these computational results add insight into the propensity of thionolactones to ring-open during copolymerizations. This is further information to add to the rROP field to aid in the goal of using thionolactones to introduce degradable linkages into commodity polymers.
Conclusion
Herein, we present the synthesis, characterization and rROP capabilities of TIC, a six-membered, sustainably-derivable thionolactone. TIC is the first thionolactone reported to readily homopolymerize without the need for copious comonomer nor repeated initiation. p(TIC) is fully degradable via sodium thiomethoxide and GC-MS analysis of the subsequent degradation products indicated a return of the small molecule monomer analog. Moreover, TIC introduces a sustained chain-end in free radical polymerization conditions by forming an S,S,O-orthoester intermediate during propagation. This development leads to the formation of block copolymers under free radical conditions by extending the lifetime of the radical and acting as a sustained chain end. DFT calculations demonstrated the kinetics and thermodynamics of the stationary points involved in the homopolymerization of TIC and showed that the ring-opening pathway is preferred over the ring retaining polymerization. TIC-co-styrene statistical copolymers, however, were found to contain ring-retained TIC; theory predicts that retention derives from a kinetic preference for addition to styrene compared to ring opening, as computed free energetics revealed that ring retention has a lower activation free energy than ring-opening during copolymerization of styrene and TIC. The introduction of a six-membered thionolactone further broadens the scope of reported rROP monomers. Experiment and theory together offer additional insight into the ring opening behavior for this emerging monomer class. Importantly, this is the first thionolactone monomer for rROP derivable from a sustainable source, introducing the possibility of divesting from petroleum-derived feedstocks in future implementations. Furthermore, the sustained chain-end proposed here shows exciting potential for introducing levels of control into free-radical polymerizations without exogenous chain transfer agents or additives.Data availability
Computational data is provided in the ESI.†Author contributions
A. M. L. is now an assistant professor at University of St. Thomas in St. Paul, Minnesota.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the NSF Center for Sustainable Polymers CHE-1901635. We acknowledge John Beumer for designing the TOC graphic. Parts of this work were carried out in the Characterization Facility, University of Minnesota, which receives partial support from the NSF through the MRSEC (Award Number DMR-2011401) and the NNCI (Award Number ECCS-2025124) programs. We thank the Minnesota Supercomputing Institute (MSI) for providing key computational resources. Thanks to Nicholas C. A. Seaton, SEM, FIB & LM Specialist, Characterization Facility, College of Science and Engineering, University of Minnesota for collection of EDX data.References
- R. Geyer, J. R. Jambeck and K. L. Law, Production, Use, and Fate of All Plastics Ever Made, Sci. Adv., 2017, 3, 25–29, DOI:10.1126/sciadv.1700782.
- D. K. Schneiderman and M. A. Hillmyer, 50th Anniversary Perspective: There Is a Great Future in Sustainable Polymers, Macromolecules, 2017, 50(10), 3733–3749, DOI:10.1021/acs.macromol.7b00293.
- D. Wan, K. Satoh, M. Kamigaito and Y. Okamoto, Xanthate-Mediated Radical Polymerization of N-Vinylpyrrolidone in Fluoroalcohols for Simultaneous Control of Molecular Weight and Tacticity, Macromolecules, 2005, 38, 10397–10405, DOI:10.1021/ma0515230.
- T. Öztürk, B. Savaş, E. Meyvacı, A. Kılıçlıoğlu and B. Hazer, Synthesis and Characterization of the Block Copolymers Using the Novel Bifunctional Initiator by RAFT and FRP Technics: Evaluation of the Primary Polymerization Parameters, J. Polym. Res., 2020, 27(3), 1–7, DOI:10.1007/s10965-020-2043-7.
- C. Barner-kowollik, M. Buback, B. Charleux, M. L. Coote, M. Drache, T. Fukuda, A. Goto, B. Klumperman, A. B. Lowe, J. B. Mcleary, G. Moad, M. J. Monteiro, R. D. Sanderson, M. P. Tonge and P. Vana, Mechanism and Kinetics of Dithiobenzoate-Mediated RAFT Polymerization. I. The Current Situation, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5809–5831, DOI:10.1002/pola.
- S. R. Mothe, J. S. J. Tan, L. R. Chennamaneni, F. Aidil, Y. Su, H. C. Kang, F. C. H. Lim and P. Thoniyot, A Systematic Investigation of the Ring Size Effects on the Free Radical Ring-Opening Polymerization (RROP) of Cyclic Ketene Acetal (CKA) Using Both Experimental and Theoretical Approach, J. Polym. Sci., 2020, 58, 1728–1738, DOI:10.1002/pol.20200210.
- J. Folini, W. Murad, F. Mehner, W. Meier and J. Gaitzsch, Updating Radical Ring-Opening Polymerisation of Cyclic Ketene Acetals from Synthesis to Degradation, Eur. Polym. J., 2020, 134, 109851, DOI:10.1016/j.eurpolymj.2020.109851.
- W. J. Bailey, Z. Ni and S.-R. Wu, Synthesis of Poly-r-Caprolactone via a Free Radical Mechanism. Free Radical Ring-Opening Polymerization of 2-Methylene-1,3-Dioxepane, J. Polym. Sci., Polym. Chem. Ed., 1982, 20, 3021–3030, DOI:10.1002/pol.1982.170201101.
- S. Agarwal, Microstructural Characterisation and Properties Evaluation of Poly (Methyl Methacrylate-Co-Ester)S, Polym. J., 2007, 39, 163–174, DOI:10.1295/polymj.PJ2006137.
- J. Y. Yuan and C. Y. Pan, “Living” Free Radical Ring-Opening Copolymerization of 4,7-Dimethyl-2-Methylene-1,3-Dioxepane and Conventional Vinyl Monomers, Eur. Polym. J., 2002, 38(10), 2069–2076, DOI:10.1016/S0014-3057(02)00085-X.
- L. F. Sun, R. X. Zhuo and Z. L. Liu, Synthesis and Enzymatic Degradation of 2-Methylene-1,3-Dioxepane and Methyl Acrylate Copolymers, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 2898–2904, DOI:10.1002/pola.10868.
- A. Tardy, J. Nicolas, D. Gigmes, C. Lefay and Y. Guillaneuf, Radical Ring-Opening Polymerization: Scope, Limitations, and Application to (Bio)Degradable Materials, Chem. Rev., 2017, 117(3), 1319–1406, DOI:10.1021/acs.chemrev.6b00319.
- T. Endo and F. Sanda, Radical Ring-Opening Polymerization, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 265–276, DOI:10.1016/B978-0-444-53349-4.00120-5.
- S. Jin and K. E. Gonsalves, A Study of the Mechanism of the Free-Radical Ring-Opening Polymerization of 2-Methylene-1,3-Dioxepane, Macromolecules, 1997, 30(10), 3104–3106, DOI:10.1021/ma961590h.
- S. Agarwal, Chemistry, Chances and Limitations of the Radical Ring-Opening Polymerization of Cyclic Ketene Acetals for the Synthesis of Degradable Polyesters, Polym. Chem., 2010, 1(7), 953–964, 10.1039/c0py00040j.
- R. A. Evans and E. Rizzardo, Free-Radical Ring-Opening Polymerization of Cyclic Allylic Sulfides, Macromolecules, 1996, 29(22), 6983–6989, DOI:10.1021/ma960573p.
- A. Evans and E. Rizzardo, Free-Radical Ring-Opening Polymerization of Cyclic Allylic Sulfides. 2. Effect of Substituents on Seven- and Eight-Membered Ring Low Shrink Monomers, Macromolecules, 2000, 33(18), 6722–6731, DOI:10.1021/ma9917646.
- M. Phelan, F. Aldabbagh, P. Zetterlund and B. Yamada, Mechanism and Kinetics of the Free Radical Ring-Opening Polymerization of Eight-Membered Cyclic Allylic Disulfide Monomers, Macromolecules, 2005, 38(6), 2143–2147, DOI:10.1021/ma047894i.
- S. Yamada and Y. Goto, Reduction of Oxygen Inhibition in Photopolymerization of Cyclic Allylic Sulfide Monomer, J. Photopolym. Sci. Technol., 2010, 23, 109–114, DOI:10.2494/photopolymer.23.109.
- J. M. J. Paulusse, R. J. Amir, R. A. Evans and C. J. Hawker, Free Radical Polymers with Tunable and Selective Bio- and Chemical Degradability, J. Am. Chem. Soc., 2009, 131(28), 9805–9812, DOI:10.1021/ja903245p.
- R. A. Smith, G. Fu, O. McAteer, M. Xu and W. R. Gutekunst, Radical Approach to Thioester-Containing Polymers, J. Am. Chem. Soc., 2019, 141(4), 1446–1451, DOI:10.1021/jacs.8b12154.
- C. Wang, S. Mavila, B. T. Worrell, W. Xi, T. M. Goldman and C. N. Bowman, Productive Exchange of Thiols and Thioesters to Form Dynamic Polythioester-Based Polymers, ACS Macro Lett., 2018, 7, 1312–1316, DOI:10.1021/acsmacrolett.8b00611.
- C. Wang, T. M. Goldman, B. T. Worrell, M. D. Alim, C. N. Bowman and M. K. Mcbride, Recyclable and Repolymerizable Thio-X Photopolymers, Mater. Horiz., 2018, 5, 1042–1046, 10.1039/c8mh00724a.
- L. Wang, G. Gu, T. Yue, W. Ren and X. Lu, Semiaromatic Poly(Thioester) from the Copolymerization of Phthalic Thioanhydride and Epoxide: Synthesis, Structure, and Properties, Macromolecules, 2019, 52, 2439–2445, DOI:10.1021/acs.macromol.9b00073.
- C. M. Plummer, P. Du, D. J. Wilson, C. Lefay, D. Gigmes and Y. Guillaneuf, Mechanistic Investigation of ε - Thiono-Caprolactone Radical Polymerization: An Interesting Tool to Insert Weak Bonds into Poly(Vinyl Esters), ACS Appl. Polym. Mater., 2021, 3, 3264–3271, DOI:10.1021/acsapm.1c00569.
- O. Ivanchenko, U. Authesserre, G. Coste, S. Mazières, M. Destarac and S. Harrisson, ε-Thionocaprolactone: An Accessible Monomer for Preparation of Degradable Poly(Vinyl Esters) by Radical Ring-Opening Polymerization, Polym. Chem., 2021, 12, 1931–1938, 10.1039/d1py00080b.
- N. M. Bingham and P. J. Roth, Degradable Vinyl Copolymers through Thiocarbonyl Addition-Ring-Opening (TARO) Polymerization, Chem. Commun., 2019, 55(1), 55–58, 10.1039/c8cc08287a.
- H. Elliss, F. Dawson, N. M. Bingham and P. J. Roth, Fully Degradable Polymer Networks from Conventional Radical Polymerization of Vinyl Monomers Enabled by Thionolactone Addition, Macromolecules, 2022, 1–15 Search PubMed.
- N. Gil, C. Thomas, R. Mhanna, J. Mauriello, R. Maury, B. Leuschel, J. Malval, J. Clément, D. Gigmes, C. Lefay, O. Soppera and Y. Guillaneuf, Thionolactone as Resin Additive to Prepare (Bio)Degradable 3D Objects via VAT Photopolymerization, Angew. Chem., Int. Ed., 2022, 61, 1–10, DOI:10.1002/anie.202117700.
- N. M. Bingham, Q. un Nisa, S. H. L. Chua, L. Fontugne, M. P. Spick and P. J. Roth, Thioester-Functional Polyacrylamides: Rapid Selective Backbone Degradation Triggers Solubility Switch Based on Aqueous Lower Critical Solution Temperature/Upper Critical Solution Temperature, ACS Appl. Polym. Mater., 2020, 2(8), 3440–3449, DOI:10.1021/acsapm.0c00503.
- M. P. Spick, N. M. Bingham, Y. Li, J. De Jesus, C. Costa, M. J. Bailey and P. J. Roth, Fully Degradable Thioester-Functional Homo- And Alternating Copolymers Prepared through Thiocarbonyl Addition-Ring-Opening RAFT Radical Polymerization, Macromolecules, 2020, 53(2), 539–547, DOI:10.1021/acs.macromol.9b02497.
- P. Galanopoulo, N. Gil, D. Gigmes, C. Lefay, Y. Guillaneuf, M. Lages, J. Nicolas, M. Lansalot and F. D'Agosto, One-Step Synthesis of Degradable Vinylic Polymer-Based Latexes via Aqueous Radical Emulsion Polymerization, Angew. Chem., Int. Ed., 2022, 1–11, DOI:10.1002/anie.202117498.
- N. Gil, B. Caron, D. Siri, J. Roche, S. Hadiouch, D. Khedaioui, S. Ranque, C. Cassagne, D. Montarnal, D. Gigmes, C. Lefay and Y. Guillaneuf, Degradable Polystyrene via the Cleavable Comonomer Approach, Macromolecules, 2022, 55, 6680–6694, DOI:10.1021/acs.macromol.2c00651.
- G. R. Kiel, D. J. Lundberg, E. Prince, K. E. L. Husted, A. M. Johnson, V. Lensch, S. Li, P. Shieh and J. A. Johnson, Cleavable Comonomers for Chemically Recyclable Polystyrene: A General Approach to Vinyl Polymer Circularity, J. Am. Chem. Soc., 2022, 144, 12979–12988, DOI:10.1021/jacs.2c05374.
- M. F. I. Rix, S. J. Higgs, E. M. Dodd, S. J. Coles, N. M. Bingham and J. Roth, Insertion of Degradable Thioester Linkages into Styrene and Methacrylate Polymers, ChemRxiv, 2022, 1–5, DOI:10.26434/chemrxiv-2022-cdt52.
- V. Rukachaisirikul, N. Khamthong, Y. Sukpondma, S. Phongpaichit, N. Hutadilok-Towatana, P. Graidist, J. Sakayaroj and K. Kirtikara, Cyclohexene, Diketopiperazine, Lactone and Phenol Derivatives from the Sea Fan-Derived Fungi Nigrospora Sp. PSU-F11 and PSU-F12, Arch. Pharmacal Res., 2010, 33(3), 375–380, DOI:10.1007/s12272-010-0305-3.
- R. Kamiki, T. Kubo and K. Satoh, Addition–Fragmentation Ring-Opening Polymerization of Bio-Based Thiocarbonyl l-Lactide for Dual Degradable Vinyl Copolymers, Macromol. Rapid Commun., 2022, 1–6, DOI:10.1002/marc.202200537.
- O. Ivanchenko, S. Mazières, S. Harrisson and M. Destarac, Lactide-Derived Monomers for Radical Thiocarbonyl Addition-Ring-Opening Copolymerisation, Polym. Chem., 2022, 13, 5525–5529, 10.1039/d2py00893a.
- S. Scheibye, J. Kristensen and S. O. Lawesson, Studies on Organophosphorus Compounds-XXVII1 1 Part XXVI See ref. 7. Synthesis of Thiono-, Thiolo- and Dithiolactones, Tetrahedron, 1979, 35(11), 1339–1343, DOI:10.1016/0040-4020(79)85027-9.
- Y. Reddi and C. J. Cramer, Mechanism and Design Principles for Controlling Stereoselectivity in the Copolymerization of CO2/Cyclohexene Oxide by Indium(III) Phosphasalen Catalysts, ACS Catal., 2021, 11(24), 15244–15251, DOI:10.1021/acscatal.1c04619.
- H. Martinez, P. Miró, P. Charbonneau, M. A. Hillmyer and C. J. Cramer, Selectivity in Ring-Opening Metathesis Polymerization of Z-Cyclooctenes Catalyzed by a Second-Generation Grubbs Catalyst, ACS Catal., 2012, 2(12), 2547–2556, DOI:10.1021/cs300549u.
- A. A. Toy, H. Chaffey-Millar, T. P. Davis, M. H. Stenzel, E. I. Izgorodina, M. L. Coote and C. Barner-Kowollik, Thioketone Spin Traps as Mediating Agents for Free Radical Polymerization Processes, Chem. Commun., 2006, 8, 835–837, 10.1039/b515561d.
- T. Junkers, G. Delaittre, R. Chapman, F. Günzler, E. Chernikova and C. Barner-Kowollik, Thioketone-Mediated Polymerization with Dithiobenzoates: Proof for the Existence of Stable Radical Intermediates in RAFT Polymerization, Macromol. Rapid Commun., 2012, 33(11), 984–990, DOI:10.1002/marc.201200128.
- T. Junkers, M. H. Stenzel, T. P. Davis and C. Barner-Kowollik, Thioketone-Mediated Polymerization of Butyl Acrylate: Controlling Free-Radical Polymerization via a Dormant Radical Species, Macromol. Rapid Commun., 2007, 28(6), 746–753, DOI:10.1002/marc.200600885.
- W. J. Bailey, S. Wu and Z. Ni, Synthesis and Free Radical Ring-Opening Polymerization of 2-Methylene-4-Phenyl-1,3-Dioxolane, Makromol. Chem., 1982, 183, 1913–1920, DOI:10.1002/macp.1982.021830811.
Footnotes |
| † Electronic supplementary information (ESI) available: Supporting figures, experimental details, and NMR, IR, DSC and SEC spectra (PDF). See DOI: https://doi.org/10.1039/d2sc06040j |
| ‡ These authors contributed equally to the research. |
| This journal is © The Royal Society of Chemistry 2023 |