
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe role of analyte concentration in accelerated reaction rates in evaporating droplets†
Casey J.
Chen
 and
Evan R.
Williams
*
and
Evan R.
Williams
*
Department of Chemistry, University of California, Berkeley, CA 94720, USA. E-mail: erw@berkeley.edu
First published on 12th April 2023
Abstract
Accelerated reactions in microdroplets have been reported for a wide range of reactions with some microdroplet reactions occurring over a million times faster than the same reaction in bulk solution. Unique chemistry at the air–water interface has been implicated as a primary factor for accelerated reaction rates, but the role of analyte concentration in evaporating droplets has not been as well studied. Here, theta-glass electrospray emitters and mass spectrometry are used to rapidly mix two solutions on the low to sub-microsecond time scale and produce aqueous nanodrops with different sizes and lifetimes. We demonstrate that for a simple bimolecular reaction where surface chemistry does not appear to play a role, reaction rate acceleration factors are between 102 and 107 for different initial solution concentrations, and these values do not depend on nanodrop size. A rate acceleration factor of 107 is among the highest reported and can be attributed to concentration of analyte molecules, initially far apart in dilute solution, but brought into close proximity in the nanodrop through evaporation of solvent from the nanodrops prior to ion formation. These data indicate that analyte concentration phenomenon is a significant factor in reaction acceleration where droplet volume throughout the experiment is not carefully controlled.
Introduction
Reaction rate acceleration in microdroplets has been widely observed for a broad range of reactions, including complexation,1 condensation,2,3 phosphorylation,4 reduction,5,6 oxidation,7,8 hydrogen–deuterium exchange,9,10 and many others.11,12 Reaction rate acceleration factors ranging between 10 and 106 have been reported.13 Many droplets are highly charged, but reaction acceleration can occur in largely uncharged droplets as well.14 A primary factor implicated in accelerated reactions is surface chemistry, which is enhanced in microdroplets owing to their high surface-to-volume ratios. Decreasing the size of the droplet can lead to increased reaction rates, consistent with reactions occurring faster at surfaces due to the higher surface-to-volume ratios.5,8,15A number of mechanisms to explain why reactions may be accelerated at surfaces have been proposed. Many reactions occur faster in the gas phase than they do in solution. In some cases, acceleration in microdroplets has been largely attributed to ion–molecule reactions in the gas phase.16,17 Reactants at the air-liquid interface of a droplet are only partially solvated and this can lead to lower reaction barriers than those of fully solvated reactants leading to rate acceleration.18–21 The rapid exchange of reactants and products between the surface and interior has been proposed to contribute to rate enhancing effects of droplet surfaces.8,22,23 A double layer model describes parallel reactions occurring at the surface and in the interior of a microdrop with free diffusion between the two regions.13,20,24 As expected, this model predicts higher reaction acceleration for small droplets and for reactants with high diffusion coefficients.13,20,24 Another model developed to understand reaction rate accelerations in micron-sized droplets at reactant concentrations above 100 μM indicates that millisecond timescales are required in order for surface reactions to contribute greatly to the reaction acceleration.18
Strong electric fields at the droplet surface may also lead to accelerated chemistry. For example, it was proposed that electric fields due to orientation of water at the droplet surface can produce water radicals, which can act as superacids or superbases,7,8,23 and lead to hydrogen peroxide formation.25,26 Computations indicate the electric field at the surface is high (∼16 MV cm−1), but insufficient to split bonds in water.27 Other experiments indicate that hydrogen peroxide is not formed in microdrops in an inert atmosphere but is produce by exposure to gaseous ozone.26 Incorporation of external gasses, such as CO2 reacting with the droplet to generate formic acid with a catalyst,6 also indicates the importance of gaseous reactants in microdroplet reactions. These reactions are enhanced at surfaces and diffusion of the initially gaseous reactant into the droplet interior can occur.24,28,29 Other factors, such as reduced pH in charged microdroplets and reagent concentration that occurs upon solvent evaporation have also been proposed as mechanisms for reaction acceleration in microdroplets.13,16
There have been several investigations into the role of analyte concentration on droplet reaction acceleration. Increasing the analyte concentration can result in either higher6,7 or lower8,15 reactant to product conversion ratios. A higher concentration of sulfone led to increased conversion to sulfonic acid, a result attributed to spontaneous oxidation at the air-liquid interface.7 In contrast, increasing the concentration of phenylhydrazine that reacts with isatin led to a lower conversion ratio.15 This observation was attributed to the surface-to-bulk concentration ratio undergoing a steep increase with decreasing concentration. In general, much of the reaction acceleration that has been observed in microdroplets for a variety of reactions under a wide range of conditions has been attributed to the role of the droplet surface.
Here, we demonstrate that the extent of reaction acceleration for a simple bimolecular reaction where surface chemistry does not appear to play a role, depends strongly on the initial reactant concentration, and an acceleration factor of 107 can be achieved primarily by increased concentration due to solvent evaporation from nanodrops.
Experimental
Charged nanodrops are formed by nano-electrospray ionization (nESI) using borosilicate theta capillaries (1.5 mm outer diameter, Sutter Instruments, Novato, CA) that were pulled using a Flaming/Brown P-87 micropipette puller (Sutter Instruments) to produce emitters with inner diameters of either 1.84 ± 0.14 μm or 684 ± 51 nm. Emitter tips were imaged using a Hitachi TM-4000 (Schaumburg, IL) microscope in the Electron Microscope Laboratory at the University of California, Berkeley. Four emitters were imaged for each emitter size. Mass spectra of ions formed by nESI were acquired using an Orbitrap Elite mass spectrometer (Thermo Fisher Scientific, San Jose, CA) using a capillary temperature of 100 °C. The theta emitters were positioned 3.0 mm from the inlet of the mass spectrometer, and a voltage of 0.7–1.2 kV was applied to platinum wires inserted into both barrels to initiate electrospray.An internal standard of either leucine-enkephalin (Leu-enk) or methionine-enkephalin (Met-enk) at a concentration of 2.0 μM was added to the solutions in each barrel in order to measure any potential differences in flow rates between the two channels in these mixing experiments. A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 2.0 μM each of Leu-enk and Met-enk at pH 3 loaded into both barrels of the theta emitters resulted in a ratio of the protonated molecular abundance of Leu-enk to Met-enk of 0.8:1.0. Any differences in flow rates between the two channels in mixing experiments were determined using the relative abundances of the protonated peptide in each channel. The relative flow rates were used to determine the concentration of reactants in the initially formed droplets from their known initial starting concentrations in each channel. The initial and mixed concentrations differed only slightly, ranging between 0% and 10%. These values, and a more detailed description of the flow calibration process are given in ESI.† Absolute overall solution flow rates were determined by measuring the masses of the theta emitters before and after electrospray for 15–30 min with the emitter in the same position and under the same conditions used in all of the experiments. To determine the contribution from evaporation, the emitters were placed in the same position in front of the instrument inlet for 15–30 min without any applied voltage. This was done to ensure equivalent temperature as a result of minor heating from the mass spectrometer interface. The mass difference was converted to a volume by using the density of water at room temperature (997.05 kg m−3).30
:1 mixture of 2.0 μM each of Leu-enk and Met-enk at pH 3 loaded into both barrels of the theta emitters resulted in a ratio of the protonated molecular abundance of Leu-enk to Met-enk of 0.8:1.0. Any differences in flow rates between the two channels in mixing experiments were determined using the relative abundances of the protonated peptide in each channel. The relative flow rates were used to determine the concentration of reactants in the initially formed droplets from their known initial starting concentrations in each channel. The initial and mixed concentrations differed only slightly, ranging between 0% and 10%. These values, and a more detailed description of the flow calibration process are given in ESI.† Absolute overall solution flow rates were determined by measuring the masses of the theta emitters before and after electrospray for 15–30 min with the emitter in the same position and under the same conditions used in all of the experiments. To determine the contribution from evaporation, the emitters were placed in the same position in front of the instrument inlet for 15–30 min without any applied voltage. This was done to ensure equivalent temperature as a result of minor heating from the mass spectrometer interface. The mass difference was converted to a volume by using the density of water at room temperature (997.05 kg m−3).30
All chemicals were obtained from Sigma-Aldrich (St. Louis, MO) except for 2,6-dichlorophenolindophenol sodium salt (DCIP) that was obtained from Fisher Scientific (Fair Lawn, NJ). All chemicals were used without further purification. All solutions were prepared using 18.2 MΩ water from a Milli-Q integral water purification system (Millipore, Billerica, MA).
Results and discussion
Effects of concentration on reaction conversion ratios
Theta emitters for nanodrop formation by electrospray ionization were prepared with tips that have an inner diameter of either 1.84 ± 0.14 μm or 684 ± 51 nm in the long dimension (Fig. 1). Two different tips sizes were used to produce nanodrops that have different initial diameters and lifetimes.31–34 These emitters were used to rapidly mix two acidified aqueous solutions (pH 3), one containing 2,6-dichlorophenolindophenol (DCIP) and the other L-ascorbic acid (L-AA) at equimolar concentrations (Scheme 1). | ||
| Fig. 1 SEM images of theta-glass emitters with the emitter oriented in the long dimension showing the two separate barrels. Four emitters were imaged for each emitter size and the average emitter diameters and standard deviations are labelled. | ||
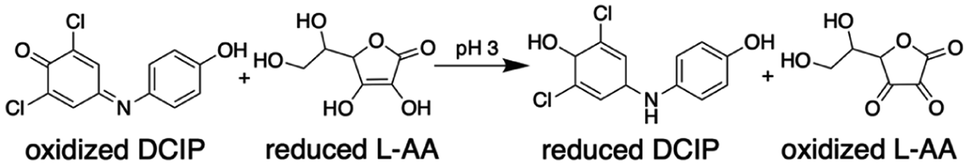 | ||
| Scheme 1 Reaction between DCIP and L-AA. | ||
Internal standards of Leu-enk and Met-enk were added to these respective solutions to measure the relative flow rates of each barrel from the relative abundances of the two protonated molecular ions (Table S1†).32–35 Differences in the relative flow rates are small, typically ranging between 0% and 10%. The initial concentrations of the reagents in data reported for mixing experiments are corrected for these minor differences in flow rates.
Solutions ranging in concentrations between 1.0 mM, close to the solubility limit for DCIP, to 10 nM were mixed using both size emitters. An equivalent concentration of each reactant was loaded into the separate barrels of the theta emitters and mass spectra were acquired. Representative mass spectra obtained at 25 nM: 25 nM and 500 μM: 500 μM concentration are shown in Fig. S2.† A conversion ratio for forming products from this reaction was obtained from eqn (1):
 | (1) |
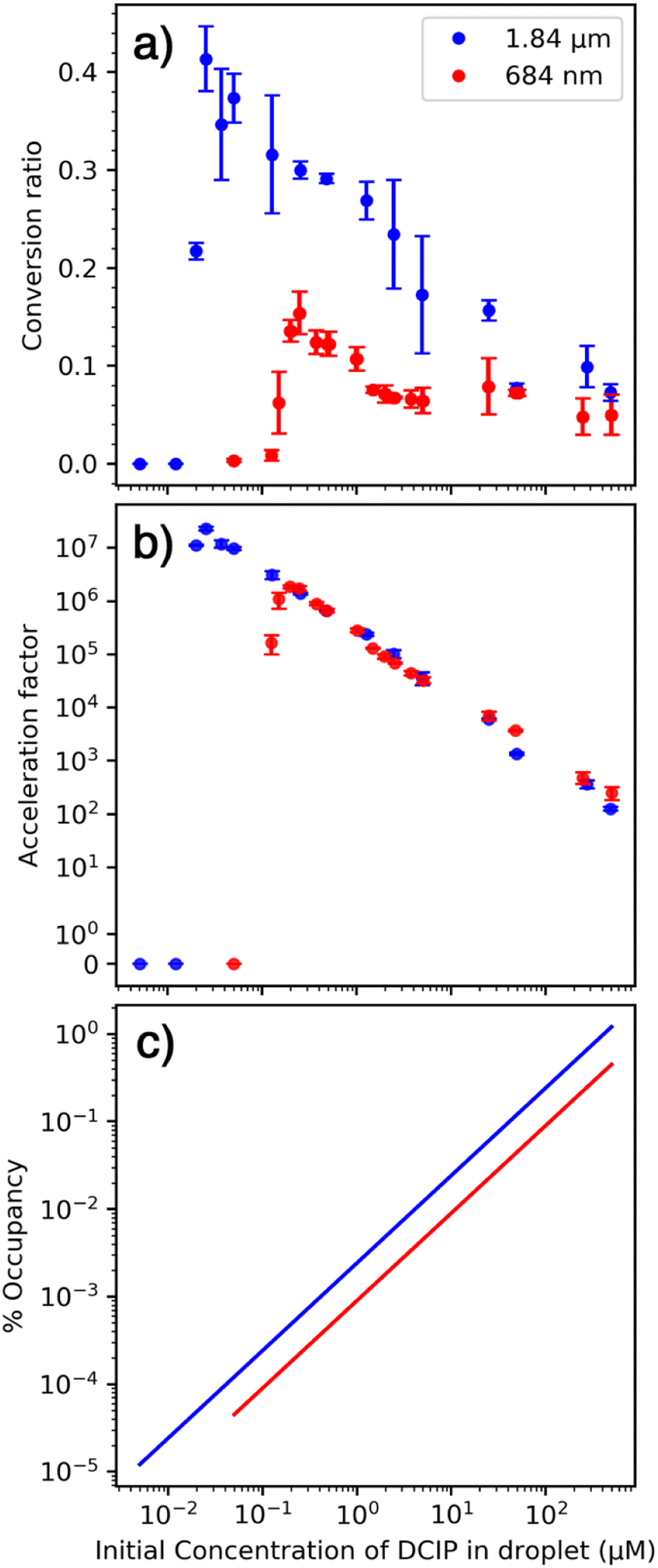 | ||
| Fig. 2 Reaction of DCIP with L-AA as a function of equimolar concentration at two droplet sizes formed by 1.84 μm (blue) and 684 nm (red) emitters showing (a) conversion ratio, (b) acceleration factor and (c) maximum percent surface occupancy. | ||
We hypothesize that the sharp drop-off in conversion ratio at low concentration is due to initial droplets containing only one or fewer reactant molecules. This occurs at a higher concentration for the smaller emitter because the initial droplet size is smaller and therefore contains fewer reactant molecules at a given solution concentration. The rapid decrease in conversion factor is consistent with a homogenous distribution of nanodrop size that is formed with these emitters. Results from Davidson et al. indicated that the initial distribution of nanodrop diameters formed from aqueous sucrose solutions produced by single barrel emitters with 1–3 μm tips was narrow and centered around ∼60 nm.36 The rapid decrease in conversion factor observed here indicates similar narrow distributions are formed with theta emitters with tip diameters below 2 μm.
Droplet lifetimes
In order to determine the reaction acceleration, the reaction time must be known. Droplet lifetimes have been estimated from larger microdrops that can be optically imaged to determine a droplet velocity, and the droplet lifetime has been estimated based on the distance to an analyzer, typically a mass spectrometer.9,10,37 However, this method may not accurately reflect the lifetimes of smaller droplets that are too small to optically image but may contribute substantially to reaction acceleration due to their higher surface-to-volume ratios and more efficient ion production.38 Formation of nanodrops by nESI leads to significant improvements in ionization efficiency from aqueous solutions and their surface-to-volume ratios are substantially higher than micron sized droplets. Thus, conclusions inferred from optical imaging of 10+ μm diameter droplets may not apply to the nanodrops formed in these and many other experiments. Moreover, nanodrops formed from 1.7 μm nESI emitters survive well into the heated metal capillary interface31 so that their lifetimes can be significantly longer than what would be the case if one assumes that ions are formed at the mass spectrometer interface.The lifetimes of aqueous nanodrops formed by theta capillaries have been determined previously by measuring the extent of protein folding that occurs upon rapid solution mixing using proteins with known folding time constants.31,32,34 This method for measuring droplet lifetimes should be well suited for determining the reaction time frame for this biomolecular reaction because the initial nanodrops size and experimental conditions are the same. This method for measuring droplet lifetimes does not rely upon imaging much larger droplets nor is it necessary to make any assumptions about where bare, unsolvated ions are formed. There is significant evidence that these unimolecular reactions that are typically done under conditions where there is one or fewer protein molecules in the droplet are not accelerated at surfaces. This appears to be true for other unimolecular reactions that occur in larger droplets as well.21
Cytochrome c in an acidified solution in one barrel was mixed with pure water in another barrel to increase the solution pH upon mixing. The resulting pH jump induces folding and the extent of folding is monitored by a change in the charge-state distribution of the protein.31,32,34,35 Results from the protein refolding experiments are shown in Fig. 3. A representative nESI mass spectrum of the acidified solution loaded into both barrels of the theta-emitter shows two distinct charge-state distributions (Fig. 3a). The charges states between 11+ and 20+ (indicated by a purple bar) are characteristic of an unfolded form or forms of this protein in solution whereas the charge states between 7+ and 10+ (indicated by a red bar) are consistent with a folded form. The population abundance of the folded form of cytochrome c in this acidified solution is determined to be 7.4% ± 1.8% from the weighted ion abundances of these two forms of the protein. When both solutions were mixed in equal volumes and the solution was loaded into both barrels of the theta emitters, the folded population is 68.4% ± 4.8% (Fig. 3b). This value represents the equilibrium distribution between the folded and unfold forms of the protein in the mixed solutions. Results from the theta emitter mixing experiments where the acidified solution containing the protein was loaded into one barrel and pure water was added to the other barrel are shown in Fig. 3c and d for the 1.84 μm and 684 nm emitters, respectively. The population of folded protein is 18.6 ± 2.6% and 11.5% ± 2.4% for the 1.84 μm and 684 nm emitters, respectively. These results show that the droplet lifetimes are insufficient to reach equilibrium with either emitter, but that the greater extent of folding with the emitter with the larger tip indicates a longer droplet lifetime. From the folding time constant of cytochrome c in unbuffered aqueous solution (114 μs),39 a reaction time corresponding to the droplet lifetime can be obtained. The droplet lifetime determined from these data are 23.0 ± 4.6 μs and 7.8 ± 2.6 μs for the 1.84 μm and 684 nm emitters, respectively. These values are slightly lower than those previously reported for theta emitters with similar size tips31 because no backing pressure that increases flow rates and droplet sizes were used in the current experiments. The two reagents in the bimolecular reaction investigated here are also in aqueous solutions, and the experiments are performed using identical emitters under the same conditions. Thus, the lifetimes of nanodrops containing the two reagents that are formed from the two different size emitters are expected to be the same as well.
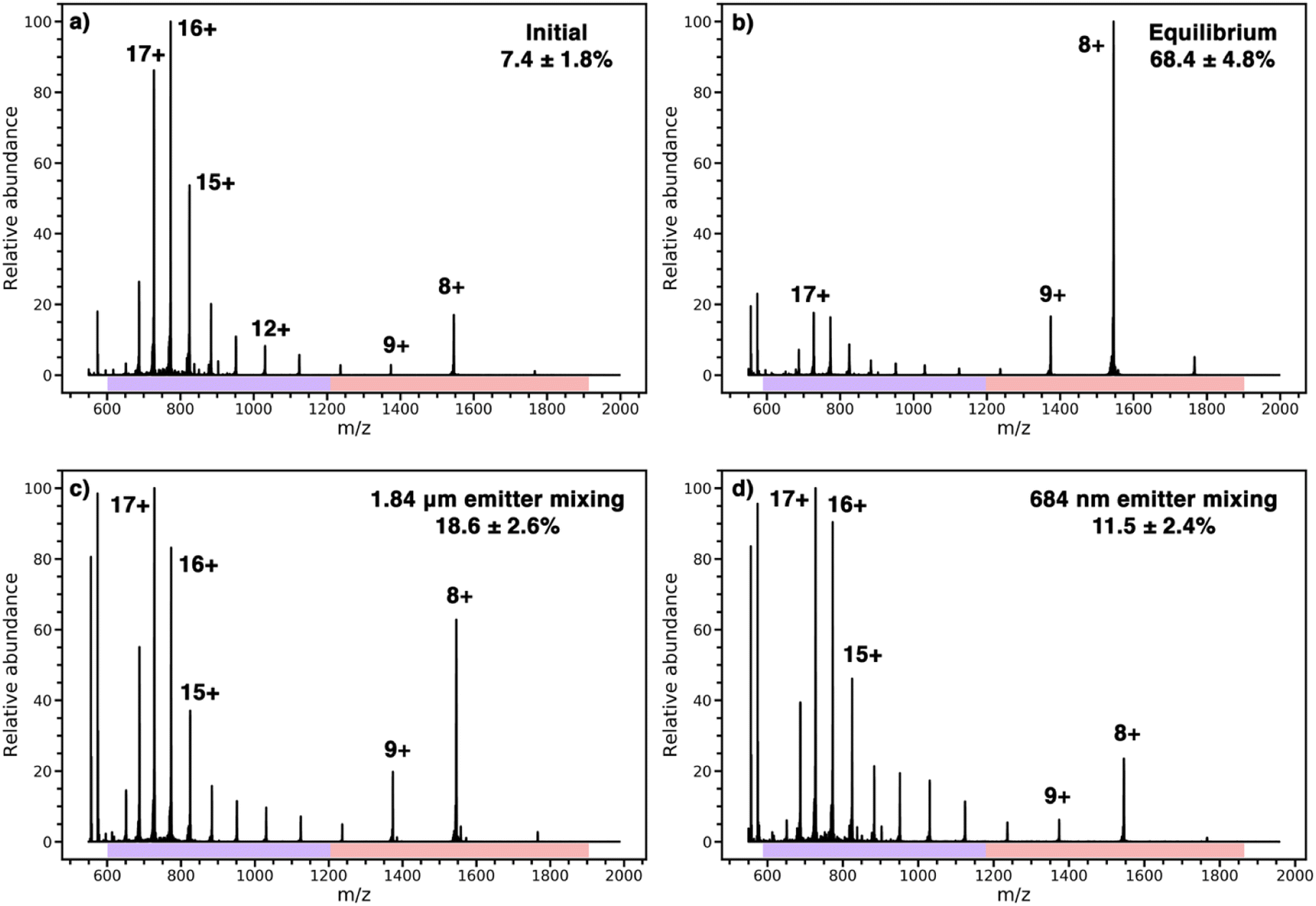 | ||
| Fig. 3 Representative mass spectra of cytochrome c used to determine the lifetime of nanodrops from the kinetics of protein refolding, (a) an initial solution of cytochrome c with Met-enk (m/z = 574) in water with 1% acetic acid (pH = 2.85), (b) a 1:1 mixture of the solution in (a) with water and Leu-enk (m/z = 556) at equilibrium (pH = 3.06), (c) theta emitter mixing of solution used in (a) with water containing Leu-enk with 1.84 μm emitters and (d) theta emitter mixing of solution used in (a) and water containing Leu-enk with 684 nm emitters. | ||
Based on a report of no apparent solvent loss from 10+ μm diameter droplets prior to the entrance to a mass spectrometer40 or minimal loss from large droplets imaged in vacuum for millisecond timescales,41 several reviewers have asserted that solvent evaporation does not occur in our experiments despite overwhelming evidence to the contrary. Charged water droplets formed by nESI with diameters ranging from a few nanometers to ∼32 nm have been trapped in either Fourier-transform ion cyclotron resonance (FT-ICR)42–44 or electrostatic ion trap based charge detection mass spectrometry (CDMS) instruments.44 Water nanodrops with diameters around 20 – 30 nm trapped for one second in an electrostatic ion trap lose 100's of kDa in mass corresponding to the continuous evaporation of many thousands of water molecules.45 The rate at which solvent evaporation occurs depends on the effective droplet temperature, which is reduced in vacuum by evaporative cooling. When ions are trapped in high vacuum, ions reach a low steady state effective temperature where heat loss by evaporative cooling is balanced by energy absorption by blackbody radiation in the low-pressure trapping region (∼10−9 Torr) of both FT-ICR MS and CDMS instruments.46 Some collisional activation also occurs in CDMS due to the high ion kinetic energy and large collisional cross sections of nanodrops with diameters of 10's of nm. The slow rate of water evaporation in ultra-high vacuum is due to the very low effective temperatures of the trapped nanodrops.43,45,47 Results from highly charged ions that are preserved in aqueous nanodrops but do not survive as bare ions in the gas phase clearly show that aqueous nanodrops can survive intact through the instrument and throughout the measurement process.48,49 However, survival of these aqueous nanodrops requires unusually “soft” interface conditions that minimize activation with rapid transfer to high vacuum where the rate of water evaporation is low. These are not conditions that are predominantly used in nESI MS, such as done here, where bare unsolvated ions are measured. Bare or unsolvated ions are achieved through energy transfer early on, in or about the electrospray interface, in the form of heated metal capillaries, strong electric fields at atmospheric or slightly reduced pressures and/or heated gasses introduced around the interface. Significant energy transfer to droplets in these atmospheric or near atmospheric conditions occurs, which substantially increases the rate of solvent evaporation and promotes bare ion formation prior to the high vacuum conditions of a mass spectrometer. Molecular dynamics simulations also provide evidence for rapid water evaporation from aqueous nanodrops when the effective temperature of the droplet is maintained at an ambient value, such as would be the case in a heated metal capillary. Simulations of a 5 nm diameter droplet indicates that full evaporation occurs in 18 ns when the temperature was maintained at 40 °C.50 This fast evaporation rate is consistent with the larger nanodrops in our experiment surviving on the low microsecond timescale.
Others have strongly asserted that bare ion formation occurs at the entrance to the mass spectrometer and thus there is no change in droplet lifetime in our experiments where different droplet sizes are produced. In our experiments, the distance between the emitter tip and the mass spectrometer is fixed at 3.0 mm. The emitter tip diameter is changed to produce droplets that have different average diameters. There is extensive evidence that smaller diameter emitters produce smaller droplets.31,34,51 There is also strong evidence that ions produced from 317 nm theta emitters are formed outside the mass spectrometer owing to their short ∼1 μs lifetime, but nanodrops formed by >1 μm emitters with lifetimes >10 μs survive long enough to enter a heated interface metal capillary.31 The temperature of a nanodrop can be affected by raising the temperature of the metal capillary to a point where the temperature of the nanodrop exceeds the melting temperature of the protein. Under these conditions, protein unfolding is energetically favorable. However, the extent of unfolding can depend on droplet size because of the different droplet lifetimes. Different droplet lifetimes can limit the time available for a chemical process, such as protein unfolding to occur, which is a kinetic effect. For example, laser heating of nanodrops outside of the mass spectrometer can induce protein unfolding in the droplets from which melting curves as a function of laser power are obtained.52 The extent of unfolding is related to nanodrop size where less unfolding has been observed for smaller nanodrops due to their shorter lifetimes and thus less time for unfolding in the droplet to occur.
Our conclusions from evaluating prior work are that (1) water evaporation from aqueous nanodrops does occur to ultimately form unsolvated gaseous ions under typical mass spectrometry operating conditions with commercial mass spectrometers, (2) smaller nanodrops are initially produced using emitters with smaller diameter tips, (3) smaller nanodrops have shorter lifetimes, and (4) the different nanodrop lifetimes can limit the kinetic time frame inside the nanodrop that can affect the extent to which either protein unfolding52 or protein folding31 occurs. Thus, we conclude that the droplet lifetimes determined from the protein folding experiments also applies to bimolecular reactions that occur in identically formed nanodrops. It is possible that the presence of the reagents may affect the physical properties of the nanodrops, especially at high initial concentrations. However, the relative lifetimes of the two different size nanodrops should be the same. Thus, while there may be a slight uncertainty in the absolute lifetimes, there is little uncertainty that the size and lifetimes of nanodrops depends on emitter tip diameter.
Effective reaction rate constant for a bimolecular reaction
In bulk solution, the concentrations of reactants typically change only due to reaction that depletes the reactant concentration. In contrast, the concentration of reactants in the nanodrops changes both due to reaction and due to water evaporation. Water evaporation leads to an increase in reactant concentration with time whereas reaction depletes the reactant concentration with time. Because the reactant concentration changes continuously throughout the nanodrop lifetime, an integrated rate law is used instead of a rate equation. An observed or “effective” rate constant in the nanodrop, knanodrop, was obtained from the integrated rate law for the bimolecular reaction, which can be simplified when the initial concentrations of the two reactants are equal (eqn (2))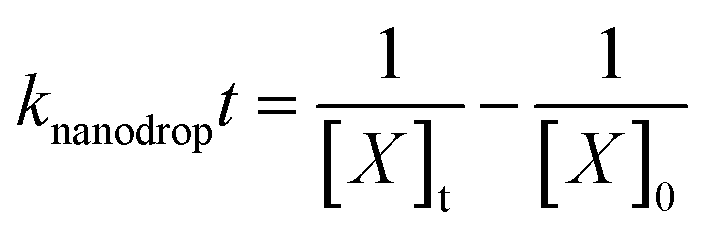 | (2) |
In order to determine the extent of mixing and reaction that can occur in the Taylor cone prior to droplet formation, the flow rates were determined from mass measurements of the nanospray emitter before and after electrospray and these values were corrected for water evaporation from the emitter.31 The total flow rate due to electrospray is approximately 36 nL min−1 and 18 nL min−1 for the 1.84 μm and 684 nm emitters, respectively (Table S4†). The Taylor cone volume was estimated as a cone with the same base diameter of the long dimension of the emitter and the height as 1.5 times this value, resulting in a volume of 2.4 fL and 0.12 fL and an analyte transit time through this region of ∼4.1 μs and ∼0.4 μs for the 1.84 μm and 684 nm emitters, respectively. The time spent in the Taylor cone is short compared to the droplet lifetime indicating that reactions in the Taylor cone do not significantly contribute to the observed acceleration factors.
Spontaneous reduction of species in aqueous nanodrops has been reported.5 To investigate whether this occurs here, solutions of DCIP at either 10 or 100 μM (pH 3) were added to one barrel and water was added to the other barrel of the emitters. No reduced product was observed. The same experiment with L-AA resulted in no oxidation product. These results indicate that both reagents are necessary for a reaction to occur and that the reaction occurs as a result of the intended bimolecular reaction. pH changes can contribute to reaction acceleration.13,16 For the reaction between DCIP and L-AA, the rate constant in solution was measured at pH 3, the same pH as our initial droplets. The rate constant in solution changes by less than 3% between pH 1 and 3.53 Thus, any acidification of the nanodrop as solvent evaporates should not significantly affect the rate for this reaction. The capillary interface temperature can affect some microdroplet acceleration factors.54 Larger nanodrops, such as ones produced by the larger emitter, survive into the interface capillary.31 To investigate the role of temperature, the capillary inlet was varied between 60 and 300 °C. No significant change in the conversion ratio was observed over this temperature range (Fig. S4†).
The role of the droplet surface
An increase in conversion ratio with smaller droplets has been used as evidence that a reaction is accelerated at the surface.2,5,15 In our experiments, the conversion ratio is lower with smaller droplets (Fig. 1a) and the acceleration factor is independent of droplet size (Fig. 1b). These experimental results strongly indicate that this reaction is not accelerated at the nanodrop surface. The initial droplet diameter depends on the diameter of the emitter tip. There is a ∼2.7-fold difference in tip diameter that should translate to a roughly 2.7-fold difference in the surface-to-volume ratios of the nanodrops formed by these two emitters. The acceleration factors (Fig. 2a) were fit to a line over the range of concentrations that these data appear linear. The ratio of acceleration factors for the two different emitter tip sizes varies from 0.73 to 1.17 over a concentration range of 0.5 μM to 50 μM. This range is much smaller than the ∼2.7 fold difference in surface-to-volume ratios. We conclude that these acceleration factors depend primarily on initial reactant concentration and not on the surface-to-volume ratio, which suggests that this reaction is not accelerated at the surface.This reaction does not lead to a significant difference in structure between the reactant and the product. Two hydrogen atoms are transferred to oxidized DCIP to produce reduced DCIP. The similarity in structures between the reactant, product, and likely transition states suggests that the reaction energetics should not be substantially affected by partial solvation that may occur at the surface.21
To further elucidate the role of the surface in these experiments, the maximum surface occupancy was (over)estimated using the average polar surface area for both reactants55,56 (∼40 Å2) and assuming all molecules are at the surface (Fig. 1c). With the smallest emitter, this value ranges from 0.00004% to 0.2%. For the larger emitters that have lower surface-to-volume ratios, the maximum surface occupancy does not exceed 2%, and is less than 0.0001% at the lowest concentration. The extraordinarily low surface occupancy indicates that the vast majority of the droplet surface remains available for reactions to occur, yet the rate acceleration factor changes by more than 104 over a range of initial concentrations. Surface occupancy increases as solvent evaporation occurs and this could be a factor as the size of the droplet shrinks. However, there should be a difference between droplets of different initial size, which is not observed (Fig. 2b). Diffusion of reactants from inside the nanodrop to the surface can occur, so all molecules have the potential to be exposed to the surface especially as the nanodrop diameter decreases due to evaporation.57 However, if this were the primary mechanism, and not reactant concentration due to evaporation, then the conversion ratio would not be expected to increase at lower concentrations. The experimental observation that the reaction acceleration factor increases by more than 104 with decreasing concentration cannot be explained by diffusion and enhanced surface reactivity.
The role of increasing reactant concentration inside a nanodrop
A range in acceleration rate factors by four orders of magnitude for the same reaction in the same initial size nanodrops may initially seem remarkable, and a value of 107 is among the highest acceleration factors reported.13 However, these results are consistent with an increase in analyte concentration due to solvent evaporation that occurs once a nanodrop is initially formed. For a bimolecular reaction, both reactant molecules must be in close proximity for a reaction to occur. Two reactant molecules coming together in dilute solution is a low probability event and hence the rate of a chemical reaction in bulk decreases with decreasing concentration of a reactant. However, the reactant concentration can increase in the nanodrops due to solvent evaporation making it more probable for two reactant molecules to come into close proximity even in initially dilute solutions. At the extreme, two molecules in a single large nanodrop are unlikely to interact because of their low probability of contact. However, interaction between two reactant molecules is nearly guaranteed when virtually all of the solvent has evaporated from the nanodrop thereby bringing the two reactant molecules into close proximity. This concentration effect leads to a large acceleration factor between a reaction in a nanodrop and that in bulk solution.If the sharp drop-off in reaction rate acceleration at lower concentrations (Fig. 1b) is due to on average fewer than two molecules per droplet, then the volume that contains two molecules can be estimated from the concentration corresponding to the midpoint of this fall-off. A volume of 8.3 × 10−2 fL and 1.7 × 10−2 fL would contain two molecules for the 1.84 μm and 684 nm emitters, respectively (Table S4†). This volume corresponds to a spherical droplet with a diameter approximately 1/3 of that of the emitter. This initial droplet size as a fraction of the tip diameter is higher than that for single bore emitters,36,58 but this value is an overestimate because of the statistical probability of having an equal number of molecules of both analytes is low with few molecules in the droplet and the possibility of analyte molecules leaving the nanodrop prior to complete solvent evaporation as a result of droplet fission or ion emission that may occur. The center divider in the theta emitters and the non-symmetrical shape may also affect the droplet formation process. These data provide additional support for our conclusion that reaction rate acceleration is highest when there are a limited number of molecules in each nanodrop, which are brought into close proximity when solvent evaporates. These results are consistent with a recent model that indicates that reactant enrichment due to solvent evaporation can lead to a few orders of magnitude in reaction acceleration and this effect is predicted to be more pronounced for smaller droplets.16
The phenomenon of analyte concentration occurs at all initial concentrations, yet the highest acceleration factors occur at the lower concentrations. The solubility limit of DCIP is around 1 mM,55 and the highest concentration of 500 μM DCIP might be expected to lead to only a 4× increase in the acceleration factor at equilibrium in bulk solution. The higher value measured here could be due to formation of a supersaturated solution owing to the very rapid solvent evaporation that may lead to increased product formation in these nanodrops. Moreover, formation of large aggregates with reactivities that may not reflect those of individual molecules in solution could occur. There are abundant protonated and sodiated homodimers of L-AA at the higher concentrations, but no heterodimers are observed at any concentration, indicating that bifurcation likely occurs with increased concentration within the nanodrop that may lead to decreased reaction efficiency at the higher initial concentrations compared to that at lower concentrations.
Conclusions
In conclusion, these experiments demonstrate the role of increasing analyte concentration due to solvent evaporation from droplets on the resulting acceleration rate factors that are measured. The acceleration rate factor for the bimolecular reaction between DCIP and L-AA ranges from 102 to 107. This value depends on the initial reagent concentration and is highest when the concentration is sufficient for at least a few molecules of each reagent to enter into each of the initially formed droplets. This wide range of acceleration factors does not appear to be related to the air–water interface at the droplet surface, but rather is primarily a result of solvent evaporation that brings widely dispersed reactants into close proximity as the droplet shrinks in size.Many factors differ between this experiment and many prior experiments on reaction acceleration in evaporating droplets. The distance from the emitter to the mass spectrometer inlet affects droplet lifetimes and droplet sizes8,10,15,23 and this effect was not investigated here. Organic solvents have been used in many prior experiments compared to the aqueous solutions used here. Solvent volatility can change droplet lifetimes and the composition of mixed solvents can vary as the more volatile solvent evaporates.59 Never-the-less, reagent concentration is expected to play a role under any condition where droplet evaporation occurs. Although reaction acceleration was demonstrated for a reaction that does not appear to be accelerated at surfaces, this effect should also play a significant role for reactions where acceleration at surfaces may also play a role.
The results presented here indicate that this analyte concentration effect in evaporating droplets is significant and can lead to acceleration factors that are among the highest that have been reported for reaction acceleration in microdroplets where droplet volume is not carefully controlled. In experiments where droplets evaporate prior to chemical analysis, such as occurs in spray ionization methods with mass spectrometry, this reactant concentration effect needs to be taken into account in order to attribute any reaction acceleration to the unusual properties of the droplet surface.
Data availability
Data for this paper, including tabulated forms of calculated values for concentrations, conversion ratios, acceleration factors, and volumes of solutions have been uploaded as part of the ESI.†Author contributions
CJC performed experiments and ERW directed the investigations. CJC and ERW designed experiments, analyzed experimental data, and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by the National Science Foundation Division of Chemistry under grant number CHE-2203907 as well as the Merck DBL SEEDS program and CALSOLV. The authors thank the staff at the University of California, Berkeley Electron Microscope Laboratory for emitter imaging assistance, and Jacob Jordan for the helpful discussions.References
- S. Banerjee and R. N. Zare, Syntheses of Isoquinoline and Substituted Quinolines in Charged Microdroplets, Angew. Chem., Int. Ed., 2015, 127, 15008–15012 CrossRef.
- N. Sahota, D. I. Abusalim, M. L. Wang, C. J. Brown, Z. Zhang, T. J. El-Baba, S. P. Cook and D. E. Clemmer, A Microdroplet-Accelerated Biginelli Reaction: Mechanisms and Separation of Isomers Using IMS-MS, Chem. Sci., 2019, 10, 4822–4827 RSC.
- T. Müller, A. Badu-Tawiah and R. G. Cooks, Accelerated Carbon-Carbon Bond-Forming Reactions in Preparative Electrospray, Angew. Chem., Int. Ed., 2012, 51, 11832–11835 CrossRef PubMed.
- Y. Ju, H. Zhang, W. Wang, Q. Liu, K. Yu, G. Kan, L. Liu and J. Jiang, Aqueous-Microdroplet-Driven Abiotic Synthesis of Ribonucleotides, J. Phys. Chem. Lett., 2022, 13, 567–573 CrossRef CAS PubMed.
- J. K. Lee, D. Samanta, H. G. Nam and R. N. Zare, Micrometer-Sized Water Droplets Induce Spontaneous Reduction, J. Am. Chem. Soc., 2019, 141, 10585–10589 CrossRef CAS PubMed.
- X. Song, Y. Meng and R. N. Zare, Spraying Water Microdroplets Containing 1,2,3-Triazole Converts Carbon Dioxide into Formic Acid, J. Am. Chem. Soc., 2022, 144, 16744–16748 CrossRef CAS PubMed.
- L. Qiu, M. D. Psimos and R. G. Cooks, Spontaneous Oxidation of Aromatic Sulfones to Sulfonic Acids in Microdroplets, J. Am. Soc. Mass Spectrom., 2022, 33, 1362–1367 CrossRef CAS PubMed.
- L. Qiu and R. G. Cooks, Simultaneous and Spontaneous Oxidation and Reduction in Microdroplets by the Water Radical Cation/Anion Pair, Angew. Chem., Int. Ed., 2022, 61, e202210765 CAS.
- E. T. Jansson, Y. H. Lai, J. G. Santiago and R. N. Zare, Rapid Hydrogen-Deuterium Exchange in Liquid Droplets, J. Am. Chem. Soc., 2017, 139, 6851–6854 CrossRef CAS PubMed.
- J. K. Lee, S. Kim, H. G. Nam and R. N. Zare, Microdroplet Fusion Mass Spectrometry for Fast Reaction Kinetics, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 3898–3903 CrossRef CAS PubMed.
- K. Iyer, J. Yi, A. Bogdan, N. Talaty, S. W. Djuric and R. G. Cooks, Accelerated Multi-Reagent Copper Catalysed Coupling Reactions in Micro Droplets and Thin Films, React. Chem. Eng., 2018, 3, 206–209 RSC.
- H. Chen, L. S. Eberlin, M. Nefliu, R. Augusti and R. G. Cooks, Organic Reactions of Ionic Intermediates Promoted by Atmospheric-Pressure Thermal Activation, Angew. Chem., Int. Ed., 2008, 47, 3422–3425 CrossRef CAS PubMed.
- Z. Wei, Y. Li, R. G. Cooks and X. Yan, Accelerated Reaction Kinetics in Microdroplets: Overview and Recent Developments, Annu. Rev. Phys. Chem., 2020, 71, 31–51 CrossRef CAS PubMed.
- R. M. Bain, C. J. Pulliam, F. Thery and R. G. Cooks, Accelerated Chemical Reactions and Organic Synthesis in Leidenfrost Droplets, Angew. Chem., Int. Ed., 2016, 55, 10478–10482 CrossRef CAS PubMed.
- B. M. Marsh, K. Iyer and R. G. Cooks, Reaction Acceleration in Electrospray Droplets: Size, Distance, and Surfactant Effects, J. Am. Soc. Mass Spectrom., 2019, 30, 2022–2030 CrossRef CAS PubMed.
- G. Rovelli, M. I. Jacobs, M. D. Willis, R. J. Rapf, A. M. Prophet and K. R. Wilson, A Critical Analysis of Electrospray Techniques for the Determination of Accelerated Rates and Mechanisms of Chemical Reactions in Droplets, Chem. Sci., 2020, 11, 13026–13043 RSC.
- M. I. Jacobs, R. D. Davis, R. J. Rapf and K. R. Wilson, Studying Chemistry in Micro-Compartments by Separating Droplet Generation from Ionization, J. Am. Soc. Mass Spectrom., 2019, 30, 339–343 CrossRef CAS PubMed.
- K. R. Wilson, A. M. Prophet, G. Rovelli, M. D. Willis, R. J. Rapf and M. I. Jacobs, A Kinetic Description of How Interfaces Accelerate Reactions in Micro-Compartments, Chem. Sci., 2020, 11, 8533–8545 RSC.
- L. Qiu, N. M. Morato, K. H. Huang and R. G. Cooks, Spontaneous Water Radical Cation Oxidation at Double Bonds in Microdroplets, Front. Chem., 2022, 10, 903774 CrossRef CAS PubMed.
- M. F. Ruiz-lo and M. T. C. Martins-Costa, Disentangling Reaction Rate Acceleration in Microdroplets, Phys. Chem. Chem. Phys., 2022, 24, 29700–29704 RSC.
- L. Qiu, Z. Wei, H. Nie and R. G. Cooks, Reaction Acceleration Promoted by Partial Solvation at the Gas/Solution Interface, ChemPlusChem, 2021, 86, 1362–1365 CrossRef CAS PubMed.
- Y. Li, X. Yan and R. G. Cooks, The Role of the Interface in Thin Film and Droplet Accelerated Reactions Studied by Competitive Substituent Effects, Angew. Chem., Int. Ed., 2016, 55, 3433–3437 CrossRef CAS PubMed.
- K. H. Huang, Z. Wei and R. G. Cooks, Accelerated Reactions of Amines with Carbon Dioxide Driven by Superacid at the Microdroplet Interface, Chem. Sci., 2021, 12, 2242–2250 RSC.
- S. Mondal, S. Acharya, R. Biswas, B. Bagchi and R. N. Zare, Enhancement of Reaction Rate in Small-Sized Droplets: A Combined Analytical and Simulation Study, J. Chem. Phys., 2018, 148, 244704 CrossRef PubMed.
- J. K. Lee, K. L. Walker, H. S. Han, J. Kang, F. B. Prinz, R. M. Waymouth, H. G. Nam and R. N. Zare, Spontaneous Generation of Hydrogen Peroxide from Aqueous Microdroplets, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 19294–19298 CrossRef CAS.
- A. Gallo Jr, N. H. Musskopf, X. Liu, Z. Yang, J. Petry, P. Zhang, S. Thoroddsen, H. Im and H. Mishra, On the Formation of Hydrogen Peroxide in Water Microdroplets, Chem. Sci., 2022, 13, 2574–2583 RSC.
- H. Hao, I. Leven and T. Head-Gordon, Can Electric Fields Drive Chemistry for an Aqueous Microdroplet?, Nat. Commun., 2022, 13, 1–8 Search PubMed.
- Y. Huang, K. M. Barraza, C. M. Kenseth, R. Zhao, C. Wang, J. L. Beauchamp and J. H. Seinfeld, Probing the OH Oxidation of Pinonic Acid at the Air-Water Interface Using Field-Induced Droplet Ionization Mass Spectrometry (FIDI-MS), J. Phys. Chem. A, 2018, 122, 6445–6456 CrossRef CAS PubMed.
- A. J. Colussi and S. Enami, Detecting Intermediates and Products of Fast Heterogeneous Reactions on Liquid Surfaces via Online Mass Spectrometry, Atmosphere, 2019, 10, 47 CrossRef CAS.
- M. Tanaka, G. Girard, R. Davis, A. Peuto and N. Bignell, Recommended Table for the Density of water between 0 °C and 40 °C Based on Recent Reports, Metrologia, 2001, 38, 301–309 CrossRef CAS.
- Z. Xia and E. R. Williams, Effect of Droplet Lifetime on Where Ions Are Formed in Electrospray Ionization, Analyst, 2019, 144, 237–248 RSC.
- D. N. Mortensen and E. R. Williams, Ultrafast (1 μs) Mixing and Fast Protein Folding in Nanodrops Monitored by Mass Spectrometry, J. Am. Chem. Soc., 2016, 138, 3453–3460 CrossRef CAS PubMed.
- D. N. Mortensen and E. R. Williams, Microsecond and Nanosecond Polyproline II Helix Formation in Aqueous Nanodrops Measured by Mass Spectrometry, Chem. Commun., 2016, 52, 12218–12221 RSC.
- D. N. Mortensen and E. R. Williams, Investigating Protein Folding and Unfolding in Electrospray Nanodrops upon Rapid Mixing Using Theta-Glass Emitters, Anal. Chem., 2015, 87, 1281–1287 CrossRef CAS PubMed.
- D. N. Mortensen and E. R. Williams, Theta-Glass Capillaries in Electrospray Ionization: Rapid Mixing and Short Droplet Lifetimes, Anal. Chem., 2014, 86, 9315–9321 CrossRef CAS PubMed.
- K. L. Davidson, D. R. Oberreit, C. J. Hogan and M. F. Bush, Nonspecific Aggregation in Native Electrokinetic Nanoelectrospray Ionization, Int. J. Mass Spectrom., 2017, 420, 35–42 CrossRef CAS.
- J. K. Lee, S. Banerjee, H. G. Nam and R. N. Zare, Acceleration of Reaction in Charged Microdroplets, Q. Rev. Biophys., 2015, 48, 437–444 CrossRef CAS.
- M. Wilm and M. Mann, Analytical Properties of the Nanoelectrospray Ion Source, Anal. Chem., 1996, 68, 1–8 CrossRef CAS PubMed.
- M. C. Ramachandra Shastry, S. D. Luck and H. Roder, A Continuous-Flow Capillary Mixing Method to Monitor Reactions on the Microsecond Time Scale, Biophys. J., 1998, 74, 2714–2721 CrossRef PubMed.
- Y. Lai, S. Sathyamoorthi, R. M. Bain and R. N. Zare, Microdroplets Accelerate Ring Opening of Epoxides, J. Am. Soc. Mass Spectrom., 2018, 29, 1036–1043 CrossRef CAS PubMed.
- C. Goy, M. A. C. Potenza, S. Dedera, M. Tomut, E. Guillerm, A. Kalinin, K. Voss, A. Schottelius, N. Petridis, A. Prosvetov, G. Tejeda, J. M. Fernández, C. Trautmann, F. Caupin, U. Glasmacher and R. E. Grisenti, Shrinking of Rapidly Evaporating Water Microdroplets Reveals Their Extreme Supercooling, Phys. Rev. Lett., 2018, 120, 015501 CrossRef CAS PubMed.
- S. Heiles, R. J. Cooper, M. J. DiTucci and E. R. Williams, Sequential Water Molecule Binding Enthalpies for Aqueous Nanodrops Containing a Mono-, Di- or Trivalent Ion and between 20 and 500 Water Molecules, Chem. Sci., 2017, 8, 2973–2982 RSC.
- R. J. Cooper, M. J. Ditucci, T. M. Chang and E. R. Williams, Delayed Onset of Crystallinity in Ion-Containing Aqueous Nanodrops, J. Am. Chem. Soc., 2016, 138, 96–99 CrossRef CAS PubMed.
- R. J. Cooper, J. T. O'Brien, T. M. Chang and E. R. Williams, Structural and Electrostatic Effects at the Surfaces of Size- and Charge-Selected Aqueous Nanodrops, Chem. Sci., 2017, 8, 5201–5213 RSC.
- C. C. Harper, D. D. Brauer, M. B. Francis and E. R. Williams, Direct Observation of Ion Emission from Charged Aqueous Nanodrops: Effects on Gaseous Macromolecular Charging, Chem. Sci., 2021, 12, 5185–5195 RSC.
- W. D. Price and E. R. Williams, Activation of Peptide Ions by Blackbody Radiation: Factors That Lead to Dissociation Kinetics in the Rapid Energy Exchange Limit, J. Phys. Chem. A, 1997, 101, 8844–8852 CrossRef CAS PubMed.
- M. J. Ditucci, C. N. Stachl and E. R. Williams, Long Distance Ion-Water Interactions in Aqueous Sulfate Nanodrops Persist to Ambient Temperatures in the Upper Atmosphere, Chem. Sci., 2018, 9, 3970–3977 RSC.
- M. J. Ditucci, S. Heiles and E. R. Williams, Role of Water in Stabilizing Ferricyanide Trianion and Ion-Induced Effects to the Hydrogen-Bonding Water Network at Long Distance, J. Am. Chem. Soc., 2015, 137, 1650–1657 CrossRef CAS PubMed.
- M. J. DiTucci and E. R. Williams, Nanometer Patterning of Water by Tetraanionic Ferrocyanide Stabilized in Aqueous Nanodrops, Chem. Sci., 2017, 8, 1391–1399 RSC.
- L. Konermann and Y. Haidar, Mechanism of Magic Number NaCl Cluster Formation from Electrosprayed Water Nanodroplets, Anal. Chem., 2022, 94, 16491–16501 CrossRef CAS.
- A. Schmidt, M. Karas and T. Dülcks, Effect of Different Solution Flow Rates on Analyte Ion Signals in Nano-ESI MS, or: When Does ESI Turn into Nano-ESI?, J. Am. Soc. Mass Spectrom., 2003, 14, 492–500 CrossRef CAS PubMed.
- T. J. El-Baba, D. R. Fuller, D. W. Woodall, S. A. Raab, C. R. Conant, J. M. Dilger, Y. Toker, E. R. Williams, D. H. Russell and D. E. Clemmer, Melting Proteins Confined in Nanodroplets with 10.6 μm Light Provides Clues about Early Steps of Denaturation, Chem. Commun., 2018, 54, 3270–3273 RSC.
- M. I. Karayannis, Comparative Kinetic Study for Rate Constant Determination of the Reaction of Ascorbic Acid with 2,6-Dichlorophenolindophenol, Talanta, 1976, 23, 27–30 CrossRef CAS PubMed.
- S. Banerjee and R. N. Zare, Influence of Inlet Capillary Temperature on the Microdroplet Chemistry Studied by Mass Spectrometry, J. Phys. Chem. A, 2019, 123, 7704–7709 CrossRef CAS PubMed.
- Ascorbic Acid, ChemSpider, Royal Society of Chemistry, CSID=10189562, http://www.chemspider.com/Chemical-Structure.10189562.html, (accessed 2022-10-24) Search PubMed.
- 2,6-Dichlorophenolindophenol, ChemSpider, Royal Society of Chemistry, CSID=10661857, http://www.chemspider.com/Chemical-Structure.10661857.html, (accessed 2022-10-24) Search PubMed.
- A. Fallah-Araghi, K. Meguellati, J. Baret, A. E. Harrak, T. Mangeat, M. Karplus, S. Ladame, C. M. Marques and A. D. Griffiths, Enhanced Chemical Synthesis at Soft Interfaces: A Universal Reaction-Adsorption Mechanism in Microcompartments, Phys. Rev. Lett., 2014, 112, 028301 CrossRef PubMed.
- A. C. Susa, Z. Xia and E. R. Williams, Small Emitter Tips for Native Mass Spectrometry of Proteins and Protein Complexes from Nonvolatile Buffers That Mimic the Intracellular Environment, Anal. Chem., 2017, 89, 3116–3122 CrossRef CAS PubMed.
- J. N. Smith, R. C. Flagan and J. L. Beauchamp, Droplet Evaporation and Discharge Dynamics in Electrospray Ionization, J. Phys. Chem. A, 2002, 106, 9957–9967 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Representative mass spectra of reaction; description and tabulated forms of calculated values for concentrations, conversion ratios, acceleration factors, and volume of solutions containing two molecules; capillary inlet temperature effects on conversion ratio. See DOI: https://doi.org/10.1039/d3sc00259d |
| This journal is © The Royal Society of Chemistry 2023 |