
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of a new fluorophore: wavelength-tunable bisbenzo[f]isoindolylidenes†
Changqing
Ye‡
a,
Rui
Huang‡
ab,
Mong-Feng
Chiou
 a,
Bo
Wang
b,
Daliang
Li
*b and
Hongli
Bao
*ac
a,
Bo
Wang
b,
Daliang
Li
*b and
Hongli
Bao
*ac
aState Key Laboratory of Structural Chemistry, Key Laboratory of Coal to Ethylene Glycol and Its Related Technology, Fujian Science & Technology Innovation Laboratory for Optoelectronic Information of China, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, 155 Yangqiao Road West, Fuzhou, Fujian 350002, P. R. China. E-mail: hlbao@fjirsm.ac.cn
bFujian Key Laboratory of Innate Immune Biology, Biomedical Research Center of South China, Key Laboratory of Optoelectronic Science and Technology for Medicine of Ministry of Education, College of Life Sciences, Fujian Normal University, Fuzhou, Fujian 350002, P. R. China. E-mail: daliangli@fjnu.edu.cn
cUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 2nd November 2023
Abstract
The creation of new functional molecules is a central task in chemical synthesis. Herein, we report the synthesis of a new type of fluorophore, bisbenzo[f]isoindolylidenes, from easily accessible dipropargyl benzenesulfonamides. Wavelength-tunable fluorophores emitting strong fluorescence of green to red light were obtained in this reaction. Late-stage modifications and incorporation of bioactive molecules into these fluorophores give rise to potential applications in biological studies. Detailed computational and experimental studies were conducted to elucidate the mechanism, and suggest a reaction sequence involving Garratt–Braverman type cyclization, isomerization, fragmentation, dimerization and oxidation.
Introduction
Human beings have been using fluorescent molecules for centuries. The earliest recorded fluorescent molecule could be matlaline, a benzofuranoid compound which was observed by the Spanish physician Nicolás Monardes in 1565 in a red sandalwood wooden cup soaked with water.1 Currently, fluorescent probes, or fluorophores are widely used to explore living systems. They have been used in the specific recognition of small signaling molecules, the interaction of proteins and in the exploration of many of the problems associated with human health.2 Small-molecule fluorescent probes are increasingly important in the imaging of living cells by fluorescence and super-resolution fluorescence microscopy.3 Fluorescein,4 coumarin,5 rhodamine,6 cyanine,7 BODIPYs,8 1,8-naphthalimides9 (Fig. 1a) and their many derivatives,10 for example, have provided a variety of tools with which the complex roles in biological research of small molecules, significant cations, anions and biological macromolecules have been investigated. Notwithstanding these classical small molecule fluorophores, development of a new family of fluorophores is a desirable and necessary challenge because they could serve as a platform for further diverse modifications which may enhance or supplement the existing fluorescent molecules in physical, chemical or biological properties.2d,11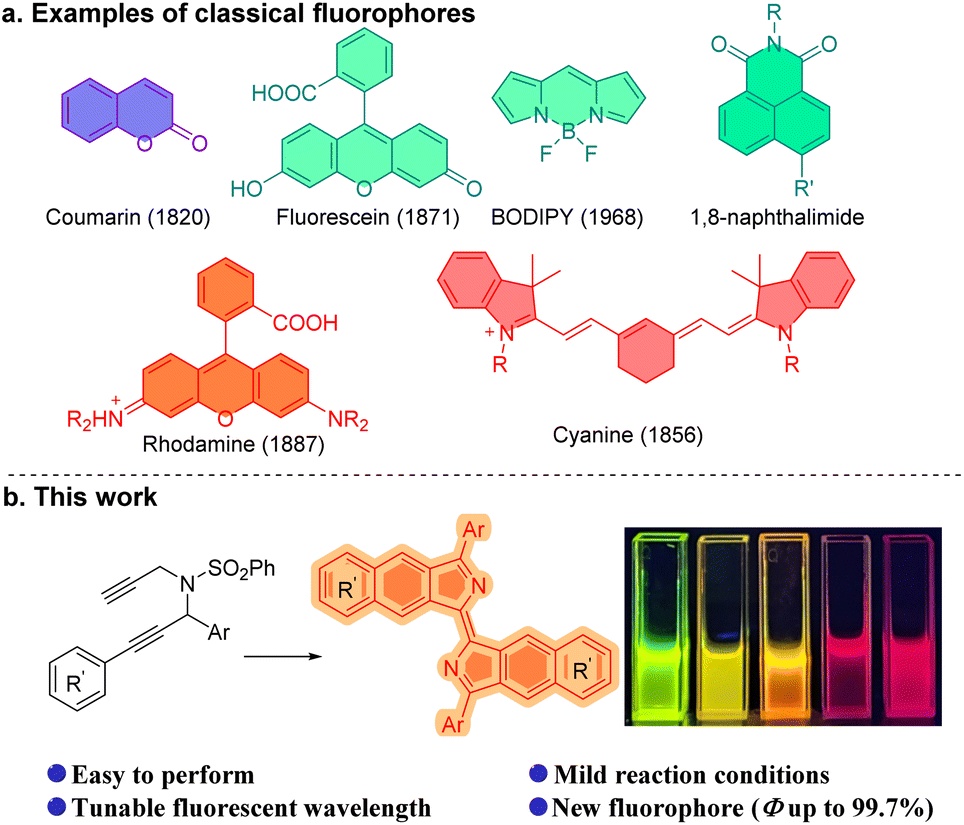 | ||
| Fig. 1 Classic fluorophores and this work. | ||
Propargylamine is a crucial intermediate in organic synthesis, particularly for propargylamine compounds that possess multiple alkynyl substitutions.12 Ye,13 Sahoo,14 and Gagosz15 developed a series of powerful methods utilizing propargyl-ynamides to afford heterocyclic compounds, such as pyrrole, pyridine, quinoline, and their derivatives. Dipropargylamine was employed by Trost et al. in the construction of the core structure of cyclic 2-vinyl-1-acyl compounds during the total synthesis of (+)-α-kainic acid.16 Zhu et al. have presented a novel method for synthesizing furodihydropyridines from tripropargylamines.17 And, bispropargylamine compounds were found to enable the synthesis of benzoisoindole.18 Herein we report the synthesis of wavelength-tunable bisbenzo[f]isoindolylidene compounds which are a new fluorophore, from dipropargyl benzenesulfonamides (Fig. 1b).
(E)-3,3′-Diphenyl-1,1′-bisbenzo[f]isoindolylidene (1), a highly fluorescent compound, was synthesized by the treatment of dipropargyl benzenesulfonamide (1s) with K2CO3 in i-PrOH at 80 °C for 24 h in air (Table S1†). This reaction can be extended to a broad scope of substituted dipropargyl benzenesulfonamides and the compatibility of various R1 substituents in the substrates was explored (Fig. 2). Both electron-donating and withdrawing substituents on the aromatic ring of R1, including alkyl (2), methylthioether (3), methoxy (4), alkenyl (5), amino (6, 7, 17), 2,6-dimethyl (8, 14, 15), bromine (9, 16), phenyl (13) and naphthyl (18) are tolerated in this cyclization reaction and produce the corresponding products in moderate yields. The influence of the R2 group was investigated and functional groups such as methoxy (10, 14), alkyl (11, 13, 16–18), alkenyl (12) and phenanthryl (15) on R2 were found to be tolerated in the reaction.
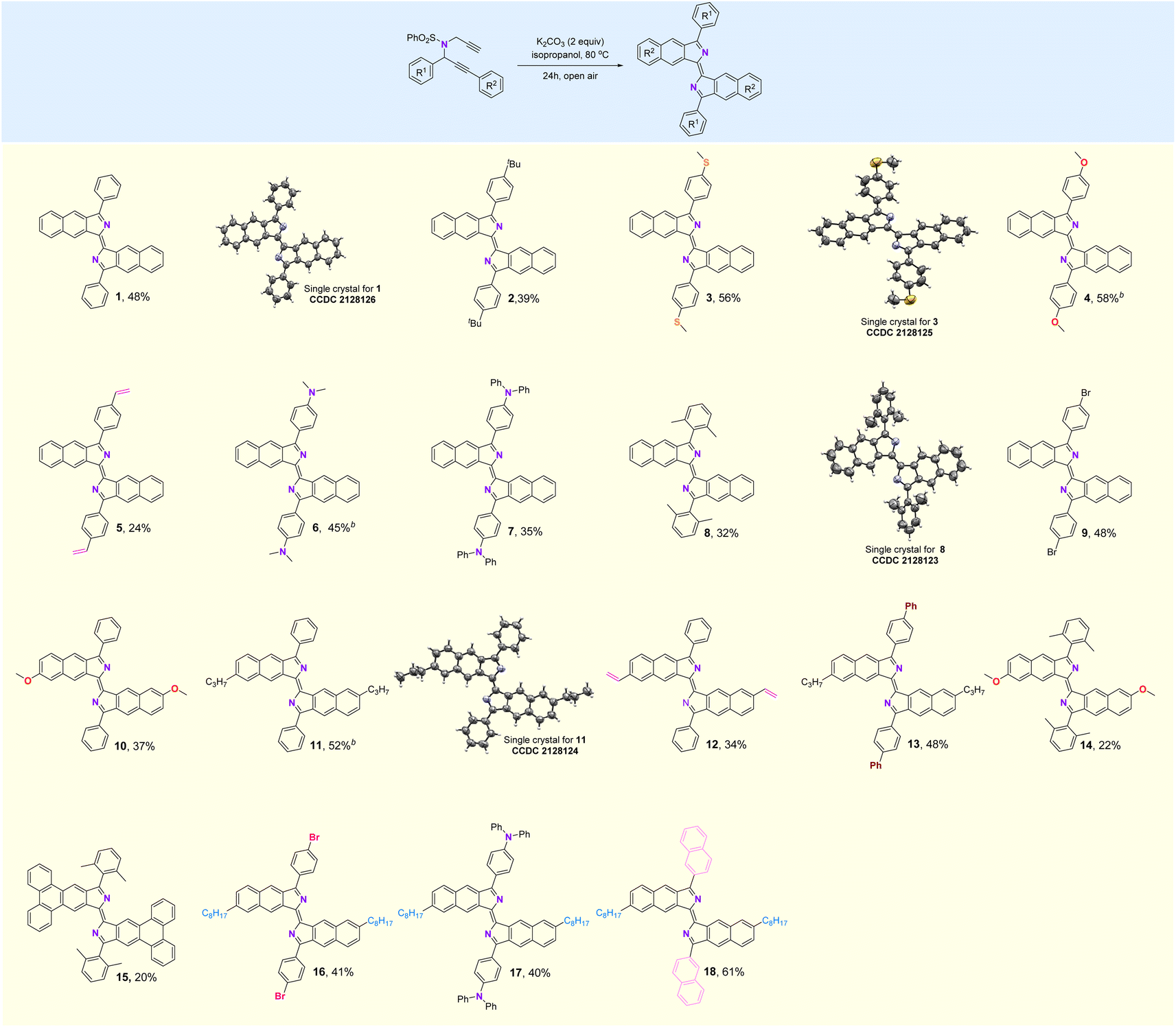 | ||
| Fig. 2 Substrate scope. aReaction conditions: dipropargyl benzenesulfonamide (0.2 mmol), K2CO3 (0.4 mmol), i-PrOH (1 mL), 80 °C, 24 h. bn-BuOH instead of i-PrOH, 110 °C. | ||
The structures of compounds 1, 3, 8, and 11 were confirmed by X-ray crystallography (Fig. 2). As shown in Fig. 3a, the crystal structure shows that compound 1 is a rigid, coplanar compound with C2h symmetry in the solid state. The packing model shows that both molecules are packed together in a J-type aggregation with a distance of ca. 3.42 Å between the neighboring molecules, indicating that the π–π surface overlap occurs (Fig. S32†). The crystal structure of 8, illustrated in Fig. 3b, reveals that the two 2,6-dimethylphenyl groups are almost perpendicular with a dihedral angle of 86°. This orientation is due to the steric effect of the two methyl groups. The packing model showed an increase in intermolecular distance to 6.28 Å due to the presence of the vertical 2,6-dimethylphenyl, which ultimately limited π-conjugation (Fig. S34†). These new fluorophores have planar and rigid structures, which limit the vibration or rotation of the free bonds leading to the effective suppression of the nonradiative transition from the excited state to the ground state, which may result in interesting photophysical properties.19
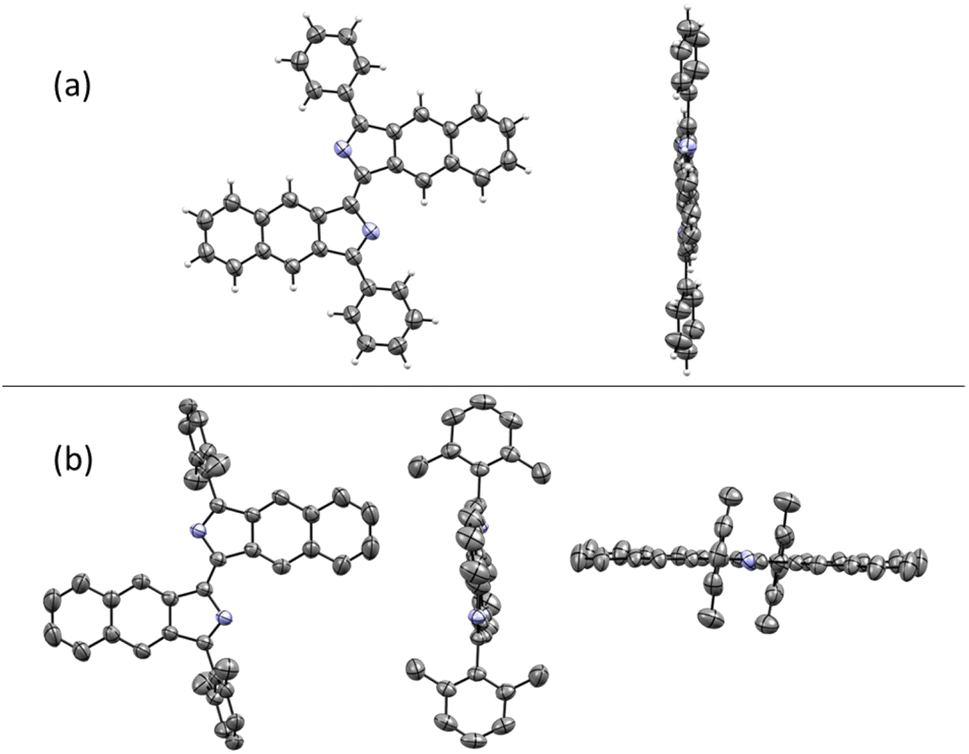 | ||
| Fig. 3 (a) Crystal structure of 1. (b) Crystal structure of 8. Hydrogen atoms are omitted for clarity. | ||
Actually, compounds 1–18 showed significant fluorescence properties in solution, and emit fluorescence at different wavelengths (Fig. 4a, b and S5–S22†). The absorption and fluorescence spectra for some selected examples were recorded in THF solvent. The samples showed absorption bands within the range of 400 to 650 nm (Fig. 4c). The maximum absorption peaks were observed at 535 nm (1, molar extinction coefficient ε = 1.93 × 104 M−1 cm−1), 590 nm (6, ε = 4.24 × 104 M−1 cm−1), and 499 nm (8, ε = 3.69 × 104 M−1 cm−1). These peaks were mainly attributed to the HOMO → LUMO transitions based on time-dependent density functional theory (TD-DFT) calculations and NTO (natural transition orbitals) analysis (Tables S2 and S3†). The emission spectra demonstrated broad emissions from the green to red region with high fluorescence quantum yields (ΦF, up to 99% for 1). The maximum emission wavelength of 1 was observed at 560 nm. Interestingly, 6 showed a red-shift in its emission spectrum upon substitution of the phenyl group on R1 with a dimethylamine group. The maximum emission wavelength of 8 was observed to be 537 nm, which was blue-shifted compared to 1. This blue-shift can be attributed to the disruption of the π-conjugation plane of the molecule caused by the perpendicular 2,6-dimethylphenyl. These results indicated that the photophysical properties of these new fluorophores can be modulated by the substituents, which offered a potential opportunity to develop a tunable fluorophore. Furthermore, we investigated the fluorescence emission of compound 1 in various solvents including ACN, DMF, THF, 1,4-dioxane, DMSO, and toluene (Fig. 4e). No significant changes were observed in other solvents, except for a 20 nm red-shift in DMSO, which indicates that the solvatochromic effect of compound 1 is not particularly pronounced. These results align with the characterization of the local excited state properties derived from computational analysis.
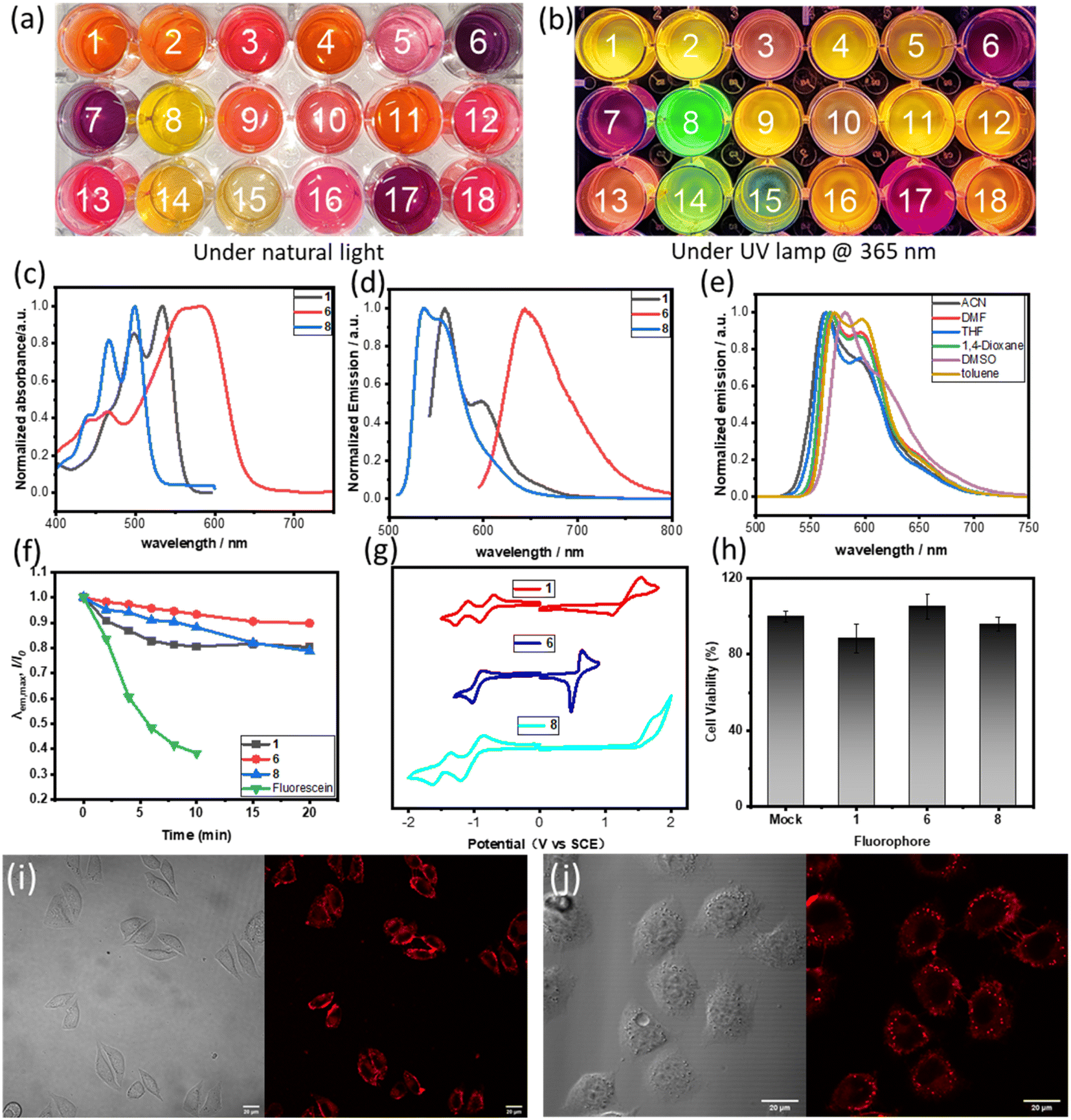 | ||
| Fig. 4 (a) Digital photos of 1–18 in THF under natural light. (b) Digital photos of 1–18 in THF under a UV lamp @ 365 nm. (c) Absorption spectrum of 1, 6 and 8 in THF (10 μM). (d) Emission spectrum (excited by the maximum absorption wavelength) of 1, 6 and 8 in THF (10 μM). (f) Comparison of the fluorescence decay rates of compounds 1, 6, 8 and fluorescein in DMSO (10 μM) under the continuous irradiation by a 532 nm laser line (200 mW cm−2). (e) Fluorescence spectra (excited by 490 nm) of compound 1 in various solvents (10 μM). (g) Cyclic voltametric curves of 1, 6 and 8 were obtained at a scan rate of 0.1 V s−1 in THF containing 0.1 M nBu4NPF6 on a glassy carbon working electrode (the saturated calomel electrode as a reference electrode). (h) Cytotoxicity of 1, 6 and 8 at 10 μm concentration for L929 cells. (i) Living cell imaging of 1 in L929 cells, bright field and red channel (ex: 543 nm, em: 550–600 nm). (j) Living cell two-photon imaging of 1 in L929 cells, bright field and red channel (2P, ex: 810 nm, em: 550–600 nm). | ||
The photostability of the new fluorophore is of paramount significance and was tested alongside compounds 1, 6, 8 and fluorescein (Fig. 4f and S26†). When subjected to irradiation with a 532 nm laser, fluorescein lost over 60% of its fluorescence intensity after just 10 minutes, while compounds 1, 6 and 8 retained over 80% of their fluorescence intensity after a longer period of 15 minutes under the same conditions. Moreover, after 15 minutes, the fluorescence intensity of these compounds only slightly decreased. This clearly demonstrates the superior photostability of the new fluorophore.
DFT calculations were conducted to investigate the impact of substituents on orbital energies. As shown in Table 1, 1 possesses HOMO and LUMO levels at −5.40 and −2.69 eV with an energy gap of 2.41 eV. The addition of a dimethylamine group on R1 results in an increase in the HOMO of 6. This leads to a smaller HOMO–LUMO gap of 2.12 eV, causing the compound to emit light at a longer wavelength (red-shift). Compared with 1, compound 8 has a larger HOMO–LUMO energy gap (2.66 eV), possibly because the introduction of 2,6-dimethyl disturbs the planarity of the structure.
| Compound | 1 | 6 | 8 | |
|---|---|---|---|---|
| a In THF. b Emission maxima upon excitation at the absorption maximum wavelength. c The optical gap, Egap, is defined as the energy corresponding to the lowest-energy absorption and estimated from corresponding spectra (Egap = 1240/λmax). d Calculations were performed at the B3LYP-D3/Def2-SVP level of theory with SMD solvation model in THF solvent. e Electrochemical data were obtained at a scan rate of 0.1 V s−1 in THF containing 0.1 M nBu4NPF6 on a glassy carbon working electrode (the saturated calomel electrode as a reference electrode). f The quasi-reversible potentials were determined by half-potential and the irreversible potentials were obtained from the intersection of the tangent line to the initial baseline. g HOMO and LUMO energy levels in eV were approximated from the first half-potential waves or the irreversible potential waves using the equation, HOMO = −(4.80 − E(Fc/Fc+) + Eox), LUMO = −(4.80 − E(Fc/Fc+) + E1red), Egap = LUMO − HOMO. | ||||
| Opticala,b,c | λ abs,max (nm) | 535 | 590 | 499 |
| λ em,max (nm) | 560 | 645 | 537 | |
| ε (M−1 cm−1) | 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 300 |
42400 |
36900 |
|
| Φ (%) | 99.7 | 34.8 | 72.2 | |
| E gap [eV] | 2.32 | 2.10 | 2.48 | |
| Calculationd | E Lumo [eV] | −2.99 | −2.69 | −2.86 |
| E Homo [eV] | −5.40 | −4.81 | −5.51 | |
| E gap (S0 → S1) [eV] | 2.41 | 2.12 | 2.65 | |
| Electrochemicale,f,g | E 2red [V] | −1.19 | — | −1.49 |
| E 1red [V] | −0.81 | −0.95 | −1.03 | |
| E 1ox [V] | 1.32 | 0.56 | 1.48 | |
| E 1Lumo [eV] | −3.39 | −3.25 | −3.17 | |
| E 1Homo [eV] | −5.52 | −4.76 | −5.68 | |
| E gap [eV] | 2.13 | 1.51 | 2.51 | |
The electrochemical properties of 1, 6 and 8 were analyzed using cyclic voltammetry (Fig. 4g and Table 1). Results showed that compounds 1 and 8 exhibit two reversible reduction waves in the test window, which can be attributed to the symmetrical structure of these compounds and their high reducibility. However, compound 6 only shows one reversible reduction wave, which may be attributed to the presence of a strong electron-donating group, dimethylamino group, making the reduction of the compound more difficult. Compounds 1 and 8 display quasi-reversible and irreversible oxidation waves, respectively. On the other hand, compound 6 demonstrates a lower oxidation potential as a result of the dimethylamino group's presence, and displays a reversible oxidation wave. Notably, the electrochemical ELUMO values of compounds 1, 6, and 8 are −3.39 eV, −3.25 eV, and −3.17 eV, respectively. The deviation from the theoretically calculated values can be attributed to the experiments on the approximation of cyclic voltammetry in solution and the use of conversion factor derived from a limited set of systems.20 In addition, the observed trend for electrochemical HOMO–LUMO gaps is consistent with that of the optical and calculated gaps.
By combining theoretical calculations, electrochemical data, and photophysical data, our study reveals that introducing a steric group on R1 disrupts the compound's planarity. This disruption leads to a decrease in the reduction potential and an increase in the oxidation potential of the molecule, and the energy gap (Egap) of the molecule increases, resulting in a blue-shift in the emitted light. When an electron-donating group is introduced on R1, it forms a D–A–D structure, which leads to a smaller Egap and red-shifted emission. On the other hand, the change of the substituent group on R2 did not significantly affect the photophysical properties.
This study showcases the potential of new fluorophores, specifically 1, 6, and 8, for cell-based imaging applications. The research was conducted using L929 cell lines as an in vitro biological model. L929 cells were incubated with 1, 6 and 8 and minimal cytotoxicity of the fluorophores was observed with a cell survival rate of over 85% (Fig. S27, S28 and S29†), and they also show good membrane permeability. Application of fluorescence imaging in living cells provided robust fluorescence intensity around 550–590 nm with 1 at a concentration of 20 μM (Fig. 4i). When excited at 810 nm, 1 exhibited two-photon (2P) physical properties (Fig. 4j), which would be beneficial to further in vivo imaging.11g Compounds 6 and 8 were investigated in cell imaging as examples of fluorescent probes with red and green light emission, and they also exhibited two-photon physical properties (Fig. S30 and S31†).
In the exploration of the feasibility of further transformations of these fluorophores, synthetic applications of the products are shown in Fig. 5. Compound 19 was obtained in 80% yield by the Miyaura boration of 9 with palladium catalysts (Fig. 5a). Compound 4 can be deprotected in the presence of 2-aminothiophenol to deliver the free phenol (21) in 82% yield. The PEG (polyethylene glycol) moiety can be introduced through an SN2 substitution reaction into the new fluorophore, leading to compounds 22 and 23. Disorders of glucose metabolism in living organisms are commonly associated with cancer, diabetes and other diseases, so the ability to effectively monitor glucose homeostasis through fluorescent glycoconjugates is critical in biological research and medicine.21 Here, fluorescent glycoconjugates (20, 25) could be obtained in 64% and 57% yields by Sonogashira coupling and a click reaction followed by hydrolytic deacetylation (Fig. 5a–c). Zidovudine is an FDA-approved drug for the treatment of AIDS, but has some toxic side effects.22 Attaching fluorophores to zidovudine will allow monitoring of its metabolic process and possibly an understanding of the relationship between its pharmacology, toxicity and efficacy. Using the click reaction, compound 26, a fluorescent derivative of zidovudine was obtained in 94% yield (Fig. 5c).
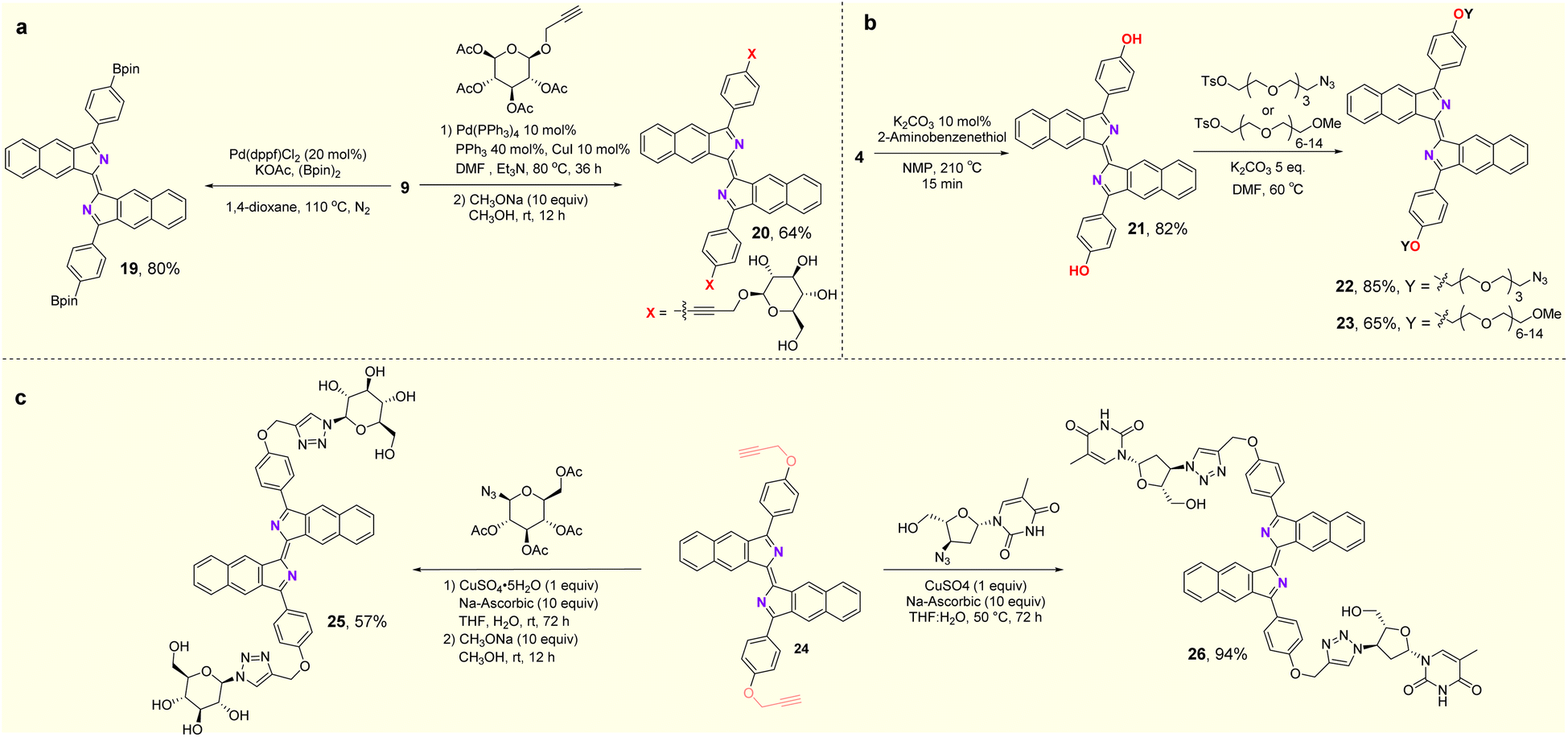 | ||
| Fig. 5 Synthetic applications. | ||
Preliminary experiments were performed to investigate the mechanism of the conversion of dipropargyl benzenesulfonamides to bisbenzo[f]isoindolylidenes (Fig. 6). A crossover experiment with two different substrates was conducted (Fig. 6a). In addition to 1 and the homodimerized product (17), a crossover product (27) was obtained in 21% yield. This result suggests that the reaction may be a stepwise process. Two deuterium labeling experiments were then carried out (Fig. 6b). A substrate (28s) with R2 = C6D5 was synthesized and subjected to the standard reaction conditions. The corresponding product (28), obtained in 51% yield, had no new additional deuterium. This suggested that migration of a deuterium atom from the phenyl ring to the isoindolyl moiety occurred, and was followed by its oxidative removal. Upon replacing the isopropyl alcohol with deuterated isopropyl alcohol (i-PrOH-d8) in the template reaction, product 29 was obtained, with two different incorporated deuterium atoms (at 55% and 88%) in both the central rings indicating the active nature of the terminal alkynyl protons and the propargyl protons which can all be exchanged with i-PrOH-d8. Two potential intermediates, obtained under different conditions were used to probe the possible reaction pathways (Fig. 6c). In contrast to the optimal reaction conditions, K2CO3 at 80 °C, compound 30 can be obtained in 30% yield from the reaction in p-xylene at room temperature with Cs2CO3 as the base, but conversion of 30 into the target product under either standard conditions or additional oxidizing conditions failed. In the absence of oxygen, compound 31 was obtained instead in 68% yield. Under the standard conditions, compound 31 can be converted into the target compound (1) in 34% yield. These two reactions suggest that rather than 30, compound 31 is more likely to be the actual reaction intermediate. When R1 = H (32s), the reaction fails to proceed under the standard conditions leaving the starting material unchanged (Fig. 6d). This result indicates that an aryl group at R1 is essential for the reaction to proceed.
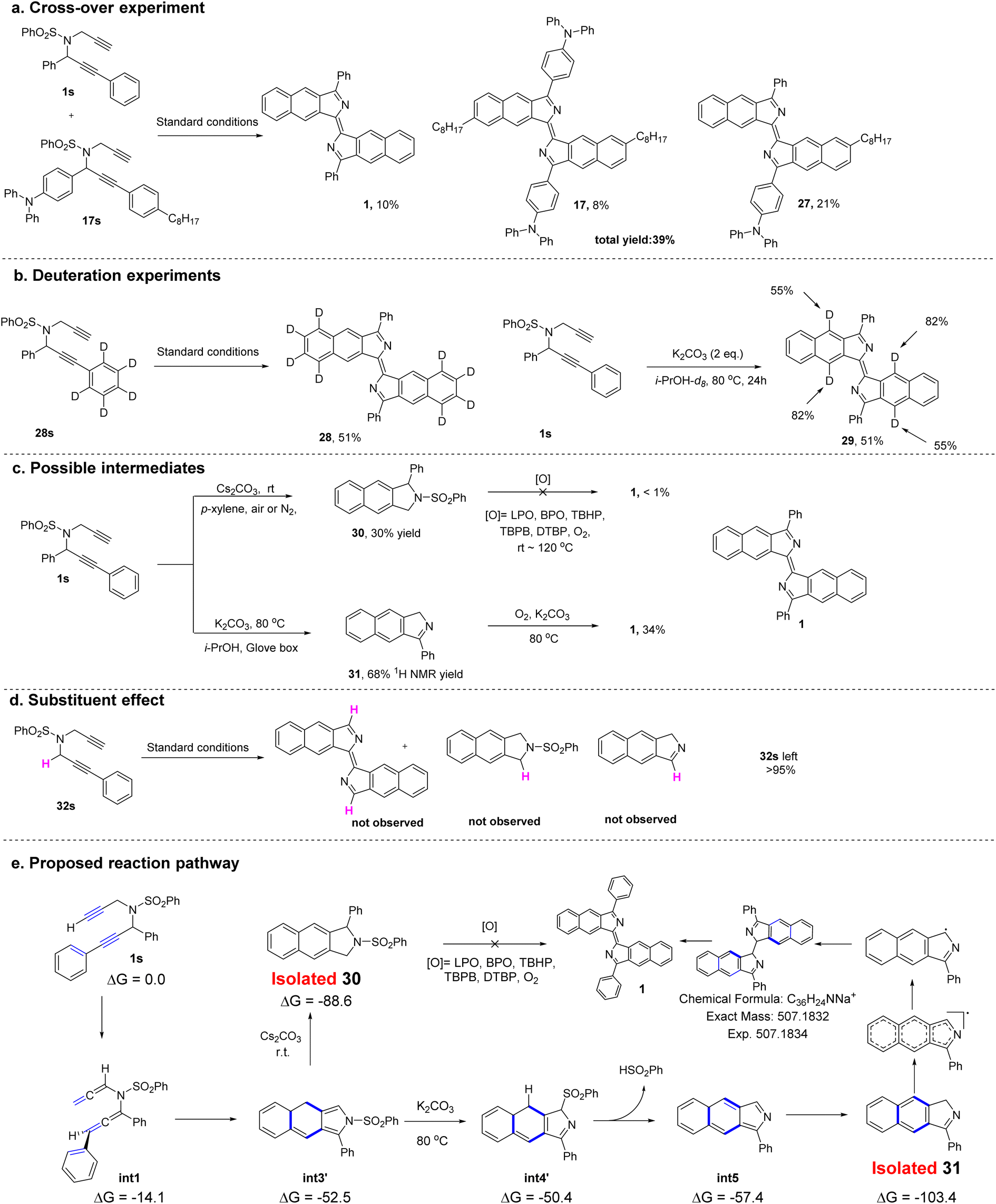 | ||
| Fig. 6 (a) Cross-over experiment. (b) Deuteration experiments. (c) Synthesis of possible intermediates and further transformations. (d) Substituent effect. (e) Proposed reaction pathway. | ||
For further understanding of the reaction mechanism, density functional theory (DFT) calculations were performed based on the experimental observations. To investigate the possible mechanism, several pathways involving a cation, a neutral species or a radical was subjected to DFT calculations (see Fig. S3 and the Computational section in the ESI for more details).† On the basis of deuteration experiments, the DFT data suggest that the cyclization pathway with assistance from potassium ions appears to be the favored pathway to form the annulated intermediate (int3 or int3′). In addition, a biradical pathway23 for the cyclization at this stage was also considered, however, it cannot be excluded.
Next, the elimination of the benzenesulfonyl group from int3 was investigated. No transition state for the direct benzenesulfonyl elimination was found, but a sequential benzenesulfonyl rearrangement and benzenesulfinic acid elimination to deliver the intermediate was calculated to be the plausible pathway. Since the intermediate (31) was isolated in experiments conducted in a glove box, the step from int5 to 31, involving a tautomeric shift of hydrogen will be a low barrier process in which the product (31) is stable with a relative free energy of −103.4 kcal mol−1 (Fig. 6e).
Based on these studies of the mechanism, a reaction pathway was proposed and is shown in Fig. 6e. The substrate (1s) initially rearranges to the diallenyl benzenesulfonamide intermediate (int1) which then cyclizes to form the annulated intermediate (int3 or int3′). Depending on the different reaction conditions, this may deliver either the byproduct (30) or the isolated intermediate (31). Finally, the intermediate (31) can dimerize to produce 1 in the presence of an oxidant.
Conclusion
In conclusion, we report the synthesis of a new type of fluorophore by an unprecedented reaction sequence from readily accessible dipropargyl benzenesulfonamides. The newly developed fluorophores exhibit several advantages, including excellent photostability, high fluorescence quantum yield (up to 99.7%) and tunable emission wavelengths. The ease of incorporation of bioactive molecules indicates the potential applications as small molecule probes in fluorescence imaging. Detailed computational and experimental studies indicate that the reaction undergoes a reaction sequence of Garratt–Braverman type cyclization, isomerization, fragmentation, radical dimerization and oxidation reactions. The new fluorophore produced in this way may serve biological research in the future.Data availability
All experimental and characterization data, as well as DFT calculation data are available in the ESI.†Author contributions
H. Bao and D. Li directed the investigations and prepared the manuscript. C. Ye and R. Huang performed the synthetic experiments and analyzed the experimental data. B. Wang performed the synthetic experiments. M.-F. Chiou completed the theoretical calculations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Professors Kuiling Ding and Xue-Long Hou of the Shanghai Institute of Organic Chemistry, Weiping Su of our institute, Haifeng Du, Yuwu Zhong, Dong Wang from the Institute of Chemistry Chinese Academy of Sciences for inspiring discussions, and Professor Daqiang Yuan from our institute for X-ray crystallographic analysis. We also thank the NSFC (Grant No. 22225107, 22301302) and Innovative Research Teams Program II of Fujian Normal University in China (IRTL1703).Notes and references
- B. Valeur and M. N. Berberan-Santos, J. Chem. Educ., 2011, 88, 731 CrossRef CAS.
- (a) J. Chan, S. C. Dodani and C. J. Chang, Nat. Chem., 2012, 4, 973 CrossRef CAS PubMed; (b) M. H. Lee, J. S. Kim and J. L. Sessler, Chem. Soc. Rev., 2015, 44, 4185 RSC; (c) H. Zhu, J. Fan, J. Du and X. Peng, Acc. Chem. Res., 2016, 49, 2115 CrossRef CAS PubMed; (d) X. Chen, F. Wang, J. Y. Hyun, T. Wei, J. Qiang, X. Ren, I. Shin and J. Yoon, Chem. Soc. Rev., 2016, 45, 2976 RSC; (e) X. Jiao, Y. Li, J. Niu, X. Xie, X. Wang and B. Tang, Anal. Chem., 2018, 90, 533 CrossRef CAS PubMed; (f) P. Gao, W. Pan, N. Li and B. Tang, Chem. Sci., 2019, 10, 6035 RSC; (g) H. Singh, K. Tiwari, R. Tiwari, S. K. Pramanik and A. Das, Chem. Rev., 2019, 119, 11718 CrossRef CAS PubMed; (h) X. Wu, W. Shi, X. Li and H. Ma, Acc. Chem. Res., 2019, 52, 1892 CrossRef CAS PubMed; (i) J.-T. Hou, K.-K. Yu, K. Sunwoo, W. Y. Kim, S. Koo, J. Wang, W. X. Ren, S. Wang, X.-Q. Yu and J. S. Kim, Chem, 2020, 6, 832 CrossRef CAS; (j) H. H. Han, H. Tian, Y. Zang, A. C. Sedgwick, J. Li, J. L. Sessler, X. P. He and T. D. James, Chem. Soc. Rev., 2021, 50, 9391 RSC; (k) X. Li, X. Liang, J. Yin and W. Lin, Chem. Soc. Rev., 2021, 50, 102 RSC; (l) Y. Chen, C. Zhu, J. Cen, Y. Bai, W. He and Z. Guo, Chem. Sci., 2015, 6, 3187 RSC.
- (a) S. J. Sahl, S. W. Hell and S. Jakobs, Nat. Rev. Mol. Cell Biol., 2017, 18, 685 CrossRef CAS PubMed; (b) G. Vicidomini, P. Bianchini and A. Diaspro, Nat. Methods., 2018, 15, 173 CrossRef CAS PubMed; (c) R. M. Dickson, A. B. Cubitt, R. Y. Tsien and W. E. Moerner, Nature, 1997, 388, 355 CrossRef CAS PubMed; (d) E. Betzig, G. H. Patterson, R. Sougrat, O. W. Lindwasser, S. Olenych, J. S. Bonifacino, M. W. Davidson, J. Lippincott-Schwartz and H. F. Hess, Science, 2006, 313, 1642 CrossRef CAS PubMed; (e) M. J. Rust, M. Bates and X. Zhuang, Nat. Methods., 2006, 3, 793 CrossRef CAS; (f) S. Samanta, Y. He, A. Sharma, J. Kim, W. Pan, Z. Yang, J. Li, W. Yan, L. Liu, J. Qu and J. S. Kim, Chem, 2019, 5, 1697 CrossRef CAS.
- F. Yan, K. Fan, Z. Bai, R. Zhang, F. Zu, J. Xu and X. Li, TrAC, Trends Anal. Chem., 2017, 97, 15 CrossRef CAS.
- H. Raunio, O. Pentikäinen and R. O. Juvonen, Int. J. Mol. Sci., 2020, 21, 4708 CrossRef CAS PubMed.
- L. D. Lavis, Annu. Rev. Biochem., 2017, 86, 825 CrossRef CAS PubMed.
- (a) G. Fei, S. Ma, C. Wang, T. Chen, Y. Li, Y. Liu, B. Tang, T. D. James and G. Chen, Coord. Chem. Rev., 2021, 447, 214134 CrossRef CAS; (b) W. Sun, S. Guo, C. Hu, J. Fan and X. Peng, Chem. Rev., 2016, 116, 7768 CrossRef CAS.
- (a) V.-N. Nguyen, J. Ha, M. Cho, H. Li, K. M. K. Swamy and J. Yoon, Coord. Chem. Rev., 2021, 439, 213936 CrossRef CAS; (b) J. Wang, C. Yu, E. Hao and L. Jiao, Coord. Chem. Rev., 2022, 470, 214709 CrossRef CAS.
- (a) R. M. Duke, E. B. Veale, F. M. Pfeffer, P. E. Kruger and T. Gunnlaugsson, Chem. Soc. Rev., 2010, 39, 3936 RSC; (b) H.-Q. Dong, T.-B. Wei, X.-Q. Ma, Q.-Y. Yang, Y.-F. Zhang, Y.-J. Sun, B.-B. Shi, H. Yao, Y.-M. Zhang and Q. Lin, J. Mater. Chem. C, 2020, 8, 13501 RSC.
- (a) Y. Yue, F. Huo, F. Cheng, X. Zhu, T. Mafireyi, R. M. Strongin and C. Yin, Chem. Soc. Rev., 2019, 48, 4155 RSC; (b) Y. Huang, Y. Zhang, F. Huo, Y. Wen and C. Yin, Sci. China: Chem., 2020, 63, 1742 CrossRef CAS.
- (a) Z. Lei, X. Li, X. Luo, H. He, J. Zheng, X. Qian and Y. Yang, Angew. Chem., Int. Ed., 2017, 56, 2979 CrossRef CAS PubMed; (b) J. Li, Y. Dong, R. Wei, G. Jiang, C. Yao, M. Lv, Y. Wu, S. H. Gardner, F. Zhang, M. Y. Lucero, J. Huang, H. Chen, G. Ge, J. Chan, J. Chen, H. Sun, X. Luo, X. Qian and Y. Yang, J. Am. Chem. Soc., 2022, 144, 14351 CrossRef CAS; (c) Y. Cheng, G. Li, Y. Liu, Y. Shi, G. Gao, D. Wu, J. Lan and J. You, J. Am. Chem. Soc., 2016, 138, 4730 CrossRef CAS PubMed; (d) Z. Wang, L. Jiang, J. Ji, F. Zhou, J. Lan and J. You, Angew. Chem., Int. Ed., 2020, 59, 23532 CrossRef CAS; (e) Y. Xiao and X. Qian, Coord. Chem. Rev., 2020, 423, 213513 CrossRef CAS; (f) X. Luo, Y. Yang and X. Qian, Chin. Chem. Lett., 2020, 31, 2877 CrossRef CAS; (g) H. M. Kim and B. R. Cho, Chem. Rev., 2015, 115, 5014 CrossRef CAS PubMed; (h) L. Wu, A. C. Sedgwick, X. Sun, S. D. Bull, X. P. He and T. D. James, Acc. Chem. Res., 2019, 52, 2582 CrossRef CAS PubMed; (i) J. Sun, H. Li, X. Gu and B. Z. Tang, Adv. Healthcare Mater., 2021, 10, e2101177 CrossRef PubMed.
- (a) V. A. Peshkov, O. P. Pereshivko, A. A. Nechaev, A. A. Peshkov and E. V. Van der Eycken, Chem. Soc. Rev., 2018, 47, 3861 RSC; (b) K. Lauder, A. Toscani, N. Scalacci and D. Castagnolo, Chem. Rev., 2017, 117, 14091 CrossRef CAS PubMed; (c) X. Xin, D. Wang, X. Li and B. Wan, Angew. Chem., Int. Ed., 2012, 51, 1693 CrossRef CAS PubMed; (d) R. L. Giles, J. D. Sullivan, A. M. Steiner and R. E. Looper, Angew. Chem., Int. Ed., 2009, 48, 3116 CrossRef CAS PubMed.
- (a) F. L. Hong, C. Y. Shi, P. Hong, T. Y. Zhai, X. Q. Zhu, X. Lu and L. W. Ye, Angew. Chem., Int. Ed., 2022, 61, e202115554 CrossRef CAS; (b) L. J. Qi, C. T. Li, Z. Q. Huang, J. T. Jiang, X. Q. Zhu, X. Lu and L. W. Ye, Angew. Chem., Int. Ed., 2022, 61, e202210637 CrossRef CAS PubMed; (c) J. J. Zhou, Y. N. Meng, L. G. Liu, Y. X. Liu, Z. Xu, X. Lu, B. Zhou and L. W. Ye, Chem. Sci., 2023, 14, 3493 RSC; (d) X. Q. Zhu, P. Hong, Y. X. Zheng, Y. Y. Zhen, F. L. Hong, X. Lu and L. W. Ye, Chem. Sci., 2021, 12, 9466 RSC; (e) C.-M. Wang, L.-J. Qi, Q. Sun, B. Zhou, Z.-X. Zhang, Z.-F. Shi, S.-C. Lin, X. Lu, L. Gong and L.-W. Ye, Green Chem., 2018, 20, 3271 RSC; (f) F. L. Hong, Y. B. Chen, S. H. Ye, G. Y. Zhu, X. Q. Zhu, X. Lu, R. S. Liu and L. W. Ye, J. Am. Chem. Soc., 2020, 142, 7618 CrossRef CAS; (g) F. L. Hong, Z. S. Wang, D. D. Wei, T. Y. Zhai, G. C. Deng, X. Lu, R. S. Liu and L. W. Ye, J. Am. Chem. Soc., 2019, 141, 16961 CrossRef CAS PubMed; (h) E. H. Huang, Y. Q. Zhang, D. Q. Cui, X. Q. Zhu, X. Li and L. W. Ye, Org. Lett., 2022, 24, 196 CrossRef CAS PubMed; (i) G. Y. Zhu, T. Y. Zhai, X. Li, C. Y. Shi, X. Q. Zhu and L. W. Ye, Org. Lett., 2021, 23, 8067 CrossRef CAS PubMed; (j) C.-Y. Shi, J.-J. Zhou, P. Hong, B.-H. Zhu, F.-L. Hong, P.-C. Qian, Q. Sun, X. Lu and L.-W. Ye, Org. Chem. Front., 2022, 9, 2557 RSC; (k) H.-J. Xu, C.-T. Li, C.-M. Chen, J. Chen, X.-Q. Zhu, B. Zhou and L.-W. Ye, Org. Chem. Front., 2023, 10, 203 RSC.
- (a) S. Dutta, R. K. Mallick, R. Prasad, V. Gandon and A. K. Sahoo, Angew. Chem., Int. Ed., 2019, 58, 2289 CrossRef CAS; (b) B. Prabagar, R. K. Mallick, R. Prasad, V. Gandon and A. K. Sahoo, Angew. Chem., Int. Ed., 2019, 58, 2365 CrossRef CAS; (c) S. Dutta, B. Prabagar, R. Vanjari, V. Gandon and A. K. Sahoo, Green Chem., 2020, 22, 1113 RSC; (d) S. Kanikarapu, M. P. Gogoi, S. Dutta and A. K. Sahoo, Org. Lett., 2022, 24, 8289 CrossRef CAS PubMed; (e) S. Nayak, N. Ghosh, B. Prabagar and A. K. Sahoo, Org. Lett., 2015, 17, 5662 CrossRef CAS; (f) B. Prabagar, S. Nayak, R. Prasad and A. K. Sahoo, Org. Lett., 2016, 18, 3066 CrossRef CAS PubMed.
- D. Campeau, A. Pommainville and F. Gagosz, J. Am. Chem. Soc., 2021, 9601–9611 CrossRef CAS PubMed.
- B. M. Trost and M. T. Rudd, Org. Lett., 2003, 5, 1467 CrossRef CAS PubMed.
- R. Wu, J. Lu, T. Cao, J. Ma, K. Chen and S. Zhu, J. Am. Chem. Soc., 2021, 143, 14916 CrossRef CAS PubMed.
- (a) D. Ghosh, S. Biswas, K. Ghosh and A. Basak, Tetrahedron Lett., 2014, 55, 3934 CrossRef CAS; (b) I. Iwai and J. Ide, Chem. Pharm. Bull., 1964, 12, 1094 CrossRef CAS PubMed.
- J. Wu, Z. Shi, L. Zhu, J. Li, X. Han, M. Xu, S. Hao, Y. Fan, T. Shao, H. Bai, B. Peng, W. Hu, X. Liu, C. Yao, L. Li and W. Huang, Adv. Opt. Mater., 2022, 10, 2102514 CrossRef CAS.
- (a) J.-L. Bredas, Mater. Horiz., 2014, 1, 17 RSC; (b) L. Qu, H. Xiao, B. Zhang, Q. Yang, J. Song, X. Zhou, Z.-X. Xu and H. Xiang, Chem. Eng. J., 2023, 471, 144709 CrossRef CAS.
- (a) B. Thomas, K. C. Yan, X. L. Hu, M. Donnier-Marechal, G. R. Chen, X. P. He and S. Vidal, Chem. Soc. Rev., 2020, 49, 593 RSC; (b) W. H. Kim, J. Lee, D. W. Jung and D. R. Williams, Sensors, 2012, 12, 5005 CrossRef CAS PubMed.
- B. N. Stretcher, J. Clin. Lab. Anal., 1991, 5, 60 CrossRef CAS.
- (a) P. Bhattacharya, M. Singha, E. Das, A. Mandal, M. Maji and A. Basak, Tetrahedron Lett., 2018, 59, 3033 CrossRef CAS; (b) S. Jana and A. Anoop, J. Org. Chem., 2016, 81, 7411 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2128123–2128126. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc04445a |
| ‡ C. Ye and R. Huang contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |