
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEvidence for and evaluation of fluorine–tellurium chalcogen bonding†
Robin
Weiss
a,
Emmanuel
Aubert
b,
Loic
Groslambert
a,
Patrick
Pale
 *a and
Victor
Mamane
*a
*a and
Victor
Mamane
*a
aLASYROC, UMR 7177, University of Strasbourg, 1 Rue Blaise Pascal, 67000 Strasbourg, France. E-mail: ppale@unistra.fr; vmamane@unistra.fr
bUniversité de Lorraine, CNRS, CRM2, F-54000 Nancy, France
First published on 5th June 2023
Abstract
In the field of noncovalent interactions, chalcogen bonding (ChB) involving the tellurium atom is currently attracting much attention in supramolecular chemistry and in catalysis. However, as a prerequisite for its application, the ChB should be studied in solution to assess its formation and, if possible, to evaluate its strength. In this context, new tellurium derivatives bearing CH2F and CF3 groups were designed to exhibit Te⋯F ChB and were synthesized in good to high yields. In both types of compounds, Te⋯F interactions were characterized in solution by combining 19F, 125Te and HOESY NMR techniques. These Te⋯F ChBs were shown to contribute to the overall JTe–F coupling constants (94–170 Hz) measured in the CH2F- and CF3-based tellurium derivatives. Finally, a variable temperature NMR study allowed us to approximate the energy of the Te⋯F ChB, from 3 kJ mol−1 for the compounds with weak Te σ-holes to 11 kJ mol−1 for Te σ-holes activated by the presence of strong electron withdrawing substituents.
Introduction
Noncovalent interactions involving halogens and more recently chalcogen atoms are increasingly of interest, especially in materials, crystal engineering, biology, in controlling molecular assembly and more recently in catalysis.1–4 Highly directional attractive interactions indeed occur between an electron-rich atom (Lewis base) and an electropositive region (σ-hole; Fig. 1)5 located at the outermost end of a σ-bond involving a halogen or chalcogen atom. In analogy with hydrogen bonds, these interactions were named halogen or chalcogen bonds (XBs or ChBs, respectively).2,6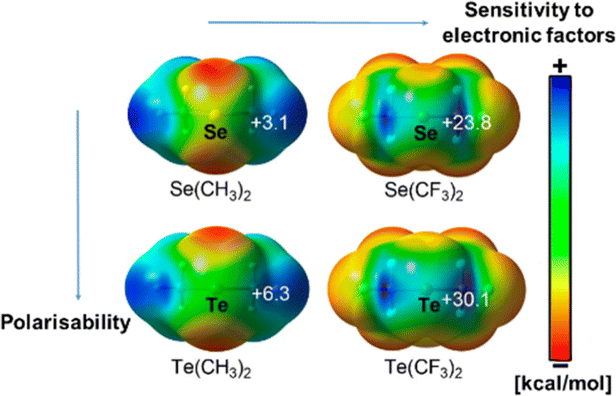 | ||
| Fig. 1 Electrostatic potential maps of selected chalcogen derivatives revealing the electropositive areas, called σ-holes. Adapted from ref. 5. | ||
It is now recognized that XBs, especially those involving an iodine atom, also play a key role in biology.3a,7 Furthermore, ChBs involving sulfur or selenium are now more frequently identified in biostructural and biochemical aspects.8 A few tellurium compounds have also found biological applications,9 but so far with no evidence of ChB involvement. On the other hand, fluorine, the most electronegative element, significantly affects the acidity or basicity, bioavailability, lipophilicity, metabolic stability and toxicity of compounds bearing it.10 Thus, fluorinated compounds increasingly impact pharma- and agro-industries. For example, 28% of newly approved drugs in the last decade (2011–2020) are fluorinated.10 With its high electronegativity, fluorine is an hydrogen bond (HB) acceptor,11 but not an XB donor unlike other halogens.12 Thus, combining both tellurium and fluorine seems worth investigating.13 However, noncovalent interactions involving fluorine as the acceptor in a ChB remains almost unknown,14 especially the Te⋯F interaction.
The extent of a σ-hole on an atom increases with its polarizability and decreases with its electronegativity (Fig. 1).15 Therefore, iodine is the most effective XB donor,16 and tellurium species should have the strongest ChB properties.17 In this respect, several studies on tellurium compounds in solution18 focused on Te⋯O,19 Te⋯N,20 Te⋯Ch,21 and Te⋯Pnictogen22 interactions (Fig. 2, top). Despite their detection in some cases in the solid state,4a,23 Te⋯F interactions have been scarcely studied in solution.24 In particular, Zhao and Gabbaï reported the recognition of a fluoride anion by 2-boronylnaphtyl telluronium; however the presence of a covalent bond was finally established with such bidentate compounds.24a
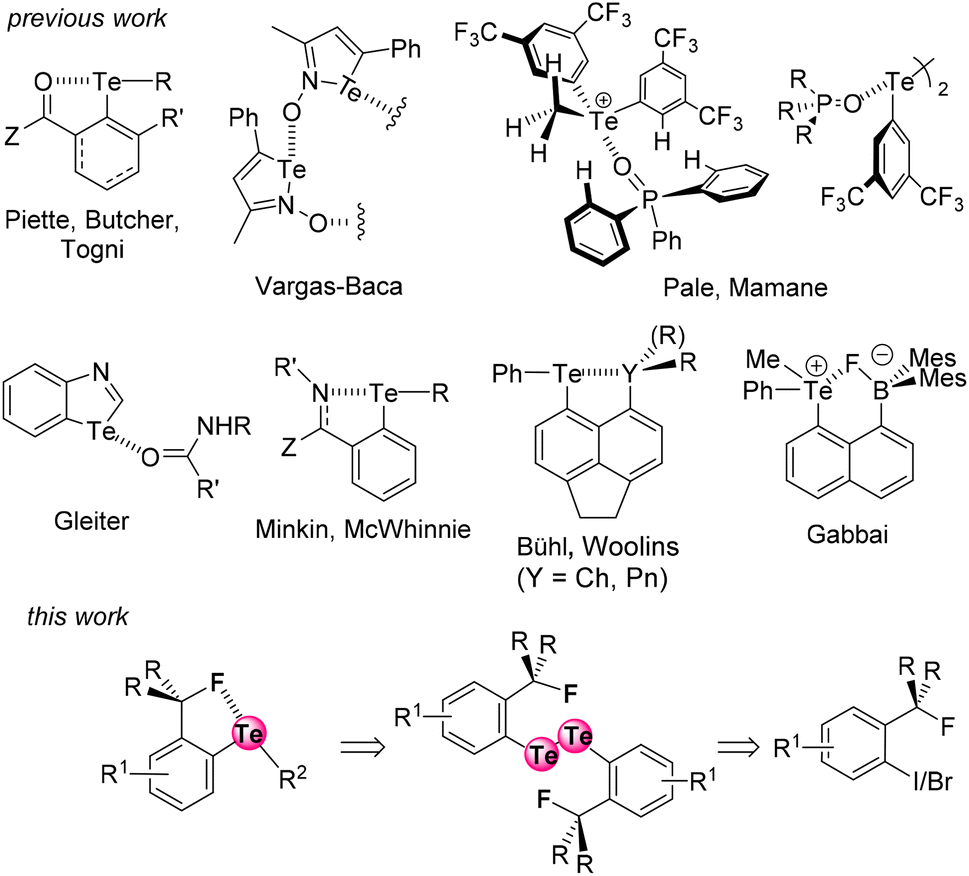 | ||
| Fig. 2 The known noncovalent interactions involving tellurium atoms, and the Te⋯F interaction described here. | ||
This context urges us to identify Te⋯F interaction and to characterize the corresponding properties.
In continuation of our recent XB and ChB studies and applications in medicinal chemistry,25 enantioseparation,26 and organocatalysis,19b,27 we have now designed two series of fluorinated ortho-tolyl telluride derivatives to identify Te⋯F interactions and to characterize their corresponding properties. We report here the first evidence of a Te⋯F interaction (ChB) in solution, as well as an evaluation of its strength (Fig. 2, bottom).
Results and discussion
Probe design
The choice of fluorinated ortho-tolyl telluride derivatives as a Te⋯F probe was motivated by three factors. First, a Te⋯F interaction should influence the rotational barrier of the fluoromethyl substituent and related groups. Second, the probe provides an opportunity to monitor the presence of rotational isomers by combining 19F and 125Te NMR, and thus to evaluate the strength of the Te⋯F interaction. Finally, Te σ-hole(s) can be tuned by using appropriate electron-withdrawing or -donating substituent(s) either on the tolyl moiety or on the other Te substituent (R1 and R2 respectively in Fig. 2, bottom left). The simplest ortho-fluoromethylphenyl telluride (CH2F series) must exhibit at least two rotamers: one (syn-CH2F) where tellurium and fluoride atoms are close to one another and a second (anti-CH2F) in which the fluoride could be far from the tellurium atom (Fig. 3A). Obviously, an intramolecular Te⋯F interaction would only exist in syn-CH2F and, if so and strong enough, this interaction could shift the conformational equilibrium towards this syn-conformer. Furthermore, the latter should exhibit different chemical shifts in 19F and 125Te NMR due to electron density transfer from F to Te depending on the Te⋯F interaction strength. Coupling constants could also be helpful because both 19F and 125Te exhibit ½ nuclear spin. In addition to the through-bond coupling constant (4JTBTe–F), a through-space contribution (1JTSTe–F) to the overall coupling constant (4JTe–F) should arise in syn-CH2F if Te⋯F interaction is present (Fig. 3B).28 Therefore, 19F and 125Te NMR monitoring could reveal such differences and provide information on the conformational equilibrium and its evolution.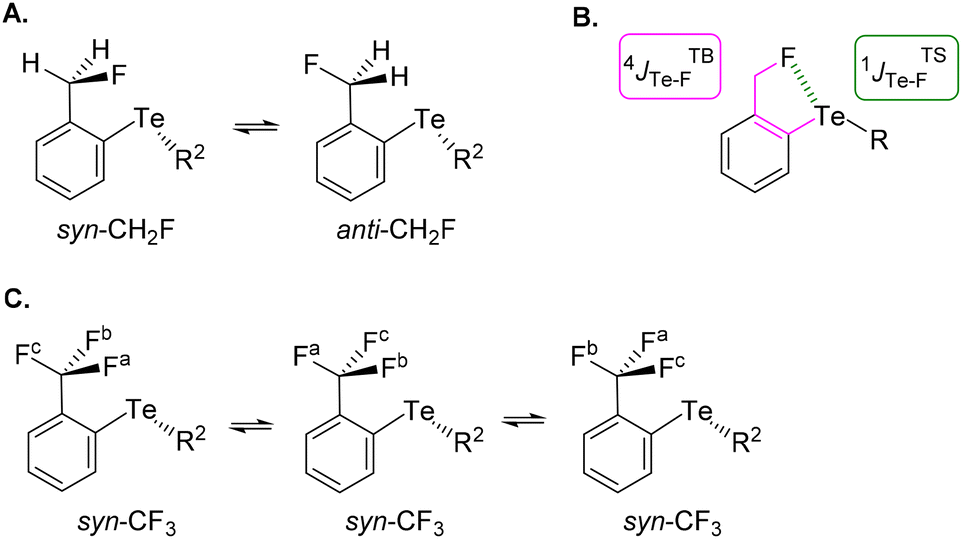 | ||
| Fig. 3 (A) The more stable syn and anti conformations for the CH2F series. (B) Through bond (TB) and through space (TS) coupling for the syn conformation in the CH2F series. (C) Permanent syn conformation in the CF3 series. | ||
In contrast, the ortho-trifluoromethylphenyl telluride (CF3 series) will always have one fluoride facing or close to the tellurium atom (Fig. 3C). Therefore, depending on the strength of the Te⋯F interaction, two possibilities could arise: this interaction could either lock the rotation of the CF3 group and thus make the fluoride atoms non-equivalent in NMR, or could be averaged over the CF3 group.
Synthesis
To address these issues, we prepared two series of ortho-tolyl tellurides bearing either a fluoromethyl or a trifluoromethyl group (Schemes 1 and 2). Starting from fluoromethyl-2-iodobenzene 1, iodine–lithium exchange followed by trapping with grey tellurium gave the corresponding lithium telluride,29 which upon quenching under air provided in high yield the corresponding diaryl ditelluride 2 (ref. 30) (Scheme 1, top). Applying the same strategy to iodo-2-trifluoromethylbenzene 3 gave the trifluoromethyl analogue 4 in similar yield.19a Upon reductive cleavage and trapping of the resulting tellurolate by electrophiles, ditelluride 2 afforded mixtures in which the expected tellurides 5 could not be detected, independent of the nature of the borohydride or electrophile (Scheme 1, top). In contrast, the treatment of ditelluride 4 with NaBH4 and subsequent electrophilic quenching readily gave the expected alkyl aryl tellurides 6a–c with good to high yields. Under these conditions, using CF2Br2 as the electrophile resulted only in the debrominated product 6d (Scheme 1, bottom).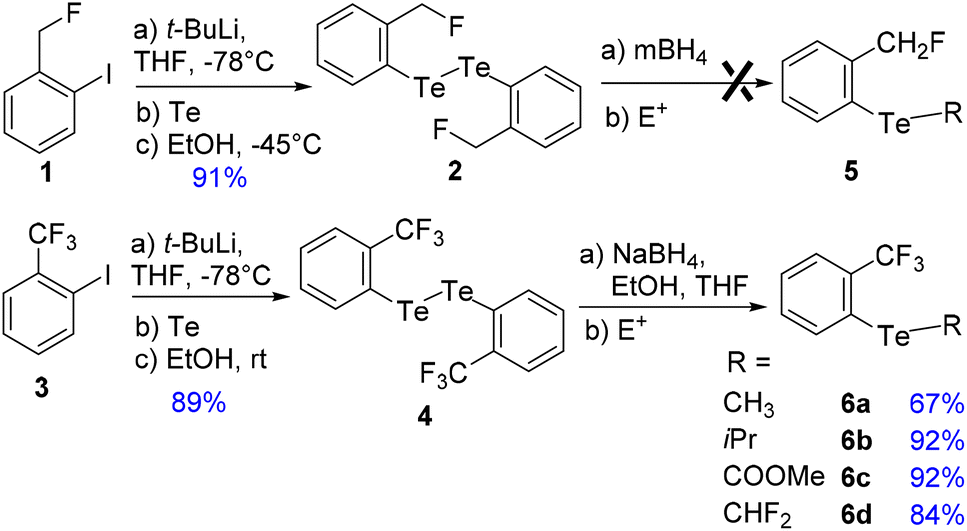 | ||
| Scheme 1 Synthesis of a series of fluorinated alkyl aryl tellurides. | ||
A more general route to both tellurides 5 and 6 was achieved by a microwave-promoted coupling reaction with various arylboronic acids (Scheme 2).31 To tune the expected ChB intensity, a series of boronic acid derivatives carrying electron-rich or electron-poor substituents was employed in this coupling to provide the corresponding diaryl tellurides 5a–e and 6e–g in good to high yields. These compounds could be stored at 4 °C for several months without degradation. Moreover, the diarylditellurides showed good stability in solution at room temperature for several days with no particular precautions. In contrast, alkyl aryl tellurides 6a–d were very sensitive to oxygen and light, causing rapid decomposition in solution with the formation of a grey metallic deposit, probably corresponding to tellurium(0).32 Unfortunately, all of the prepared tellurides 5 and 6 were viscous oils that could not be crystallized.
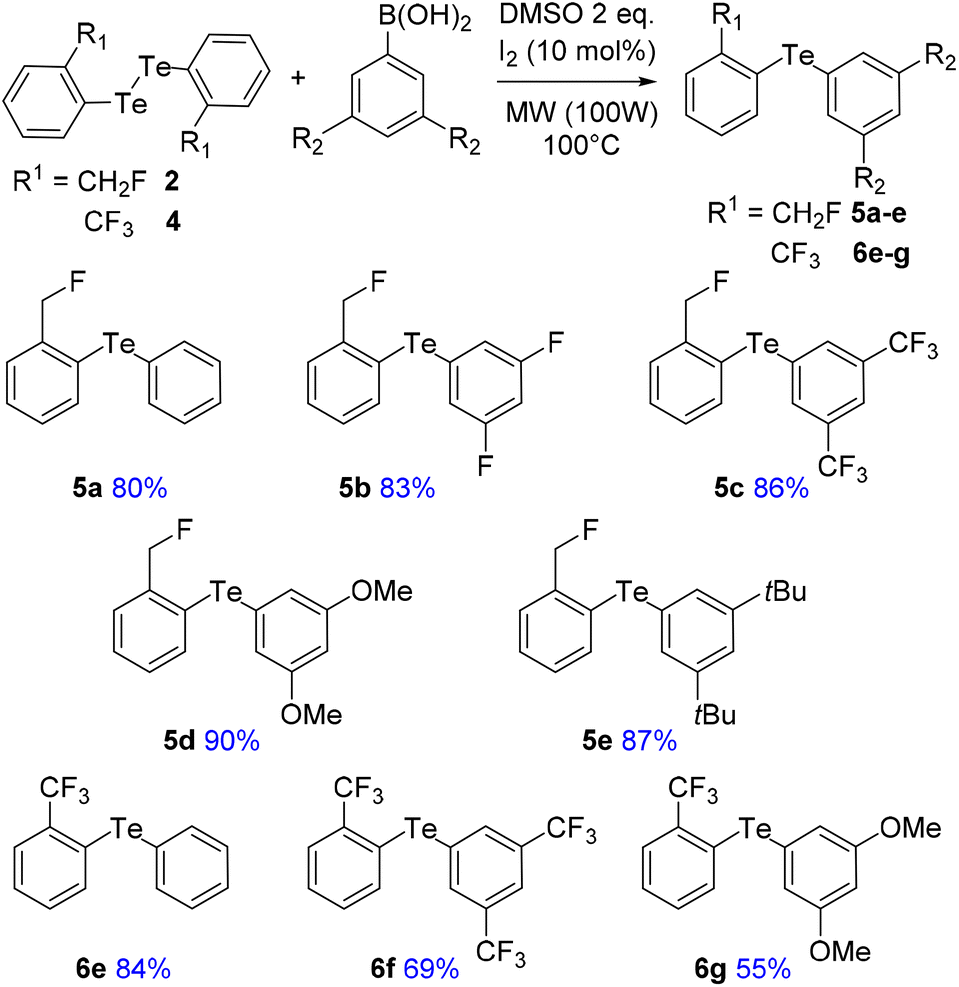 | ||
| Scheme 2 Synthesis of a series of fluorinated diaryl tellurides. | ||
Electrostatic surface potential (ESP) analysis
For ESP analysis, compounds 5a, 5c, 6e, and 6f were chosen as representative members of respectively the CH2F and CF3 series. Their molecular geometries were optimized by DFT (B3LYP/Def2TZVPP, see ESI† Section IV.B for details). As anticipated for the CH2F series, the syn-CH2F and anti-CH2F conformations were found to be the most stable ones among all possible conformations for both 5a and 5c (see ESI Fig. S10 and Table S8†). Coherently, only one stable conformation was detected for 6e and 6f (Fig. 3C, S10 and Table S9†). The Vs,max extrema were then characterized for each conformation of 5a and 5c (see ESI, Tables S8 and S9 and Fig. S11†). Interestingly, only one σ-hole was detected in the syn conformers in the elongation of the bond between Te and the aromatic group bearing CH2F. The other σ-hole that was expected to be facing the fluorine atom could not be characterized. The latter σ hole could be detected in the anti conformer, however. These results suggest a possible through-space electron delocalization from fluorine to a tellurium σ-hole in the syn conformer. This hypothesis was further supported by the topological analysis of the DFT calculated electron density, where a bond path is observed between F and Te atoms with a small but non-negligible delocalization index of about 0.065 (see ESI Fig. S12 and Table S10†). Similar data were obtained with compounds 6e and 6f (see ESI Table S10†).Te⋯F interaction in solution
The mono- and tri-fluorotolyl tellurides 5a–e and 6a–g, as well as their ditelluride precursors 2 and 4, were then studied in solution, using 19F and 125Te NMR to probe the presence or absence of σ-hole Te⋯F interactions (see spectra in ESI, Section V†).The trifluorotolyl tellurium derivatives 6a–g exhibit similar chemical shifts in 19F NMR (−61 ± 1 ppm), but a large shift range in 125Te NMR (369–795 ppm) (Table 1). The latter are nevertheless typical of divalent tellurides,33 as observed in the 19F and 125Te NMR spectra of 4 (−60.5 ppm and 438.1 ppm, respectively). The 125Te signal appears as a quadruplet, indicating a 4JTe–F coupling with the three fluorine atoms of the trifluoromethyl group. This quadruplet also reveals the magnetic equivalence of these fluorine atoms, and thus the free rotation of the trifluoromethyl group at room temperature and even at −80 °C. Interestingly, these 4JTe–F coupling constants (94–154 Hz, Table 1) are far lower than those observed for covalent Te–F bonds (∼950 Hz).24,34 Nevertheless, they are two to four times higher than 3JTe–F coupling values in the 2,6-difluorophenylditelluride 7 (45 Hz), used for comparison purposes. Such a counter-intuitive difference was observed in various fluorinated compounds35 and in HBs involving fluorine.36 The underlying phenomenon was ascribed to through-space orbital overlap,37 and non-linearly correlated to the distance of the interacting atoms, and thus to the strength of the interaction.35b The latter could be assessed from the good linear correlation between 19F chemical shifts (δF) and 4JTe–F coupling constants (Fig. 4). This correlation revealed that more shielded δF correspond to higher 4JTe–F values, and therefore to stronger interactions.
|
|
|||||
|---|---|---|---|---|---|
| Tellurium (R) | δ Te (ppm) | δ F (ppm) | 3 J Te–F (Hz) | 4 J Te–F (Hz) | 5 J C–F (Hz) |
| a Conditions: tellurium species (5–20 mM) in CDCl3 at 25 °C. b The product decomposed during the acquisition of the 125Te NMR spectrum. c Measured on the 19F spectrum. | |||||
| 4 | 438.1 | −60.5 | — | 170.2 | — |
| 6a (CH3) | 369.4 | −62.0 | — | 153.3 | 2.98 |
| 6b (iPr) | 720.7 | −60.7 | — | 99.5 | 1.74 |
| 6c (COOMe) | 795.5 | −60.0 | — | 107.4 | 2.87 |
| 6d (CF2H) | -b | −60.3 | — | 95.9c | 2.67 |
| 6e (Ph) | 728.2 | −61.8 | — | 162.5 | — |
| 6f (3,5-(CF3)2Ph) | 779.5 | −61.1 | — | 146.9 | — |
| 6g (3,5-(MeO)2Ph) | 760.1 | −61.8 | — | 167.5 | — |
| 7 | 215.6 | −86.2 | 45.5 | — | — |
| 8 | — | −63.8 | — | — | — |
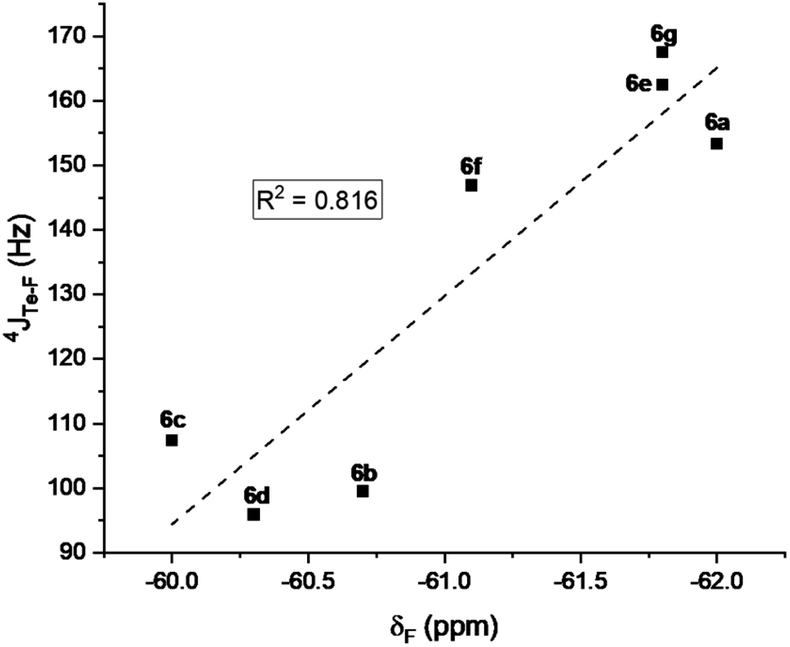 | ||
| Fig. 4 Correlation between the 19F NMR chemical shifts of the tellurides 6a–g and 4JTe–F coupling constants. | ||
Furthermore, the 19F NMR chemical shifts of 4 and 6a–g (−61 ± 1 ppm) were higher than that of trifluorotoluene 8 (−63.8 ppm), independent of the Te substituent. This deshielding is mostly due to the presence of tellurium and indicates a modification of fluorine polarization, as expected from an interaction between the F lone pair and Te σ-holes. These combined observations strongly suggest the presence of noncovalent Te⋯F interactions in this series of Te compounds.
Unexpected observations in 13C NMR corroborate this possibility. The alkyl aryl tellurides 6a–d exhibit what appears as a 5JC–F coupling between the Te-bound alkyl, aryl or carboxylate carbon bonded to the tellurium atom and the fluorine atoms (Fig. 5). Despite the large number of bonds separating them and the presence of the tellurium atom, the observed coupling constants were unexpectedly high (1.74–2.98 Hz; see Table 1). These values are usually typical of 3JC–F couplings.38 Such coupling39 could be due to noncovalent Te⋯F interactions, and if so, the Te σ-hole involved should impose a configuration in which the Te substituent would be aligned with the Te⋯F interaction in an anti position (see the drawing in Fig. 6).
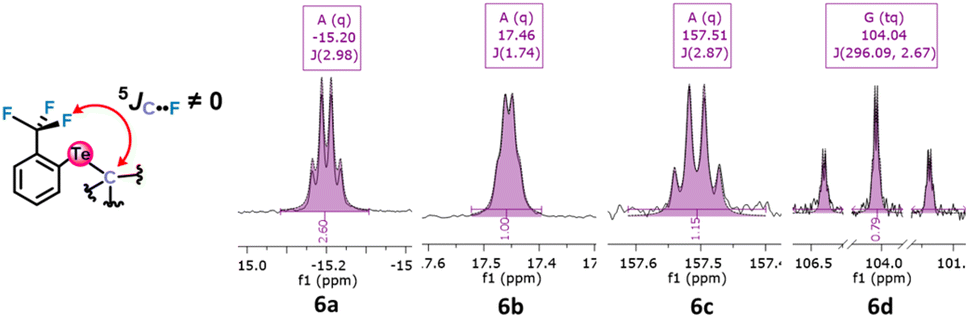 | ||
| Fig. 5 Fragments of tellurides 6a–d13C NMR spectra revealing 5JC–F due to coupling between the carbon bonded to the tellurium atom and the fluorine atoms. | ||
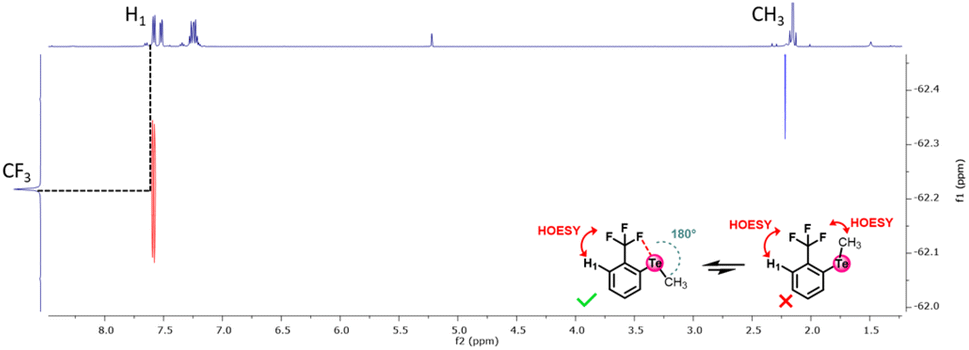 | ||
| Fig. 6 HOESY spectrum of telluride 6a in CD2Cl2. | ||
To verify this hypothesis, a 1H–19F HOESY NMR experiment was performed with the methyl-substituted telluride 6a (Fig. 6). This experiment revealed that the methyl is not close to the CF3 group, whereas the latter is close to the ortho-proton of the phenyl moiety. Therefore, the methyl group should be oriented away from the CF3 group in solution. Such observations further corroborate the presence of a Te⋯F interaction.
The monofluorotolyl tellurides 5a–e were then investigated under the same NMR conditions (Table 2). 125Te NMR chemical shifts (345–645 ppm) were again in the classical range for divalent tellurides.33 As for the CF3 series, the 19F NMR chemical shifts of 2 and 5a–e (−204 ± 1 ppm) were higher than that of the reference compound 9 (−206.1 ppm). A long-range 4JTe–F coupling between the fluorine and tellurium atoms was also observed as a doublet in this series, with similar values (104.6–129.6 Hz). The latter are nevertheless lower than in the CF3 series, as are the 125Te chemical shifts (see 5avs.6e, 5cvs.6f, 5dvs.6g and 2vs.4). Both facts further support the presence of a Te⋯F σ-hole-based interaction for the same reason.
|
|
|||
|---|---|---|---|
| Tellurium (R) | δ Te (ppm) | δ F (ppm) | 4 J Te–F (Hz) |
| a Conditions: tellurium species (20 mM) in CDCl3 at 25 °C. | |||
| 2 | 345.6 | −203.2 | 129.6 |
| 5a (H) | 596.4 | −205.3 | 110.4 |
| 5b (F) | 644.8 | −204.3 | 108.7 |
| 5c (CF3) | 645.1 | −202.3 | 127.0 |
| 5d (OMe) | 622.8 | −205.3 | 108.8 |
| 5e (tBu) | 594.3 | −205.5 | 108.5 |
| 9 | — | −206.1 | — |
Because the deepness of a σ-hole is directly affected by adjacent electron-withdrawing or -donating substituent(s) (see Fig. 1), we attempted to tune Te σ-holes by selecting typical substituents at the meta position of the Te aryl moiety (5b–evs.5a) in this more structurally homogeneous series.
Rewardingly, the electronic effect (σm) of these meta substituents could clearly be correlated to 125Te chemical shifts (Fig. 7A). Moreover, 19F chemical shifts correlated with the 4JTe–F coupling constants (Fig. 7B) and showed that the more electron-withdrawing substituent induced the largest 4JTe–F coupling, as expected for a σ-hole-based interaction. These effects confirmed that the 4JTe–F value represents a measure of the strength of the noncovalent Te⋯F interaction.
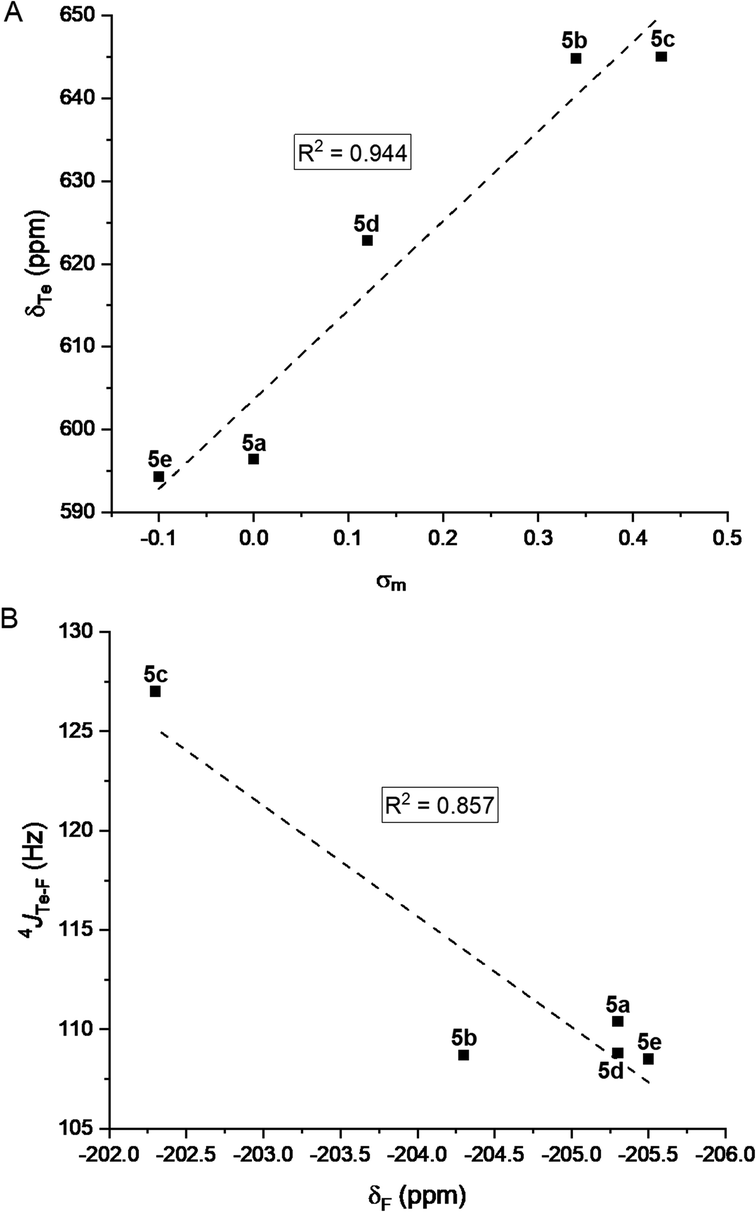 | ||
| Fig. 7 (A) Correlation between the 125Te NMR chemical shifts of the tellurides 5a–e and Hammett parameter (σm). (B) Correlation between 4JTe–F coupling constants and 19F NMR chemical shifts. | ||
Contribution of the Te⋯F ChB to the overall 4JTe–F
Two different studies were performed in order to confirm the role of the Te σ-hole in the Te⋯F interaction and to estimate the ChB contribution to the overall coupling constant.In the first study, the lithium tellurolate intermediate 10-TeLi, where the negative charge on Te was expected to reduce the involvement of its σ-hole in the Te⋯F interaction, was prepared (Scheme 3). The characterization of 10-TeLi by NMR and in particular the determination of the Te–F coupling constant should provide an approximate assessment of the through-space coupling due to chalcogen bonding.
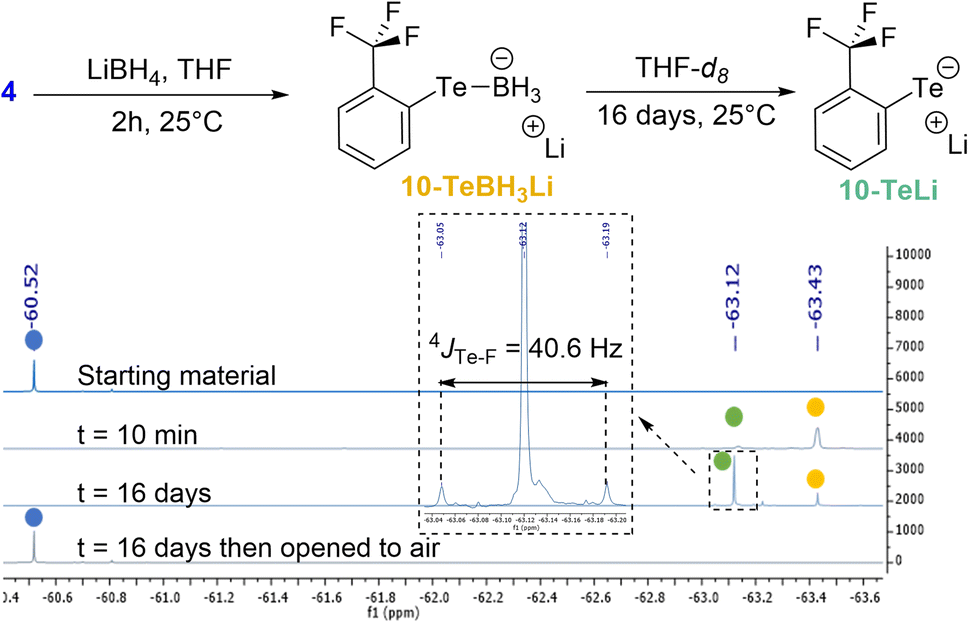 | ||
| Scheme 3 Preparation of lithium tellurolate 10-TeLi and 19F NMR monitoring (color code: 4 in blue, 10-TeBH3Li in yellow and 10-TeLi in green). | ||
Lithium tellurolate 10-TeLi was obtained by slow decomposition of 10-TeBH3Li. The latter was generated by the reaction of ditelluride 4 with an excess of LiBH4 in THF.40 The decomposition of 10-TeBH3Li to 10-TeLi was monitored by 19F NMR in THF-d8 (Scheme 3 and ESI† for full characterization). Almost full conversion was observed after 16 days with complete disappearance of the boron signal of 10-TeBH3Li in 11B NMR and the appearance of a new 125Te signal for 10-TeLi (see the ESI†).41 Moreover, ditelluride 4 was recovered by oxidation of 10-TeLi after opening the NMR tube to air. These observations proved that 10-TeLi was formed after 16 days. The Te–F coupling constant of 40.6 Hz measured for 10-TeLi was much lower than the 4JTe–F values observed for tellurides 6a–g (Table 1). Such a large difference between the telluride and the tellurolate should at least partly reveal the through-space contribution and thus indicates a contribution of the Te⋯F ChB to the overall 4JTe–F in compounds 6a–g.
Similarly, we attempted to form the analogous tellurolate 11-TeLi but all attempts failed, probably because it dimerized to tellurocine 12 (Scheme 4A).30
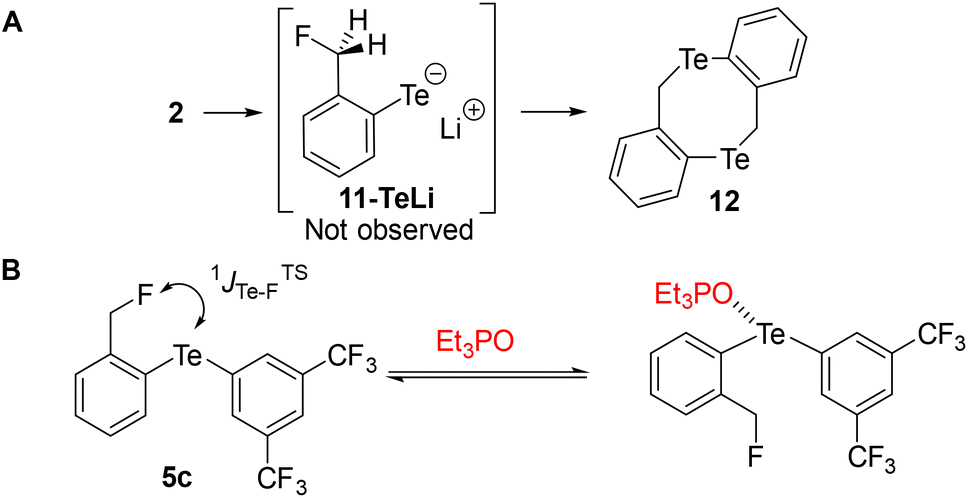 | ||
| Scheme 4 (A) Attempted preparation of tellurolate 11-TeLi. (B) Competition experiments between 5c and Et3PO. | ||
In the second study, the 4JTe–F evolution of tellurides in the presence of Et3PO Lewis base was investigated by NMR. The tellurides were expected to form an intermolecular Te⋯O interaction in competition with the intramolecular Te⋯F ChB with a direct influence on 4JTe–F (Scheme 4B). This strategy was tested on telluride 5c with the strongest Te⋯F ChB, and to avoid a possible HB between Et3PO and CDCl3, competition experiments were performed in deuterated cyclohexane.19a Large variations of 4JTe–F were obtained after the addition of 1, 5 and 10 equiv. of Et3PO (Δ4JTe–F = −7.8, −24.3 and −32.8 Hz, respectively) and a plateau (∼33 Hz) was reached between 10 and 20 equiv. of Lewis base (see the ESI† for details).
At this plateau, the intramolecular Te⋯F interaction was completely replaced by the intermolecular Te⋯O interaction. Therefore, the difference of about 33 Hz should reflect the contribution of the Te⋯F ChB to the overall 4JTe–F value.
This strategy was unsuccessful when applied to telluride 6f. Indeed, no modification of 4JTe–F was observed after the addition of 1 equiv. of Et3PO and only small variations were observed when 5 or 10 equiv. were employed (Δ4JTe–F = −3.7 and −5.9 Hz, respectively) (see the ESI† for details). These variations represent only 2.4 and 3.9% of the 4JTe–F value before the addition of the Lewis base, suggesting that the Te⋯F interaction is strong enough in telluride 6f to outweigh the competition from an intermolecular Lewis base such as Et3PO.
Evaluating the strength of Te⋯F interaction in solution for compounds 5
As determined above, the measured 4JTe–F coupling may reflect the noncovalent Te⋯F interaction strength, but its value is averaged over the various conformations. Based on our conformational analysis, the syn and anti conformers are the major ones; we can reasonably assume a two state equilibrium model in which the 4JTe–F coupling results from the relative proportion of each conformation contribution 4JTe–F(syn) and 4JTe–F(anti) (eqn (1); χsyn is a temperature-dependent variable representing the relative population of the syn conformer).| 4JTe–F = χsyn(4JTe–F(syn)) + (1 − χsyn)(4JTe–F(anti)) | (1) |
This expression can be inserted into eqn (2), which defines the anti–syn equilibrium constant KA to give eqn (3).
| KA = χsyn/(1 − χsyn) | (2) |
| KA = (4JTe–F − 4JTe–F(anti))/(4JTe–F(syn) − 4JTe–F) | (3) |
The Gibbs free energy of this equilibrium is thus expressed as a function of coupling constants and of temperature (eqn (4)).
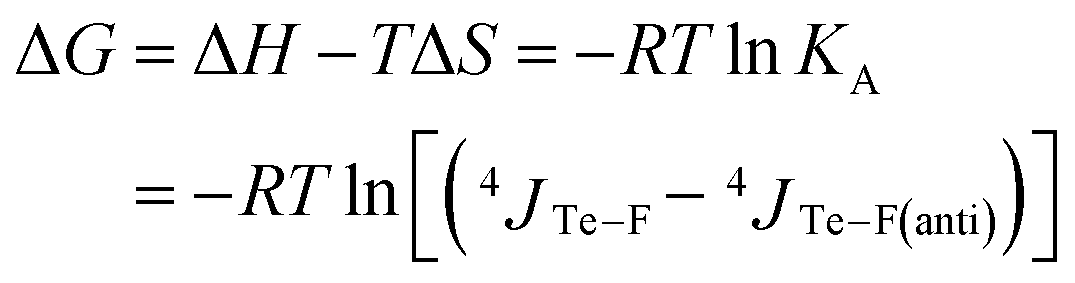 | (4) |
To evaluate the 4JTe–F values as a function of the conformation, relativistic DFT calculations were performed on each conformer. These calculations provided large negative values for 4JTe–F (−249 ± 4 Hz for 5a–c; −164 ± 2 Hz for 6e and 6f) and a much smaller positive value for 4JTe–F(anti) (+19–20 Hz) (see ESI Section IV.D, Table S11†). Surprisingly, a related study with fluoroseleno compounds suggested that 4JTe–F(anti) was approximately zero because no Se⋯F coupling was detected in selenolate 11-SeLi, which was considered to be in an anti conformation due to electronic repulsion.14 This could not be confirmed on the analogous tellurolate 11-TeLi because of its instability (Scheme 4A).30 We nevertheless performed relativistic DFT calculations for 11-TeLi, along all the C–C–C–F torsion angles from the anti to the syn conformer. The resulting data showed that 4JTe–F(anti) remains relatively constant (20 ± 10 Hz) from the anti to a 90° arrangement and then sharply decreases to −130 Hz for the syn conformer (see ESI Section IV.D and Fig. S13†). All of the computed data showed that 4JTe–F(anti) is close to 20 Hz, independent of the telluronium structure, the tellurolate 11-TeLi, the ditelluride 2 and the monotellurides 5a and 5c (see ESI Section IV.D, Fig. S13 and Table S11†).
To obtain experimental data on 4JTe–F, and in order to assess thermodynamic parameters, typical mono- and tri-fluorotolyl tellurium derivatives (2, 4, 5a, 6e, 5c and 6f) were examined by variable temperature (VT) NMR from 233 to 308 K (Fig. 8 and ESI, Section III.A and B†). The trifluoro compounds were studied as models with permanent syn conformations in order to verify the non-dependency of 4JTe–F(syn) on temperature.
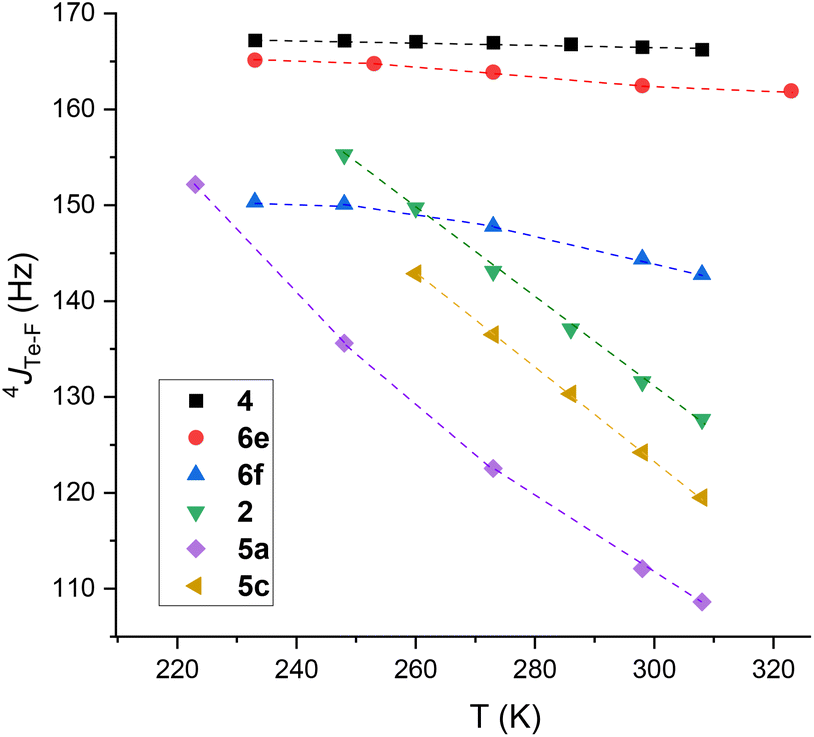 | ||
| Fig. 8 Evolution of 4JTe⋯F of trifluorotolyl (2, 6e and 6f) and monofluorotolyl (4, 5a and 5c) derivatives as a function of temperature. | ||
Low variations of 4JTe–F were obtained for the trifluorotolyl derivatives (variation for 4: 0.7%; 6e: 2.6%; 6f: 6.9%) proving that 4JTe–F is almost independent of temperature (see ESI Section III.A† for complete data). In contrast, the monofluorotolyl compounds (2, 5a and 5c) showed large variations (Δ4JTe–F up to 44 Hz), as expected from temperature-dependent modification of the anti–syn equilibrium. Interestingly, the highest value was observed for the 3,5-bis(trifluoromethyl)phenyl derivative 5c that exhibits the largest Te σ-hole (Fig. 8 and ESI, Section III.B†). Similarly, δF displayed a net deshielding upon decreasing the temperature for the monofluorotolyl derivatives (ΔδF ∼ 4 ppm, Tables S4–S6 in ESI†), but a slight shielding for the trifluorotolyl analogs (ΔδF ∼ 0.5 ppm) (Tables S1–S3 in ESI†). Such effects are in line with an increased electron density transfer from an F lone pair to the Te σ-hole in the monofluorotolyl series, as expected from increasing predominance of the syn conformer.
Based on the results at low temperature, the enthalpy of the anti–syn equilibrium seems to be mostly driven by the Te⋯F interaction. Therefore, this enthalpy can be estimated from the energy of the Te⋯F interaction using eqn (5):
| ΔH ≈ ETe⋯F | (5) |
From the experimental VT NMR data, the ΔH and ΔS of eqn (4) were determined as follows. Assuming a two state model, we first determined the values of 4JTe–F(anti) and 4JTe–F(syn) that give the best linearity of −RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnKA as a function of T. Due to poor convergence, one of the two parameters was fixed while refining the second one. We thus chose to solve eqn (4) for two different fixed values of 4JTe–F(anti), namely −20 Hz and +20 Hz because these values represent the average theoretical calculations for 4JTe–F(anti) (see above) and because positive and negative coupling constants were calculated for non-syn conformations. The optimal value of 4JTe–F(syn) was then determined, leading to a linear fit and values of ΔH and ΔS that were obtained as intervals corresponding to the positive and negative estimated values of 4JTe–F(anti) (see ESI Section III.C and Table S7†). The estimated values of ΔH and ΔS were then used to calculate ETe⋯F and ΔG298 (Table 3). For compounds 2 and 5c, the obtained negative ΔG298 values for the anti ⇌ syn equilibrium indicated a favorable shift towards the syn conformer, and thus confirmed the preference in solution for the conformer exhibiting a σ-hole Te⋯F interaction. In contrast, the positive value of ΔG298 obtained for compound 5a indicated that the anti conformer is the major component in solution at room temperature. This result revealed the strong influence of the presence of electron-withdrawing substituents, which increase the deepness of the Te σ-hole, and thus the Te⋯F interaction, as recently demonstrated for related σ-hole interactions involving tellurium atoms.19a,b
lnKA as a function of T. Due to poor convergence, one of the two parameters was fixed while refining the second one. We thus chose to solve eqn (4) for two different fixed values of 4JTe–F(anti), namely −20 Hz and +20 Hz because these values represent the average theoretical calculations for 4JTe–F(anti) (see above) and because positive and negative coupling constants were calculated for non-syn conformations. The optimal value of 4JTe–F(syn) was then determined, leading to a linear fit and values of ΔH and ΔS that were obtained as intervals corresponding to the positive and negative estimated values of 4JTe–F(anti) (see ESI Section III.C and Table S7†). The estimated values of ΔH and ΔS were then used to calculate ETe⋯F and ΔG298 (Table 3). For compounds 2 and 5c, the obtained negative ΔG298 values for the anti ⇌ syn equilibrium indicated a favorable shift towards the syn conformer, and thus confirmed the preference in solution for the conformer exhibiting a σ-hole Te⋯F interaction. In contrast, the positive value of ΔG298 obtained for compound 5a indicated that the anti conformer is the major component in solution at room temperature. This result revealed the strong influence of the presence of electron-withdrawing substituents, which increase the deepness of the Te σ-hole, and thus the Te⋯F interaction, as recently demonstrated for related σ-hole interactions involving tellurium atoms.19a,b
| Compound | ΔH ≈ ETe⋯F (kJ mol−1) | ΔS (J K−1 mol−1) | ΔG298 (kJ mol−1) |
|---|---|---|---|
| a See ESI, Table S7 for full data. | |||
| 2 | −7.6 to −6.3 | −21.3 to −15.5 | −1.2 to −1.7 |
| 5a | −4.4 to −3.0 | −20.7 to −17.0 | +1.6 to +2.1 |
| 5c | −10.6 to −9.1 | −29.5 to −22.4 | −1.8 to −2.4 |
This effect is also reflected by the more negative entropy of compound 5c compared to 5a (−26.0 vs. −18.9 J K−1 mol−1). Here again, the increased deepness of the Te σ-hole strengthens the Te⋯F interaction, which in turn increases the proportion of the syn conformer and reduces the molecular degree of freedom. The energies of these Te⋯F interactions are modest (−3 to −11 kJ mol−1), and slightly lower than the well-known Te⋯Te interactions.42 The Te⋯F interactions described here are thus in the lower range of the already known noncovalent interactions involving a tellurium atom (Fig. 9).
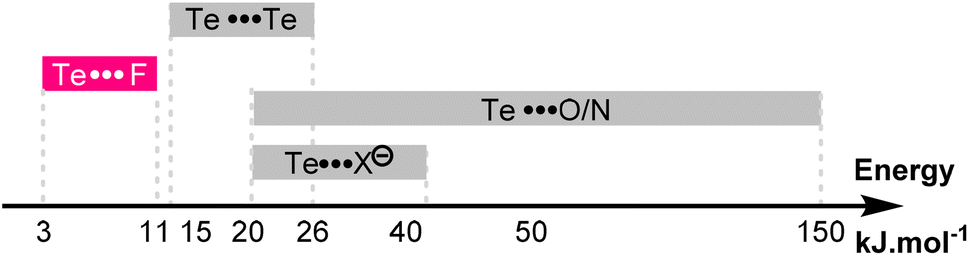 | ||
| Fig. 9 Energy range of the known noncovalent interactions involving the tellurium atom and of the Te⋯F interaction reported here. | ||
Conclusions
In conclusion, after having prepared new fluoro- and trifluoro tellurium derivatives, we demonstrated the presence of noncovalent Te⋯F interactions through the σ-holes on tellurium and their importance in solution as indicated by 19F and 125Te NMR studies. The various effects reported here established the presence of ChB between Te and F as schematically shown in Fig. 3B.By variable temperature experiments, we were even able to evaluate the intensity of such noncovalent Te⋯F interactions (−3 to −11 kJ mol−1). The identification of noncovalent Te⋯F interactions reported here should provide new opportunities in organic chemistry, especially in organocatalysis, in drug design and in biology. Further work in these areas is underway in our group.
Data availability
Experimental and computational data associated with this work are provided in the accompanying ESI.†Author contributions
RW and LG carried out the experiments. EA performed the computational studies. RW, PP and VM designed the project. PP and VM co-wrote the manuscript. VM co-wrote and edited the ESI.† RW and EA co-wrote the ESI† and contributed to writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the International Center Frontier Research in Chemistry (icFRC), the LabEx CSC (ANR-10-LABX-0026 CSC) and the French National Research Agency (ANR-21-CE07-0014). RW thanks the LabEx CSC, Strasbourg, for a PhD fellowship and LG thanks the ANR for a PhD fellowship. We thank the EXPLOR mesocenter for providing access to their computing facility (project 2021CPMXX2483) and the NMR service (B. Vincent) of the LeBel Federation FR2010 for the easy access to the spectrometers and for advice. EA thanks Dr Axel Gansmüller for fruitful discussions.References
- (a) P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2013, 15, 11178–11588 RSC; (b) A. Bauzá, T. J. Mooibroek and A. Frontera, ChemPhysChem, 2015, 16, 2496–2517 CrossRef PubMed.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- For recent developments in XB applications in biology and in organic chemistry, see: (a) G. Berger, P. Frangville and F. Meyer, Chem. Commun., 2020, 56, 4970–4981 RSC; (b) R. L. Sutar and S. M. Huber, ACS Catal., 2019, 9, 9622–9639 CrossRef CAS.
- ChB has been sparingly investigated in the last decade, see: (a) P. Scilabra, G. Terraneo and G. Resnati, Acc. Chem. Res., 2019, 52, 1313–1324 CrossRef CAS PubMed; (b) N. Biot and D. Bonifazi, Coord. Chem. Rev., 2020, 413, 213243 CrossRef CAS; (c) R. Hein and P. D. Beer, Chem. Sci., 2022, 13, 7098 RSC.
- A. Frontera and A. Bauza, Int. J. Mol. Sci., 2022, 23, 4188 CrossRef CAS PubMed.
- C. B. Aakeröy, D. L. Bryce, G. R. Desiraju, A. Frontera, A. C. Legon, F. Nicotra, K. Rissanen, S. Scheiner, G. Terraneo, P. Metrangolo and G. Resnati, Pure Appl. Chem., 2019, 91, 1889–1892 CrossRef.
- (a) M. R. Scholfield, C. M. Vander Zanden, M. Carter and P. S. Ho, Protein Sci., 2013, 22, 139–152 CrossRef CAS PubMed; (b) P. S. Ho, Future Med. Chem., 2017, 4, 637–640 CrossRef PubMed.
- (a) M. Iwaoka and N. Isozumi, Molecules, 2012, 17, 7266–7283 CrossRef CAS PubMed; (b) K. Kriz, J. Fanfrik and M. Lepsik, ChemPhysChem, 2018, 19, 2540–2548 CrossRef CAS PubMed.
- E. R. T. Tiekink, Dalton Trans., 2012, 41, 6390–6395 RSC.
- (a) P. Shah and A. D. Westwell, J. Enzyme Inhib. Med. Chem., 2007, 22, 527–540 CrossRef CAS PubMed; (b) Fluorine in Medicinal Chemistry and Chemical Biology, ed. I. Ojima, Wiley-Blackwell, 2009 Search PubMed.
- W. Pietrus, R. Kafel, A. J. Bojarski and R. Kurczab, Molecules, 2022, 27, 1005–1019 CrossRef CAS PubMed.
- (a) The actual presence of XBs with the F atom is still a matter of debate; see ref. 2 and: P. Metrangolo, J. S. Murray, T. Pilati, P. Politzer, G. Resnati and G. Terraneo, Cryst. Growth Des., 2011, 11, 4238–4246 CrossRef CAS; (b) K. Eskandari and M. Lesani, Chem.–Eur. J., 2015, 21, 4739–4746 CrossRef CAS PubMed; (c) S. Scheiner, J. Phys. Chem. A, 2020, 124, 7290–7299 CrossRef CAS PubMed; (d) S. A. Harry, S. Vemulapalli, T. Dudding and T. Lectka, J. Org. Chem., 2022, 13, 8413–8419 CrossRef PubMed.
- K. Grollier, A. Taponard and T. Billard, Eur. J. Org. Chem., 2020, 2020, 6943–6954 CrossRef CAS.
- For a study on the Se–F interaction, see: M. Iwaoka, H. Komatsu, T. Katsuda and S. Tomoda, J. Am. Chem. Soc., 2002, 124, 1902–1909 CrossRef CAS PubMed.
- Pauling electronegativity of halogens: F 4.0, Cl 3.0, Br 2.8, and I 2.5, and of chalcogens: O 3.5, S 2.5, Se 2.4, and Te 2.1.
- P. Politzer, P. Lane, M. C. Concha, Y. Ma and J. S. Murray, J. Mol. Model., 2007, 13, 305–311 CrossRef CAS PubMed.
- B. Zhou and F. P. Gabbai, Chem. Sci., 2020, 11, 7495–7500 RSC.
- P. Pale and V. Mamane, ChemPhysChem, 2023, 24, e202200481 CrossRef CAS PubMed.
- (a) R. Weiss, E. Aubert, L. Groslambert, P. Pale and V. Mamane, Chem.–Eur. J., 2022, 28, e202200395 CAS; (b) R. Weiss, E. Aubert, P. Pale and V. Mamane, Angew. Chem., Int. Ed., 2021, 60, 19281–19286 CrossRef CAS PubMed; (c) H. Nishiyama, F. Zheng, S. Inagi, H. Fueno, K. Tanaka and I. Tomita, Polym. Chem., 2020, 11, 4693–4698 RSC; (d) S. Mehrparvar, C. Wölper, R. Gleiter and G. Haberhauer, Angew. Chem., Int. Ed., 2020, 59, 17154–17161 CrossRef CAS PubMed; (e) L. Chen, J. Xiang, Y. Zhao and Q. Yan, J. Am. Chem. Soc., 2018, 140, 7079–7082 CrossRef CAS PubMed; (f) E. Pietrasiak and A. Togni, Organometallics, 2017, 36, 3750–3757 CrossRef CAS; (g) P. C. Ho, P. Szydlowski, J. Sinclair, P. J. W. Elder, J. Kübel, C. Gendy, L. M. Lee, H. Jenkins, J. F. Britten, D. R. Morim and I. Vargas-Baca, Nat. Commun., 2016, 7, 11299 CrossRef CAS PubMed; (h) P. R. Prasad, K. Selvakumar, H. B. Singh and R. J. Butcher, J. Org. Chem., 2016, 81, 3214–3226 CrossRef CAS PubMed; (i) M. Baiwir, G. Llabres, J. Denoel and J.-L. Piette, Mol. Phys., 1973, 25, 1–7 CrossRef CAS.
- (a) T. Glodde, Y. V. Vishnevskiy, L. Zimmermann, H.-G. Stammler, B. Neumann and N. W. Mitzel, Angew. Chem., Int. Ed., 2021, 60, 1519–1523 CrossRef CAS PubMed; (b) M. Hejda, D. Duvinage, E. Lork, R. Jirásko, A. Lyčka, S. Mebs, L. Dostál and J. Beckmann, Organometallics, 2020, 39, 1202–1212 CrossRef CAS; (c) I. D. Sadekov, V. I. Minkin, A. V. Zakharov, A. G. Starikov, G. S. Borodkin, S. M. Aldoshin, V. V. Tkachev, G. V. Shilov and F. J. Berry, J. Organomet. Chem., 2005, 690, 103–116 CrossRef CAS; (d) A. G. Maslakov, W. R. McWhinnie, M. C. Perry, N. Shaikh, S. L. W. McWhinnie and T. A. Hamor, J. Chem. Soc., Dalton Trans., 1993, 619–624 RSC.
- (a) M. Bühl, F. R. Knight, A. Křístková, I. M. Ondík, O. L. Malkina, R. A. M. Randall, A. M. Z. Slawin and J. D. Woollins, Angew. Chem., Int. Ed., 2013, 52, 2495–2498 CrossRef PubMed; (b) F. R. Knight, L. M. Diamond, K. S. Athukorala Arachchige, P. Sanz Camacho, R. A. M. Randall, S. E. Ashbrook, M. Bühl, A. M. Z. Slawin and J. D. Woollins, Chem.–Eur. J., 2015, 21, 3613–3627 CrossRef CAS PubMed; (c) M. W. Stanford, F. R. Knight, K. S. A. Arachchige, P. S. Camacho, S. E. Ashbrook, M. Bühl, A. M. Z. Slawin and J. D. Woollins, Dalton Trans., 2014, 43, 6548–6560 RSC.
- (a) A. Nordheider, E. Hupf, B. A. Chalmers, F. R. Knight, M. Bühl, S. Mebs, L. Chęcińska, E. Lork, P. S. Camacho, S. E. Ashbrook, K. S. Athukorala Arachchige, D. B. Cordes, A. M. Z. Slawin, J. Beckmann and J. D. Woollins, Inorg. Chem., 2015, 54, 2435–2446 CrossRef CAS PubMed; (b) T. G. Do, E. Hupf, A. Nordheider, E. Lork, A. M. Z. Slawin, S. G. Makarov, S. Yu. Ketkov, S. Mebs, J. D. Woollins and J. Beckmann, Organometallics, 2015, 4, 5341–5360 CrossRef.
- K. T. Mahmudov, A. V. Gurbanov, V. A. Aliyeva, M. F. C. Guedes da Silva, G. Resnati and A. J. L. Pombeiro, Coord. Chem. Rev., 2022, 464, 214556 CrossRef CAS.
- (a) H. Zhao and F. P. Gabbai, Nat. Chem., 2010, 2, 984–990 CrossRef CAS PubMed; (b) J. Y. C. Lim, I. Marques, A. L. Thompson, K. E. Christensen, V. Felix and P. D. Beer, J. Am. Chem. Soc., 2017, 139, 3122–3133 CrossRef CAS PubMed.
- A. Dessì, P. Peluso, R. Dallocchio, R. Weiss, G. Andreotti, M. Allocca, E. Aubert, P. Pale, V. Mamane and S. Cossu, Molecules, 2020, 25, 2213 CrossRef PubMed.
- (a) P. Peluso, V. Mamane, A. Dessì, R. Dallocchio, E. Aubert, C. Gatti, D. Mangelings and S. Cossu, J. Chromatogr. A, 2020, 1616, 460788 CrossRef CAS PubMed; (b) P. Peluso, C. Gatti, A. Dessì, R. Dallocchio, R. Weiss, E. Aubert, P. Pale, S. Cossu and V. Mamane, J. Chromatogr. A, 2018, 1567, 119–129 CrossRef CAS PubMed; (c) R. Dallocchio, A. Dessì, M. Solinas, A. Arras, S. Cossu, E. Aubert, V. Mamane and P. Peluso, J. Chromatogr. A, 2018, 1563, 71–81 CrossRef CAS PubMed; (d) P. Peluso, V. Mamane, R. Dallocchio, A. Dessì, R. Villano, D. Sanna, E. Aubert, P. Pale and S. Cossu, J. Sep. Sci., 2018, 41, 1247–1256 CrossRef CAS PubMed; (e) P. Peluso, V. Mamane, E. Aubert, A. Dessì, R. Dallocchio, A. Dore, P. Pale and S. Cossu, J. Chromatogr. A, 2016, 1467, 228–238 CrossRef CAS PubMed.
- (a) R. Weiss, E. Aubert, P. Peluso, S. Cossu, P. Pale and V. Mamane, Molecules, 2019, 24, 4484 CrossRef CAS PubMed; (b) V. Mamane, P. Peluso, E. Aubert, R. Weiss, E. Wenger, S. Cossu and P. Pale, Organometallics, 2020, 39, 3936–3950 CrossRef CAS; (c) R. Weiss, T. Golisano, P. Pale and V. Mamane, Adv. Synth. Catal., 2021, 363, 4779–4788 CrossRef CAS; (d) E. Aubert, A. Doudouh, E. Wenger, B. Sechi, P. Peluso, P. Pale and V. Mamane, Eur. J. Inorg. Chem., 2022, 2022, e202100927 CrossRef CAS; (e) L. Groslambert, A. Padilla-Hernandez, R. Weiss, P. Pale and V. Mamane, Chem.–Eur. J., 2023, 29, e202203372 CrossRef CAS PubMed.
- (a) For a general review on the importance of through-space spin-spin coupling in NCI, see: J.-C. Hierso, Chem. Rev., 2014, 114, 4838–4867 CrossRef CAS PubMed; (b) For a recent study using 19F NMR to probe the XB through the determination of through-bond 1JC–F couplings, see: B. Jimmink, D. Sethio, L. Turunen, D. von der Heiden and M. Erdélyi, J. Am. Chem. Soc., 2021, 143, 10695–10699 CrossRef CAS PubMed.
- (a) H. B. Singh, N. Sudha, A. A. West and T. A. Hamor, J. Chem. Soc., Dalton Trans., 1990, 907–913 RSC; (b) For an overview, see: H. B. Singh and G. Mugesh, Acc. Chem. Res., 2002, 35, 226–236 CrossRef PubMed.
- R. Weiss, Y. Cornaton, H. Khartabil, L. Groslambert, E. Hénon, P. Pale, J.-P. Djukic and V. Mamane, ChemPlusChem, 2022, 87, e202100518 CAS.
- S. Saba, J. Rafique and A. L. Braga, Adv. Synth. Catal., 2015, 357, 1446–1452 CrossRef CAS.
- A. Ouchi, T. Hyugano and C. Liu, Org. Lett., 2011, 11, 4870–4873 CrossRef PubMed.
- A. Panda and H. B. Singh, NMR of Organoselenium and Organotellurium Compounds, PATAI'S Chemistry of Functional Groups, John Wiley & Sons, Ltd, 2013 Search PubMed.
- A. Hammerl, T. M. Klapötke, B. Krumm and M. Scherr, Z. Anorg. Allg. Chem., 2007, 633, 1618–1626 CrossRef CAS.
- (a) F. B. Mallory, E. D. Luzik Jr, C. W. Mallory and P. J. Carroll, J. Org. Chem., 1992, 57, 366–370 CrossRef CAS; (b) F. B. Mallory, C. W. Mallory, K. E. Butler, M. B. Lewis, A. Q. Xia, E. D. Luzik Jr, L. E. Fredenburgh, M. M. Ramanjulu, Q. N. Van, M. M. Francl, D. A. Freed, C. C. Wray, C. Hann, M. Nerz-Stormes, P. J. Carroll and L. E. Chirlian, J. Am. Chem. Soc., 2000, 122, 4108–4116 CrossRef CAS.
- S. Grzesiek, F. Cordier, V. Jaravine and M. Barfield, Prog. Nucl. Magn. Reson., 2004, 45, 275–300 CrossRef CAS.
- L. Ernst and K. Ibrom, Angew. Chem., Int. Ed., 1995, 34, 1881–1882 CrossRef CAS.
- For comparison, see the nJC–F coupling in trifluorotoluene (Hz): 1JC–F = 272; 2JC–F = 32; 3JC–F = 4 ; 4JC–F = 1; 5JC–F = 0.
- C. Otake, T. Namba, H. Tabata, K. Makino, K. Hirano, T. Oshitari, H. Natsugari, T. Kusumi and H. Takahashi, J. Org. Chem., 2021, 86, 4638–4645 CrossRef CAS PubMed.
- R. Ramalakshmi, K. Saha, D. K. Roy, B. Varghese, A. K. Phukan and S. Ghosh, Chem.–Eur. J., 2015, 21, 17191–17195 CrossRef CAS PubMed.
- The chemical shift for 10-Li (δTe = 105.8 ppm) is in the same range as for the known PhTeLi (δTe = 133.9 ppm), see: M. A. Banks, O. T. Beachley, H. J. Gysling and H. R. Luss, Organometallics, 1990, 9, 1979–1982 CrossRef CAS.
- R. Gleiter, G. Haberhauer, D. B. Werz, F. Rominger and C. Bleiholder, Chem. Rev., 2018, 118, 2010–2041 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterisation data and NMR spectra; VT NMR spectra and coupling constants; computed conformational analysis and Vs,max data; computed NMR data; methodology for the determination of thermodynamic parameters. See DOI: https://doi.org/10.1039/d3sc00849e |
| This journal is © The Royal Society of Chemistry 2023 |