
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRadiometallation and photo-triggered release of ready-to-inject radiopharmaceuticals from the solid phase†
Dariusz
Śmiłowicz
a,
Shawn
Eisenberg
a,
Shin Hye
Ahn
 a,
Angus J.
Koller
a,
Philip P.
Lampkin
b and
Eszter
Boros
*a
a,
Angus J.
Koller
a,
Philip P.
Lampkin
b and
Eszter
Boros
*a
aDepartment of Chemistry, Stony Brook University, 100 Nicolls Road, Stony Brook, NY 11794, USA. E-mail: eszter.boros@stonybrook.edu
bDepartment of Chemistry, University of Wisconsin-Madison, Madison, WI 53705, USA
First published on 17th April 2023
Abstract
The efficient, large-scale synthesis of radiometallated radiopharmaceuticals represents an emerging clinical need which, to date, is inherently limited by time consuming, sequential procedures to conduct isotope separation, radiochemical labeling and purification prior to formulation for injection into the patient. In this work, we demonstrate that a solid-phase based, concerted separation and radiosynthesis strategy followed by photochemical release of radiotracer in biocompatible solvents can be employed to prepare ready-to-inject, clinical grade radiopharmaceuticals. Optimization of resin base, resin loading, and radiochemical labeling capacity are demonstrated with 67Ga and 64Cu radioisotopes using a short model peptide sequence and further validated using two peptide-based radiopharmaceuticals with clinical relevance, targeting the gastrin-releasing peptide and the prostate specific membrane antigen. We also demonstrate that the solid-phase approach enables separation of non-radioactive carrier ions Zn2+ and Ni2+ present at 105-fold excess over 67Ga and 64Cu by taking advantage of the superior Ga3+ and Cu2+ binding affinity of the solid-phase appended, chelator-functionalized peptide. Finally, a proof of concept radiolabeling and subsequent preclinical PET-CT study with the clinically employed positron emitter 68Ga successfully exemplifies that Solid Phase Radiometallation Photorelease (SPRP) allows the streamlined preparation of radiometallated radiopharmaceuticals by concerted, selective radiometal ion capture, radiolabeling and photorelease.
Introduction
Radiopharmaceuticals enable non-invasive patient diagnosis and development of personalized treatment strategies for a vast number of clinical patients globally.1–3 Consequently, there is growing demand to develop scalable, versatile, rapid, high-yielding chemical syntheses of radiopharmaceuticals.4,5 Radionuclides for nuclear medicine applications require purification following their nuclear synthesis to separate them from the target material, before they can be employed in radiosynthesis and purification prior to dose preparation.6,7 Second, due to the short isotope half-life of many such radioisotopes of interest, radiochemical synthesis needs to be fast, simple and modular, while producing the target radiopharmaceutical in high chemical yield, purity and molar activity.8,9 To date, this is achieved by a time-consuming, multi-step process that results in decay of a significant fraction of the radionuclide prior to its use for the patient.10,11As a consequence, radiosynthesis strategies that could condense multiple steps into a simple, timesaving one-pot procedure are desirable. We argued that isotope capture, separation, and tracer synthesis should feasibly occur at once, followed by efficient release of the target radiopharmaceutical in a ready-to-inject form.12 This is inherently difficult to implement, especially for the radiolabeling with metal-ions and metalloids, which are usually separated under highly acidic conditions (2–12 M HCl)13,14 from their parent nuclide using ion-exchange chromatography but subsequently require much milder, aqueous radiosynthesis conditions for stable chelate formation.15–17 Specifically, bifunctional chelators, peptides and biomolecules utilized as disease-specific targeting vectors typically require a pH range of 4–8 to form stable complexes.18–22
To enable the concerted separation and labeling with radiometals, we proposed the immobilization of a radiopharmaceutical synthesis precursor comprised of a chelator linked peptide appended to a water-compatible solid-support with a photo-labile amino acid. We drew inspiration from solid-phase synthesis approaches of metal-based drugs and the clinical, solid-phase based radiosynthesis 131I-MIBG,23,24 and recently reported photochemical radiosynthesis strategies. Photochemistry is an emerging radiosynthesis tool that produces no significant in-solution impurities or waste materials while remaining compatible with high-throughput and automation procedures prevalent in the routine production of radiopharmaceuticals.25,26 Indeed, photochemical radiosynthesis has previously enabled bond-formation strategies that produce radiofluorinated arenes using photoredox catalysis,27 photoreactive substrate mediated substitution with radiofluoroalkenes28 and photochemical protein ligation of radiometal chelates (Fig. 1A).29
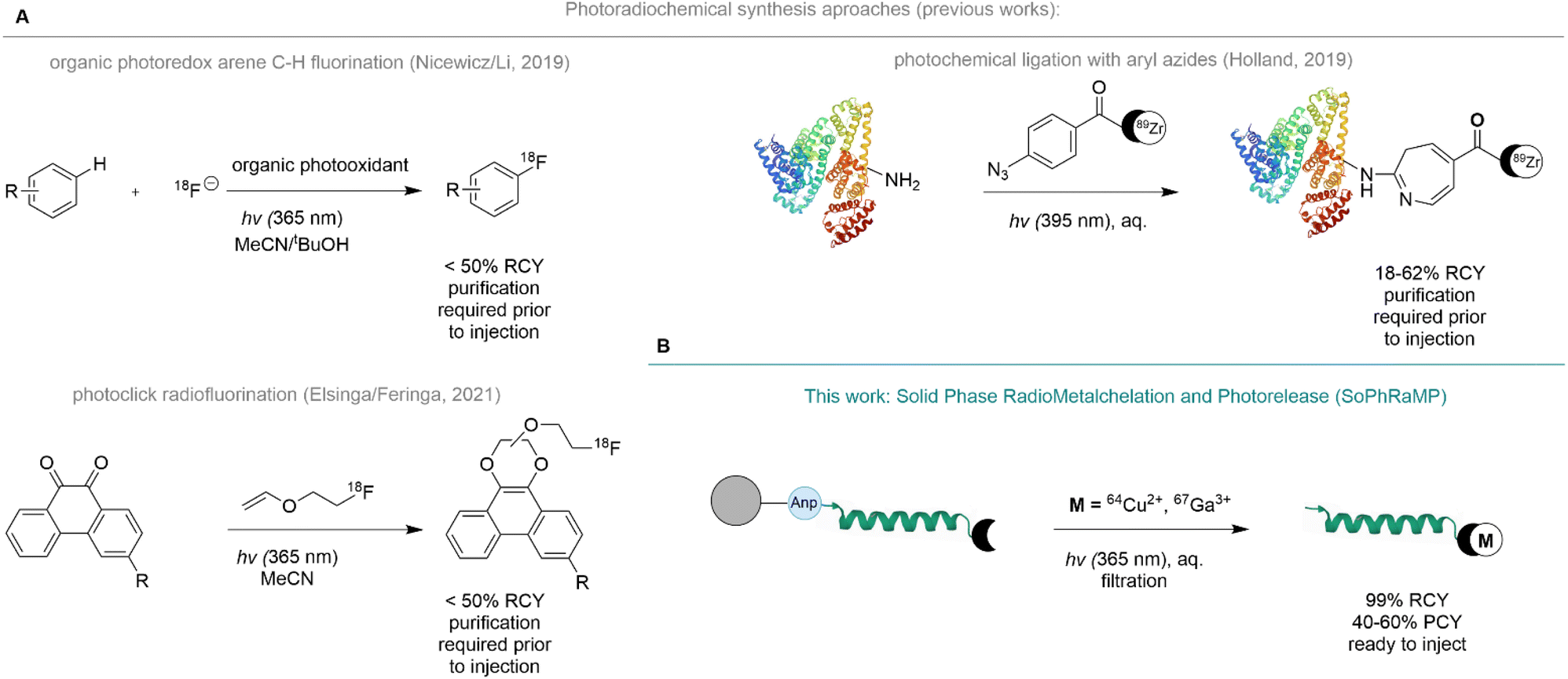 | ||
| Fig. 1 Photo-triggered radiolabeling strategies. (A) Prior work relies on multi-step preparation of radiopharmaceuticals and employs photochemistry as means for selective bond formation. (B) Our approach utilizes photochemical bond cleavage as means to release the ready-made radiopharmaceutical into solution directly amenable to in vivo injection. | ||
Here, we report the development, optimization, and validation of Solid Phase Radiometallation and Photorelease (SPRP), a modular radiosynthesis approach of radiometal-based radiopharmaceuticals. The already broadly applied, photochemically triggered release of peptides from solid support using 3-amino-3-(2-nitrophenyl)propionic acid (Anp)30–32 has been extensively validated for applications ranging from on-bead analysis of peptide sequences using MALDI to the selective capture of Cr6+ ions in drinking water.33 Other accounts report successful implementation of the photochemical cleavage of linkers and core peptides for concerted synthesis applications.34–36 We argued that the traceless nature of the photochemical amide bond cleavage of the Anp amino acid was especially well suited to the selective release of a radiolabeled construct into a biocompatible liquid phase such as phosphate buffered saline (PBS) with 0–20% EtOH additive (Fig. 1B).
To this end, we designed a short, chelator-linked model tri-peptide to optimize resin attachment, radiolabeling volume, activity scope and photocleavage with radioactive Positron Emission Tomography (PET) or Single Photon Emission Computed Tomography (SPECT) radionuclides 67Ga, 68Ga and 64Cu. Subsequently, we synthesized two chelator-linked peptides with defined, clinically relevant peptide sequences using a pre- and post-synthetic resin linkage strategy. We demonstrated that efficient radiochemical labeling of both constructs was feasible under previously established conditions. Additionally, we show that SPRP can efficiently exploit the differences in pH-dependent speciation of metal ion chelation to selectively capture 67Ga and 64Cu isotopes in presence of a 5-orders of magnitude excess of stable Zn2+ and Ni2+ isotopes. As a proof-of-concept, we (1) demonstrate that SPRP efficiently produces a 68Ga-labeled radiopharmaceutical with successful subsequent validation in a preclinical animal model and (2) performs favorably in comparison with in-solution radiolabeling strategies using unprocessed, large 68Ge/68Ga generator eluates.
Results and discussion
Initial validation and optimization
To probe, validate and optimize the SPRP concept, we hypothesized that a short model peptide sequence appended to various, water compatible resin materials could serve as an ideal surrogate for future proof-of-concept radiopharmaceuticals. Initial screening criteria for this phase included: feasibility of synthetic approach and resin attachment, radiochemical purity of the labeled, photoreleased product, as well as the influence of reaction volume, reaction time and distance to the photon source on radiochemical and photocleavage yield.We prepared the model peptide NOTA-Aca-Q-W (10). The peptide included a chelator 1,4,7-triazacyclononane-triacetate (NOTA) linked via a short aminocaproic (Aca) linker to two amino acids for enhanced solubilization (Gln) and visualization (Trp) of the corresponding product following Anp-mediated photochemical release from the solid support. To enable selective, orthogonal deprotection and post-synthetic loading onto various resin materials, we pursued a two-resin synthesis approach: 3-amino-3-(2-nitrophenyl)propionic acid (Anp) as a photosensitive moiety was synthesized on 2-chlorotrityl resin (CTC) using standard solid-phase peptide synthesis (SPPS) strategies. We linked using an aminocaproic acid linker with the orthogonally bis-protected NOTA chelator to afford protected NOTA(tBu)2-Aca-Q-W-ANP-H following mild, acid mediated resin bond cleavage. The terminal carboxylic acid of this construct was activated with N-hydroxysuccinimide (NHS) in solution, followed by removal of orthogonal protecting groups to produce NHS-Anp-10. Following reverse-phase chromatographic purification, NHS-Anp-10 was loaded onto three different water-compatible amine functionalized resins: tentagel, agarose-based and anion-exchange resin (Fig. 2B). Successful resin loading of Anp-10 was confirmed by photocleavage at 365 nm followed by spectroscopic quantitation and mass spectrometric confirmation of the desired NOTA-Aca-Q-W (10) product (see ESI†).
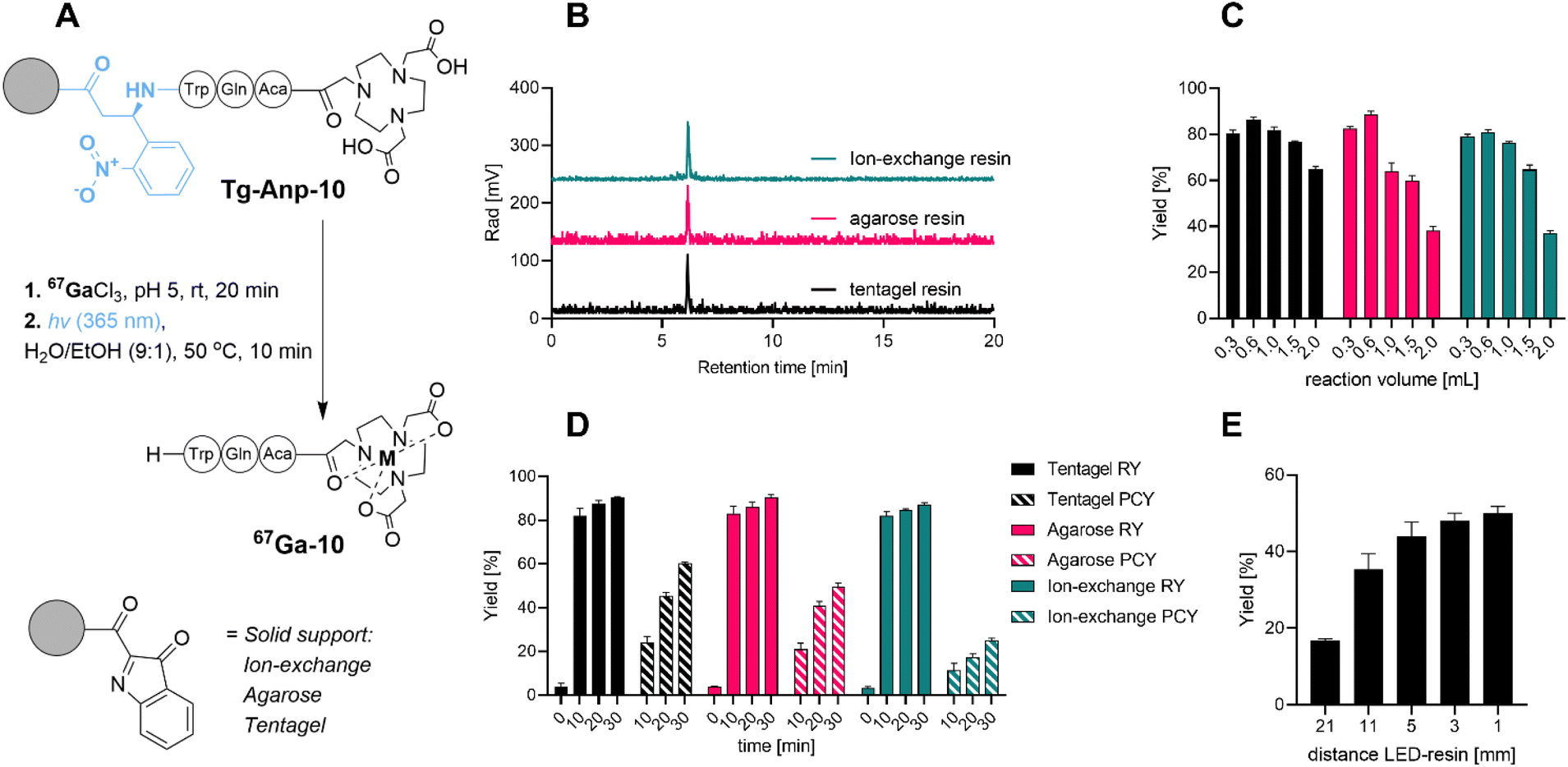 | ||
| Fig. 2 (A) Reaction scheme of prototype solid phase model construct designed for 67/68Ga isotopes, 67Ga(NOTA)-Q-W-NH2 model peptide using 3 different resin material approaches (anion-exchange, agarose-based and tentagel). (B) Analytical radio HPLC chromatogram of crude filtrate following photocleavage showing >99% purity of the desired product with all resin materials tested. (C) Radiochemical and photocleavage in dependence of reaction volume and reaction times for different resin materials (D) indicate that tentagel resin shows most robust radiolabeling and photocleavage performance (10 nmol peptide), while all resins exhibit negligible non-specific binding (time 0). (E) Photocleavage yield in dependence of LED photon source to reaction vessel distance for 10 min photocleavage. RY: radiolabeling yield. PCY: photochemical cleavage yield. Yields reported as an average of n = 3 reactions. | ||
With these compounds in hand, we conducted a first validation of the SPRP strategy. As a surrogate for the clinical PET isotope 68Ga (t1/2 = 1.1 h), we utilized the longer-lived SPECT isotope 67Ga (t1/2 = 79 h) to probe and optimize reaction parameters. Batch radiolabeling followed by photocleavage (Fig. 2A) was conducted with 50 μCi of 67Ga at room temperature using typical radiolabeling conditions at pH 5 (10 mM sodium acetate). To determine radiolabeling yields, we monitored retention of radioactivity on the resin versus filtered solution at different time points (Fig. 2D). High radiolabeling yields (RY, defined herein as activity retained on the resin following filtration for SPRP experiments, or isolated radiolabeled product obtained by direct, in-solution radiolabeling) were observed for all solid support materials within 10 min. Non-functionalized resin exhibited negligible non-specific binding of activity below 5% (ESI,†Fig. 2D, time point t = 0 min). Further extension of the solid-phase radiolabeling time resulted in only marginally increased radiolabeling yield, giving >90% RY within 30 min for all solid supports.
We next probed the selective photocleavage of the radiolabeled peptide 67Ga-10 from solid support. Following removal of the radiolabeling solution and buffer, the resin was suspended in 300 μL 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 EtOH/water and photocleavage was initiated using a custom 3D printed photoreactor with 3 light-emitting diode (LED) 365 nm photon sources, previously validated for in solution photochemistry and outfitted with a syringe bed (Fig. S1†).37 All photocleavage reactions were conducted using a shaker bed to ascertain uniform mixing and irradiation of the suspended solid support. This additional reaction step was quantified separately for its yield, described as photocleavage yield (PCY). PCY was determined by activity eluted from the resin following photocleavage, divided by initially resin-retained (chelated) radioactivity. 10 min photocleavage gave moderate photochemical yield (PCY) of approximately 20%, with photochemical yields increasing following longer irradiation times of 20 to 30 minutes, albeit results varied significantly in the dependence of the solid support: agarose and anion exchange resins performed moderately, while tentagel resin providing most efficient photorelease (60% after 30 minutes). Separation of solid support via filtration produced the desired product 67Ga-NOTA-Aca-Q-W (67Ga-10) with >99% radiochemical purity (Fig. 2D). We subsequently also probed distance effects to the LED light source and found that photocleavage yields after 10 minutes could be dramatically increased to 50% if a distance of 5 mm or less was used between reaction vessel and LED (for further detail see ESI†).
:9 EtOH/water and photocleavage was initiated using a custom 3D printed photoreactor with 3 light-emitting diode (LED) 365 nm photon sources, previously validated for in solution photochemistry and outfitted with a syringe bed (Fig. S1†).37 All photocleavage reactions were conducted using a shaker bed to ascertain uniform mixing and irradiation of the suspended solid support. This additional reaction step was quantified separately for its yield, described as photocleavage yield (PCY). PCY was determined by activity eluted from the resin following photocleavage, divided by initially resin-retained (chelated) radioactivity. 10 min photocleavage gave moderate photochemical yield (PCY) of approximately 20%, with photochemical yields increasing following longer irradiation times of 20 to 30 minutes, albeit results varied significantly in the dependence of the solid support: agarose and anion exchange resins performed moderately, while tentagel resin providing most efficient photorelease (60% after 30 minutes). Separation of solid support via filtration produced the desired product 67Ga-NOTA-Aca-Q-W (67Ga-10) with >99% radiochemical purity (Fig. 2D). We subsequently also probed distance effects to the LED light source and found that photocleavage yields after 10 minutes could be dramatically increased to 50% if a distance of 5 mm or less was used between reaction vessel and LED (for further detail see ESI†).
We also probed other parameters with possible influence on the reaction time and yield. For instance, resin-based chemistry typically requires swelling of the resin prior to conducting chemical reactions and extensive washing steps prior to subsequent chemical reactions. However, when we compared performance of dry resin, we found that pre-conditioning by swelling and post-labeling step washing protocols had no significant influence on the observed radiochemical and photochemical cleavage yields (see ESI Fig. S39†). This is advantageous as it further reduces the time required to conduct radiolabeling with the SPRP approach. We concluded that the sample peptide affixed to tentagel resin performed consistently best among all tested resin materials with robust radiolabeling and subsequent photochemical cleavage.
SPRP for clinical tracer synthesis
We next sought to confirm the feasibility of SPRP by conducting the concerted radiosynthesis of established and clinically translated pharmaceuticals. To this end, we constructed two peptide sequences: bombesin (BBN), targeting gastrin-releasing peptide receptor overexpressed in prostate, glioma and gastric cancers, as well as a modified version of a peptide targeting the prostate-specific membrane antigen (PSMA). Both peptides have been used for the diagnostic imaging of disease in a clinical setting using gallium and copper isotopes,38,39 and therefore represented ideal constructs to further probe the feasibility of SPRP.The synthesis of tentagel appended BBN-NOTA and PSMA-NOTA was conducted using two distinct strategies (Fig. 3): direct, sequential resin loading (A) as well as a post-synthetic, terminal resin loading strategy (B). While the direct resin loading strategy was straightforward and produced the product in high purity following photocleavage from tentagel, the post-synthetic resin loading strategy (which had already been successfully applied for compound 10, vide supra) remains the strategy of choice for lower yielding solid phase syntheses that require post-synthetic purification using chromatography, as well as chemically sensitive sequences such as proteins which are not compatible with comparatively harsh, acidic deprotection conditions typical for SPPS. Ultimately, both synthetic approaches were successful in providing the Anp-linked precursors 19 and 31 on tentagel resin in >99% chemical purity.
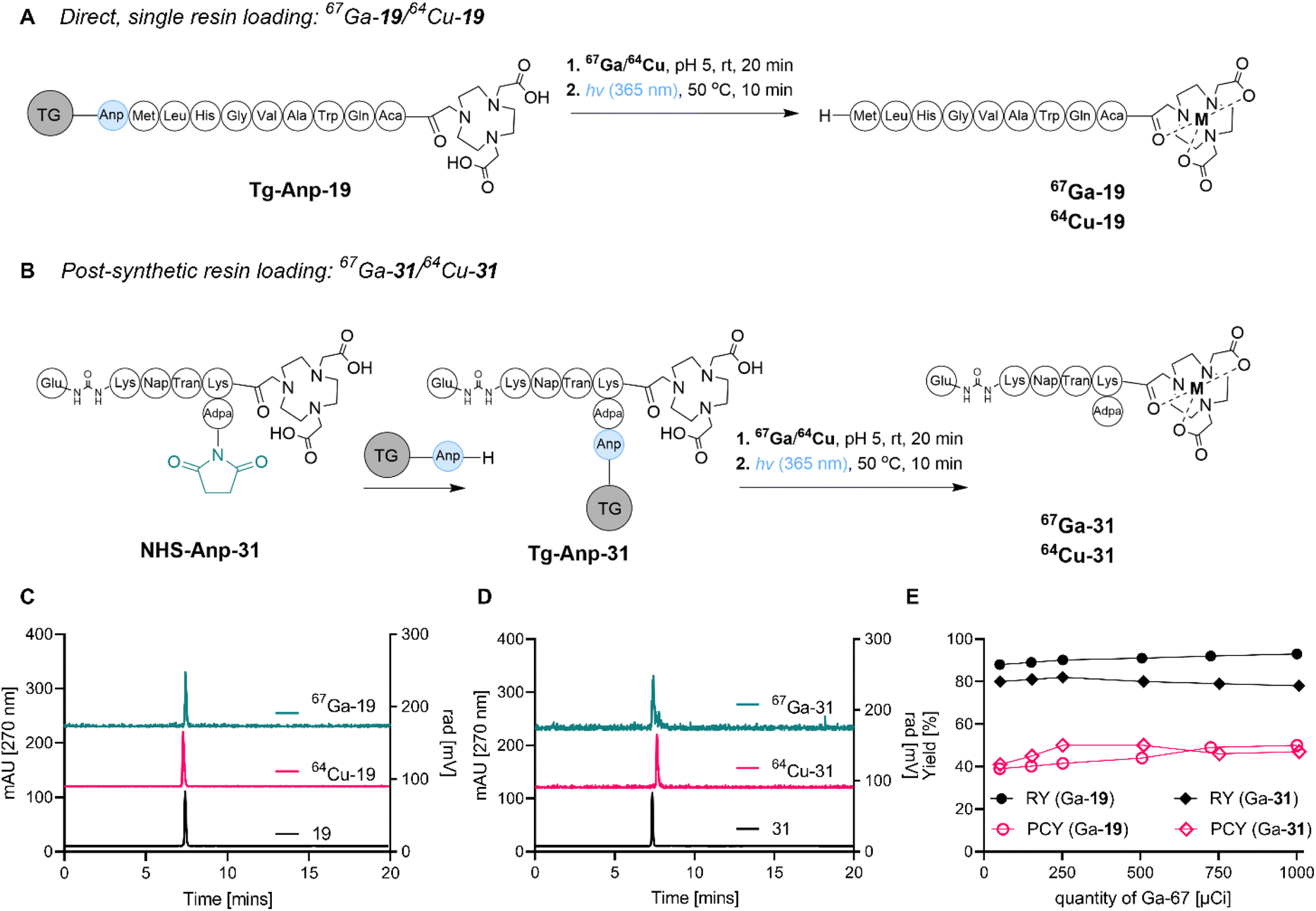 | ||
| Fig. 3 (A) Direct, single-resin loading strategy for 64Cu/67Ga-19, a peptide targeting the gastrin-releasing peptide receptor. (B) Post-synthetic resin loading strategy for 64Cu/67Ga-31, a peptide targeting the prostate-specific membrane antigen. (C) Analytical radio-HPLC chromatogram of crude filtrates following photocleavage showing >99% purity of the desired product 64Cu/67Ga-19. (D) Analytical radio HPLC chromatogram of crude filtrates following photocleavage showing >99% purity of the desired product 64Cu/67Ga-31. (E) Activity-dependent radiochemical and photocleavage yields for 67Ga-31 and 67Ga-19 demonstrate robust scalability. RY: radiolabeling yield. PCY: photochemical cleavage yield. | ||
We subsequently tested radiochemical labeling efficiency, radiochemical purity and photocleavage efficiency with two radionuclides, 67Ga and 64Cu. The direct solid-phase radiolabeling to produce 67Ga-31 resulted in high radiolabeling yield (>85%) at pH 5.0 and incubation at room temperature, without the need for any organic solvent additives. Similarly, high radiolabeling yield (>88%) was obtained for radiolabeling with 64Cu using the same conditions, with radiolabeling yields remaining >80% even after only 20 minutes reaction time. In accordance with optimized photocleavage conditions from 67Ga-10, we determined that while 60 min photoirradiation produced a photocleavage yield of 60% for 67Ga-31 and 51% for 64Cu-31, the 10 min protocol resulted in 40% for 67Ga-31 and 41% for 64Cu-31 (Fig. 3). Overall, 64Cu-19 and 67Ga-19 were synthesized in 15% over-all yield and >99% radiochemical purity. Yield and purity were scalable, achieving molar activities of 0.1 mCi nmol−1 with both isotopes (Fig. 3E).
Concerted ion separation and radiolabeling
As stated within the introduction section, we posited that the SPRP method is especially well suited for concerted ion separation/trace impurity removal and radiolabeling. To probe this hypothesis, we investigated the tolerance of radiochemical labeling yields to the presence of excess non-radioactive metal ion, which could arise from residual parent target material or decayed, stable isotope in solution (Fig. 4A). Presence of excess 64Ni with 64Cu (irradiation target and product) and excess 68Zn with 68Ga (non-radioactive decay product and parent isotope) can affect radiochemical labeling yields in the production of high molar activity radiopharmaceuticals; in fact, the build-up of excess 68Zn following extended usage of the 68Ge/68Ga generator is one of the main contributors of the generator systems' limited lifetime in clinical radiopharmacies.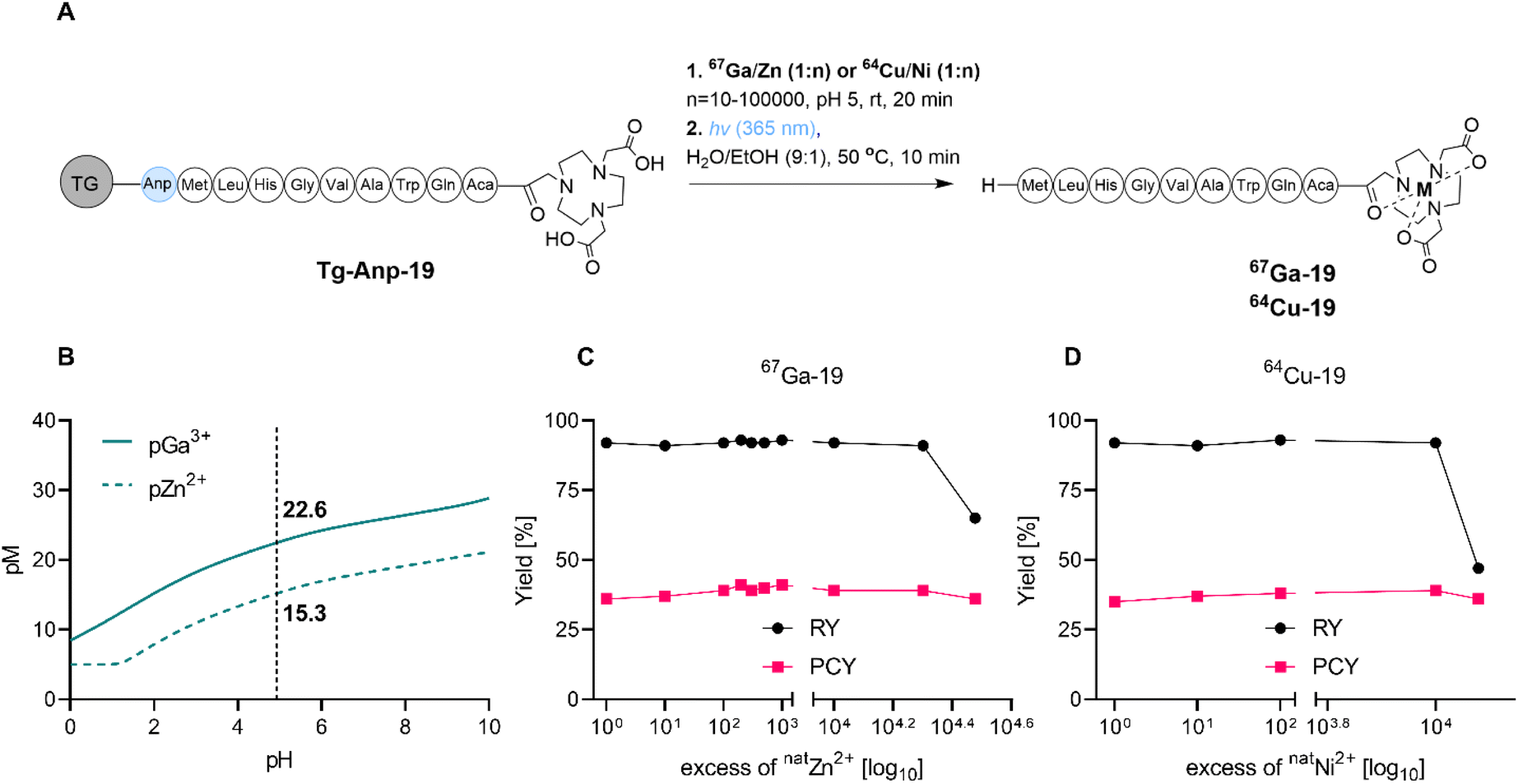 | ||
| Fig. 4 (A) Schematic description of concerted ion separation and radiolabeling procedure with variable non-radioactive carrier concentration. (B) pH dependence of relative pM values of NOTA for Ga3+ and Zn2+; at radiolabeling pH and 100:1 ligand to metal ratio, pM values are 22.6 (pGa) and 15.3 (pZn) respectively. (C) Radiolabeling and photocleavage yields for the SPRP synthesis of 67Ga-19 in presence of increasing amounts of non-radioactive natZn carrier. (D) Radiolabeling and photocleavage yields for the SPRP synthesis of 64Cu-19 in presence of increasing amounts of natNi carrier. | ||
The efficient on-resin separation of metal ions requires the resin material to exhibit different binding affinities for the ions of interest.40,41 For the NOTA chelator, the Irving–Williams series predicts higher binding affinity of the chelator for Cu2+ over Ni2+, as well as enhanced affinity for Ga3+ over Zn2+ due to the difference in electrostatic interactions.42,43 Under typical radiochemical labeling conditions that employ a 1000:1 ligand to metal ratio, pM values can predict binding affinities at relevant pH values (Fig. 4B and S40†). Indeed, at pH 5, NOTA exhibits a pNi of 12.2, a pCu of 16.3, a pZn of 15.3 and a pGa of 22.6. This indicates that separation of Ni and Cu, as well as Zn and Ga, should be feasible due to a >4 orders of magnitude difference in affinity of NOTA to the metal ions of interest. Indeed, when we conducted radiochemical labeling experiments in presence of increasing amounts of non-radioactive carrier for both isotopes, we observed that radiolabeling yields were only affected once the binding affinity difference was exceeded (104–105 excess carrier relative to radionuclide, Fig. 4D and E and Tables S8–S10†). Quantification of trace metal ion content using ICP-OES indicates that Zn elutes in the loading fraction as long as the resin's binding capacity is not outcompeted. We note that use of a chelator with greater ion selectivity than NOTA will further enhance separation capacity of SPRP and prove especially useful for the separation of long-lived, radioactive contaminants from complex parent isotope mixtures.
Optimized radiolabeling with 68Ga
To validate SPRP as an efficient strategy for preparation of radiopharmaceuticals, we prepared and validated the 68Ga-PSMA derivative 68Ga-31 with 68Ga sourced from a commercial 68Ge/68Ga-generator. To date, two 68Ga-based PET tracers have reached FDA approval and utilize 68Ga provided predominantly from the generator; the comparably large elution volumes require fractionation and concentration of the eluted activity using ion-exchange chromatography to ascertain high molar activity (MA) radiolabeling in solution.44,45 Following radiolabeling, the product is further purified with solid phase extraction to remove unbound 68Ga. These steps are both time consuming and can lead to additional loss of radioactivity during separation and transfer. With SPRP, these steps can be omitted, as concentration of activity, removal of trace metal impurities and unbound 68Ga is achieved during the solid-phase radiolabeling step.We prepared 68Ga-31 by adaptation of our optimized protocol (Fig. 5A). In brief, generator eluate was collected and buffered to pH 5 using 1 M NaOAc buffer and loaded directly onto the functionalized resin TG-Anp-31. Radiolabeling was conducted at room temperature with 1.05 mCi 68Ga using 10 nmol peptide on resin. The radiolabeling step proceeded with a high radiochemical (>80%) and moderate photocleavage yield of 30% to produce the desired product 68Ga-31 with >99% radiochemical purity and at 0.2 mCi nmol−1 molar activity within 40 minutes of generator elution (Fig. 5A and B). No further purification or separation was performed, and the filtrate was used directly for subsequent in vitro and in vivo experiments. Synthesis of 68Ga-31 using SPRP produced molar activities comparable to those obtained using automated synthesis procedures for clinically utilized 68Ga-radiopharmaceuticals.46–48 Subsequent validation with cell binding assays (Fig. 5C), PET-CT imaging (Fig. 5A, right) and biodistribution analysis (Fig. 5D) demonstrated appropriate, selective binding to cells expressing the PSMA target, with no appreciable off-target binding or uptake indicative of free 68Ga. Conclusively, 68Ga-31 synthesized using SPRP produces preclinically viable radiopharmaceuticals with excellent target localization and no significant off-target uptake.
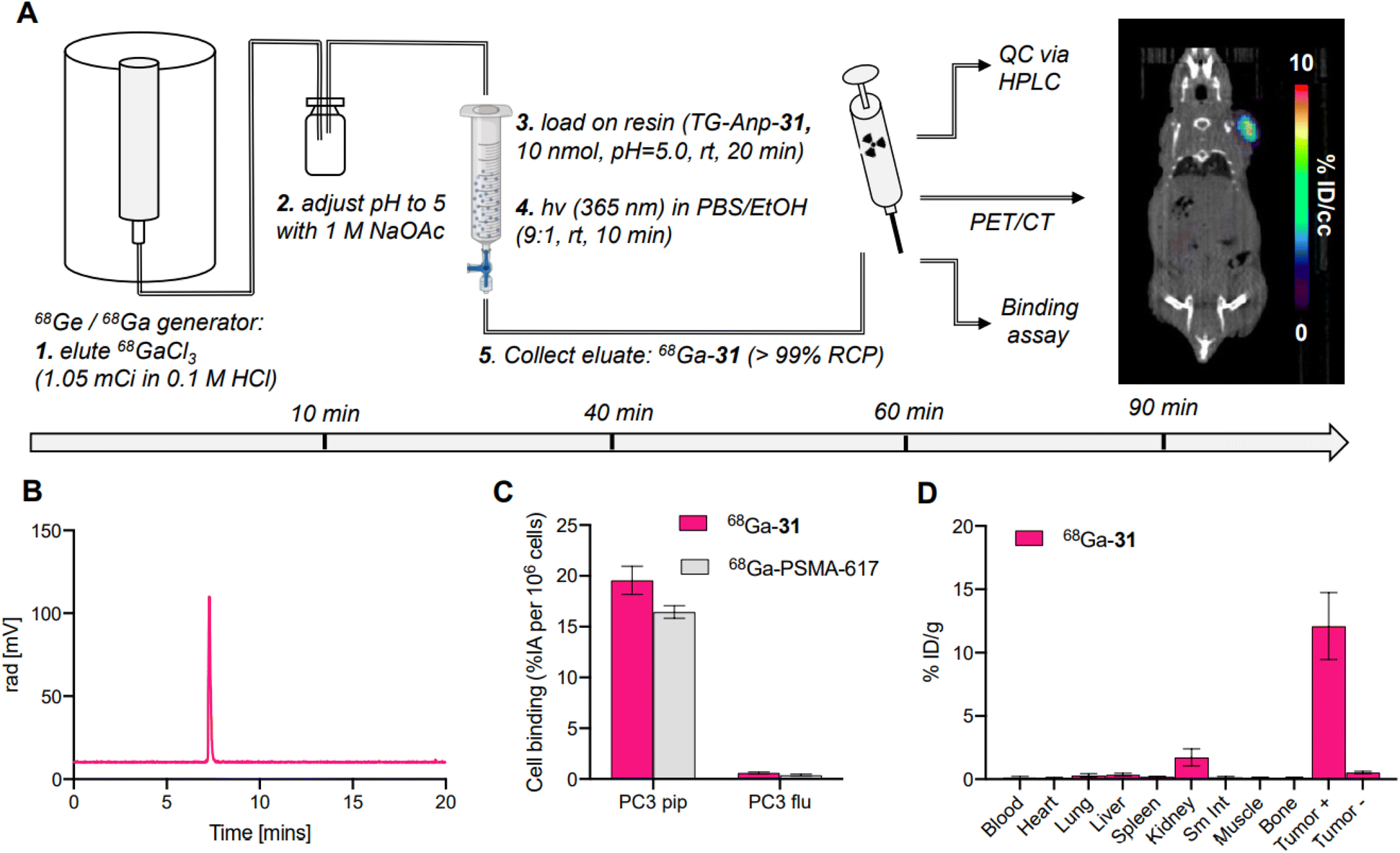 | ||
| Fig. 5 (A) Schematic description of 68Ga radiolabeling procedure and corresponding in vivo studies along with timeline; far right image shows coronal PET-CT image slice acquired 90 min post injection with 68Ga-31 produced using SPRP in a PSMA+/− mouse xenograft model; activity localizes predominantly in the PSMA+ PC-3 xenograft on the right shoulder. (B) Analytical radio HPLC chromatogram of crude filtrate following photocleavage showing >99% radiochemical purity (RCP) of the desired product 68Ga-31. (C) In vitro cell binding data for 68Ga-31 and 68Ga-PSMA-617 as the control. (D) Biodistribution data 2 hours after injection of 68Ga-31 in PC-3 PiP/Flu tumor-bearing mice (n = 5). | ||
Comparison with in-solution 68Ga labeling
While a successful in vivo validation of SPRP with 68Ga eluted from a typical, commercial generator is encouraging, it is important to validate the performance of the solid-phase method in the context of clinically validated compounds and radiolabeling methods. To this end, we selected to compare the performance of TG-Anp-31 with that of PSMA-617, a well-established precursor for radiochemical labeling with various isotopes, including 68Ga and 177Lu for the diagnostic and therapeutic management of prostate cancer.49,50 A common challenge of 68Ga-tracer production from generator eluate is the issue of decreasing activity per volume of eluate as parent 68Ge decays on aging generators, which ultimately limits the time that corresponding generators can be utilized in the clinic. Potential solutions to this issue have been developed and include fractioned generator elution, secondary concentration of eluate using ion-exchange chromatography and purification of radiolabeling solutions using C18-reverse-phase chromatography (if the 68Ga-tracer is sufficiently lipophilic to not co-elute with unreacted 68Ga-chloride).51–54 These methods all require additional time, handling, and processing of the eluted activity, which reduces the maximum possible attainable, ready-to-inject dose of the radiopharmaceutical and can increase radioactive exposure to the operator.55We conducted a head-to-head comparison of the SPRP approach (Fig. 6A, top) with in solution radiolabeling (Fig. 6A, bottom) using an “aged” generator with a usage time of >200 days after installation. At time of experiments, produced a maximum of 6 mCi in a single, 5 mL elution fraction or 2 mCi by collection of 1 mL fractions (fractioned elution).55 Following adjustment of pH, radiolabeling was conducted using both approaches. As anticipated, in-solution labeling in large reaction volumes does not proceed beyond 72% or 62% respectively (Fig. 6B and Table S9,† chromatographic traces shown in Fig. S43 and S44†), whereas SPRP retains high radiolabeling yields essentially independent of the reaction volume (Fig. 6B and Table S10†). Subsequent purification steps result in a terminal, non-decay corrected yield of 32% for the fractioned elution method (22% non-fractioned) at the end of synthesis. However, SPRP produced considerably improved, non-decay corrected yields of 50% and 52% respectively (non-fractioned and fractioned elution, Fig. 6C and Table S10†). Both procedures produce comparable decay-corrected (d.c.) yields: 59% (d.c.) for in solution labeling, 70% (d.c.) for SPRP – these values are comparable to published, automated synthesis procedures.56 However, effective product quantity of the target radiopharmaceutical differs by a factor of 2 using manual synthesis and purification, which was required for the solution-phase labeling due to low radiochemical purity. Additionally, considering that a typical 68Ga-PSMA-617 dose for clinical PET imaging is 1.8–8 mCi,50 only the herein described SPRP method would be able to produce sufficient clinical product from an “aged” generator using both fractioned and non-fractioned elution protocols. Lower activity injections at 1.3 mCi and below have demonstrated that dose reduction is not feasible without a negative impact on image quality and lesion detectability.57 In context of recent supply chain issues involving the 68Ge isotope, this could represent a promising approach to extend the lifetime of currently installed clinical 68Ge/68Ga generator systems.
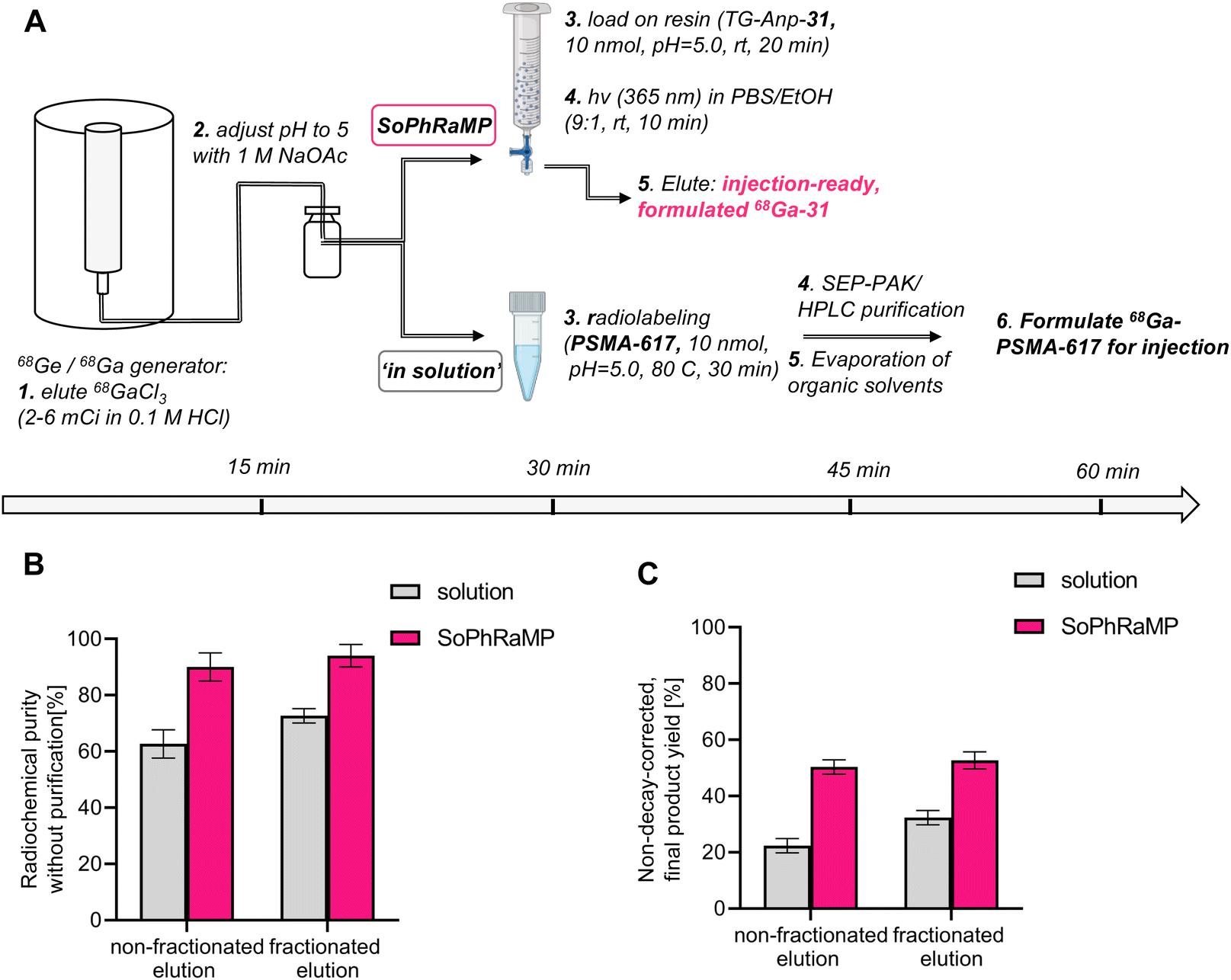 | ||
| Fig. 6 (A) Schematic description of 68Ga radiolabeling procedure along with timeline for synthesis of injection-ready 68Ga-31 using SPRP procedure and 68Ga-PSMA-617 using standard in solution protocol followed by secondary purification. (B) Radiochemical purity of crude labeling solution comparing solid phase vs. in-solution radiolabeling. (C) Non-decay corrected, end-of radiosynthesis yields for solid phase vs. in-solution radiolabeling. Yields reported as an average of n = 3 reactions. | ||
Conclusions
We have successfully developed and validated a novel, solid-phase approach to the synthesis of radiopharmaceuticals that employ radiometals and radiometalloids. This method, termed SPRP, utilizes selective capture of the radionuclide by a solid-phase appended peptide chelator conjugate. Solid phase resin material, radiochemical labeling and photochemical cleavage conditions were optimized using a model peptide sequence, followed by further validation using full-length targeted peptide sequences and radiochemical labeling with gallium and copper radionuclides. We successfully demonstrated that the capture by chelation allows simultaneous radiochemical labeling and selective removal of excess trace (metal) impurities, even from large stock volumes. Subsequently, photochemical cleavage from the resin provides target radiopharmaceuticals in a ready-to-inject form with >95% radiochemical purity and excellent in vivo performance in a preclinical mouse model. A subsequent head-to-head comparison with in-solution labeling using large reaction volumes directly obtained from a 68Ga-generator demonstrates the potential of the SPRP method to enhance clinical tracer production.Future efforts are focused on improving the thus far comparatively low-yielding photocleavage step, combined with adaptation of SPRP for microfluidic systems and exploration of radiochemical labeling with other isotopes of imminent biomedical interest. We hypothesize that the implementation of a microfluidics platform will not only further shorten the time required for synthesis, but also increase yields for individual reaction steps. The SPRP approach has the potential to accelerate and simplify selective metallation beyond the radiochemical synthesis of radiopharmaceuticals and provide a versatile platform technology for the development of a large class of metal-based pharmaceuticals.
Experimental section
Materials and methods
All starting materials were purchased from Acros Organics, Alfa Aesar, Millipore Sigma or TCI America and used without further purification. Fmoc-protected amino acids were purchased from Bachem. NOTA-bis(t-Bu ester) compound was obtained from Macrocyclics. Agarose beads, amine functionalized, were purchased from NANOCS company. AmberLite® FPA66 Anion Exchange Resin free base and TentaGel™ S-NH2 resins were purchased from Millipore Sigma. 67Ga-citrate was received from Jubilant Radiopharma. 64CuCl2 was produced and received from the University of Wisconsin, Medical Physics Dept., Madison, WI. Mass spectrometry: high-resolution ESI mass spectrometry and MALDI-TOF MS was carried out at the Stony Brook University, Center for Advanced Study of Drug Action (CASDA). Inductively coupled plasma spectroscopy (ICP) was performed on an Agilent Technologies ICP-OES (Model 5110). A 10-point standard with respect to gallium or copper was used and lines of best fit were found with R2 of 0.999. UV-vis spectra were collected with a NanoDrop 1 C instrument (AZY1706045). High-Performance Liquid Chromatography (HPLC): semi-preparative HPLC was carried out using a Shimadzu HPLC-20AR equipped with a binary gradient, pump, UV-vis detector, and manual injector on a Phenomenex Luna C18 column (250 mm × 21.2 mm, 100 Å, AXIA packed). Method A (preparative purification method). A = 0.1% TFA in water, B = 0.1% TFA in MeCN. Gradient: 0–5 min: 95% A; 5–24 min: 5–95% B gradient. Analytical HPLC analysis was carried out using a Shimadzu HPLC-20AR equipped with a binary gradient, pump, UV-vis detector, autoinjector, and PMT/NaI-1′′ remote radio-detector (for medium energy gammas) on a Phenomenex Luna C18 column (150 mm × 3 mm, 100 Å). Method B (analytical HPLC analysis). A = 0.1% TFA in water, B = 0.1% TFA in MeCN with a flow rate of 0.8 mL min−1, UV detection at 220 and 270 nm. Gradient 0–2 min: 5% B; 2–14 min 5–95% B; 14–16 min 95% B; 16–16.5 min 95–5% B; 16.5–20 min 5% B or with Method C (radioanalysis). A = 0.1% TFA in water, B = 0.1% TFA in MeCN with a flow rate of 0.8 mL min−1. Gradient 0–2 min: 5% B; 2–14 min 5–95% B; 14–16 min 95% B; 16–16.5 min 95–5% B; 16.5–20 min 5% B.Synthesis of NOTA-Aca-Q-W-NH2 model peptide
The synthesis of the NOTA-Aca-Q-W-NH2 peptide (10) was carried out manually on a 0.2 mmol scale using a 2-chlorotrityl chloride resin (200 mg, 1.0 mmol g−1) (Scheme S1†). 2-Chlorotrityl resin (CTC) was swollen in DCM (3 mL) and DMF (3 mL) for 1 min three times each. (S)-3-(Fmoc-amino)-3-(2-nitrophenyl)propionic acid (Anp) (312 mg, 0.60 mmol) was coupled to the resin using N,N-diisopropylethylamine (DIEA) (139 μL, 0.80 mmol) in DCM within 12 h. The Fmoc group was subsequently removed by treatment of the resin with 20% piperidine in DMF (3 mL) for 20 min. Subsequent amino acids were coupled in the same manner until the short sequence (QW) was assembled. The peptide was elongated by coupling the Fmoc-Aca-OH (283 mg, 0.80 mmol) using PyBOP (208 mg, 0.40 mmol) as the coupling reagent in the presence of DIEA (139 μL, 0.80 mmol) within 24 h in DMF. The Fmoc group from N-terminus was removed by treatment of the resin with solution of 20% piperidine in DMF for 20 min. Next, NOTA-bis(t-Bu ester) (166 mg, 0.40 mmol) was coupled to the free amine using PyBOP (208 mg, 0.40 mmol) as coupling reagent in the presence of DIEA (139 μL, 0.80 mmol) within 6 h. The resin-appended protected NOTA-Aca-Q-W-Anp (Anp-10′) peptide (200 mg, 0.20 mmol) was treated with a 1% TFA in DCM to selectively cleave the peptide from solid phase. Subsequently, the NHS active ester of NOTA-Aca-Q-W-Anp (NHS-Anp-10′) was synthesized by addition of excess of NHS (460 mg, 4.0 mmol) and EDC (621 mg, 4.0 mmol) as the coupling reagent in the presence of DIEA (1.40 mL, 8.00 mmol) within 16 h. After coupling, the activated peptide was treated with a mixture of TFA/TIS/H2O (95%/2.5%/2.5%) for 5 h in order to deprotect the side chain of the peptide. Compound NHS-Anp-10 was purified by reverse-phase semi-preparative HPLC and subsequently re-appended to tentagel resin, agarose-based resin or ion-exchange resin. To this end, each resin (100 mg, 0.26 mmol g−1) was swollen in DCM and DMF. Next, the compound NHS-Anp-10 (27 mg, 0.03 mmol) was coupled to three types of resin using DIEA (18 μL, 0.11 mmol) in DMF within 12 h. Eventually, the resin was washed, dried and stored until use. Synthesis of targeting peptides 19 and 31 is described in details in ESI.† General methods of quantitation of ligand in solution following cleavage from resin materials using conventional TFA cleavage or photocleavage were conducted using a spectrophotometric and quantitative chromatographic method described in the ESI and Fig. S5 and S6.†:2.5:2.5, v/v/v) at room temperature for 6 h. Then the resin was removed and the residual TFA solution transferred to a 15 mL falcon tube and an ice cooled mixture of Et2O/hexane (10 mL; 1:1 v/v) was added to precipitate the peptide. The supernatant was removed after centrifugation, and the peptide pellets were washed twice with Et2O/hexane. The crude peptide precipitate was re-dissolved in MeCN/H2O (1:1, v/v) and lyophilized, followed by purification via semi-preparative HPLC (Method A). Pure fractions were collected and characterized by analytical HPLC and MALDI-TOF MS.
Synthesis of complexes with Ga and Cu
The resin-appended peptide, TG-Anp-10 (1.0 mg, 100 nmol) was washed with 3 mL of DCM, DMF, H2O and 10 mM NaOAc buffer (pH = 5.0) three times each for 1 min. Next, the aqueous stock solutions of Ga3+ or Cu2+ were prepared and an aliquot of the resin corresponding to one equivalent was added and reacted with the resin-appended peptide for at room temperature for 12 h in pH 5 NaOAc buffer to achieve full complexation. The metal-peptide conjugates were subsequently photocleaved and the formation of the complexed molecule was affirmed with analytical HPLC and mass spectrometry (ESI-MS and MALDI-TOF). The complex conjugates were then purified via semi-preparative HPLC.Radiolabeling with 67Ga
The resin-bound peptide, NOTA-Aca-Q-W-NH2 (0.1 mg, 10 nmol) was washed with 3 mL of DCM, DMF, H2O and 10 mM NaOAc buffer (pH = 5.0) three times each for 1 min. Eventually, the resin was stored in 10 mM NaOAc buffer until use. 67Ga-citrate was purchased from Jubilant Radiopharma at an average molar activity of 5 mCi mL−1. The 67Ga-citrate solution was first converted to 67GaCl3 using an established solid-phase extraction protocol.58 The resulting average molar activity (MA) of the obtained 67Ga-chloride solution used for solid-phase radio-labelling was 1.1 mCi mL−1. For radio-labelling of TG-Anp-10, a 10 μL aliquot containing 56 μCi of 67GaCl3 was added to the resin-bound conjugate (10 nmol, 300 μL) in 10 mM NaOAc buffer. The pH of the solution was adjusted to 5.0. Radiolabeling was complete after 20 minutes at room temperature. The radiolabeled conjugates were photocleaved for 10 min and characterized with radio-HPLC.Radiolabeling with 64Cu
The 64Cu isotope stock solution (copper-64 chloride, pH = 2) was produced at the University of Wisconsin-Madison, Medical Physics Dept., Madison, WI. The 64Cu-chloride was obtained at an average molar activity of 230 μCi μL−1. For radiolabeling of TG-Anp-19 or TG-Anp-31, a 15 μL aliquot containing 50 μCi of 64CuCl2 was added to the resin-bound ligand (10 nmol, 300 μL) in 10 mM of sodium acetate (NaOAc) buffer. The pH of the solution was adjusted to 5.0. Radiolabeling was complete within 20 minutes at room temperature. The radiolabeled conjugates were photocleaved for 10 min and characterized with radio-HPLC.Radiolabeling with 68Ga
The 68Ga-chloride was obtained from a 30 mCi Ga/Ge generator (Eckert & Ziegler) at an average molar activity of 10 mCi mL−1. For radiolabeling of TG-Anp-31, a 1.0 mL aliquot containing 1.05 mCi of 68GaCl3 was added to the resin-bound conjugate (10 nmol, 300 μL) in 1.0 M of sodium acetate (NaOAc) buffer. The pH of the solution was adjusted to 5.0. Radiolabeling was completed after 20 minutes at room temperature. The radiolabeled conjugate was photocleaved for 10 min and characterized with radio-HPLC.Photocleavage procedure
A 3D printed, open source photoreactor was employed for all photocleavage procedures (for more detail see Lampkin et al.).37 The appropriate Anp-linked resin was suspended in 300 μL 1:9 EtOH/water in a filter frit endowed plastic syringe. The syringe was subsequently placed inside the photoreactor and reacted under gentle agitation and cooling with the built-in fan and photoirradiation with a 365 nm LED light source. After photocleavage, the product was filtered and characterized by HPLC (or radio-HPLC) HR-ESI-MS or MALDI-TOF MS.
Radiolabeling of fractionated/non-fractionated elution with 68Ga
For radiolabeling with non-fractionated elution, 6 mCi of 68Ga-chloride was obtained from a Ga/Ge generator (Eckert & Ziegler) in 5 mL. The 68Ga-chloride solution in 0.1 M HCl, at an average molar activity of 1.2 mCi mL−1, was pH adjusted to 5 with 1 M NaOAc buffer solution (1 mL) and added to the TG-Anp-31 (2.0 mg, 10 nmol), resin-bound conjugate. Radiolabeling was complete after 20 minutes at room temperature. The radiolabeled conjugate was photocleaved for 10 min and characterized with radio-HPLC. Rt (Method C): 7.40 min. MA: 0.6 mCi nmol−1, RY: 88.0%, PCY: 57.2%, RCP: 90%. For radiolabeling in solution, the 68Ga-chloride at the same average molar activity was used. The PSMA-617 ligand (10 nmol) was synthesized as reported previously.59 Radiolabeling was carried out at 80 °C for 30 min, followed by sep-pak purification and solvent evaporation and resuspension in PBS for formulation. Rt (Method C): 7.65 min. MA: 0.6 mCi nmol−1, RY: 22.3%, RCP: 62.7%.For radiolabeling with fractionated elution, 2.0 mCi of 68Ga-chloride was obtained by fractioned elution of the 68Ga/68Ge generator (Eckert & Ziegler) in a 1 mL fraction. The 68Ga-chloride solution was eluted in 0.1 M HCl, at an average molar activity of 2.0 mCi mL−1. Following adjustment of pH by addition of 1 M NaOAc buffer solution (0.25 mL), the mixture was loaded onto a plastic reaction vessel containing TG-Anp-31 (2.0 mg, 10 nmol) resin-bound conjugate. Radiolabeling was complete after 20 minutes at room temperature. The radiolabeled conjugate was subsequently photocleaved in 10 min and characterized with radio-HPLC. Rt (Method C): 7.40 min. MA: 0.2 mCi nmol−1, RY: 90.0%, PCY: 58.6%, RCP: 94%. For radiolabeling of PSMA-617 ligand (10 nmol) in solution, the 68Ga-chloride at the same average molar activity was used. The radiolabeling was carried out at 80 °C for 30 min. Radiolabeling was carried out at 80 °C for 30 min, followed by sep-pak purification and solvent evaporation and resuspension in PBS for formulation. Rt (Method C): 7.65 min. MA: 0.2 mCi nmol−1, RY: 32.3%, RCP: 72.7%.
Excess metal experiments
The resin-bound peptide, TG-Anp-19 (0.1 mg, 10 nmol) was washed with 3 mL of DCM, DMF, H2O and 10 mM NaOAc buffer (pH = 5.0) three times each for 1 min. For radio-labelling of TG-Anp-10 with 67Ga, a 10 μL aliquot containing 57 μCi of 67GaCl3 was pre-mixed with natural ZnSO4 salt in ratio 1:n (n = 10–10000) and eventually the mixture was added to the resin-bound conjugate (10 nmol, 300 μL) in 10 mM of sodium acetate (NaOAc) buffer. For radiolabeling of TG-Anp-10 with 64Cu, a 15 μL aliquot containing 50 μCi of 64Cu was pre-mixed with natural NiSO4 salt in ratio 1:n (n = 10–10000) and eventually the mixture was added to the resin-bound conjugate (10 nmol, 300 μL) in 10 mM of sodium acetate (NaOAc) buffer. Radiolabeling was complete after 10 minutes at room temperature. Finally, the labeling solution was removed followed by resuspension in EtOH/H2O and irradiation at 365 nm. After photocleavage, the product was filtered and characterized by radio-HPLC. The concentration of free Zn2+ was determined by ICP-OES measurements for 250–30,000 equivalent excess.
Calculation of pH dependent pM values of NOTA
Fig. 4B details the determination of pM in the dependence of pH. Plots of the variation of pM values with pH were computed using previously reported stability constants of ligand protonation and metal complexation of Ga3+,60 Zn2+, Cu2+, and Ni2+.61 Values were computed using HySS62 HYSS potentiometric data fitting software with [Mn+] = 10−6 M, and [NOTA3−] = 10−4 M, and pM defined as: pM = −log([Mfreen+]) at the pH of interest.Cell binding assay
Cells were maintained in DMEM with 5% FBS at 37 °C and 5% CO2. PC-3 PiP or PC-3 Flu cells were seeded in 24-well plates (∼5 × 105 cells in 2 mL standard growth medium/well) allowing adhesion and growth overnight. The cells were washed twice with PBS, prior to the addition of DMEM cell culture medium (950 μL per well) and the corresponding radioligand 68Ga-31 (50 μCi, 50 μL per well, diluted in saline containing 0.05% bovine serum albumin). The well plates were incubated for 90 min at 37 °C and 5% CO2. To determine the uptake of radioligand, the cells were washed three times with ice-cooled PBS. All cell samples were lysed by addition of NaOH (1 M, 1 mL) to each well and the radioactivity was quantified by gamma counting and expressed as % IA per 106 cells.Biodistribution study
All animal experiments were conducted according to the guidelines of the Institutional Animal Care and Use Committee (IACUC) at Stony Brook Medicine. Male NCr nude mice (12 weeks, Taconic Biosciences) were implanted subcutaneously on the right shoulder with 0.9–1.0 × 106 PC-3 PiP cells (PSMA(+)) and on the left shoulder with 0.5–0.6 × 106 PC-3 Flu cells (PSMA(−)). When the tumors reached 80–100 mm3, the mice were anesthetized with isoflurane and injected with 72–104 μCi of 68Ga-31 at 0.2 mCi nmol−1 molar activity using a tail vein catheter. At 90 min, mice were sacrificed, and select organs were harvested. Radioactivity was quantified by gamma counting, and the radioactivity associated with each organ was expressed as % ID per g.Data availability
Experimental data is described in the ESI,† including raw spectroscopic data.Author contributions
Experiments were designed by EB, experiments were conducted by contributions from authors. The manuscript was written and edited by all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
EB acknowledges the Gordon & Betty Moore foundation for generous support of this work through the 2020 Moore Fellowship and the National Institutes of Health (R01EB032349-01A1). PL acknowledges NSF grant CHE-1904940 (Gellman) for support.Notes and references
- I. Velikyan, A. Wennborg, J. Feldwisch, H. Lindman, J. Carlsson and J. Sörensen, Am. J. Nucl. Med. Mol. Imaging., 2016, 6, 135 CAS.
- H. Hong, L. Zhang, F. Xie, R. Zhuang, D. Jiang, H. Liu, J. Li, H. Yang, X. Zhang, L. Nie and Z. Li, Nat. Commun., 2019, 10, 989 CrossRef PubMed.
- S. M. Okarvi and I. AlJammaz, EJNMMI Res., 2019, 9, 88 CrossRef PubMed.
- B. A. Vaughn, S. H. Ahn, E. Aluicio-Sarduy, J. Devaraj, A. P. Olson, J. Engle and E. Boros, Chem. Sci. J., 2020, 11, 333–342 CAS.
- J. T. Mattila, W. Beaino, P. A. Maiello, M. T. Coleman, A. G. White, C. A. Scanga, J. L. Flynn and C. J. Anderson, J. Immunol., 2017, 199(2), 806–815 CrossRef CAS PubMed.
- Z. Talip, C. Favaretto, S. Geistlich and N. P. Meulen, Molecules, 2020, 25, 966 CrossRef CAS PubMed.
- S. Pal, S. Chattopadhyay, M. Das and M. Sudersanan, Appl. Radiat. Isot., 2006, 64, 1521–1527 CrossRef CAS PubMed.
- J. Wang and R. M. van Dam, Mol. Imaging, 2020, 19, 1–21 Search PubMed.
- B. J. Nelson, J. D. Andersson, F. Wuest and S. Spreckelmeyer, EJNMMI Radiopharm. Chem., 2022, 7, 1–26 CrossRef PubMed.
- R. Chakravarty and A. Dash, Cancer Biother. Radiopharm., 2012, 27, 621–641 CrossRef CAS PubMed.
- R. Mikolajczak, S. Huclier-Markai, C. Alliot, F. Haddad, D. Szikra, V. Forgacs and P. Garnuszek, EJNMMI Radiopharm. Chem., 2021, 6, 1–40 CrossRef PubMed.
- Q. Zheng, H. Xu, H. Wang, W. H. Du, N. Wang, H. Xiong, Y. Gu, L. Noodleman, K. B. Sharpless, G. Yang and P. Wu, J. Am. Chem. Soc., 2021, 143, 3753–3763 CrossRef CAS PubMed.
- N. P. van der Meulen, M. Bunka, K. A. Domnanich, C. Muller, S. Haller, C. Vermeulen, A. Turler and R. Schibli, Nucl. Med. Biol., 2015, 42, 745–751 CrossRef CAS PubMed.
- F. Alves, V. H. P. Alves, S. J. C. Do Carmo, A. C. B. Neves, M. Silva and A. J. Abrunhosa, Mod. Phys. Lett. A, 2017, 32, 1740013 CrossRef CAS.
- G. Sugiura, H. Kühn, M. Sauter, U. Haberkorn and W. Mier, Molecules, 2014, 19, 2135–2165 CrossRef PubMed.
- I. F. Chaple, H. A. Houson, A. Koller, A. Pandey, E. Boros and S. E. Lapi, Nucl. Med. Biol., 2022, 108–109, 16–23 CrossRef CAS PubMed.
- L. Wharton, C. Zhang, H. Yang, J. Zeisler, V. Radchenko, C. Rodríguez-Rodríguez, M. Osooly, B. O. Patrick, K.-S. Lin, F. Bénard, P. Schaffer and C. Orvig, Bioconjugate Chem., 2022, 33, 505–522 CrossRef CAS PubMed.
- A. A. Larenkov, A. G. Makichyan and V. N. Iatsenko, Molecules, 2021, 26, 6371 CrossRef CAS PubMed.
- E. Eppard, M. Wuttke, P. L. Nicodemus and F. Rösch, J. Nucl. Med., 2014, 55, 1023–1028 CrossRef CAS PubMed.
- E. W. Price, B. M. Zeglis, J. F. Cawthray, C. F. Ramogida, N. Ramos, J. S. Lewis, M. J. Adam and C. Orvig, J. Am. Chem. Soc., 2013, 135, 12707–12721 CrossRef CAS PubMed.
- A. J. Koller, S. Saini, I. F. Chaple, M. A. Joaqui-Joaqui, B. M. Paterson, M. T. Ma, P. J. Blower, V. C. Pierre, J. R. Robinson, S. E. Lapi and E. Boros, Angew. Chem., Int. Ed., 2022, e202201211, DOI:10.1002/anie.202201211.
- H. Yang, J. J. Wilson, C. Orvig, Y. Li, D. S. Wilbur, C. F. Ramogida, V. Radchenko and P. Schaffer, J. Nucl. Med., 2022, 63, 5–13 CrossRef CAS PubMed.
- G. Vaidyanathan and M. R. Zalutsky, Appl. Radiat. Isot., 1993, 44, 621–628 CrossRef CAS PubMed.
- L. J. Brown, D. R. Bouvet, S. Champion, A. M. Gibson, Y. Hu, A. Jackson, I. Khan, N. Ma, N. Millot, H. Wadsworth and R. C. Brown, Angew. Chem., Int. Ed., 2007, 46, 941–944 CrossRef CAS PubMed.
- B. P. Burton-Pye, I. Radivojevic, D. McGregor, I. M. Mbomekalle, W. W. Lukens Jr and L. C. Francesconi, J. Am. Chem. Soc., 2011, 133, 18802–18815 CrossRef CAS PubMed.
- M. Patra, L. S. Eichenberger, G. Fischer and J. P. Holland, Angew. Chem., Int. Ed., 2019, 58, 1928–1933 CrossRef CAS PubMed.
- W. Chen, Z. Huang, N. E. Tay, B. Giglio, M. Wang, H. Wang, Z. Wu, D. A. Nicewicz and Z. Li, Science, 2019, 364, 1170–1174 CrossRef CAS PubMed.
- Y. Fu, H. Helbert, N. A. Simeth, S. Crespi, G. B. Spoelstra, J. M. van Dijl, M. van Oosten, L. R. Nazario, D. van der Born, G. Luurtsema, W. Szymanski, P. H. Elsinga and B. L. Feringa, J. Am. Chem. Soc., 2021, 143, 10041–10047 CrossRef CAS PubMed.
- D. F. Earley, A. Guillou, S. Klingler, R. Fay, M. Gut, F. d'Orchymont, S. Behmaneshfar, L. Reichert and J. P. Holland, J. Am. Chem. Soc., 2022, 2, 646–664 CAS.
- D. I. Bryson, W. Zhang, W. K. Ray and W. L. Santos, Mol. Biosyst., 2009, 5, 1070–1073 RSC.
- S.-n. Nishimura, N. Hokazono, Y. Taki, H. Motoda, Y. Morita, K. Yamamoto, N. Higashi and T. Koga, ACS Appl. Mater. Interfaces, 2019, 11, 24577–24587 CrossRef CAS PubMed.
- X. Liang, S. Vézina-Dawod, F. Bédard, K. Porte and E. Biron, Org. Lett., 2016, 18, 1174–1177 CrossRef CAS PubMed.
- A. S. Knight, E. Y. Zhou, J. G. Pelton and M. B. Francis, J. Am. Chem. Soc., 2013, 135, 17488–17493 CrossRef CAS PubMed.
- A. Ajayaghosh and V. R. Pillai, Tetrahedron Lett., 1995, 36, 777–780 CrossRef CAS.
- M. Hurevich, J. Kandasamy, B. M. Ponnappa, M. Collot, D. Kopetzki, D. T. McQuade and P. H. Seeberger, Org. Lett., 2014, 16, 1794–1797 CrossRef CAS PubMed.
- N. Bindman, R. Merkx, R. Koehler, N. Herrman and W. A. Van Der Donk, Chem. Commun., 2010, 46, 8935–8937 RSC.
- P. P. Lampkin, B. J. Thompson and S. H. Gellman, Org. Lett., 2021, 23, 5277–5281 CrossRef CAS PubMed.
- J. Zhang, D. Li, L. Lang, Z. Zhu, L. Wang, P. Wu, G. Niu, F. Li and X. Chen, J. Nucl. Med., 2016, 57, 9–14 CrossRef CAS PubMed.
- A. Afshar-Oromieh, A. Malcher, M. Eder, M. Eisenhut, H. Linhart, B. Hadaschik, T. Holland-Letz, F. Giesel, C. Kratochwil and S. Haufe, Eur. J. Nucl. Med. Mol. Imaging, 2013, 40, 486–495 CrossRef CAS PubMed.
- L. E. McInnes, S. E. Rudd and P. S. Donnelly, Coord. Chem. Rev., 2017, 352, 499–516 CrossRef CAS.
- J. Šimeček, P. Hermann, H. J. Wester and J. Notni, ChemMedChem, 2013, 8, 95–103 CrossRef PubMed.
- V. c. Kubíček, Z. Bohmova, R. Ševčíková, J. Vaněk, P. e. Lubal, Z. Polakova, R. Michalicová, J. Kotek and P. Hermann, Inorg. Chem., 2018, 57, 3061–3072 CrossRef PubMed.
- J. Simecek, M. Schulz, J. Notni, J. Plutnar, V. Kubicek, J. Havlickova and P. Hermann, Inorg. Chem., 2012, 51, 577–590 CrossRef CAS PubMed.
- P. Caroli, S. P. Colangione, U. De Giorgi, G. Ghigi, M. Celli, E. Scarpi, M. Monti, V. Di Iorio, A. Sarnelli and G. Paganelli, Biomedicines, 2020, 8, 536–547 CrossRef CAS PubMed.
- C. I. Lang, P. Döring, R. Gäbel, P. Vasudevan, H. Lemcke, P. Müller, J. Stenzel, T. Lindner, M. Joksch and J. Kurth, Cells, 2020, 9, 1358–1373 CrossRef CAS PubMed.
- S. Spreckelmeyer, M. Balzer, S. Poetzsch and W. Brenner, EJNMMI Radiopharm. Chem., 2020, 5, 1–10 CrossRef PubMed.
- C. Da Pieve, M. Costa Braga, D. R. Turton, F. A. Valla, P. Cakmak, K.-H. Plate and G. Kramer-Marek, Molecules, 2022, 27, 675 CrossRef CAS PubMed.
- M. Nader, D. Valla, C. Vriamont, J. Masset, A. Pacelli, K. Herrmann and F. Zarrad, Nucl. Med. Biol., 2022, 110–111, 37–44 CrossRef CAS PubMed.
- H. Zhu, Q. Xie, N. Li, H. Tian, F. Liu and Z. Yang, J. Radioanal. Nucl. Chem., 2016, 309, 575–581 CAS.
- A. Afshar-Oromieh, H. Hetzheim, C. Kratochwil, M. Benesova, M. Eder, O. C. Neels, M. Eisenhut, W. Kübler, T. Holland-Letz and F. L. Giesel, J. Nucl. Med., 2015, 56, 1697–1705 CrossRef CAS PubMed.
- L. Calderoni, A. Farolfi, D. Pianori, E. Maietti, V. Cabitza, A. Lambertini, G. Ricci, S. Telo, F. Lodi and P. Castellucci, J. Nucl. Med., 2020, 61, 716–722 CrossRef CAS PubMed.
- M. Benešová, M. Schäfer, U. Bauder-Wüst, A. Afshar-Oromieh, C. Kratochwil, W. Mier, U. Haberkorn, K. Kopka and M. Eder, J. Nucl. Med., 2015, 56, 914–920 CrossRef PubMed.
- C. Liu, T. Liu, N. Zhang, Y. Liu, N. Li, P. Du, Y. Yang, M. Liu, K. Gong and X. Yang, Eur. J. Nucl. Med. Mol. Imaging, 2018, 45, 1852–1861 CrossRef CAS PubMed.
- X. Duan, Z. Cao, H. Zhu, C. Liu, X. Zhang, J. Zhang, Y. n. Ren, F. Liu, X. Cai and X. Guo, Eur. J. Nucl. Med. Mol. Imaging, 2022, 49, 1030–1040 CrossRef CAS PubMed.
- J. Kleynhans, S. Rubow, J. le Roux, B. Marjanovic-Painter, J. R. Zeevaart and T. Ebenhan, J. Labelled Compd. Radiopharm., 2020, 63, 553–563 CAS.
- C. W. Wichmann, U. Ackermann, S. Poniger, K. Young, B. Nguyen, G. Chan, J. Sachinidis and A. M. Scott, J. Labelled Compd. Radiopharm., 2021, 64, 140–146 CrossRef CAS PubMed.
- I. Rauscher, W. P. Fendler, T. A. Hope, A. Quon, S. G. Nekolla, J. Calais, A. Richter, B. Haller, K. Herrmann and W. A. Weber, J. Nucl. Med., 2020, 61, 189–193 CrossRef CAS PubMed.
- A. Pandey, D. Smilowicz and E. Boros, Chem. Sci., 2021, 12, 14546–14556 RSC.
- D. Śmiłowicz, D. Schlyer, E. Boros and L. Meimetis, Mol. Pharm., 2022, 19, 3217–3227 CrossRef PubMed.
- J. Simecek, M. Schulz, J. Notni, J. Plutnar, V. Kubicek, J. Havlickova and P. Hermann, Inorg. Chem., 2012, 51, 577–590 CrossRef CAS PubMed.
- V. Kubicek, Z. Bohmova, R. Sevcikova, J. Vanek, P. Lubal, Z. Polakova, R. Michalicova, J. Kotek and P. Hermann, Inorg. Chem., 2018, 57, 3061–3072 CrossRef CAS PubMed.
- L. Alderighi, P. Gans, D. Peters, A. Sabatini, A. Ienco and A. Vacca, Coord. Chem. Rev., 1999, 184, 311–318 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, HRMS, MALDI-TOF and HPLC results; characterization of complexes, radiolabeling data and PET/CT imaging results. See DOI: https://doi.org/10.1039/d2sc06977f |
| This journal is © The Royal Society of Chemistry 2023 |