
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic duality of indolyl 1,3-heteroatom transposition†
Yujin
Lee
,
Yun Seung
Nam
,
Soo Young
Kim
,
Jeong Eun
Ki
and
Hong Geun
Lee
 *
*
Department of Chemistry, Seoul National University, 1, Gwanak-ro, Gwanak-gu, Seoul 08826, South Korea. E-mail: hgleee@snu.ac.kr
First published on 16th June 2023
Abstract
A novel mechanistic duality has been revealed from the indolyl 1,3-heteroatom transposition (IHT) of N-hydroxyindole derivatives. A series of in-depth mechanistic investigations suggests that two separate mechanisms are operating simultaneously. Moreover, the relative contribution of each mechanistic pathway, the energy barrier for each pathway, and the identity of the primary pathway were shown to be the functions of the electronic properties of the substrate system. Based on the mechanistic understanding obtained, a mechanism-driven strategy for the general and efficient introduction of a heteroatom at the 3-position of indole has been developed. The reaction developed exhibits a broad substrate scope to provide the products in various forms of the functionalised indole. Moreover, the method is applicable to the introduction of both oxygen- and nitrogen-based functional groups.
Introduction
Indole is one of the most important heterocyclic motifs found in natural and synthetic compounds of pharmacological importance.1 Although the native form of the bicyclic heterocycle acts as a centrally active scaffold, an even greater degree of structural diversity and associated bioactivity could be acquired by dehydrogenative/dearomative incorporation of a heteroatom at the 3-position (Fig. 1A).2 Therefore, the preparation of the C3-heterofunctionalised structures, in the form of an indole (A), indolenine (B), or indoline (C) has been an important subject of investigation. In the rich history of their synthetic development, the vast majority of approaches have relied on the intrinsic nucleophilicity of indole at the 3-position to react with an electrophilic source of heteroatoms (Fig. 1B(a)).3 More recently, alternative strategies that exploit the nucleophilic sources of heteroatoms under oxidative conditions (Fig. 1B(b)) or radical precursors (Fig. 1B(c)) have emerged as useful toolkits to embellish the indolyl framework.4,5 Despite these remarkable advances, however, general access to all three types of products, i.e., A–C, has yet to be established, primarily because of the highly reactive nature of the products and the chemical compatibility of the reagents used for the preparation of each type of product. | ||
| Fig. 1 (A) Indole compounds with a heteroatom substituent at 3-position; (B) conventional synthetic strategies towards C3-functionalized indole derivatives. | ||
An entirely orthogonal approach that can address these synthetic challenges is the indolyl 1,3-heteroatom transposition (IHT, Fig. 1B(d)). The rearrangement reaction of the N-hydroxyindole derivatives, originally reported by Sundberg and further expanded by Hamana and Somei, exploits the readily accessible N-hydroxyindole as the pivotal starting point that can undergo the C3-oxygenation reaction with the intermediacy of the corresponding ester, phosphonate, or sulfonate.6 Over time, the IHT strategy and its variants have shown their competence as effective toolkits for the functionalisation of the 3-position of indole derivatives together with other related heterocycles.7,8 However, the lack of understanding of the mechanistic details of the system hampers the broader application of the synthetic strategy for indole functionalisation, especially with regard to the deliverable types of heteroatoms.
Analogous rearrangements of N-oxyenamines have been reported in numerous settings with extensive mechanistic research.9,10 On the other hand, the mechanistic details of the IHT reaction in particular have not been systematically surveyed. Because of the unique electronic nature of the N-hydroxyindolyl framework and the involvement of a dearomatisation process in the course of the reaction, we speculated that a special feature might be in action during the IHT reaction. Consequently, in order to establish the IHT reaction as a general synthetic strategy for the functionalisation of the indole derivatives, the identification of the corresponding mechanism has been pursued.
Herein, we report an in-depth investigation to identify the unique mechanistic basis of the IHT process. Extensive 18O isotope labelling experiments revealed the unprecedented mechanistic duality of the IHT reaction. A key substituent effect of the system was also found, which became the basis of the electronically driven rate enhancement strategy for the IHT reaction. Based on the acquired mechanistic understanding, a general and efficient synthetic platform could be established for the oxygenation or amidation at the 3-position of an indole to produce all possible forms of products, i.e., indole, indolenine and indoline.
Result and discussion
Electronically driven rate enhancement of the IHT reaction
A key observation was serendipitously made when the N-hydroxytryptamine derivative 1a underwent the IHT reaction at room temperature via acylation with carboxylic acids with different electronic properties (Scheme 1). When n-hexanoic acid (2a) or unsubstituted benzoic acid (2b) was used as the coupling partner, only direct O-acylation products were obtained (2-Int). However, as the arene portion of the acyl donor became more electron-deficient, the rearrangement products began to form as a mixture with the acylation intermediate (2c–2e). Finally, the use of pentafluorobenzoic acid gave the rearrangement product as an exclusive product of the reaction with a 53% yield (2f).11 The results clearly demonstrate the strong correlation of the reaction efficiency with the electron deficiency of the substituent at the 2′-position, suggesting that the inductive facilitation is responsible for the observed rate enhancement. Accordingly, the observed facilitation of the reaction prompted us to carry out a systematic analysis of the mechanism of the IHT reaction with a special focus on the electronics of the 2′-postion.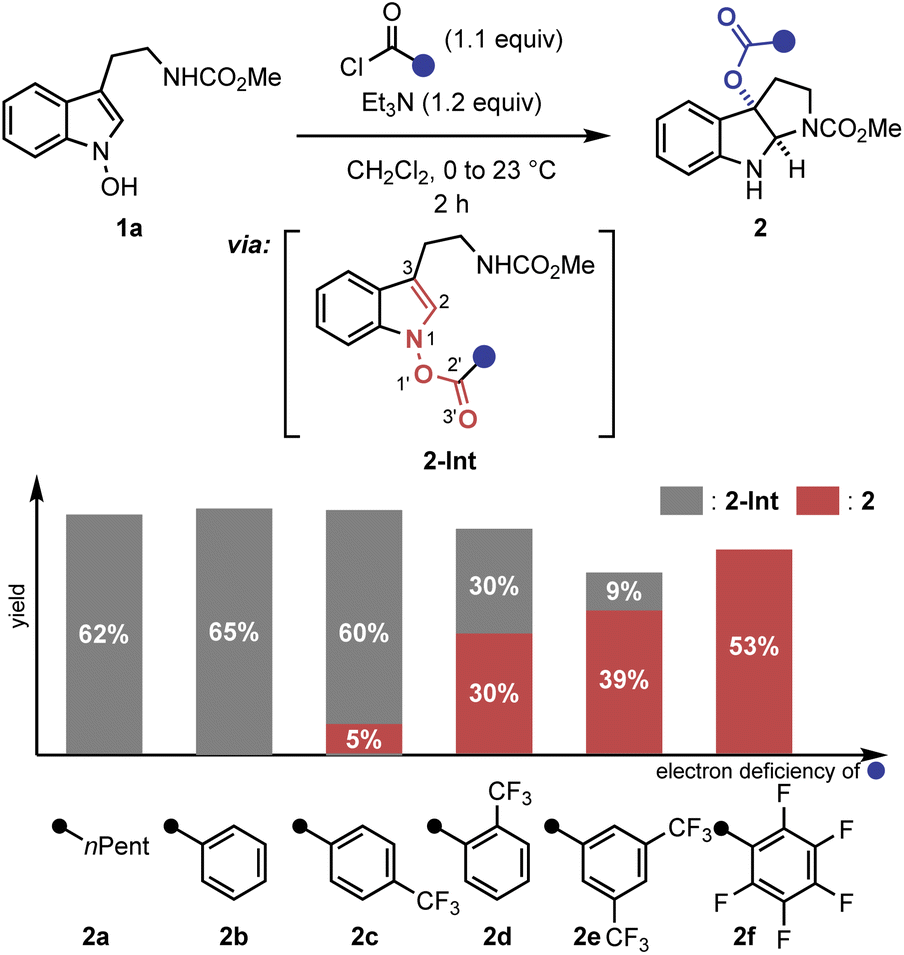 | ||
| Scheme 1 Identification of the 2′-substituent effect in the IHT reaction. Reactions performed with benzoyl chloride (1.1 equiv.) and Et3N (1.2 equiv.) in CH2Cl2 (0.05 M) at 0 °C to 23 °C for 2 h on a 0.1–0.3 mmol scale. Yields of the isolated product are reported and the ratio of 2-Int and 2 was determined by 1H-NMR. | ||
The plausible mechanism of the transformation from the functionalised N-hydroxyindole starting material (I) to the rearrangement product (V) can be divided into three different pathways (Fig. 2).12 Firstly, a mechanism involving the early formation of the C–O bond to give either a diradical (II) or a zwitterionic (II′) intermediate was conceived (path a, associative mechanism). Alternatively, a classical concerted pericyclic pathway with varying degrees of synchronicity can be postulated (path b, concerted mechanism). This particular case includes pathways that involve some degree of charge separation in the transition state. The last possibility proceeds through the initial cleavage of the N–O bond of the starting material (I) to form either a radical pair (IV) or an ion pair intermediate (IV′) (path c, dissociative mechanism). Of the three major candidates, the associative pathway (path a) is highly unlikely, because of the lability of the N–O bond (∼57 kcal mol−1) compared to that of the newly formed C–O bond (80–90 kcal mol−1).13,14 Accordingly, we have tried to assess the mechanism with special focus on the concerted (path b) and the dissociative (path c) mechanisms.
 | ||
| Fig. 2 Mechanistic possibilities for the IHT reaction. | ||
Mechanistic investigation I: 18O-labelling studies
The mechanistic investigation began with isotope labelling studies of the carbonyl oxygen of the IHT reaction precursors, the acyl N-hydroxyindoles, with the 18O atom (Fig. 3). With the position-specific placement of the 18O atom in the carbonyl group of the IHT precursor (18O-1), the intervention of a specific pathway can be traced (Fig. 3A).15 The involvement of the concerted pathway should result in the exclusive migration of 18O to the 3-position of pyrroloindoline (18O-2a), the involvement of the dissociative pathway should produce a mixture of products: 18O-2a and 18O-2b, in which the 18O labelling is evenly distributed between the C3-oxygen and the carbonyl oxygen. Based on these assumptions, a series of 18O-enriched indolyl benzoates with different electron-withdrawing capacities were prepared (Fig. 3B). Benzoates with phenyl (18O-1-A), 3-bromo-4-fluorophenyl (18O-1-B), and pentafluorophenyl (18O-1-C) groups were independently exposed to synthetically relevant rearrangement reaction conditions for each precursor. The products subsequently underwent N-alkylation and ester cleavage to give 3-hydroxypyrroloindoline products 18O-4, which were subjected to high-resolution mass spectrometry (HRMS) analysis to determine the degree of the 18O enrichment.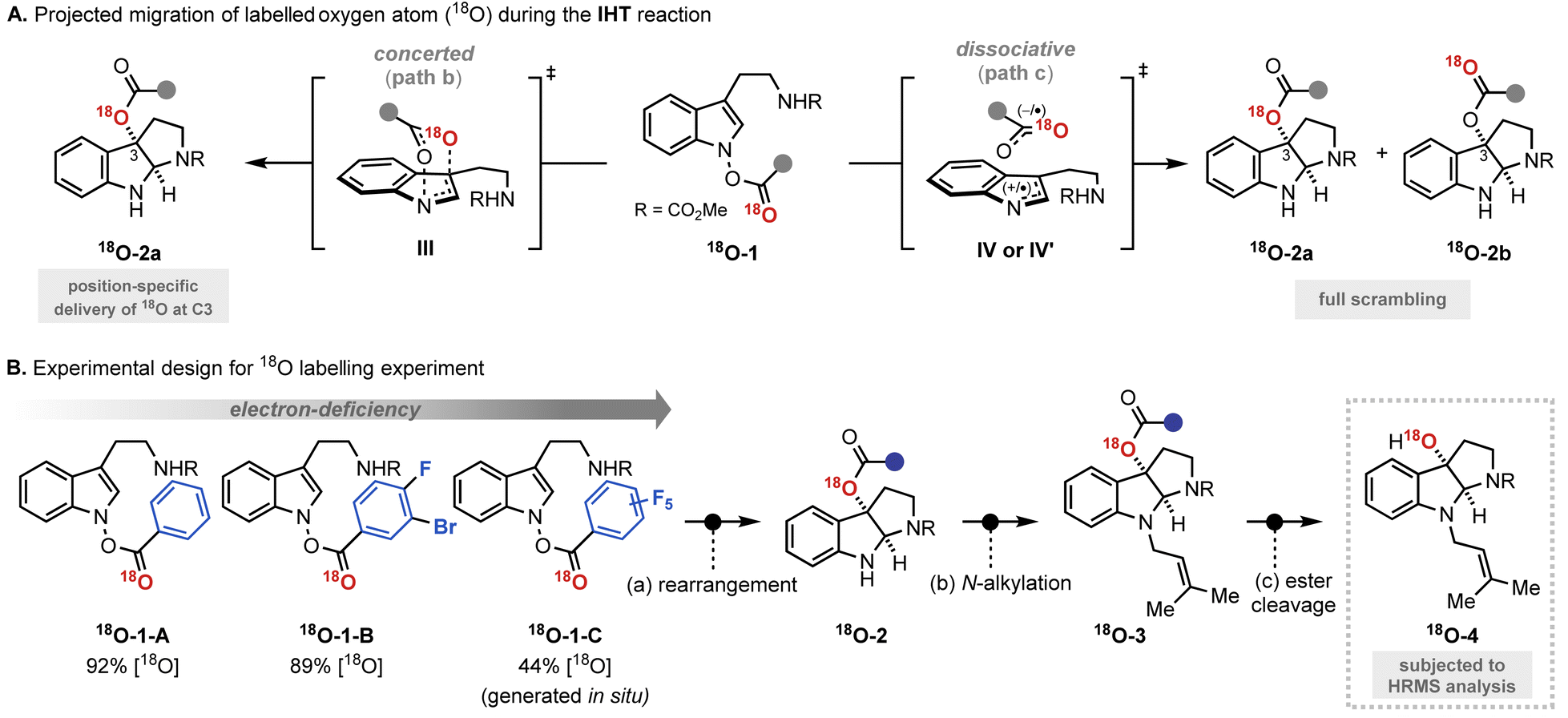 | ||
| Fig. 3 Mechanistic investigation with 18O labelling experiment. (A) Projected migration of labelled oxygen atom (18O) during the IHT reaction; (B) experimental design for 18O labelling experiment. | ||
The experimental results revealed a number of unique mechanistic features of the rearrangement process (Fig. 4A). Most importantly, the rearrangement reactions featured only partial conservation with respect to the position of the 18O labelling. The expected completely position-specific delivery (path b) or full scrambling (path c) of 18O was not observed in any of the cases tested. Based on the report that even the most asynchronous concerted pathway exhibits complete preservation of the isotopically labelled atom during the analogous rearrangement process, the contribution of a single border-line mechanism cannot explain the observed experimental outcome.16 Thus, the isotope labelling experiment indicates the involvement of at least two independent reaction mechanisms, path b and path c.
 | ||
| Fig. 4 The result of the 18O labelling experiment. (A) Effect of the electronic properties of the system; (B) quantitative analysis to determine the involvement of each pathway. | ||
The second and even more remarkable characteristics of the rearrangement process were identified using a quantitative analysis. Assuming path b and path c are primarily operating for the IHT process, the relative contribution of each pathway could be determined (Fig. 4B). With the relative degrees of participation for path b and path c denoted as x and y, respectively, the ratio of x![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :y could be derived from the ratio of 18O-4 and 16O-4 in the final product (see ESI† for details). The calculated values, x and y, revealed that the contribution of each pathway was directly affected by the electronic nature of the substituent at the 2′-position. As benzoate became more electron deficient, a greater degree of isotope mixing was observed, indicating an increased level of involvement for the dissociative pathway (path c). More importantly, the major pathway with the dominant contribution to the product formation changed from the concerted pericyclic pathway (path b) to the stepwise pathway (path c). It is believed that the inductive stabilisation of the fragment pair is responsible for the observed preference, although the exact structural identity of the pair is unclear at this point. Also, kinetic facilitation by the weakening of the N–O bond during the formation of the fragment pair (path c) could be more effective than that in the pericyclic process (path b).
:y could be derived from the ratio of 18O-4 and 16O-4 in the final product (see ESI† for details). The calculated values, x and y, revealed that the contribution of each pathway was directly affected by the electronic nature of the substituent at the 2′-position. As benzoate became more electron deficient, a greater degree of isotope mixing was observed, indicating an increased level of involvement for the dissociative pathway (path c). More importantly, the major pathway with the dominant contribution to the product formation changed from the concerted pericyclic pathway (path b) to the stepwise pathway (path c). It is believed that the inductive stabilisation of the fragment pair is responsible for the observed preference, although the exact structural identity of the pair is unclear at this point. Also, kinetic facilitation by the weakening of the N–O bond during the formation of the fragment pair (path c) could be more effective than that in the pericyclic process (path b).
The observed mechanistic duality in the IHT reaction was particularly notable in several ways. The idea of the participation of multiple mechanisms in a [3,3]-sigmatropic rearrangement was found by the pioneering contributions of Houk, Doering, and Gajewski.17 The chameleon characteristic of the mechanism is expressed as a nature in which the single operating mechanism shifts from one type to another by the electronic perturbation of the system. In IHT, however, the overall transformation is supported by the coexistence of two independent mechanisms, in which their weighted average can be shifted based on the electronics of the system. Such a unique mechanistic duality of a [3,3]-sigmatropic rearrangement has been reported only in specially substituted O-benzoyl aniline N-oxides, wherein the isotope scrambling pathway was detected as an insignificant side pathway that contributes minimally to product formation.10g In the current case, the special mechanistic duality is inherently present in the general indolyl framework. Furthermore, either of the mechanistic pathways can be the dominant pathway depending on the electronics of the substrate, suggesting that special engineering of the system should be possible.
The third and final thing to note regarding the observed characteristics is the overall rate enhancement. The change in the electronics at the 2′-position not only altered the composition of the contributing mechanism but also lowered the activation energy required for both pathways. While 18O-1-A is incapable of generating the rearrangement product at room temperature, the pentafluorophenyl variant 18O-1-C smoothly gave the product with a 1:3 ratio of the concerted and stepwise mechanisms. Both processes could be operated at room temperature in the case of 18O-1-C, indicating a significant reduction in the required activation energies. Therefore, the facilitated product formation could be observed in both the mechanistic pathways in action.
Next, the dependence of the mechanism on the electronic properties of the indole back bone was examined (Fig. 5). When an analogous set of 18O labelling experiments were performed with tryptamine derivatives carrying a 5-bromo substituent (σp = 0.26), the substrate underwent the IHT reaction primarily by a concerted pathway (path b). Regardless of the electronic properties of the benzoyl group, the contribution of the dissociative pathway (path c) was shown to be minimal. In addition, the new system required either a longer reaction time (18O-1-D) or an elevated reaction temperature (18O-1-E) to fully consume the reactant. The observed 18O labelling result, together with the diminished reaction kinetics, was particularly consistent with the reduced contribution of the dissociative pathway, where the fragmented indolyl intermediate should suffer from electronic destabilisation (path c).
 | ||
| Fig. 5 The influence of electronic modification on the indole backbone. | ||
Mechanistic investigation II: additional mechanistic studies
Additional experiments were performed to further delineate the mechanistic details of the IHT reaction (Fig. 6). Firstly, a crossover experiment was conducted using 2b-Int and 2b′-Int (Fig. 6A). Under standard conditions, no appreciable amount of the crossover product was observed. The absence of the crossover products reinforces the primary involvement of the concerted mechanism in this particular system (path b). However, the possibility of rapid recombination of dissociated fragments cannot be excluded (path c). Subsequently, a series of trapping experiments were carried out in the presence of various radical scavengers to identify the nature of the fragments involved (Fig. 6B).18 Under the standard C–O bond-forming conditions, no significant decrease in yield was observed for 1a. However, based on the HRMS analysis, a small amount of the trapping product was detected in the presence of 1,1-diphenylethylene, suggesting the involvement of a radical intermediate. Taken together, the formation of the radical pair that undergoes rapid recombination is considered as a non-significant yet contributing factor for the dissociative mechanism (path c).19 | ||
| Fig. 6 Additional mechanistic studies for IHT reaction. (A) Crossover experiment; (B) radical trap experiment. | ||
Next, to further substantiate the mechanistic duality of the IHT reaction, indolyl N-carbamate substrates were utilised (Fig. 7). We hypothesised that whereas the direct operation of the concerted pathway should generate a C3-oxygenation product, the engagement of a dissociative mechanism should result in the C3-amination product via a rapid decarboxylation of the carbamate anion.20 Indeed, the indolyl N-phenyl carbamate 2g-Int successfully gave a 2:1 mixture of the C3-oxygenation (2g) and C3-amination (2h) products, indicating the presence of two independently operating mechanisms.21
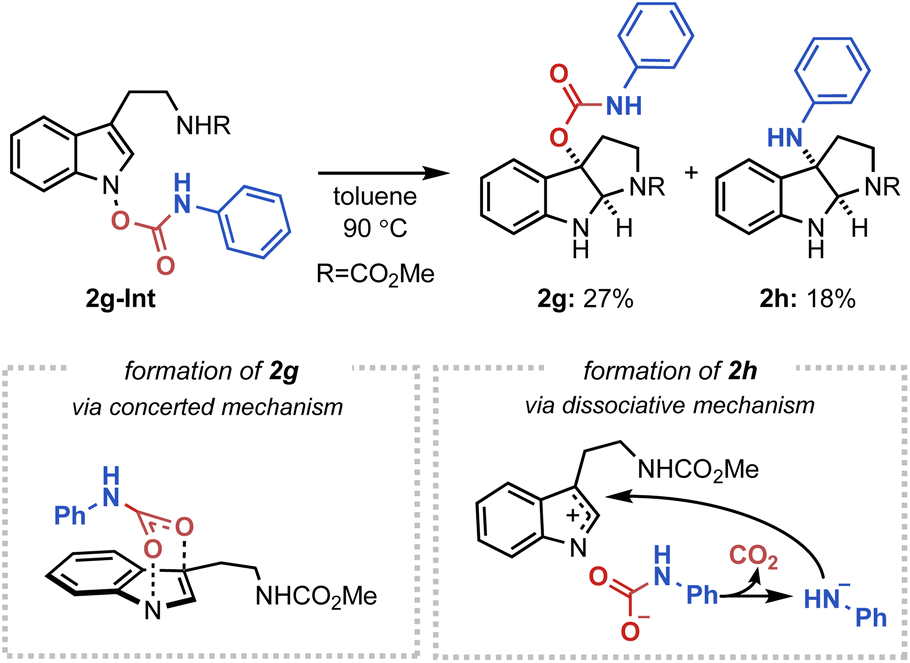 | ||
| Fig. 7 The IHT reaction of indolyl N-carbamates. | ||
The conclusions drawn from the previous experimental investigations are shown in Fig. 8. Firstly, two separate mechanisms, a concerted and a dissociative mechanism, operate simultaneously in the IHT reaction system. Secondly, the degree of mechanistic contribution of each pathway can be shifted by modifying the electronic properties of the system, particularly at the 2′ position, and it is important to note that either mechanistic pathway can be the dominant process for product formation. Thirdly, in the course of the electronic modification, the level of the activation energy can be changed for both pathways of the IHT reaction. Fourthly, the identity of the fragment pair in the dissociative pathway is unclear.
 | ||
| Fig. 8 Mechanistic highlights of the IHT reaction. | ||
The electronic responsiveness consistently observed in the IHT reaction can be rationalised from both the concerted and dissociative mechanistic points of view (Fig. 9). In the case of the concerted mechanism, the observed reactivity trend can be explained by frontier molecular orbital theory (Fig. 9A).22 The energy level of the highest occupied molecular orbital (HOMO), which is mainly located in the indole framework, can be increased by incorporation of electron-donating groups on the indole core. On the other hand, the energy level of the lowest unoccupied molecular orbital (LUMO), which lies primarily in the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of the benzoate can be lowered by installing electron-withdrawing groups next to the CO bond. Thus, electronic polarisation within the substrate should reduce the HOMO–LUMO gap. In the case of the dissociation mechanism, the increased electron density on the indolyl backbone and the electron-withdrawing nature of the benzoate should facilitate the formation of the indolyl cation and benzoate anion by heterolytic cleavage of the N–O bond (Fig. 9B). However, the basis for the influence of the electronic polarisation in favour of the radical pair formation by N–O homolysis is unclear.
O bond of the benzoate can be lowered by installing electron-withdrawing groups next to the CO bond. Thus, electronic polarisation within the substrate should reduce the HOMO–LUMO gap. In the case of the dissociation mechanism, the increased electron density on the indolyl backbone and the electron-withdrawing nature of the benzoate should facilitate the formation of the indolyl cation and benzoate anion by heterolytic cleavage of the N–O bond (Fig. 9B). However, the basis for the influence of the electronic polarisation in favour of the radical pair formation by N–O homolysis is unclear.
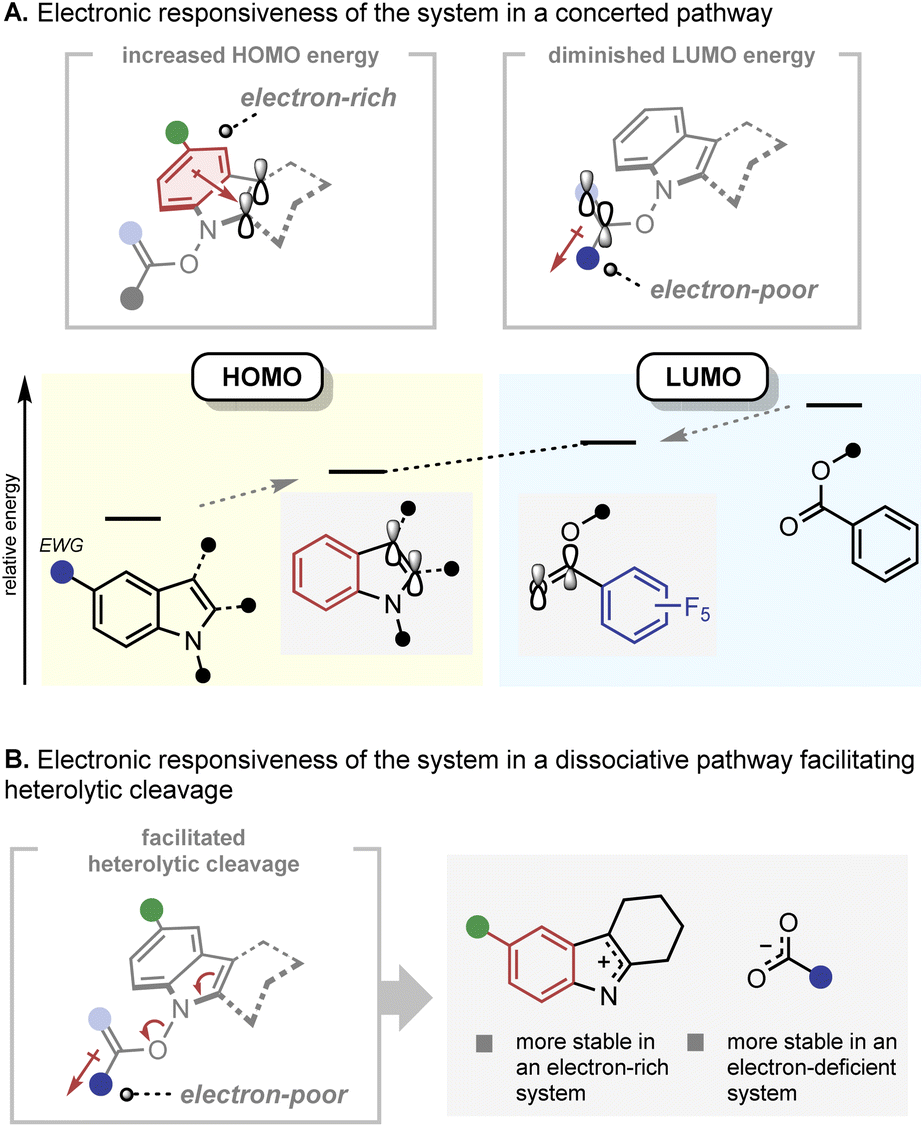 | ||
| Fig. 9 Mechanistic rationale of the IHT reaction. (A) Electronic responsiveness of the system in a concerted pathway; (B) electronic responsiveness of the system in a dissociative pathway. | ||
C3-acyloxylation of indoles via a facilitated IHT reaction
The established mechanistic understanding has served as a general rate enhancing element of the IHT process, improving the synthetic efficiency of the reaction. Firstly, the accelerated reactivity could be utilised as an efficient synthetic platform to provide various forms of the oxygenated indole products under mild conditions (Scheme 2). A delicate indolenine product could be smoothly formed at ambient temperature (2i). Also, various pyrroloindolines could be obtained using the developed method (2j–2n). In agreement with the mechanistic proposal, however, a higher reaction temperature was required for electronically under-activated cases (2j–2l). In addition, the formation of the 3-acyl indole products containing a range of C2 substituents, including primary and secondary alkyl groups, could be realised in a highly straightforward manner (2o–2q). It should be noted that an identical reaction with an acetyl or benzoyl group failed to yield the 3-acyl indole products, indicating the importance of electronic activation at the 2′-position.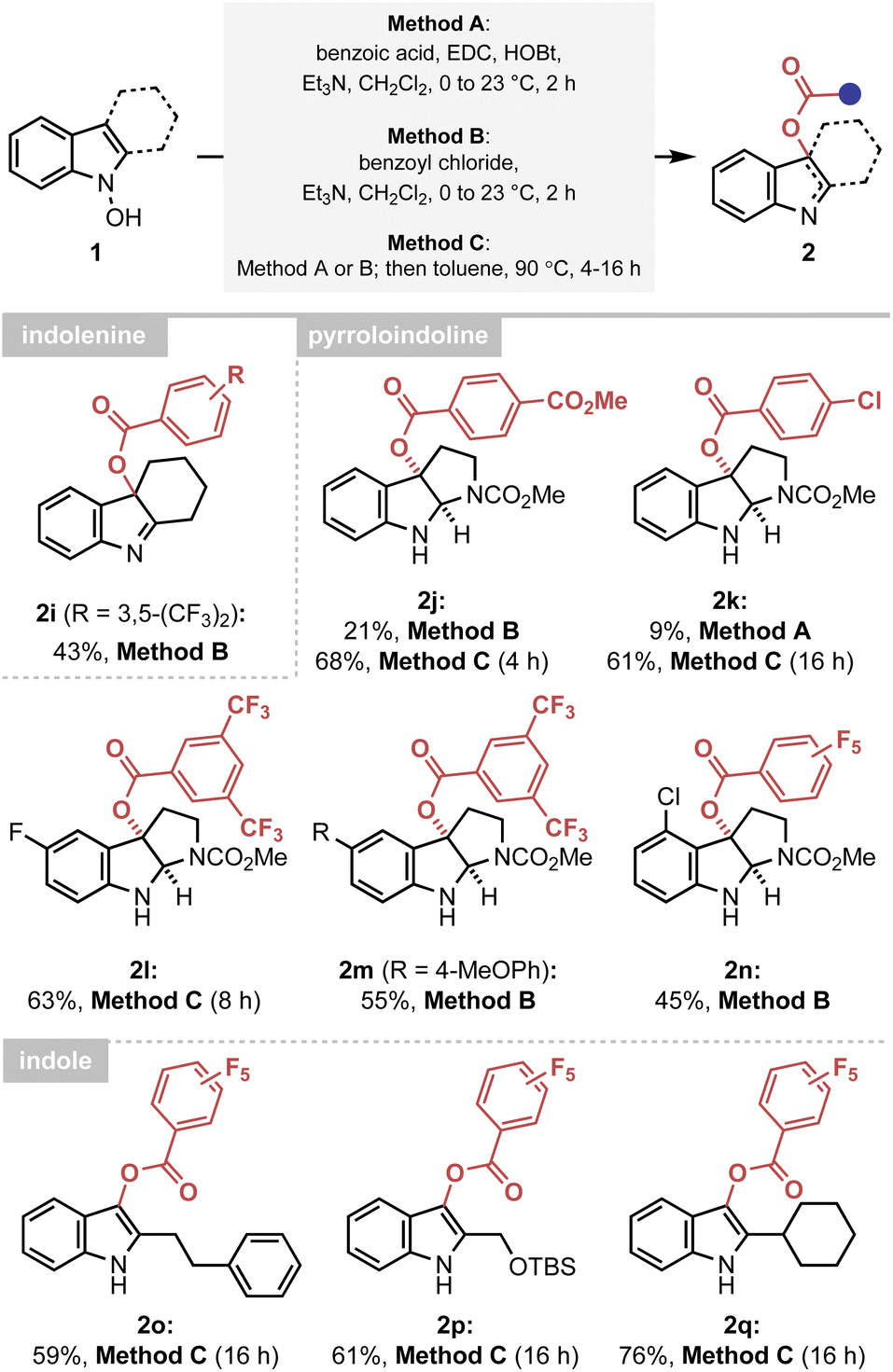 | ||
| Scheme 2 Synthesis of C-oxygenated indole derivatives via a facilitated IHT reaction. Method A: reactions performed with benzoyl chloride (1.1 equiv.) and Et3N (1.2 equiv.) in CH2Cl2 (0.05 M) at 0 °C to 23 °C for 2 h on a 0.1–0.3 mmol scale. Method B: reactions performed with benzoic acid (1.0 equiv.), EDC HCl (1.1 equiv.), HOBt (1.1 equiv.) and Et3N (2.2 equiv.) in CH2Cl2 (0.05 M) at 0 °C to 23 °C for 2 h on a 0.2–0.6 mmol scale. Method C: reactions performed via Method A or B at 23 °C for the indicated time on a 0.1–0.7 mmol scale. After filtration through silica gel, the solvent was exchanged to toluene (0.05 M) and the system was heated to 90 °C. Yields of the isolated product are reported. EDC HCl = 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride, HOBt = 1-hydroxybenzotriazole. | ||
Extension to the amidation reactions
The results obtained from the accelerated oxygenation reaction led us to the new hypothesis that an identical inductive rate enhancement strategy can facilitate other types of rearrangements in the indolyl frameworks. Thus, we attempted to extend the developed strategy to introduce a nitrogen atom at the 3-position of the indole via the IHT process. Among the various molecular architectures eligible for the desired C–N bond formation, the trichloroacetimidate group, a well-known system for the 1,3-allylic O/N transposition reaction reported by Overman and Carpenter, has been chosen to realise the facilitated amidative IHT.23 The trichloroacetimidate system could be easily accessed by the reaction of N-hydroxyindole with trichloroacetonitrile in the presence of a catalytic amount of triethylamine (see ESI† for the optimisation of the reaction parameters). Ultimately, the generated system induced a transposition from the N1–O1′ bond to the C3–N3′ bond in an extremely efficient manner.After securing the optimal conditions, the applicability of the amidative IHT reaction was evaluated (Scheme 3). Firstly, starting from N-hydroxytryptamines, the method was utilised to prepare a diverse array of functionalised pyrroloindolines (3a–3o). Functional groups with varying electronic properties could be installed at the 5-position of the indole framework without significantly affecting the synthetic efficiency (3a–3i). In addition, various substituents at the 4-, 6-, or 7-positions were also tolerated (3j–3l). Also, the reaction could be mediated in an indole system that exhibits steric congestion near the reaction centre (3m). Finally, the interception of the initially formed imine intermediate could also be realised by a pendant amide (3n) or a substituted carbamate (3o).
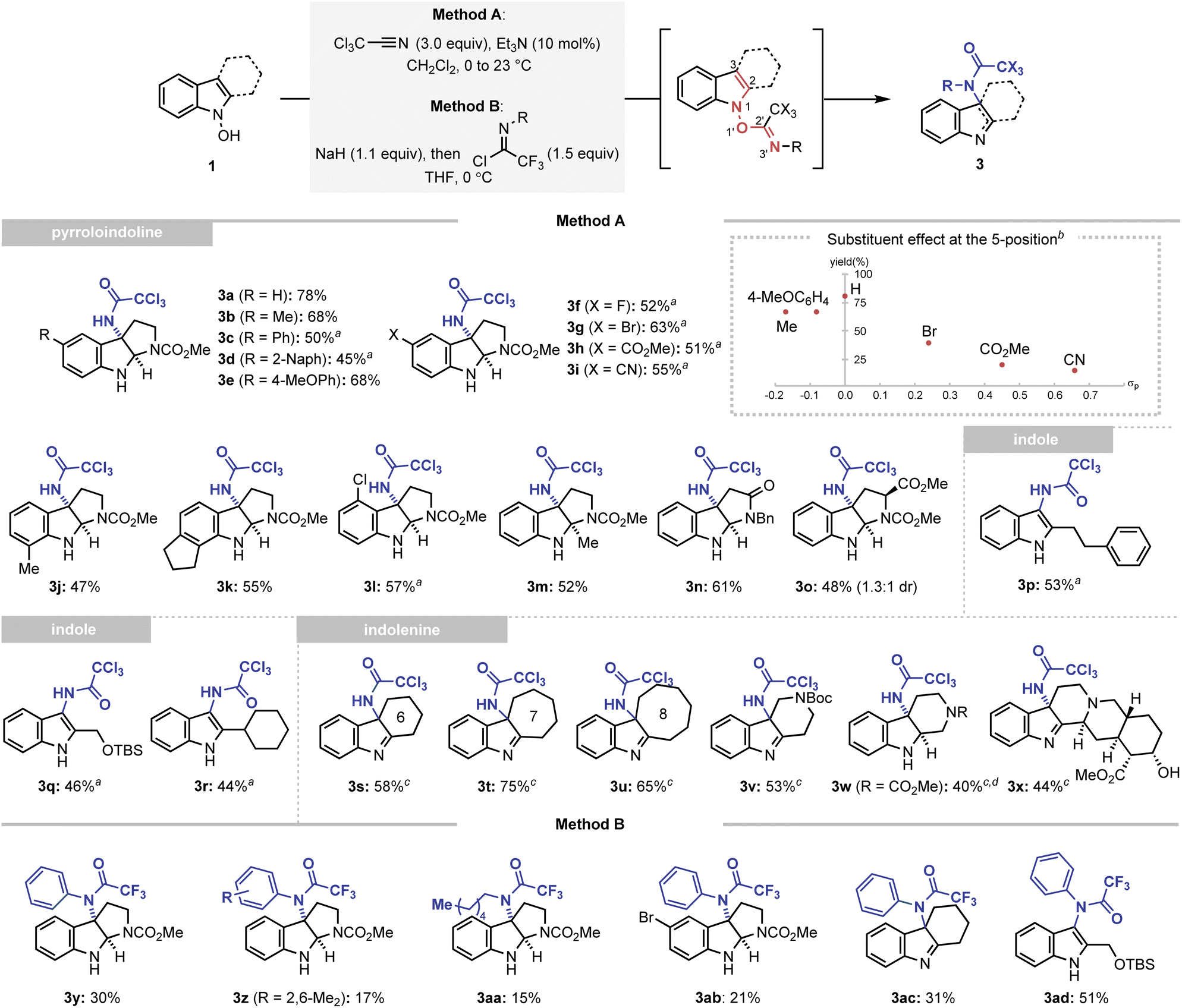 | ||
| Scheme 3 Synthesis of C-amidated indole derivatives via a facilitated IHT reaction. Method A: reactions performed with trichloroacetonitrile (3.0 equiv.) and Et3N (0.1 equiv.) in CH2Cl2 (0.05 M) at 0 °C to 23 °C for 3 h on a 0.1–0.7 mmol scale. Method B: reactions performed with imidoyl chloride (1.5 equiv.) and NaH (1.1 equiv.) in THF (0.05 M) at 0 °C for 1 h on a 0.1–0.9 mmol scale. aThe reaction performed with trichloroacetonitrile (3.0 equiv.) and Et3N (0.1 equiv.) in DCE (0.05 M) at 23 °C to 90 °C for 2 h on a 0.1–0.3 mmol scale. bReactions performed using Method A. cReactions performed at 0 °C instead of 0 °C to 23 °C. dYield of the isolated product after reduction with NaBH4 in MeOH. Yields of the isolated product are reported. | ||
To further corroborate the dependence of the reaction efficiency on the electronics of the indole framework, N-hydroxytryptamines bearing C5 substituents with varying electronic properties were subjected to the C3-amidation conditions without thermal activation (inset of Scheme 3). The product yields were plotted against the Hammett constant of the corresponding substituent, and a generally negative reactivity was noted as the C5 substituent became more electron-withdrawing. This observation is consistent with the participation of an intermediate with a positive charge partitioned in the indolyl part, such as intermediate IV′ (Fig. 2).
Expanding the scope of the developed reaction to indole scaffolds without an internal nucleophile provided a new opportunity to access the unexplored structural diversity based on indoles or indolenine. The 3-amidated indoles were conveniently synthesised using the developed strategy (3p–3r). Tricyclic indolenine with various carbocycles or heterocycles were formed in a straightforward manner (3s–3w). Finally, the method was applied to the diversification of a complex natural product yohimbine without the use of protection groups, which demonstrated the synthetic robustness of the strategy (3x).24
An even higher level of structural diversity could be attained with the use of trifluoroimidoyl chloride as a coupling partner (Method B). The presence of an even more electron-withdrawing trifluoromethyl group enabled the installation of an additional carbon-based substituent at the C3-nitrogen atom of the pyrroloindoline (3y-3ab), indolenine (3ac), and indole (3ad) products.
Next, we assessed the practicality of the developed method (Scheme 4). Firstly, the reaction scale was conveniently increased to the gram-scale without a significant loss of efficiency, demonstrating the robustness of the system (Scheme 4A). In addition, the conversion of trichloromethyl acetamide to the corresponding amine was realised in a single step via simple acidic hydrolysis, allowing for future conversion to a variety of amine derivatives (Scheme 4B). Finally, the preparation of a key intermediate for the alkaloid natural product, psychotriasine, was achieved in a highly convergent way (Scheme 4C). The introduction of a nitrogen atom at 3-position, formation of pyrroloindoline, and installation of an ortho-bromophenyl group were realised in a single step. After the straightforward removal of the trifluoroacetamide group, the key intermediate for psychotriasine was acquired.3m,4k,25
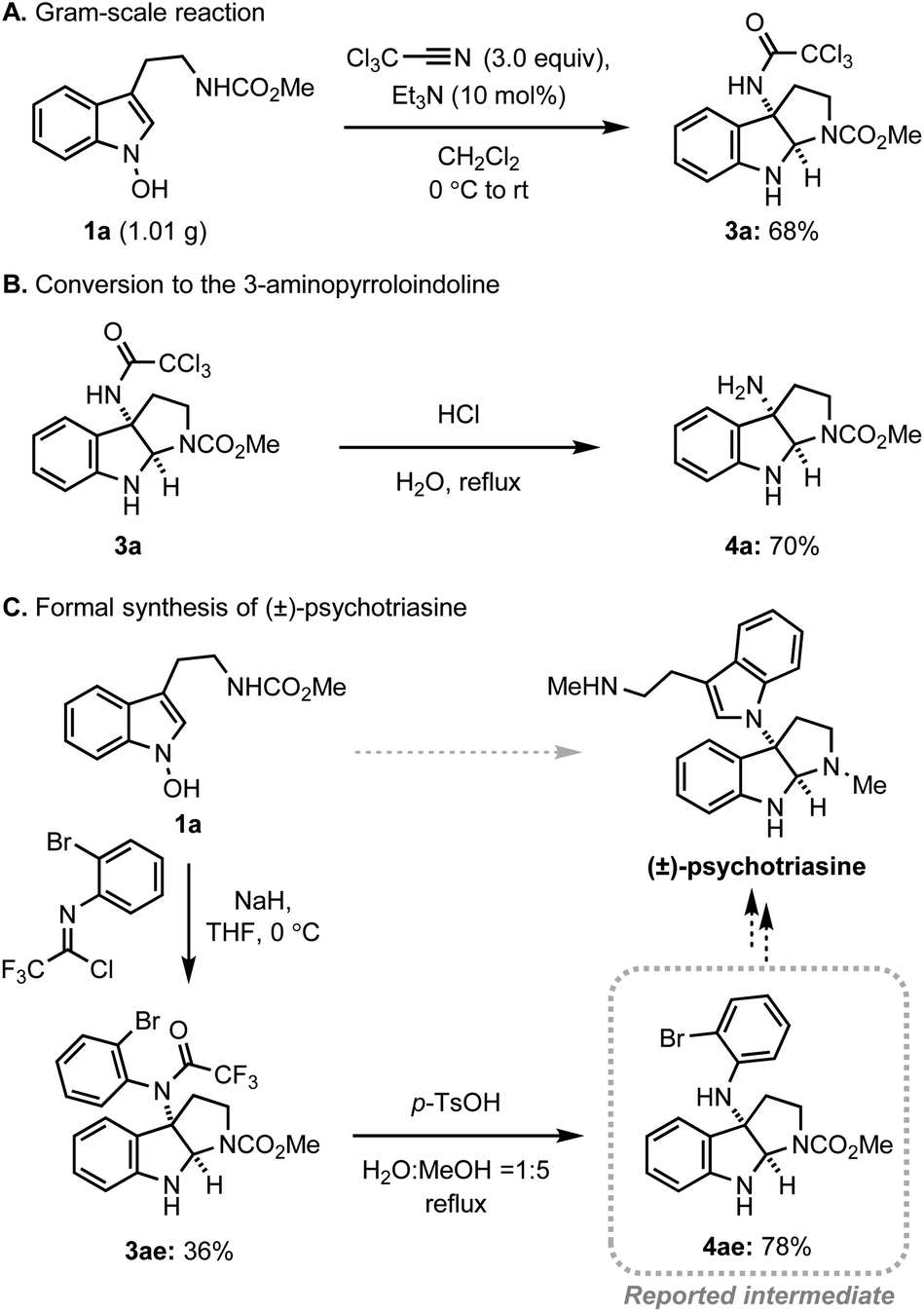 | ||
| Scheme 4 Practicality and versatility of the amidative IHT reaction. | ||
Conclusions
In summary, a detailed investigation of the mechanistic features of the 1,3-heteroatom transposition of N-hydroxyindole has been conducted. Extensive mechanistic studies, including isotope labelling experiments, indicate that the reaction proceeds through two concurrently operating reaction pathways, a concerted mechanism and a dissociative mechanism, the weighted average of which can be shifted as a function of the electronic nature of the system. Furthermore, the electronic perturbation can induce the lowering of the activation barrier for both pathways at different rates, thereby changing the major mechanistic pathway for product formation.The identification of the mechanistic nature of the reaction has enabled a mechanism driven reaction design for an efficient method for the C3-hetero-functionalisation of indoles via facilitated IHT. The success of the developed strategy stems from the exploitation of a critical electronic effect of the system, particularly at the 2′-position. The mild conditions could be applied to access numerous C3-acyloxylated or C3-amidated indole products in an effortless manner. As a result, sensitive and/or complex products in the form of indole, indolenine, or indoline could be conveniently prepared. From a synthetic point of view, the developed method should provide reliable access to diversified structures derived from indole frameworks.
Data availability
Supplementary data supporting this article are available in the ESI,† which include general synthetic procedures, mechanistic experimental details and compound characterisation with spectral data.Author contributions
H. G. L. initiated and supervised the project. Y. L. designed the experiments and performed the reaction optimisation for the preliminary results. Y. S. N. performed optimisation of the reaction utilizing imidoyl chlorides. Y. L., Y. S. N., S. Y. K., and J. E. K. carried out the synthetic experiments and Y. L. conducted the mechanistic investigations. All the authors analysed and discussed the experiment data. H. G. L. and Y. L. prepared the manuscript, and Y. L. and Y. S. N. prepared the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean Government (MSIT) (grant no. 2021R1F1A1064141), and MSIT (grant no. 2022R1A2C1012469). The authors thank H. N. Bae (Seoul National University National Center for Inter-University Research Facilities) for assistance with the NMR spectroscopy experiments. They also thank C. R. Choi (Department of Chemistry, Seoul National University), H. K. Lee (the Organic Chemistry Research Center, Sogang University), and D. H. Lee (National Instrumentation Center for Environmental Management, Seoul University) for the HRMS analysis.Notes and references
- (a) Y. Ban, Y. Murakami, Y. Iwasawa, M. Tsuchiya and N. Takano, Med. Res. Rev., 1988, 8, 231–308 CrossRef CAS; (b) R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed; (c) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed; (d) Y. Wan, Y. Li, C. Yan, M. Yan and Z. Tang, Eur. J. Med. Chem., 2019, 183, 111691 CrossRef CAS PubMed; (e) D. S. Seigler, in Plant Secondary Metabolism, Springer, 1998, pp. 628–654 CrossRef.
- (a) A. H. Sandtorv, Adv. Synth. Catal., 2015, 357, 2403–2435 CrossRef CAS; (b) S. Liu, F. Zhao, X. Chen, G.-J. Deng and H. Huang, Adv. Synth. Catal., 2020, 362, 3795–3823 CrossRef CAS; (c) F.-T. Sheng, J.-Y. Wang, W. Tan, Y.-C. Zhang and F. Shi, Org. Chem. Front., 2020, 7, 3967–3998 RSC; (d) K. Urbina, D. Tresp, K. Sipps and M. Szostak, Adv. Synth. Catal., 2021, 363, 2723–2739 CrossRef CAS; (e) H. Abou-Hamdan, C. Kouklovsky and G. Vincent, Synlett, 2020, 31, 1775–1788 CrossRef CAS.
- For reviews, see: (a) S. P. Roche, J.-J. Youte Tendoung and B. Tréguier, Tetrahedron, 2015, 71, 3549–3591 CrossRef CAS; (b) A. Hałuszczuk, N. Babul, Ł. Nierzwicki and W. Przychodzeń, Eur. J. Org. Chem., 2019, 2019, 4411–4416 CrossRef; For selected examples of C–O bond formation, see: (c) H. Takayama, K. Misawa, N. Okada, H. Ishikawa, M. Kitajima, Y. Hatori, T. Murayama, S. Wongseripipatana, K. Tashima, K. Matsumoto and S. Horie, Org. Lett., 2006, 8, 5705–5708 CrossRef CAS PubMed; (d) M. Movassaghi, M. A. Schmidt and J. A. Ashenhurst, Org. Lett., 2008, 10, 4009–4012 CrossRef CAS PubMed; (e) F. Kolundzic, M. N. Noshi, M. Tjandra, M. Movassaghi and S. J. Miller, J. Am. Chem. Soc., 2011, 133, 9104–9111 CrossRef CAS; (f) S. Han and M. Movassaghi, J. Am. Chem. Soc., 2011, 133, 10768–10771 CrossRef CAS; For selected examples of C–N bond formation, see: (g) P. S. Baran, C. A. Guerrero and E. J. Corey, Org. Lett., 2003, 5, 1999–2001 CrossRef CAS PubMed; (h) A. Padwa, A. C. Flick, C. A. Leverett and T. Stengel, J. Org. Chem., 2004, 69, 6377–6386 CrossRef CAS PubMed; (i) T. Newhouse and P. S. Baran, J. Am. Chem. Soc., 2008, 130, 10886–10887 CrossRef CAS PubMed; (j) T. Newhouse, C. A. Lewis, K. J. Eastman and P. S. Baran, J. Am. Chem. Soc., 2010, 132, 7119–7137 CrossRef CAS; (k) Y.-Q. Zhang, Y.-A. Yuan, G.-S. Liu and H. Xu, Org. Lett., 2013, 15, 3910–3913 CrossRef CAS; (l) Z. Shen, Z. Xia, H. Zhao, J. Hu, X. Wan, Y. Lai, C. Zhu and W. Xie, Org. Biomol. Chem., 2015, 13, 5381–5384 RSC; (m) C. Liu, J.-C. Yi, Z.-B. Zheng, Y. Tang, L.-X. Dai and S.-L. You, Angew. Chem., Int. Ed., 2016, 55, 751–754 CrossRef CAS; (n) X. Ma, J. J. Farndon, T. A. Young, N. Fey and J. F. Bower, Angew. Chem., Int. Ed., 2017, 56, 14531–14535 CrossRef CAS; (o) G. X. Ortiz, B. N. Hemric and Q. Wang, Org. Lett., 2017, 19, 1314–1317 CrossRef CAS PubMed.
- For reviews, see: (a) M. Bandini, Org. Biomol. Chem., 2013, 11, 5206–5212 RSC; (b) A. Cerveri and M. Bandini, Chin. J. Chem., 2020, 38, 287–294 CrossRef CAS; For selected examples of C–O bond formation, see: (c) K. Liu, P. Wen, J. Liu and G. Huang, Synthesis, 2010, 2010, 3623–3626 CrossRef; (d) K. Liu, S. Tang, P. Huang and A. Lei, Nat. Commun., 2017, 8, 775 CrossRef; (e) Y.-Z. Cheng, Q.-R. Zhao, X. Zhang and S.-L. You, Angew. Chem., Int. Ed., 2019, 58, 18069–18074 CrossRef CAS; For selected examples of C–N bond formation, see: (f) P. S. Baran and R. A. Shenvi, J. Am. Chem. Soc., 2006, 128, 14028–14029 CrossRef CAS PubMed; (g) D. Lubriks, I. Sokolovs and E. Suna, J. Am. Chem. Soc., 2012, 134, 15436–15442 CrossRef CAS PubMed; (h) P. K. Prasad, R. G. Kalshetti, R. N. Reddi, S. P. Kamble and A. Sudalai, Org. Biomol. Chem., 2016, 14, 3027–3030 RSC; (i) K. Watanabe and K. Moriyama, J. Org. Chem., 2018, 83, 14827–14833 CrossRef CAS; (j) H. Tanaka, N. Ukegawa, M. Uyanik and K. Ishihara, J. Am. Chem. Soc., 2022, 144, 5756–5761 CrossRef CAS PubMed; For selected examples of unified strategy towards various C-heteroatom bond formations, see: (k) E. C. Gentry, L. J. Rono, M. E. Hale, R. Matsuura and R. R. Knowles, J. Am. Chem. Soc., 2018, 140, 3394–3402 CrossRef CAS; (l) J. Wu, Y. Dou, R. Guillot, C. Kouklovsky and G. Vincent, J. Am. Chem. Soc., 2019, 141, 2832–2837 CrossRef CAS PubMed; (m) R. Cui, J. Ye, J. Li, W. Mo, Y. Gao and H. Chen, Org. Lett., 2020, 22, 116–119 CrossRef CAS PubMed.
- For selected examples of C–O bond formation, see: (a) Y. Zhou, G. Chen, C. Li, X. Liu and P. Liu, Synth. Commun., 2018, 48, 2912–2922 CrossRef CAS; (b) M. Wang, Y. Yang, L. Yin, Y. Feng and Y. Li, J. Org. Chem., 2021, 86, 683–692 CrossRef CAS PubMed; (c) T. Tomakinian, R. Guillot, C. Kouklovsky and G. Vincent, Angew. Chem., Int. Ed., 2014, 53, 11881–11885 CrossRef CAS PubMed; For selected examples of C–N bond formation, see: (d) N. Fu, G. S. Sauer, A. Saha, A. Loo and S. Lin, Science, 2017, 357, 575–579 CrossRef CAS PubMed; (e) K. Liu, W. Song, Y. Deng, H. Yang, C. Song, T. Abdelilah, S. Wang, H. Cong, S. Tang and A. Lei, Nat. Commun., 2020, 11, 3 CrossRef PubMed; (f) M.-X. He, Y.-Z. Wu, Y. Yao, Z.-Y. Mo, Y.-M. Pan and H.-T. Tang, Adv. Synth. Catal., 2021, 363, 2752–2756 CrossRef CAS.
- (a) R. Sundberg, J. Org. Chem., 1965, 30, 3604–3610 CrossRef CAS; (b) T. Nagayoshi, S. Saeki and M. Hamana, Heterocycles, 1977, 6, 1666–1674 CrossRef CAS; (c) T. Nagayoshi, S. Saeki and M. Hamana, Chem. Pharm. Bull., 1981, 29, 1920–1926 CrossRef CAS; (d) T. Nagayoshi, S. Saeki and M. Hamana, Chem. Pharm. Bull., 1984, 32, 3678–3682 CrossRef CAS; (e) M. Somei, T. Kawasaki, Y. Fukui, F. Yamada, T. Kobayashi and H. Aoyama, Heterocycles, 1992, 34, 1877–1884 CrossRef CAS; (f) Y. Fukui and M. Somei, Heterocycles, 2001, 55, 2055–2057 CrossRef CAS; (g) M. Somei, K. Noguchi and F. Yamada, Heterocycles, 2001, 55, 1237 CrossRef CAS; (h) Y. Fukui, T. Kobayashi, T. Kawasaki, F. Yamada and M. Somei, Heterocycles, 2019, 99, 465–483 CrossRef CAS PubMed.
- (a) M. P. Duarte, R. F. Mendonça, S. Prabhakar and A. M. Lobo, Tetrahedron Lett., 2006, 47, 1173–1176 CrossRef CAS; (b) Z. Chen and Q. Wang, Org. Lett., 2015, 17, 6130–6133 CrossRef CAS PubMed; (c) T. Tomakinian, C. Kouklovsky and G. Vincent, Synlett, 2015, 26, 1269–1275 CrossRef CAS; (d) M. Shevlin, N. A. Strotman and L. L. Anderson, Synlett, 2021, 32, 197–201 CrossRef CAS; (e) N. H. Nguyen, S. M. Oh, C.-M. Park and S. Shin, Chem. Sci., 2022, 13, 1169–1176 RSC; (f) M. Bera, H. S. Hwang, T.-W. Um, S. M. Oh, S. Shin and E. J. Cho, Org. Lett., 2022, 24, 1774–1779 CrossRef CAS.
- For selected examples in other related heterocycles, see: (a) H. J. Shrives, J. A. Fernández-Salas, C. Hedtke, A. P. Pulis and D. J. Procter, Nat. Commun., 2017, 8, 14801 CrossRef; (b) Z. He, H. J. Shrives, J. A. Fernández-Salas, A. Abengózar, J. Neufeld, K. Yang, A. P. Pulis and D. J. Procter, Angew. Chem., Int. Ed., 2018, 57, 5759–5764 CrossRef CAS PubMed.
- For review, see: A. A. Tabolin and S. L. Ioffe, Chem. Rev., 2014, 114, 5426–5476 CrossRef CAS PubMed.
- For selected examples with mechanistic studies, see: (a) S. Oae, T. Kitao and Y. Kitaoka, J. Am. Chem. Soc., 1962, 84, 3359–3362 CrossRef CAS; (b) T. Koenig, J. Am. Chem. Soc., 1966, 88, 4045–4049 CrossRef CAS; (c) R. Bodalski and A. R. Katritzky, J. Chem. Soc. B, 1968, 831–838 RSC; (d) G. Tisue, M. Grassmann and W. Lwowski, Tetrahedron, 1968, 24, 999–1006 CrossRef; (e) D. Gutschke and A. Heesing, Chem. Ber., 1973, 106, 2379–2394 CrossRef CAS; (f) S. Oae, T. Sakurai, H. Kimura and S. Kozuka, Chem. Lett., 1974, 3, 671–674 CrossRef; (g) S. Oae and T. Sakurai, Tetrahedron, 1976, 32, 2289–2294 CrossRef CAS; (h) N. Binding and A. Heesing, Chem. Ber., 1983, 116, 1822–1833 CrossRef CAS; (i) A. J. Castellino and H. Rapoport, J. Org. Chem., 1984, 49, 4399–4404 CrossRef CAS; (j) M. Bosco, R. Dalpozzo, G. Bartoli, G. Palmieri and M. Petrini, J. Chem. Soc., Perkin Trans., 1991, 2, 657–663 RSC; (k) Q. Liu, C.-S. Zhang, H. Sheng, D. Enders, Z.-X. Wang and X.-Y. Chen, Org. Lett., 2020, 22, 5617–5621 CrossRef CAS PubMed.
- Reactions with extremely electron deficient acyl donors, such as trifluoroacetic anhydride, resulted in extensive decomposition to produce a mixture of untractable products.
- The pathway involving [1,3]-sigmatropic shift has been ruled out due to the high activation energy originating from the unfavorable orbital alignment. For reviews and selected examples of [1,3]-sigmatropic shift, see: (a) C. G. Nasveschuk and T. Rovis, Org. Biomol. Chem., 2008, 6, 240–254 RSC; (b) I. Nakamura and M. Terada, Tetrahedron Lett., 2019, 60, 689–698 CrossRef CAS; (c) S. Oae, T. Kitao and Y. Kitaoka, Tetrahedron, 1963, 19, 827–832 CrossRef CAS; (d) S. Hou, X. Li and J. Xu, J. Org. Chem., 2012, 77, 10856–10869 CrossRef CAS PubMed; (e) I. Nakamura, M. Owada, T. Jo and M. Terada, Org. Lett., 2017, 19, 2194–2196 CrossRef CAS PubMed.
- Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC press, 2007 Search PubMed.
- For selected examples with mechanistic consideration in related systems, see: (a) N. Çelebi-Ölçüm, Y.-H. Lam, E. Richmond, K. B. Ling, A. D. Smith and K. N. Houk, Angew. Chem., Int. Ed., 2011, 50, 11478–11482 CrossRef PubMed; (b) J. Sączewski, J. Fedorowicz, M. Gdaniec, P. Wiśniewska, E. Sieniawska, Z. Drażba, J. Rzewnicka and Ł. Balewski, J. Org. Chem., 2017, 82, 9737–9743 CrossRef PubMed.
- For selected examples of 18O labeling experiment for mechanistic investigation, see: (a) P. Mauleón, J. L. Krinsky and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 4513–4520 CrossRef; (b) B.-L. Lu, Y. Wei and M. Shi, Chem.–Eur. J., 2010, 16, 10975–10979 CrossRef CAS PubMed; (c) A. Sekar Kulandai Raj and R.-S. Liu, Adv. Synth. Catal., 2020, 362, 2517–2522 CrossRef CAS Also, see, ref. 10a–h..
- M. Agirre, S. Henrion, I. Rivilla, J. I. Miranda, F. P. Cossío, B. Carboni, J. M. Villalgordo and F. Carreaux, J. Org. Chem., 2018, 83, 14861–14881 CrossRef CAS PubMed.
- For selected examples, see: (a) J. J. Gajewski, J. Am. Chem. Soc., 1979, 101, 4393–4394 CrossRef CAS; (b) J. J. Gajewski and N. D. Conrad, J. Am. Chem. Soc., 1979, 101, 6693–6704 CrossRef CAS; (c) J. J. Gajewski and K. E. Gilbert, J. Org. Chem., 1984, 49, 11–17 CrossRef CAS; (d) W. V. E. Doering and Y. Wang, J. Am. Chem. Soc., 1999, 121, 10112–10118 CrossRef CAS; (e) W. V. E. Doering and Y. Wang, J. Am. Chem. Soc., 1999, 121, 10967–10975 CrossRef; (f) D. A. Hrovat, B. R. Beno, H. Lange, H.-Y. Yoo, K. N. Houk and W. T. Borden, J. Am. Chem. Soc., 1999, 121, 10529–10537 CrossRef CAS; (g) K. A. Black, S. Wilsey and K. N. Houk, J. Am. Chem. Soc., 2003, 125, 6715–6724 CrossRef CAS PubMed; (h) W. T. Borden, in Theory and Applications of Computational Chemistry, ed. C. E. Dykstra, G. Frenking, K. S. Kim and G. E. Scuseria, Elsevier, Amsterdam, 2005, pp. 859–873 Search PubMed; (i) D. V. Vidhani, M. E. Krafft and I. V. Alabugin, J. Am. Chem. Soc., 2016, 138, 2769–2779 CrossRef CAS PubMed; (j) D. V. Vidhani and I. V. Alabugin, J. Org. Chem., 2019, 84, 14844–14853 CrossRef CAS PubMed.
- TEMPO could not be used with N-hydroxyindole (1a) due to the rapid decomposition of 1a when exposed to TEMPO. However, indolyl benzoate (2a), which is stable at room temperature, could be safely isolated and subjected to the rearrangement conditions in the presence of TEMPO without decomposition. The result was almost identical to the result using 1,1-diphenylethylene. No noticeable yield loss was observed and only a trace of product trapped by TEMPO was detected by HRMS.
- (a) D. A. Braden, E. E. Parrack and D. R. Tyler, Coord. Chem. Rev., 2001, 211, 279–294 CrossRef CAS; (b) J. T. Barry, D. J. Berg and D. R. Tyler, J. Am. Chem. Soc., 2016, 138, 9389–9392 CrossRef CAS PubMed; (c) J. T. Barry, D. J. Berg and D. R. Tyler, J. Am. Chem. Soc., 2017, 139, 14399–14405 CrossRef CAS PubMed.
- (a) E. F. da Silva and H. F. Svendsen, Ind. Eng. Chem. Res., 2006, 45, 2497–2504 CrossRef CAS; (b) N. McCann, D. Phan, D. Fernandes and M. Maeder, Int. J. Greenhouse Gas Control, 2011, 5, 396–400 CrossRef CAS; (c) M. Gupta and H. F. Svendsen, Int. J. Greenhouse Gas Control, 2020, 98, 103061 CrossRef CAS.
- The possibility of an involvement of the C=N tautomer of the carbamate for the generation of compound 2p was excluded, since the C=N tautomer is highly unfavored compared to the corresponding carbamate. See: (a) D. Xing and D. Yang, Org. Lett., 2010, 12, 1068–1071 CrossRef CAS PubMed; (b) U. K. Das, A. Kumar, Y. Ben-David, M. A. Iron and D. Milstein, J. Am. Chem. Soc., 2019, 141, 12962–12966 CrossRef CAS PubMed.
- (a) F. C. Gozzo, S. A. Fernandes, D. C. Rodrigues, M. N. Eberlin and A. J. Marsaioli, J. Org. Chem., 2003, 68, 5493–5499 CrossRef CAS PubMed; (b) S. Debnath and S. Mondal, Comput. Theor. Chem., 2014, 1046, 42–48 CrossRef CAS; (c) V. Aviyente, H. Y. Yoo and K. N. Houk, J. Org. Chem., 1997, 62, 6121–6128 CrossRef CAS.
- For reviews, see: (a) L. E. Overman and N. E. Carpenter, Organomet. React., 2005, 1–107 CAS; (b) H. Nomura and C. J. Richards, Chem.–Asian J., 2010, 5, 1726–1740 CrossRef CAS PubMed; (c) R. A. Fernandes, P. Kattanguru, S. P. Gholap and D. A. Chaudhari, Org. Biomol. Chem., 2017, 15, 2672–2710 RSC.
- The stereochemical outcome at the 3-position was tentatively assigned on the basis of the analogous rarrangement in the yohimbine system. See ref. 6f.
- (a) Q. Li, T. Xia, L. Yao, H. Deng and X. Liao, Chem. Sci., 2015, 6, 3599–3605 RSC; (b) A. A. Adhikari and J. D. Chisholm, Org. Lett., 2016, 18, 4100–4103 CrossRef CAS PubMed; (c) S. Gallego, P. Lorenzo, R. Alvarez and A. R. de Lera, Tetrahedron Lett., 2017, 58, 210–212 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc00716b |
| This journal is © The Royal Society of Chemistry 2023 |