
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeparan sulfate glycomimetics via iterative assembly of “clickable” disaccharides†
Cangjie
Yang‡
a,
Yu
Deng‡
a,
Yang
Wang
a,
Chaoshuang
Xia
 b,
Akul Y.
Mehta
c,
Kelly J.
Baker
c,
Anuj
Samal
a,
Putthipong
Booneimsri
ad,
Chanthakarn
Lertmaneedang
ad,
Seung
Hwang
a,
James P.
Flynn
a,
Muqing
Cao
a,
Chao
Liu
a,
Alec C.
Zhu
a,
Richard D.
Cummings
c,
Cheng
Lin
b,
Udayan
Mohanty
*a and
Jia
Niu
*a
b,
Akul Y.
Mehta
c,
Kelly J.
Baker
c,
Anuj
Samal
a,
Putthipong
Booneimsri
ad,
Chanthakarn
Lertmaneedang
ad,
Seung
Hwang
a,
James P.
Flynn
a,
Muqing
Cao
a,
Chao
Liu
a,
Alec C.
Zhu
a,
Richard D.
Cummings
c,
Cheng
Lin
b,
Udayan
Mohanty
*a and
Jia
Niu
*a
aDepartment of Chemistry, Boston College, Chestnut Hill, Massachusetts 02467, USA. E-mail: udayan.mohanty@bc.edu; jia.niu@bc.edu
bCenter for Biomedical Mass Spectrometry, Boston University School of Medicine, Boston, MA 02118, USA
cDepartment of Surgery, Beth Israel Deaconess Medical Center, Harvard Medical School, Brookline Ave, Boston, MA 02215, USA
dDepartment of Chemistry, Faculty of Science, Chulalongkorn University, Bangkok, 10330, Thailand
First published on 28th February 2023
Abstract
Heparan sulfate (HS) glycosaminoglycans are widely expressed on the mammalian cell surfaces and extracellular matrices and play important roles in a variety of cell functions. Studies on the structure–activity relationships of HS have long been hampered by the challenges in obtaining chemically defined HS structures with unique sulfation patterns. Here, we report a new approach to HS glycomimetics based on iterative assembly of clickable disaccharide building blocks that mimic the disaccharide repeating units of native HS. Variably sulfated clickable disaccharides were facilely assembled into a library of mass spec-sequenceable HS-mimetic oligomers with defined sulfation patterns by solution-phase iterative syntheses. Microarray and surface plasmon resonance (SPR) binding assays corroborated molecular dynamics (MD) simulations and confirmed that these HS-mimetic oligomers bind protein fibroblast growth factor 2 (FGF2) in a sulfation-dependent manner consistent with that of the native HS. This work established a general approach to HS glycomimetics that can potentially serve as alternatives to native HS in both fundamental research and disease models.
Introduction
As a major class of glycosaminoglycan polysaccharides, heparan sulfates (HS), are essential components of the cell surface glycocalyx and extracellular matrix that play important roles in cell proliferation, differentiation, migration, and cell–cell interaction.1 HS consist of uronic acid-(1 → 4)-D-glucosamine disaccharide repeating units that are sulfated at various positions. Variable patterns of N-sulfate, O-sulfate and N-acetyl substitutions on the polymeric backbone of HS give rise to a high degree of sequence complexity, which in turn enables active modulation of the sulfate-dependent interactions of HS with a plethora of signaling proteins.2,3 Many HS-binding proteins employ a distinct sequence of positively charged residues to engage with HS, and correspondingly, require specific sulfation patterns of HS to achieve selectivity among the complex glycome.To understand the structure–activity relationships of the sulfation patterns in HS, Seeberger,4,5 Linhardt,6,7 Liu,8,9 Boons,10,11 Weigel,12 DeAngelis,13 Hung,14,15 Huang,16–18 and others19–24 have developed chemical and chemoenzymatic synthetic approaches to well-defined HS oligosaccharides that have significantly advanced our understanding of HS biology. Nevertheless, the chemical synthesis of HS oligosaccharides often requires exquisite synthetic route design and skillful carbohydrate chemistry manipulations, resulting in high production cost and scalability challenges. Chemoenzymatic synthesis has emerged as a scalable method to access HS oligosaccharides, but the requirements for engineered enzymes and large quantities of activated substrates often render the synthesis expensive and technically demanding.
Compared to the native HS structures, HS-mimetic oligomers and polymers require less efforts to synthesize and provide comparable activities in many functional assays, making them highly useful tools to study HS biology. For example, the work by Hsieh–Wilson25,26 and Godula27,28 demonstrated that the repeated display of sulfated disaccharides along a polymer backbone possesses activities similar to the native HS in anticoagulation, stem cell differentiation, and chemokine response. However, most existing HS-mimetic polymers incorporate sulfated oligosaccharides as pendant groups and do not allow precision control of the sulfation patterns, limiting their utility in the investigation into functional roles of sulfation patterns in HS. Herein, we developed a series of “clickable” disaccharides that resemble the repeating unit of HS and can be rapidly assembled into sequence-defined glycomimetic oligomers via the copper(I)-catalyzed alkyne–azide cycloaddition (CuAAC) click chemistry (Fig. 1). These clickable disaccharides consist of an iduronic acid (IdoA)-(β1 → 4)-glucosamine (GlcN) core structure and an azide and a triisopropyl silyl (TIPS)-protected alkyne end groups, with various positions modified by the sulfate group (Fig. 1a). We have shown that a variety of HS-mimetic oligomer structures with defined sulfation patterns could be facilely synthesized following efficient solution-phase iterative assembly and their sequences could be independently verified by tandem mass spectrometry (Fig. 1b). A microarray consisting of HS-mimetic oligomers with various sulfation patterns was fabricated to examine their ability to bind fibroblast growth factor-2 (FGF2), a model protein with well-established sulfation-dependent interactions with HS.29,30 Molecular dynamics (MD) simulation of the glycomimetic oligomers bound to FGF2 corroborated the binding assay results, confirming that these HS-mimetic oligomers bind FGF2 in a sulfation-dependent fashion similar to the native HS oligosaccharides. Taken together, our studies suggest that the clickable disaccharides are facile tools for investigating the functional roles of sulfation in diverse physiological processes involving HS.
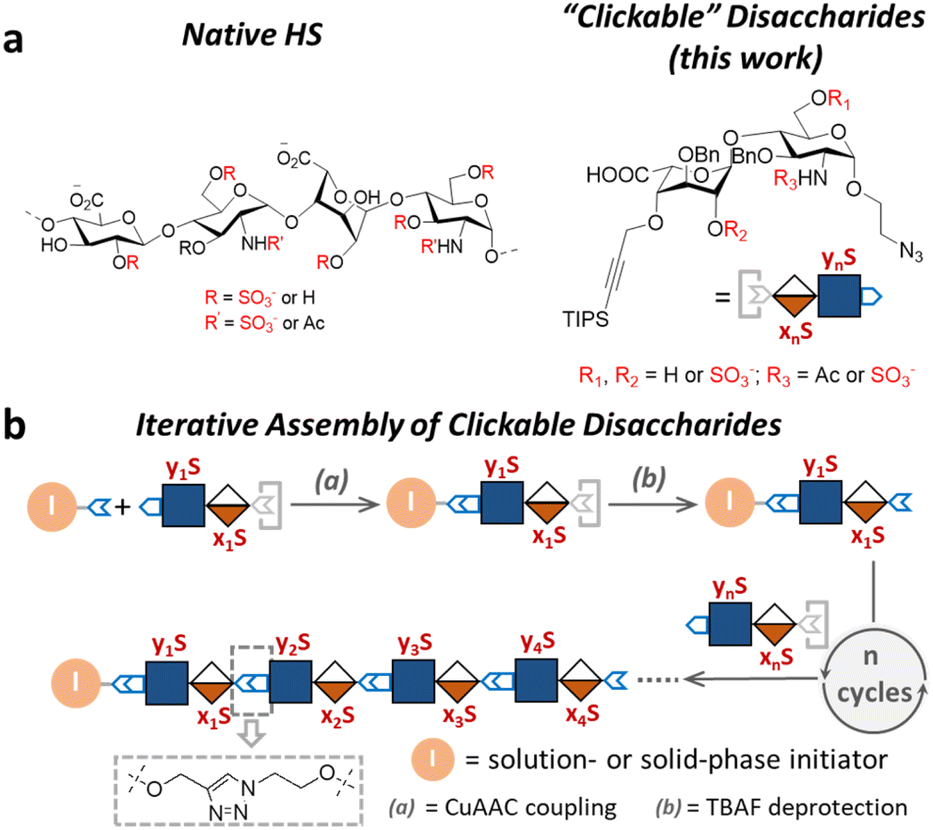 | ||
| Fig. 1 Overview of this work. (a) Chemical structures of natural HS and the HS glycomimetics developed in this work. (b) The iterative assembly of the clickable disaccharides via CuAAc click chemistry. | ||
Results and discussion
Design and synthesis of clickable disaccharides
The development of chemically defined HS mimetics has been stymied by the difficulties in preparing large collections of these compounds with tunable chain length and precisely controlled sulfation patterns. Inspired by the chemical synthesis of polypeptides31 and nucleic acids32via solid-phase and solution-phase iterative synthesis, we envision that a synthetic approach to HS-mimetic oligomers could be realized by connecting modular disaccharide building blocks using the CuAAC click chemistry, due to its mild reaction condition, high coupling efficiency, and excellent tolerance to a broad scope of functionalities.33 In particular, CuAAC coupling can be performed in the late stage of the synthesis with minimal impacts on the structural integrity of the oligomers generated from the iterative synthesis. To this end, we designed the partially protected IdoA-(β1 → 4)-GlcN disaccharides with sulfation at specific hydroxyl and amino groups and an azide and a TIPS-alkyne group at C1 and C4 positions, respectively (Fig. 1). Non-sulfated hydroxyl groups were protected by benzyl ether (Bn) groups. As such, these disaccharide building blocks could be iteratively assembled after the TIPS group is removed via tetra-n-butylammonium fluoride (TBAF) after each coupling step.We began the synthesis of the “clickable” disaccharide building blocks using commercially available diacetone glucose and N-acetylglucosamine starting materials (Fig. 2). Specifically, 1,6-anhydro β-(L)-idopyranose was synthesized from diacetone glucose in six steps.34 While our initial attempt to install triisopropyl silane (TIPS) protected alkyne using TIPS-protected propargyl bromide was unsuccessful due to ester migration, an alternative route from TIPS-protected propargylic trichloroacetimidate successfully afforded anhydrosugar 2. Acetolytic ring opening of 2 provided the 1,6-diacetate, which was converted into thioglycoside 3 in a combined yield of 38% over 3 steps. After the removal of the C6 acetyl group, 4 was oxidized and esterified into thioglycoside methyl ester 5 as an iduronic acid donor in 57% overall yield over 3 steps. To access the glucosamine acceptors, glycosylation of (D)-N-acetylglucosamine 6 with the 2-chloroethanol followed by sequential acetylation and azidation yielded the 1-azidoethyl compound in 65% overall yield. The followed installation of 4,6-benzylidene acetal and benzyl ether furnished 7 in 77% yield. After the benzylation on C4 hydroxyl and the removal of the 4,6-benzylidene protection in 7, the 6-hydroxyl was selectively protected by levulinic acid to afford N-acetylglucosamine acceptor 9 in 61% overall yield. To prepare the glucosamine acceptor for N-sulfation, we deacetylated 7 and protected the C2 amine with 2,2,2-trichloroethoxycarbonyl (Troc) group to arrive at 12 in 86% yield over two steps. Tin(II)-catalyzed benzylation of 12 under acidic conditions followed by the removal of 4,6-benzylidene protection produced the glucosamine acceptor 13 with Troc-protected amine in 31% overall yield. Lastly, glycosylation of acceptor 9 or 13 by thioglycoside donor 5 generated β-linked core disaccharide 10 and 14 in 65% and 76% yields, respectively.
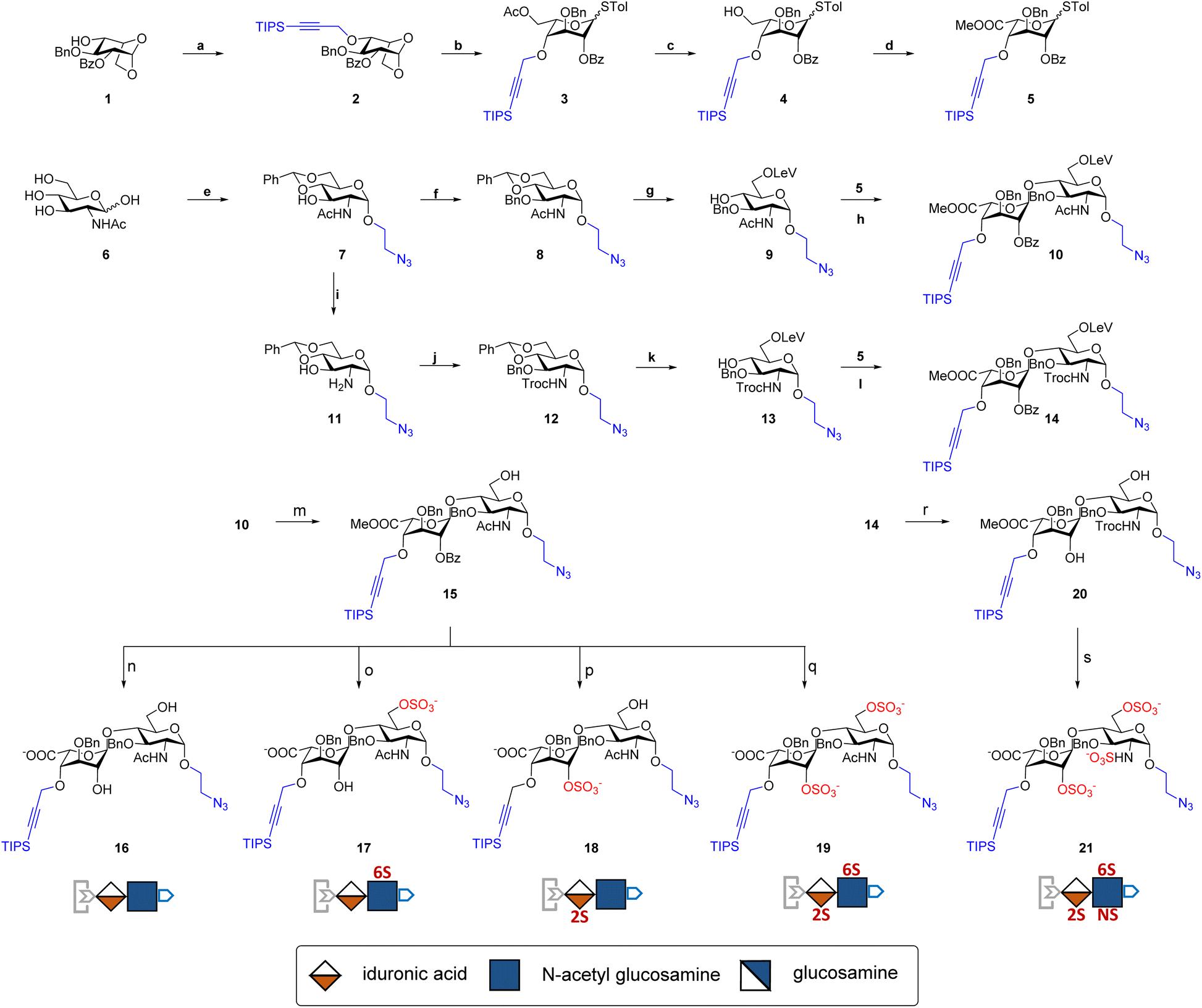 | ||
| Fig. 2 Synthesis of clickable disaccharides. Reagents and conditions: (a) TIPSCHCCH2ONHCCl3, TfOH, molecular sieve 4 Å, 45 °C, 46%; (b): (i) Cu(OTf)2, acetic anhydride, (ii) TolSH, TfOH, DCM, 82% in two steps; (c) AcCl, DCM/MeOH, 92%; (d): (i) TEMPO, BAIB, DCM/H2O, (ii) Me2SO4, acetone, 0 °C, 62%; (e): (i) ClCH2CH2OH, AcCl, 70 °C, 65%, (ii) acetic anhydride, pyridine, quant, (iii) NaN3, DMF, 60 °C, quant, (iv) NaOMe, MeOH, quant, (v) benzaldehyde dimethyl acetal, TsOH, DMF, 65 °C, 77%; (f) BnBr, NaH, DMF, 90%; (g): (i) TFA, DCM/H2O, 30 °C, 74%, (ii) Lev acid, EDC, DMAP, DCM, 91%; (h) TIS, TfOH, molecular sieve 4 Å, 69%; (i) KOH, dioxane/2-methoxyethanol, 120 °C; (j) TrocCl, sat. NaHCO3, THF, 86% in two steps, (k) (i) BnOCNHCCl3, Sn(OTf)2, Et2O, molecular sieve 4 Å, 62%; (ii) TFA, DCM/H2O, 30 °C, 66%; (iii) Lev acid, EDC, DMAP, DCM, 76%; (l) TIS, TfOH, molecular sieve 4 Å, 76%; (m) hydrazine acetate, MeOH/THF, quant; (n): (i) LiOH, MeOH, 0 °C, (ii) NaOH, MeOH/H2O, 80% in 2 steps; (o) (i) SO3·Py, DMF, 87%; (ii) LiOH, MeOH/H2O, 0 °C, (iii) NaOH, MeOH/H2O, 93% in 2 steps; (p): (i) MeONa, MeOH, 88%, (ii) Lev acid, EDC, DMAP, DCM, 83%, (iii) SO3·Py, DMF, 77%, (iv) LiOH, NaOH, THF/H2O, 87%; (q): (i) MeONa, MeOH, 88%, (ii) SO3·Py, DMF, 90%, (iii) LiOH, MeOH/H2O, 93%; (r): (i) hydrazine acetate, THF/H2O, (ii) MeONa, MeOH, 66%; (s) (i) SO3·Py, DMF, 89%, (ii) LiOH, MeOH/H2O, (iii) SO3·Py, NaOH, TEA, DMF, 79% in two steps. | ||
The orthogonal protecting groups in the core disaccharide 10 provided the opportunity for regioselective deprotection and sulfation at various positions. Through protecting group manipulation and O-sulfation, non-sulfated disaccharide 16 and differentially sulfated disaccharides (17−19) were readily obtained. Similarly, trisulfated disaccharide building block 21 was generated from 20via sequential deprotection and O- and N-sulfation. Importantly, disaccharides 17–19 and 21 were confirmed to be compatible with CuAAC coupling and TBAF deprotection conditions (Fig. S1†). Taken together, variably sulfated clickable disaccharides were efficiently prepared and validated for an iterative coupling–deprotection procedure, making them promising building blocks for the synthesis of sequence-defined HS-mimetic oligomers.
Iterative assembly of clickable disaccharides
Next, we explored the iterative assembly of the clickable disaccharides into sequence-defined HS-mimetic oligomers. Starting from a benzyl chloroformate (Cbz)-protected propargyl amine initiator, successive CuAAC coupling and TBAF deprotection were performed in several iterations until the desired length of the oligomer was obtained (Fig. 3a). In each cycle, a unique disaccharide building block was incorporated to generate a sequence-defined oligomer with a specific sulfation pattern. The product of each coupling and deprotection step was isolated by flash silica chromatography and confirmed by liquid chromatography/electrospray ionization mass spectrometry (LC-ESI-MS) before the next cycle. Using 6-O-sulfated disaccharide 17 as a model building block, we constructed a model octasaccharide S34 in an overall yield of 16% in eight steps after isolation by semi-prep HPLC (80% average yield in each step) (Fig. S2†).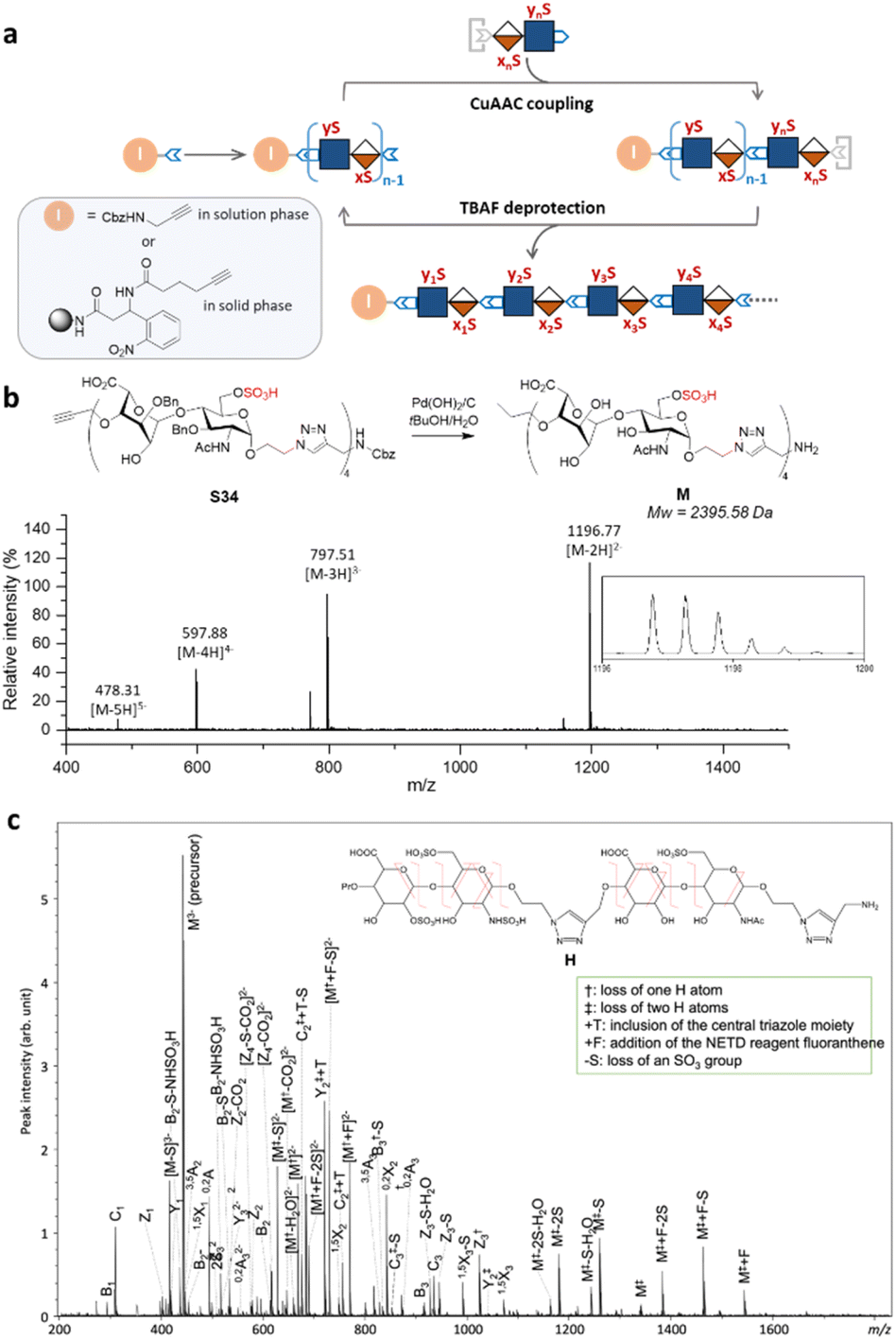 | ||
| Fig. 3 Assembly and MS/MS sequencing of the HS-mimetic oligomer. (a) Solution-phase assembly of variably sulfated clickable disaccharides. (b) LC-ESI-MS analysis of a model HS-mimetic octasaccharide oligomer M after the hydrogenolysis of oligomer S34 generated from solution-phase assembly. (c) NETD MS/MS sequencing of oligomer H allowed for confident assignment of the sulfation pattern. | ||
The Bn-protected oligomers from the solution-phase were deprotected via hydrogenolysis by Pd(OH)2/C in tert-butanol/water (1/1, v/v) in high efficiency, producing the HS-mimetic oligomers in excellent yields. For example, hydrogenolysis of S34 afforded fully deprotected octasaccharide M in 70% yield (Fig. 3b). Following the synthetic procedures established in the model studies, we constructed a library of 14 HS-mimetic tetrasaccharides and octasaccharides with various sulfation patterns (A–N) (Fig. 4a).
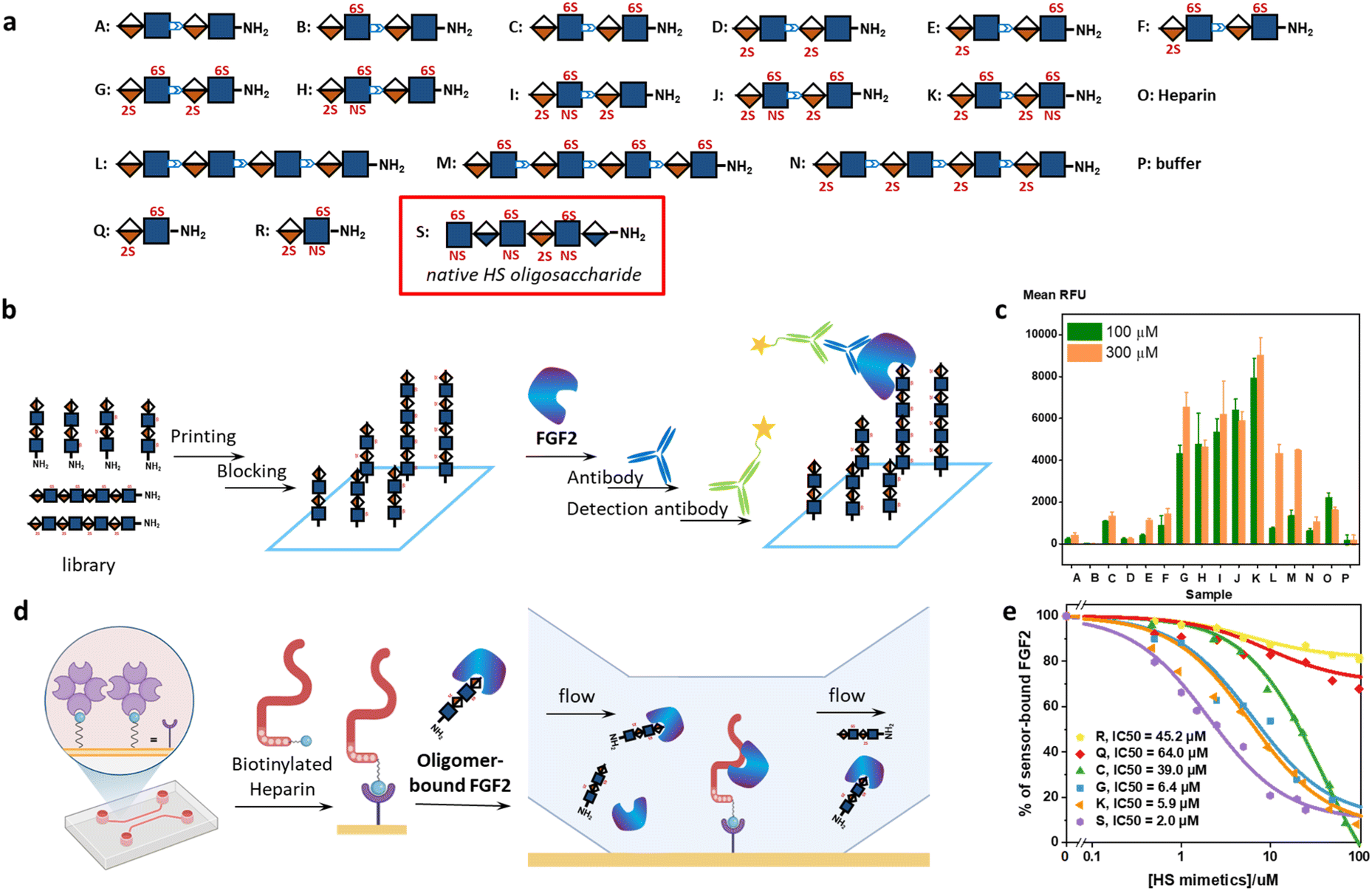 | ||
| Fig. 4 Characterization of the interactions between the HS-mimetic oligomers and FGF2. (a) Sequences of the HS-mimetic oligomer library. A native HS hexasaccharide is used as a control in the SPR competitive binding assay. (b) Workflow of the glycan microarray experiment. (c) Binding profile of the HS-mimetic oligomer library microarray at 100 μM and 300 μM (n = 4). The concentration of FGF2 was 5 μg mL−1. P: buffer only. (d) Workflow of the SPR experiment. (e) FGF2 binding of the HS-mimetic oligomers and native HS hexasaccharide in competition with the immobilized heparin in the SPR experiment. The results are fitted to the competitive inhibition model. | ||
The sequencing ability of the HS-mimetic oligomers is important for future combinatorial library synthesis and high-throughput screening. To this end, the sequence of the synthetic HS glycomimetics were independently verified by tandem mass spectrometry (MS/MS). The conventional collision-induced dissociation (CID) spectrum was dominated by sulfate loss and unable to produce informative tandem mass spectra for reliable sequencing due to the lability of the sulfate group during collisional excitation (Fig. S3†). To overcome this challenge, negative electron transfer dissociation (NETD) was performed on the tetrasaccharide oligomer H (Fig. 3c).35,36 Encouragingly, NETD generated complete series of glycosidic fragments, including all B-, C-, Y-, and Z-ions. Product ions at each cleavage site were detected without sulfate loss, allowing confident assignment of the sulfate modifications at the residue level. Localization of the sulfate modifications within each residue could also be achieved by identifying the mass difference between key cross-ring fragments and their corresponding glycosidic fragment. For example, the presence of a 3,5A2 ion (m/z 451.022) and a 0,2X2 ion (m/z 845.181) provides conclusive evidence for assignment of the 6-O-sulfation and N-sulfation at the second GlcN residue. The presence of the triazole linker does not appear to interfere with the structural characterization, as no fragmentation within the triazole moiety was observed in the NETD spectrum.
Protein-binding studies
We used glycan microarray and surface plasmon resonance (SPR) to analyze the interactions between the HS-mimetic oligomers and FGF2. Glycan microarrays have become a key glycomic technology for systematically evaluating the structure–activity relationships of complex carbohydrates.37 We fabricated microarrays of the HS-mimetic oligomers by reacting their terminal amino groups with N-hydroxysuccinimide (NHS)-activated glass slides (Fig. 4b). The microarray of HS-mimetic oligomers was incubated with FGF2 in the Tris buffer (20 mM Tris–HCl pH 7.4, 150 mM NaCl, 2 mM CaCl, 2 mM MgCl) containing Tween-20 (0.05%) and BSA (1%), and binding was indirectly observed using unlabeled anti-FGF2 antibody and a goat-anti-mouse IgG 635 detection antibody. Fluorescence signal at excitation wavelength 635 nm was measured and the average signal for replicate spots (n = 4) was calculated as the mean RFU. Encouragingly, the HS-mimetic oligomers exhibited sulfation-dependent binding to FGF2 similar to the native HS (Fig. 4c). It was observed that, at both 100 μM and 300 μM of HS-mimetic oligomers, FGF2 bound tetrasaccharide oligomers G–K comparably but to a greater extent than the oligomers with lower sulfation density. Specifically, the oligomers with N-sulfation, 2-O-, and 6-O-sulfation (e.g., J and K) exhibited higher fluorescent intensity than those carrying only 2-O- or 6-O-sulfation (e.g., C, D, M, or N) in the microarray experiment using fluorescently labeled FGF2. To rule out the avidity effect in which multiple surface-bound HS-mimetic oligomers in proximity bind to the same FGF2 protein, the FGF2-oligomer interactions were further investigated using an SPR competition assay (Fig. 4d). Biotinylated heparin was immobilized onto the streptavidin-functionalized SPR sensor chip, to which a solution containing a fixed concentration of FGF2 and various concentrations of a specific HS-mimetic oligomer was injected and allowed to flow through the sensor chip. The HS-mimetic oligomer competes with the immobilized heparin and inhibits its binding to FGF2, resulting in decreased responses in the SPR sensorgrams. The half maximal inhibitory concentration (IC50) of the HS-mimetic oligomers against the FGF2 binding by heparin could be determined by fitting the concentration-dependent SPR responses to a competitive inhibition model (see ESI Methods†).38 Consistent with the microarray results, oligomers with higher sulfation, e.g., K (IC50 = 5.9 μM) and G (IC50 = 6.4 μM), exhibited higher affinity to FGF2 than the ones with lower sulfation, e.g., C (IC50 = 39.0 μM) (Fig. 4e and S4†). Notably, the IC50 of K is only slightly lower than a native HS hexasaccharide S, GlcNS(6S)-GlcA-GlcNS(6S)-IdoA(2S)-GlcNS(6S)-GlcA (IC50 = 2.0 μM). Furthermore, the IC50 value of K is also consistent with the FGF2 affinity of an analogous native IdoA-GlcNS(6S)-IdoA(2S)-GlcNS(6S) tetrasaccharide reported by Boons et al. (KD = 1.2 μM).10 In contrast, disaccharide Q and R are insufficient to inhibit the FGF2 binding by heparin: not only their IC50 values are much higher (IC50 = 45.2 μM for trisulfated R; IC50 = 64.0 μM for disulfated Q), the maximal inhibition for Q and R were also lower than 30% of the total binding. Taken together, the structure–activity relationship demonstrated by the HS-mimetic oligomers in FGF2 recognition is consistent with that of the native HS oligosaccharides.10,39,40Molecular dynamics simulations
While the CuAAC coupling chemistry enables highly efficient iterative assembly in the solution phase, it involves incorporation of a non-natural triazole spacer between adjacent disaccharide units. To study how the spacer in HS-mimetic oligomers affects their ability to replicate behavior of native HS, we performed molecular dynamics (MD) simulations of compound K (Fig. 5a) and compared it with its native counterpart, a IdoA(2S)-GlcNAc(6S)-IdoA(2S)-GlcNS(6S) tetrasaccharide. To model the compounds, we employed a combination of the GLYCAM 06j41,42 and generalized Amber force fields43 in the OpenMM software package44 (refer to ESI† for details). We analyzed four major polymer properties to compare the behavior of HS mimetic with native HS: (1) glycosidic linkages; (2) ring-flipping states; (3) end-to-end distance; and (4) ion atmosphere. For this, we used the crystallographic conformation of an FGF2-bound HS tetramer (PDB: 1BFB45) as a point of reference. In addition, we performed simulations of FGF2 binding to demonstrate that mimetic HS can reproduce the sulfate-dependent binding behavior of the native polymer.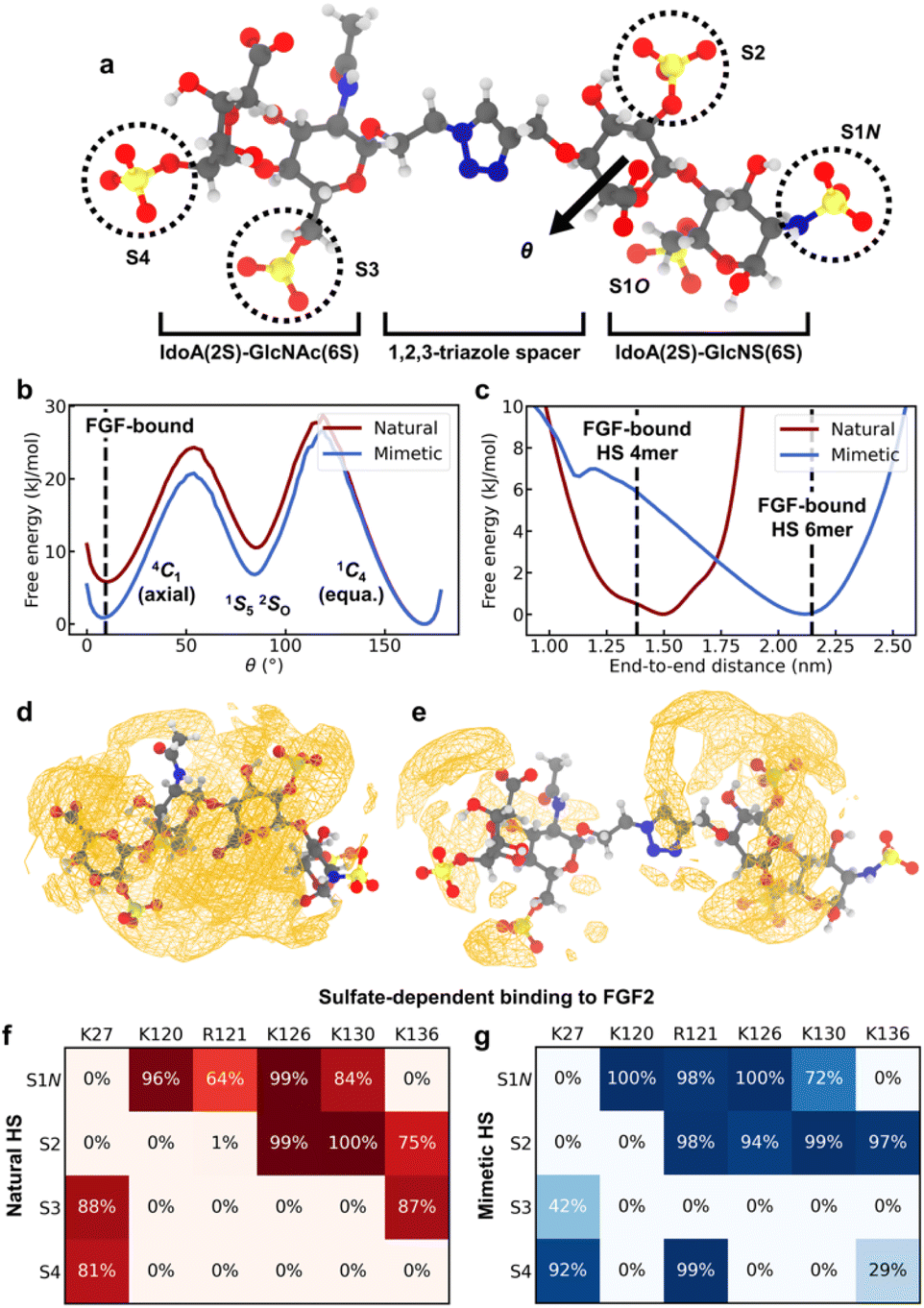 | ||
| Fig. 5 MD simulation results. (a) Representative configuration of compound K, with the sulfate groups on each monosaccharide residue labeled. Atom color code: H – white, C – gray, N – blue, O – red, S – yellow. (b) Free energy of ring flipping for central IdoA(2S) (adjacent to spacer). θ is the Cremer–Pople parameter50 for monitoring ring flipping between chair conformers. The 1C4 conformer corresponds to 2-O-sulfate adopting the equatorial geometry, while 4C1 corresponds to the axial geometry. (c) Free energy of end-to-end distances, defined as the distance between C4 of terminal IdoA(2S) and C1 of terminal GlcNS(6S). Dashed lines give the crystallographic distances for FGF2-bound tetramer and hexamer structures (PDB: 1BFB and 1BFC). (d) Spatial distribution function (SDF) of Na+, a representative counterion, around native HS. Orange wireframe shows the 1.2 M isosurface for Na+ concentration. (e) SDF of Na+ ions around compound K, with the 1.2 M isosurface shown. (f and g) Interactions between the sulfate groups of K or the native HS tetrasaccharide and the amino acids of the FGF2 heparin binding domain. (f) Probability for sulfate groups to be in contact with positively charged residues when the native HS tetrasaccharide binds to FGF2. A sulfate is defined to be in contact with a residue if the distance between any non-hydrogen atoms is less than 0.3 nm. (g) Probability of contact with positively charged residues when compound K binds to FGF2. | ||
Rotation about glycosidic linkages are important for determining the long-chain conformation of HS polymers.46 For K, we find that the flexibility of the GlcNAc(6S)-IdoA(2S) linkage is greatly enhanced due to the triazole spacer (Fig. S5a†). In contrast, conformations of adjacent linkages are identical between the native HS tetrasaccharide and K (Fig. S5b and c†), indicating they are unaffected by the spacer.
Ring flipping is also key for deciding HS conformation and can affect protein binding.47 The ring of central IdoA(2S) has a thermodynamically preferred chair conformer (1C4) (Fig. 5b), consistent with previous studies.47,48 In K, the less favored chair conformer (4C1) is stabilized by ∼5 kJ mol−1 relative to the native HS tetrasaccharide. The lower energy of this conformer (4C1) in the mimetic is due to its spacer, which creates a spatial buffer between residues and mitigates unfavorable interactions in the axial geometry. Based on the crystal structure (PDB: 1BFB) reported by Faham, et al.,45 HS tetramer binds to FGF2 mainly through two adjacent sulfate groups, one of which is on a central IdoA(2S) residue. Interestingly, in order for FGF2-binding to occur, IdoA(2S) must adopt the less favored ring conformer (Fig. 5b). Since the triazole spacer in mimetic HS stabilizes the less favored conformer, this suggests that the free-energy cost of binding due to ring flipping is lowered in the mimetic. Meanwhile, ring flipping for other residues is similar, in terms of free energy, between native HS tetrasaccharide and K (Fig. S6†).
The overall polymer conformation of HS can be characterized by measuring its end-to-end distance. The preferred end-to-end distance of native HS tetrasaccharide is 1.5 nm, close to the FGF2-bound distance (Fig. 5c). In contrast, the most favorable distance for K is 2.1 nm. However, the flexibility of the triazole spacer enables K to adopt folded conformations where its polymer length is close to the FGF2-bound distance, at a free-energy cost of ∼6 kJ mol−1. On the other hand, the preferred end-to-end distance of K is closest in length to HS hexamer (PDB: 1BFC45) when bound to FGF2 (Fig. 5c).
Protein binding in HS occurs through its sulfate groups,49 which requires desolvation of counterions. We analyzed Na+ solvation around native HS tetrasaccharide and compound K through the potential of mean force (PMF). The PMF value at a given distance from a sulfate group is the free energy change associated with moving an Na+ ion from bulk solution to that distance. The PMF between sulfate groups and their surrounding Na+ solvation shell is greater in the native HS tetrasaccharide than in K (Fig. S7†). This suggests that desolvation of Na+ ions around sulfate groups can occur more easily in the mimetic than in native HS. This effect can be attributed to the greater polymer length, and hence smaller linear charge density, of K. Accordingly, we find the counterion density around most regions of compound K to be smaller than that around native HS (Fig. 5d and e). Furthermore, the average excess number51 of Na+ ions surrounding native HS is greater than that around the mimetic by 0.4 (Fig. S7†). This is due to the stronger interaction of native HS tetrasaccharide, compared to K, with its surrounding Na+ ion environment.
Finally, we compared the sulfate-dependent binding behavior of native HS tetrasaccharide and K by performing representative simulations of these two oligomers binding to FGF2. Based on the crystal structure (PDB: 1BFB45), HS tetramer binds to the positively charged domain of FGF2 containing Lys120, Arg121, Lys126, and Lys130. For the native HS tetrasaccharide, we reproduced this interaction in our simulations (Fig. S8a†) and find that the polymer remains stably bound to FGF2 (Fig. S8c†). Specifically, sulfate groups S1N and S2 (Fig. 5a) show the most extensive binding to the positively charged domain (Fig. 5f and S8a†), consistent with the crystallographic binding configuration (Table S1†). Sulfates S3 and S4 (Fig. 5a) can also interact via Lys27 and Lys136, though to a lesser degree. Likewise, K binds to FGF2 stably (Fig. S8d†). This binding occurs predominantly through S1N and S2 (Fig. 5g and S8b†), the same as in the native HS tetrasaccharide and similar to the crystal structure (Table S1†). In addition, K adopts a partly folded conformation (Fig. S8b†), enabled by the flexibility of the triazole spacer. This conformation facilitates partial interactions of S3 with Lys27 and binding of S4 to both Lys27 and Arg121 (Fig. 5g and S8b†). These lesser variations in S3/S4 interactions are the main point of difference between the bound structures of K and native HS tetrasaccharide. Overall, these results show that the HS mimetic is capable of reproducing the sulfate-dependent binding behavior of its native counterpart.
The simulation results also provide further insights on the sulfate-mediated binding of HS-mimetic oligomers. From the protein-binding studies by microarray and SPR, we find that oligomers G–K show strong binding affinity to FGF2 (Fig. 4a and c). With the exception of G, these mimetic oligomers all possess N- and 2-O-sulfation on adjacent residues. This is consistent with our simulations, which indicate that the predominant interactions of the glycomimetic oligomers with the positively charged domain of FGF2 occurs through adjacent N- and 2-O-sulfates (i.e., S1N and S2 in Fig. 5g). The importance of these two sulfation positions is also observed in native HS (Fig. 5e).39,40,45 In addition, the strong binding oligomers (G–K) have the highest sulfation density, with 4–5 sulfate groups across four monosacharride units. The simulated binding conformation of K illustrates how up to four of these sulfates can simultaneously participate in interactions with FGF2 (Fig. 5g and S8b†), which explains the contribution of the additional sulfate groups in G–K. Binding by these additional sulfate groups is also seen in native HS tetrasaccharides (Fig. 5e).45
Overall, our simulations suggest that the greater flexibility and weaker ion interactions of the HS-mimetic oligomers can compensate for the effects of the triazole spacer. This can lead to a high degree of similarity in protein binding between HS-mimetic oligomers and native HS, as shown by their sulfate-dependent interactions with FGF2.
Conclusions
In conclusion, we developed a new approach to HS glycomimetics based on iterative assembly of clickable disaccharide building blocks. Mimicking the disaccharide repeating units of native HS, these clickable disaccharides can undergo solution-phase iterative assembly via click chemistry to generate sequence-defined glycooligomers with variable sulfation patterns. To demonstrate the versatility of this general approach, we constructed a library of HS-mimetic oligomers with defined sulfation patterns that can be readily sequenced by tandem mass spectrometry. Microarray and SPR experiments confirmed that, despite incorporating non-natural triazole spacers, these HS-mimetic oligomers bind FGF2 in a sulfation-dependent manner consistent with that of the native HS. This structure–function relationship was further confirmed by MD simulations of the HS-mimetic tetramer and the corresponding native HS tetrasaccharide. As a future direction, we envision that the combinatorial assembly of modular HS disaccharide structures via flexible non-natural linkers will allow access to sulfation patterns and conformations both within native HS and beyond. Such an ability makes the method presented herein readily applicable to generating glycomimetic oligomers capable of interacting with other HS-binding proteins. In parallel to our work, Huang and coworkers demonstrated that the amidation chemistry widely used in peptide synthesis can also be used to construct HS-mimetic oligomers with diverse sulfation patterns.52 Collectively, these results established general approaches to HS glycomimetics that can serve as promising alternatives to native HS in functional studies in both fundamental research and disease models.Data availability
MD simulation files are included in the electronic attachment (HS_simulation_files.zip); all trajectory data are available on request. All other data, including the experimental data and characterizations of the new compounds, are available in the manuscript or the ESI.†Author contributions
CY, YD and JN conceived and designed the project and wrote the manuscript. JN oversaw the project. CY, YD, PB, CL, SH, JF, MC, AS, CL, and ACZ synthesized the clickable disaccharides and the HS glycomimetics. YW and UM performed the computational studies. AYM, KJB, and RDC performed the microarray experiment. CX and CL conducted the MS/MS analysis of the glycomimetic oligomers. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a startup fund from Boston College to J. N. and the NIH Director's New Innovator Award (1DP2HG011027-01) to J. N. We gratefully acknowledge the support of the National Center for Functional Glycomics (NCFG) at Beth Israel Deaconess Medical Center funded by the National Institute for General Medical Sciences (R24GM137763) at NIH. NMR characterizations in this work were supported by the NIH-S10 (1S10OD026910-01A1) and the NSF-MRI (CHE-2117246). The content of this work is solely the responsibility of the authors and does not necessarily represent the official views of the NIGMS or the NIH.Notes and references
- D. L. Rabenstein, Heparin and heparan sulfate: structure and function, Nat. Prod. Rep., 2002, 19, 312–331 RSC.
- H. Lortat-Jacob, A. Grosdidier and A. Imberty, Structural diversity of heparan sulfate binding domains in chemokines, Proc. Natl. Acad. Sci., 2002, 99, 1229–1234 CrossRef CAS PubMed.
- A. Yayon, M. Klagsbrun, J. D. Esko, P. Leder and D. M. Ornitz, Cell surface, heparin-like molecules are required for binding of basic fibroblast growth factor to its high affinity receptor, Cell, 1991, 64, 841–848 CrossRef CAS PubMed.
- J. L. de Paz, C. Noti and P. H. Seeberger, Microarrays of synthetic heparin oligosaccharides, J. Am. Chem. Soc., 2006, 128, 2766–2767 CrossRef CAS PubMed.
- S. Eller, M. Collot, J. Yin, H. S. Hahm and P. H. Seeberger, Automated solid-phase synthesis of chondroitin sulfate glycosaminoglycans, Angew. Chem., Int. Ed., 2013, 52, 5858–5861 CrossRef CAS PubMed.
- S. Y. Kim, F. Zhang, D. A. Harris and R. J. Linhardt, Structural Features of Heparin and Its Interactions With Cellular Prion Protein Measured by Surface Plasmon Resonance, Front. Mol. Biosci., 2020, 7, 594497 CrossRef CAS PubMed.
- B. F. Cress, U. Bhaskar, D. Vaidyanathan, A. Williams, C. Cai, X. Liu, L. Fu, V. M-Chari, F. Zhang, S. A. Mousa, D. J. S, M. A. G. Koffas and R. J. Linhardt, Heavy heparin: a stable isotope-enriched, chemoenzymatically-synthesized, poly-component drug, Angew. Chem., Int. Ed., 2019, 131, 6023–6027 CrossRef.
- Y. Xu, S. Masuko, M. Takieddin, H. Xu, R. Liu, J. Jing, S. A. Mousa, R. J. Linhardt and J. Liu, Chemoenzymatic synthesis of homogeneous ultralow molecular weight heparins, Science, 2011, 334, 498–501 CrossRef CAS PubMed.
- Z. Wang, P.-H. Hsieh, Y. Xu, D. Thieker, E. J. E. Chai, S. Xie, B. Cooley, R. J. Woods, L. Chi and J. Liu, Synthesis of 3-O-sulfated oligosaccharides to understand the relationship between structures and functions of heparan sulfate, J. Am. Chem. Soc., 2017, 139, 5249–5256 CrossRef CAS PubMed.
- C. Zong, A. Venot, X. Li, W. Lu, W. Xiao, J.-S. L. Wilkes, C. L. Salanga, T. M. Handel, L. Wang, M. A. Wolfert and G.-J. Boons, Heparan sulfate microarray reveals that heparan sulfate–protein binding exhibits different ligand requirements, J. Am. Chem. Soc., 2017, 139, 9534–9543 CrossRef CAS.
- S. Arungundram, K. Al-Mafraji, J. Asong, F. E. Leach III, I. J. Amster, A. Venot, J. E. Turnbull and G.-J. Boons, Modular synthesis of heparan sulfate oligosaccharides for structure− activity relationship studies, J. Am. Chem. Soc., 2009, 131, 17394–17405 CrossRef CAS PubMed.
- E. N. Harris and P. H. Weigel, The ligand-binding profile of HARE: hyaluronan and chondroitin sulfates A, C, and D bind to overlapping sites distinct from the sites for heparin, acetylated low-density lipoprotein, dermatan sulfate, and CS-E, Glycobiology, 2008, 18, 638–648 CrossRef CAS PubMed.
- T. A. Kane, C. L. White and P. L. DeAngelis, Functional characterization of PmHS1, a Pasteurella multocida heparosan synthase, J. Biol. Chem., 2006, 281, 33192–33197 CrossRef CAS.
- Y.-P. Hu, Y.-Q. Zhong, Z.-G. Chen, C.-Y. Chen, Z. Shi, M. M. L. Zulueta, C.-C. Ku, P.-Y. Lee, C.-C. Wang and S.-C. Hung, Divergent synthesis of 48 heparan sulfate-based disaccharides and probing the specific sugar–fibroblast growth factor-1 interaction, J. Am. Chem. Soc., 2012, 134, 20722–20727 CrossRef CAS.
- Y.-P. Hu, S.-Y. Lin, C.-Y. Huang, M. M. L. Zulueta, J.-Y. Liu, W. Chang and S.-C. Hung, Synthesis of 3-O-sulfonated heparan sulfate octasaccharides that inhibit the herpes simplex virus type 1 host–cell interaction, Nat. Chem., 2011, 3, 557–563 CrossRef CAS PubMed.
- S. B. Dulaney, Y. Xu, P. Wang, G. Tiruchinapally, Z. Wang, J. Kathawa, M. H. El-Dakdouki, B. Yang, J. Liu and X. Huang, Divergent synthesis of heparan sulfate oligosaccharides, J. Org. Chem., 2015, 80, 12265–12279 CrossRef CAS PubMed.
- N. J. Pawar, L. Wang, T. Higo, C. Bhattacharya, P. K. Kancharla, F. Zhang, K. Baryal, C. X. Huo, J. Liu and R. J. Linhardt, Expedient synthesis of core disaccharide building blocks from natural polysaccharides for heparan sulfate oligosaccharide assembly, Angew. Chem., Int. Ed., 2019, 131, 18750–18756 CrossRef.
- B. Yang, K. Yoshida, Z. Yin, H. Dai, H. Kavunja, M. H. El-Dakdouki, S. Sungsuwan, S. B. Dulaney and X. Huang, Chemical synthesis of a heparan sulfate glycopeptide: Syndecan-1, Angew. Chem., Int. Ed., 2012, 124, 10332–10336 CrossRef.
- B. Kuberan, M. Z. Lech, D. L. Beeler, Z. L. Wu and R. D. Rosenberg, Enzymatic synthesis of antithrombin III–binding heparan sulfate pentasaccharide, Nat. Biotechnol., 2003, 21, 1343–1346 CrossRef CAS PubMed.
- U. Lindahl, J.-p. Li, M. Kusche-Gullberg, M. Salmivirta, S. Alaranta, T. Veromaa, J. Emeis, I. Roberts, C. Taylor and P. Oreste, Generation of “Neoheparin” from E. coli K5 Capsular Polysaccharide, J. Med. Chem., 2005, 48, 349–352 CrossRef CAS PubMed.
- M. D. Burkart, M. Izumi, E. Chapman, C.-H. Lin and C.-H. Wong, Regeneration of PAPS for the enzymatic synthesis of sulfated oligosaccharides, J. Org. Chem., 2000, 65, 5565–5574 CrossRef CAS PubMed.
- J. Zhao, Y. Zhu, X. Song, Y. Xiao, G. Su, X. Liu, Z. Wang, Y. Xu, J. Liu and D. Eliezer, 3-O-Sulfation of Heparan Sulfate Enhances Tau Interaction and Cellular Uptake, Angew. Chem., Int. Ed., 2020, 59, 1818–1827 CrossRef CAS PubMed.
- B. Wu, N. Wei, V. Thon, M. Wei, Z. Yu, Y. Xu, X. Chen, J. Liu, P. G. Wang and T. Li, Facile chemoenzymatic synthesis of biotinylated heparosan hexasaccharide, Org. Biomol. Chem., 2015, 13, 5098–5101 RSC.
- Y. Chen, Y. Li, H. Yu, G. Sugiarto, V. Thon, J. Hwang, L. Ding, L. Hie and X. Chen, Tailored design and synthesis of heparan sulfate oligosaccharide analogues using sequential one-pot multienzyme systems, Angew. Chem., Int. Ed., 2013, 125, 12068–12072 CrossRef.
- G. J. Sheng, Y. I. Oh, S.-K. Chang and L. C. Hsieh-Wilson, Tunable heparan sulfate mimetics for modulating chemokine activity, J. Am. Chem. Soc., 2013, 135, 10898–10901 CrossRef CAS PubMed.
- Y. I. Oh, G. J. Sheng, S. K. Chang and L. C. Hsieh-Wilson, Tailored glycopolymers as anticoagulant heparin mimetics, Angew. Chem., Int. Ed., 2013, 125, 12012–12015 CrossRef.
- M. L. Huang, R. A. Smith, G. W. Trieger and K. Godula, Glycocalyx remodeling with proteoglycan mimetics promotes neural specification in embryonic stem cells, J. Am. Chem. Soc., 2014, 136, 10565–10568 CrossRef CAS PubMed.
- M. R. Naticchia, L. K. Laubach, E. M. Tota, T. M. Lucas, M. L. Huang and K. Godula, Embryonic stem cell engineering with a glycomimetic FGF2/BMP4 co-receptor drives mesodermal differentiation in a three-dimensional culture, ACS Chem. Biol., 2018, 13, 2880–2887 CrossRef CAS PubMed.
- W. L. McKeehan, F. Wang and M. Kan, The heparan sulfate–fibroblast growth factor family: diversity of structure and function, Prog. Nucleic Acid Res. Mol. Biol., 1997, 59, 135–176 Search PubMed.
- L. Pellegrini, Role of heparan sulfate in fibroblast growth factor signalling: a structural view, Curr. Opin. Struct. Biol., 2001, 11, 629–634 CrossRef CAS PubMed.
- R. Merrifield, Automated Synthesis of Peptides: Solid-phase peptide synthesis, a simple and rapid synthetic method, has now been automated, Science, 1965, 150, 178–185 CrossRef CAS.
- M. H. Caruthers, Gene synthesis machines: DNA chemistry and its uses, Science, 1985, 230, 281–285 CrossRef CAS PubMed.
- H. C. Kolb, M. Finn and K. B. Sharpless, Click chemistry: diverse chemical function from a few good reactions, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- S.-C. Hung, X.-A. Lu, J.-C. Lee, M. D.-T. Chang, S.-l. Fang, T.-c. Fan, M. M. L. Zulueta and Y.-Q. Zhong, Synthesis of heparin oligosaccharides and their interaction with eosinophil-derived neurotoxin, Org. Biomol. Chem., 2012, 10, 760–772 RSC.
- Y. Huang, X. Yu, Y. Mao, C. E. Costello, J. Zaia and C. Lin, De novo sequencing of heparan sulfate oligosaccharides by electron-activated dissociation, Anal. Chem., 2013, 85, 11979–11986 CrossRef CAS PubMed.
- H. Hu, Y. Huang, Y. Mao, X. Yu, Y. Xu, J. Liu, C. Zong, G.-J. Boons, C. Lin and Y. Xia, A computational framework for heparan sulfate sequencing using high-resolution tandem mass spectra, Mol. Cell. Proteomics, 2014, 13, 2490–2502 CrossRef CAS PubMed.
- X. Song, J. Heimburg-Molinaro, R. D. Cummings and D. F. Smith, Chemistry of natural glycan microarrays, Curr. Opin. Chem. Biol., 2014, 18, 70–77 CrossRef CAS PubMed.
- S. Cochran, C. P. Li and V. Ferro, A surface plasmon resonance-based solution affinity assay for heparan sulfate-binding proteins, Glycoconjugate J., 2009, 26, 577–587 CrossRef CAS PubMed.
- S. Guimond, M. Maccarana, B. Olwin, U. Lindahl and A. Rapraeger, Activating and inhibitory heparin sequences for FGF-2 (basic FGF). Distinct requirements for FGF-1, FGF-2, and FGF-4, J. Biol. Chem., 1993, 268, 23906–23914 CrossRef CAS.
- M. Ishihara, Y. Kariya, H. Kikuchi, T. Minamisawa and K. Yoshida, Importance of 2-O-sulfate groups of uronate residues in heparin for activation of FGF-1 and FGF-2, J. Biochem., 1997, 121, 345–349 CrossRef CAS PubMed.
- K. N. Kirschner, A. B. Yongye, S. M. Tschampel, J. González-Outeiriño, C. R. Daniels, B. L. Foley and R. J. Woods, GLYCAM06: a generalizable biomolecular force field. Carbohydrates, J. Comput. Chem., 2008, 29, 622–655 CrossRef CAS PubMed.
- A. Singh, M. B. Tessier, K. Pederson, X. Wang, A. P. Venot, G.-J. Boons, J. H. Prestegard and R. J. Woods, Extension and validation of the GLYCAM force field parameters for modeling glycosaminoglycans, Can. J. Chem., 2016, 94, 927–935 CrossRef CAS PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, Development and testing of a general amber force field, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- P. Eastman, J. Swails, J. D. Chodera, R. T. McGibbon, Y. Zhao, K. A. Beauchamp, L.-P. Wang, A. C. Simmonett, M. P. Harrigan and C. D. Stern, OpenMM 7: Rapid development of high performance algorithms for molecular dynamics, PLoS Comput. Biol., 2017, 13, e1005659 CrossRef PubMed.
- S. Faham, R. Hileman, J. Fromm, R. Linhardt and D. Rees, Heparin structure and interactions with basic fibroblast growth factor, Science, 1996, 271, 1116–1120 CrossRef CAS PubMed.
- J. Berglund, T. Angles d'Ortoli, F. Vilaplana, G. Widmalm, M. Bergenstråhle-Wohlert, M. Lawoko, G. Henriksson, M. Lindström and J. Wohlert, A molecular dynamics study of the effect of glycosidic linkage type in the hemicellulose backbone on the molecular chain flexibility, Plant J., 2016, 88, 56–70 CrossRef CAS.
- P.-H. Hsieh, D. F. Thieker, M. Guerrini, R. J. Woods and J. Liu, Uncovering the relationship between sulphation patterns and conformation of iduronic acid in heparan sulphate, Sci. Rep., 2016, 6, 1–8 CrossRef PubMed.
- B. M. Sattelle, S. U. Hansen, J. Gardiner and A. Almond, Free energy landscapes of iduronic acid and related monosaccharides, J. Am. Chem. Soc., 2010, 132, 13132–13134 CrossRef CAS.
- M. C. Meneghetti, A. J. Hughes, T. R. Rudd, H. B. Nader, A. K. Powell, E. A. Yates and M. A. Lima, Heparan sulfate and heparin interactions with proteins, J. R. Soc., Interface, 2015, 12, 20150589 CrossRef PubMed.
- D. Cremer and J. Pople, General definition of ring puckering coordinates, J. Am. Chem. Soc., 1975, 97, 1354–1358 CrossRef CAS.
- A. Wang, M. Levi, U. Mohanty and P. C. Whitford, Diffuse ions coordinate dynamics in a ribonucleoprotein assembly, J. Am. Chem. Soc., 2022, 144, 9510–9522 CrossRef CAS PubMed.
- J. Zhang, L. Liang, W. Yang, S. Ramadan, K. Baryal, C.-X. Huo, J. Bernard, J. Liu, L. Hsieh-Wilson, F. Zhang, R. J. Linhardt and X. Huang, Expedient Synthesis of a Library of Heparan Sulfate-Like “Head-to-Tail” Linked Multimers for Structure and Activity Relationship Studies, Angew. Chem., Int. Ed., 2022, 61, e202209730 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc00260h |
| ‡ These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |