
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dimethyl carbonate synthesis from CO2 and methanol over CeO2: elucidating the surface intermediates and oxygen vacancy-assisted reaction mechanism†
Dragos
Stoian
 abe,
Toshiyuki
Sugiyama
c,
Atul
Bansode
af,
Francisco
Medina
b,
Wouter
van Beek
e,
Jun-ya
Hasegawa
c,
Akira
Nakayama
*cd and
Atsushi
Urakawa
*af
abe,
Toshiyuki
Sugiyama
c,
Atul
Bansode
af,
Francisco
Medina
b,
Wouter
van Beek
e,
Jun-ya
Hasegawa
c,
Akira
Nakayama
*cd and
Atsushi
Urakawa
*af
aInstitute of Chemical Research of Catalonia (ICIQ), The Barcelona Institute of Science and Technology (BIST), Av. Països Catalans 16, 43007 Tarragona, Spain
bDepartment of Chemical Engineering, University Rovira i Virgili, Av. Països Catalans 26, 43007 Tarragona, Spain
cInstitute for Catalysis, Hokkaido University, Sapporo 001-0021, Japan
dDepartment of Chemical System Engineering, The University of Tokyo, Tokyo 113-8656, Japan. E-mail: nakayama@chemsys.t.u-tokyo.ac.jp
eThe Swiss-Norwegian Beamlines (SNBL) ESRF – The European Synchrotron Radiation Facility, BP 220, F-38043 Grenoble, France
fCatalysis Engineering, Department of Chemical Engineering, Delft University of Technology, Van der Maasweg 9, 2629 HZ Delft, The Netherlands. E-mail: A.Urakawa@tudelft.nl
First published on 21st November 2023
Abstract
Surface intermediate species and oxygen vacancy-assisted mechanism over CeO2 catalyst in the direct dimethyl carbonate (DMC) synthesis from carbon dioxide and methanol are suggested by means of transient spectroscopic methodologies in conjunction with multivariate spectral analysis. How the two reactants, i.e. CO2 and methanol, interact with the CeO2 surface and how they form decisive surface intermediates leading to DMC are unraveled by DFT-based molecular dynamics simulation by precise statistical sampling of various configurations of surface states and intermediates. The atomistic simulations and uncovered stability of different intermediate states perfectly explain the unique DMC formation profile experimentally observed upon transient operations, strongly supporting the proposed oxygen vacancy-assisted reaction mechanism.
Introduction
The increasing interest in dimethyl carbonate (DMC) has been witnessed in the last decades for its usefulness: polar solvent for paints and coatings, methylating and carbonylating agent in organic synthesis, electrolytes in Li-ion batteries, and an appropriate substitute for methyl tert-butyl ether (MTBE) as a fuel additive owing to its high oxygen content.1–7 While one of the major synthesis paths of DMC is via the reaction of methanol with phosgene (COCl2), nowadays the industrially recognized processes involve either oxidative carbonylation reactions using CO (Enichem, Ube and Bayer AG), trans-esterification reactions of organic carbonates with methanol (Asahi Kasei), or a two-step urea methanolysis method developed by Catalytic Distillation Technologies, Inc.8 Despite these improvements, it can be easily understood why a safer, more eco-efficient and economic pathway for DMC synthesis is desired. The development of the direct reaction between CO2 and short chain alcohols, particularly methanol (MeOH) and ethanol (EtOH), for the synthesis of organic carbonates such as DMC or diethyl carbonate (DEC) (Scheme 1, DMC synthesis) has drawn great attention in the light of urged CO2 chemical fixation, green chemistry (high atom efficiency processes), and safety by replacing the highly toxic and corrosive phosgene molecule by CO2 as carbonyl source. | ||
| Scheme 1 Direct DMC synthesis from CO2 and methanol. | ||
In the last 20 years, various homogeneous and heterogeneous catalysts were reported for the direct carboxylation reaction of methanol using CO2. Heterogeneous catalysts are more widely investigated and different materials have been tested under both batch and continuous operation. Among them, zirconia (ZrO2), ceria (CeO2), and ZrO2–CeO2 solid solutions9–11 have been reported as the most effective catalysts in the direct DMC synthesis, while other catalyst materials afforded no or very little formation of DMC.12 Generally, the presence of both acidic and basic sites is required for the activation of CO2 and methanol. Indeed, the aforementioned reported active catalysts possess both the acidic and basic properties.13 Nevertheless, in practice the reaction is highly thermodynamically limited and the DMC yield can be enhanced only up to about 1% even under thermodynamically favourable high-pressure conditions (ca. 400 bar).14 Recently, a drastic improvement in DMC yield (>90%) was reported by Tomishige et al. over CeO2 as the unique catalyst through in situ dehydration where 2-cyanopyridine (2-CP) serves as organic dehydrating agent to remove the water byproduct (Scheme 1). CeO2 functions as catalyst in both DMC synthesis and dehydration reaction optimally at ca. 120 °C. This strategy has been demonstrated for batch as well as continuous operations.14–17 It is noteworthy that the rate determining step of the DMC synthesis is unaltered by the presence of 2-CP. This implies that understanding the simpler reaction system without the dehydrating agent would be of direct relevance in understanding more complex yet more practical system with the dehydrating agent.16
The unique catalytic performance of CeO2 in this reaction may originate from the synergy between its acid-base and redox properties. Izumi et al. studied the reducibility of CeO2 promoted with Cu (0.1 to 0.5 wt%) by X-ray absorption spectroscopy (XAS) after high temperature reduction treatment (400 °C) and subsequent CO2 adsorption. They suggested that the partial reduction of Ce4+ sites to Ce3+ may be beneficial for DMC formation.18 Also, Aresta et al. correlated by ex situ X-ray photoelectron spectroscopy (XPS) the origin of catalyst deactivation, thus the activity, with the oxidation state of Ce.19,20 In a more recent study, Li et al. synthesized Zr-doped CeO2 nanorods, and they linked the DMC production rate to the highest number of oxygen vacancies (i.e. extracted from ex situ Raman and XPS measurements; versus bare CeO2) that allows an increased CO2 adsorption rate.21 Despite all these indications, the contribution and importance of the redox properties or surface defects of CeO2 in the reaction has not been proven under working (operando) reaction conditions. Particularly, the reaction is commonly performed at relatively low temperatures (100–150 °C) where the reduction of surface and bulk CeO2 is unlikely to occur according to temperature programmed reduction studies22–24 and the detection of electronic structure change of Ce, if any, is expected to be challenging. Furthermore, no operando spectroscopic investigation has been reported bridging the type of surface species present under reaction conditions with DMC formation by simultaneous detection of the product concentration.
Following this background, in this work we elucidate the surface chemical intermediate, at its energetically resting state, leading to DMC formation by means of operando diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) coupled with multivariate spectral analysis25–27 which enables unreferenced spectral separations (ESI, Fig. S1†). Furthermore, through density functional theory (DFT) calculations, XAS and Raman spectroscopy, the critical roles of surface oxygen vacancies of CeO2 at the rate limiting step are uniquely unravelled.
Results and discussions
Fig. 1 presents time-resolved operando DRIFT spectra and DMC concentration profile under a periodic concentration change of methanol (16.5 vol% in He, the first half period of 128 s) and CO2 (the second half period of 128 s) performed at 120 °C (averaged over 8 periods to improve S/N). The MS signal (Fig. 1, left) shows that DMC was formed under the studied condition. The production level of DMC increased upon switching from CO2 to methanol, reaching a constant level after ca. 40 s. Interestingly, the DMC production was boosted by ca. 4 times upon switching from methanol to CO2 and then gradually ceased with time, almost completely under the flow of CO2. The stable activity after 40 s is at the same level as that of the steady-state activity of DMC formation by passing MeOH + CO2 (ESI, Fig. S3†). This means that MeOH vs. CO2 perturbation creates favourable condition for DMC formation, enhancing the formation rate by 4 times compared to the steady-state activity. The differential DRIFT spectra in the low frequency region from 950 to 1800 cm−1 (Fig. 1, right; the last spectrum of the period, i.e., at the end of the CO2 period, was used as the background) shows the characteristic C–O stretching vibrations of bridged and terminal methoxy species appearing at ca. 1060 and 1120 cm−1, respectively,10 as positive bands (in red) when the atmosphere was switched to methanol. Likewise, the absorbance of the methoxy bands (hence the concentration of methoxy species) for the MeOH + CO2vs. CO2 experiment is ca. 4 time less compared to the case of MeOH vs. CO2, well in-line with the semi-quantitative analysis of the MS profiles and the observation from Fig. 1, left which tells us that methanol adsorption needs to take place first in order to activate the DMC formation (ESI, Fig. S4† for more detailed discussion).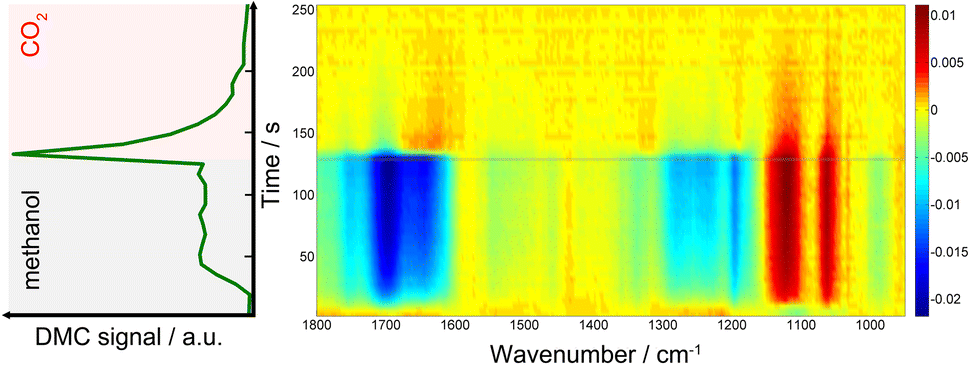 | ||
| Fig. 1 (left) MS signal of DMC (m/z = 59) and (right) time-resolved DRIFT spectra taken during methanol (the first half period) vs. CO2 (the second half period) concentration perturbation experiment performed at 120 °C at 7 ml min−1. The time-axes (y-axis) of both figures are identical. The DRIFT spectra were calculated taking the last spectrum in the CO2 atmosphere as background. | ||
At the same time, several negative bands characteristic of carbonates/bicarbonates were observed in the region from 1150–1800 cm−1 under the flow of MeOH. According to literature, these bands originate from carboxylate and protonated carboxylate species on the surface of the catalyst (1695 cm−1), bridged (bi)carbonates (1235 and 1645 cm−1), and monodentate and bidentate carbonates (1336, 1458 cm−1 and 1282, 1548 cm−1, respectively).10,28–31 The results clearly indicate that carbonates/bicarbonates formed under CO2 atmosphere were replaced (thus the bands appear negative) by the methoxy species under methanol atmosphere, and vice versa under CO2 atmosphere. At first glance, the DRIFT spectra do not show signatures of transient surface species whose concentration is similar to that of DMC, except a few slightly positive signals at ca. 1310 and 1630 cm−1 appearing upon switching to CO2. This sort of ambiguousness in spectral analysis is often encountered and arises from overlapping peaks and also impossibility to deconvolute the spectral contributions of different chemical species due to the unavailability of proper reference spectra of the chemical species that are only present/detected under reaction conditions and/or under transient conditions.
To overcome this limitation, multivariate spectral analysis was employed to disentangle overlapping peaks into the spectra of “kinetically pure” components (i.e., surface species). Besides the spectral separation, it yields conveniently the corresponding concentration profiles of the components. Provided that the data quality is good in terms of S/N, the multivariate spectral analysis can overpower another famous technique used for detection of minor species, i.e., modulation excitation spectroscopy (MES, using phase sensitive detection), especially when there is a high degree of spectral overlap and species with extremely different kinetics are involved. For more details and comparison between the multivariate spectral analysis and MES, the reader is redirected elsewhere.26,32–34Fig. 2 shows the results of the multivariate spectral analysis on the DRIFT spectra shown in Fig. 1. For comparison, the results obtained via MES analysis are included in the ESI, Fig. S5.† The analysis identified three kinetically distinct components. Most strikingly, the analysis could separate the spectral component (Fig. 2, green) with the concentration profile perfectly matching with that of DMC (Fig. 1, left). The other two components show characteristic features of (i) methoxy species (Fig. 2, black) and (ii) carbonate/bicarbonate species (Fig. 2, red). The corresponding concentration profiles of these components (Fig. 2, right) obviously reflect the atmosphere of the gas phase.
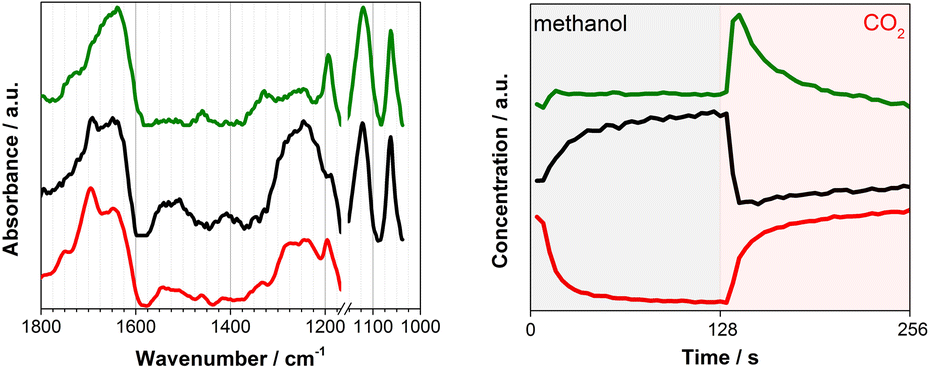 | ||
| Fig. 2 (left) Three components spectra and (right) the corresponding concentration profiles obtained by multivariate spectral analysis applied on the DRIFT spectra shown in Fig. 1 (the first half is methanol and the second half is CO2 atmosphere). To ease the data analysis and improve component separation, the spectral analysis has been performed on two separate regions of methoxy (below 1170 cm−1) and carbonates (above 1180 cm−1). Both spectra and concentration profiles are normalized for clarity. | ||
In situ infrared and Raman studies reported by Tomishige, Bell and their co-workers over ZrO2 materials suggested the formation of monomethyl carbonate (MMC, CH3O–COO–Zr) as possible intermediate species through the reaction between methoxy species (CH3O−) and CO2.35–37MMC further reacts with methanol (methoxy) to afford DMC. Later, it was indicated that the reaction proceeds via the same intermediate (i.e.MMC) over CeO2 catalysts.10,16 There are also reports supporting the existence of a different intermediate like carbomethoxide (CH3OCO–Ce) species as suggested by Wang et al.28 The spectral characteristics of the captured intermediate (Fig. 2, green) whose concentration profile resembles that of gas phase DMC generally show the mixed features of methoxy (1060 and 1120 cm−1) and (bi)carbonates (e.g. 1200–1800 cm−1), although they are somehow different especially in the region of 1200–1600 cm−1 where the characteristic bands of MMC are expected to appear according to the literature.10,20,28,38 More precisely, Lavalley et al. assigned the two bands we observed at ca. 1330 and 1460 cm−1 to a coupling between the bending mode of CH3 and the stretching vibration of OCO.39 This clearly indicates that the captured intermediate is MMC or species alike. The spectral features of the intermediate (Fig. 2, green) in the methoxy region appear similarly to those of methoxy species (Fig. 2, black). A closer look verifies that they are similar but there are noticeable differences especially in the band of terminal methoxy at ca. 1120 cm−1 of the intermediate, showing a broadening and a small shift towards lower vibrational frequency (ESI, Fig. S2†). This implies that the terminal methoxy has reacted or is interacting with the CO2 molecule trapped on the surface, resulting in the red shift. Also, intriguingly the species responding to the gas-phase methanol concentration (Fig. 2, black) show the spectral features of (bi)carbonates in the region 1200–1800 cm−1. This is indicative of the formational correlation of methoxy species with specific surface (bi)carbonate species. The dissociation of MeOH into MeO and H is known10 to take place upon methanol adsorption over CeO2 and this surface adsorbed H may lead to more pronounced formation of bicarbonates from carbonates (see the discussion below for DFT calculations) and the spectral feature of the methoxy species may reflect the accompanied formation of bicarbonate surface species since the multivariate spectral analysis cannot disentangle spectral components behaving kinetically identical. A similar discussion for the high frequency region (above ca. 2000 cm−1) is presented in the ESI (Fig. S6 and S7†) including the difference DRIFT spectra and the corresponding multivariate spectral analysis results.
Under similar transient conditions we looked into the change in the oxidation state of Ce by XAS at Ce K and L3-edges and structural changes by Raman spectroscopy. Under oxidation (O2) and reduction (H2) treatment at high temperature (350 °C), the multivariate spectral analysis on the XAS and Raman data could clearly extract the redox features of CeO2 (ESI, Fig. S8–S10†), whereas these redox features could not be observed at the reaction temperature of 100–150 °C which was obviously too low for the redox to take place as expected. Although we could not detect spectral changes in the bulk-sensitive XAS under the transient reaction conditions of MeOH vs. CO2 flow, there was a clear change in the F2g Raman active mode at ca. 460 cm−1, corresponding to the symmetric breathing of O2− atoms vibrations around the Ce4+ cations (Fig. 3). The width and position of this band is known to be extremely sensitive to any structural disorder of the O-sublattice.40 Multivariate spectral analysis on the Raman data disentangled the two spectral components with contrasting concentration profiles: one increasing upon CO2 exposure which is obviously assigned to the band associated with the Ce4+ state (Fig. 3, Component 1, red), and the other increasing with methanol with the band feature slightly red-shifted and broadened (Fig. 3, Component 2, black). These changes are in accordance with the reports on CeO2-based materials under reduction–oxidation cycles.41–43 Yoshimura et al. use the changes in Raman-allowed F2g mode (i.e. shifts to lower energies, and the line shape getting progressively asymmetric with the crystal size decrease) in a study about the identification of defects in ceria-based nanocrystals by UV resonance Raman spectroscopy.44 This subtle change took place on the surface of the material and therefore could not be measured by XAS. Likewise, the change was less obvious when a longer wavelength excitation laser (785 nm) was used in Raman since in this case we sample more bulk of the material (ESI, Fig. S11†). Given the magnitude of the spectral change and the mild reaction conditions it becomes rather difficult to state the existence of a complete Ce4+ ⇔ Ce3+ cycle despite a recent publication by Tomishige et al. which reported the redox properties of CeO2 catalysts in organic reactions (i.e. synthesis of imines from alcohols and amines) at temperature as low as 30 °C.45 In literature, Mullins et al. probed the surface sites of CeO2 nanocrystals with well-define surface planes via methanol adsorption, and they observed slight reduction of the rods (110) and cubes (100) surfaces at room temperature by a UV Raman study. In addition, surface science reports on methanol adsorption over thin-film model CeO2 catalyst support the reduction phenomenon occurring in the topmost oxide layers even at ambient or sub-ambient temperatures (Mullins et al. and Skala et al.).46–48 Based on what has been reported, the creation of a nonstoichiometric CeO2−δ surface via the formation of some surface and subsurface O-vacancies (i.e. defective surface) is possible and explains the Raman results. Such defective surface formation would lead to a more electron deficient or more electron rich CeO2 state depending on the gas atmosphere. Glatzel et al. have previously invoked this picture of an electron sponge for CeO2 nanoparticles studied by HERFD-XAS.49 They found an increase of the interatomic distances between Ce and O during the catalytic decomposition of hydrogen peroxide while stating that the redox partner is not a local Ce3+ site, but the electron density that is received and released during the reaction is delocalized over the atoms of the nanoparticles. Furthermore, the emergence of two bands in the 2000–2250 cm−1 region of the DRIFT spectra that can be linked to Ce3+ surface sites formation induced by methanol adsorption over CeO2 can further support the Raman results and thus methanol adsorption-induced Ce3+ formation (ESI, Fig. S6†) which is important for the enhanced DMC formation (Fig. 1, left, Fig. S3†).
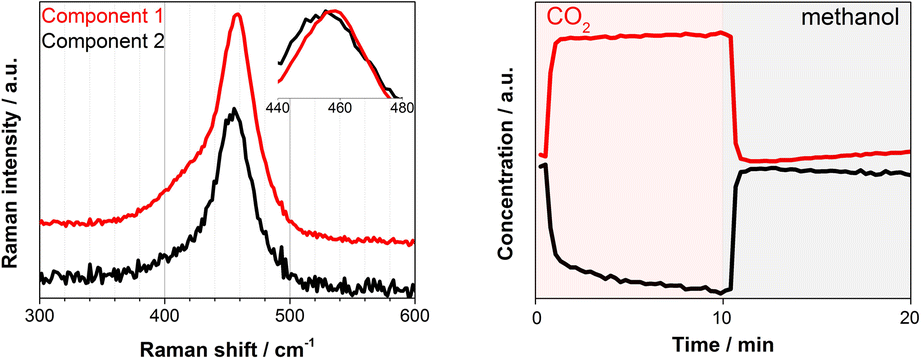 | ||
| Fig. 3 Multivariate spectral analysis applied for F2g band of CeO2 during DMC synthesis experiment (CO2vs. methanol) at 140 °C. The Raman measurement was performed with a 532 nm laser (green). Both spectra (left) and concentration (right) profiles are normalized for clarity. | ||
The mechanistic investigation by DFT calculations strongly supports our experimental observations and clarifies atomistic and electronic insights. The computational details are described in ESI.† While methanol adsorption occurs preferentially via the dissociation of the molecule into CH3O− and H+, the favoured and most stable adsorption state of CO2 is represented by a monodentate carbonate species involving a covalent bond between the C atom of the CO2 molecule and surface O atoms belonging to CeO2 (see ESI, Fig. S12† for adsorption structures).
Here, we considered the two reaction pathways for DMC formation, one starting from an adsorbed CO2 molecule that is a monodentate carbonate species, CO2(m) (Path A), and the other starting from the dissociative adsorption of a methanol molecule (Path B), as shown in Fig. 4. In Path A, this monodentate structure is attacked by the methoxy species (CH3O−), leading to the formation of surface monomethyl carbonate species (MMCS). After removal of the hydroxyl group by accepting a proton, an intermediate species (INT_1) is formed by releasing a water molecule. Subsequently, another methoxy moiety attacks the carbon atom, forming an intermediate structure INT_2, and then the carbon and surface oxygen bond is broken to release a DMC molecule. In Path B, a CO2 molecule is inserted into the adsorbed methoxy species, leading to the formation of monomethyl carbonate (MMC) species. This MMC species is nucleophilically attacked by the surface oxygen atom, which results in the formation of MMCS by accepting a proton from nearby methanol molecule. The subsequent reaction pathway from MMCS is the same as that in Path A.
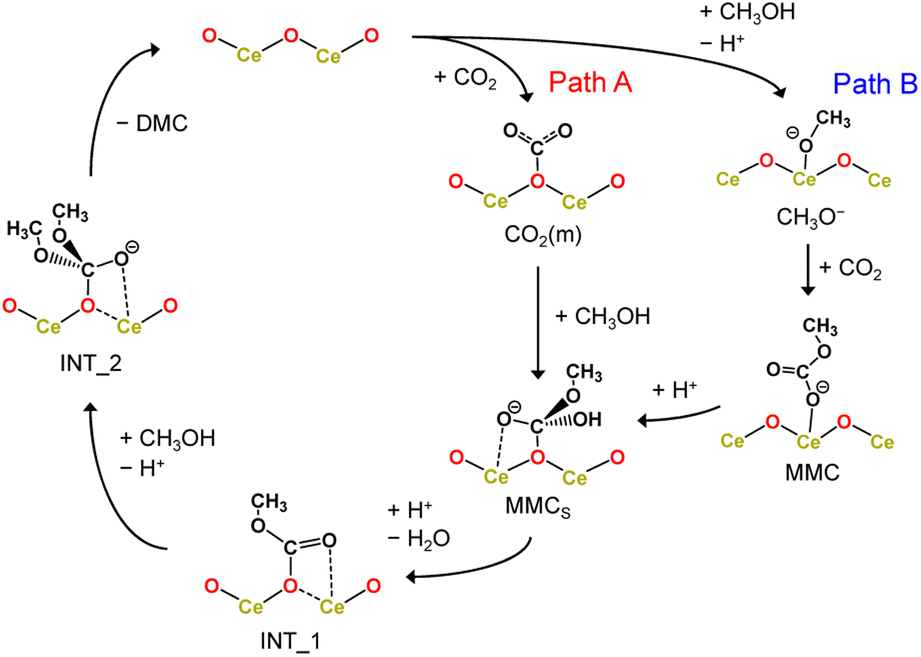 | ||
| Fig. 4 Reaction mechanism of DMC formation from CO2 and methanol. Path A starts with an adsorbed CO2 molecule while Path B starts from the dissociative adsorption of a methanol molecule. | ||
Fig. 5 shows the free energy profiles along the reaction pathways. In the first step of Path A, the CO2 molecule is adsorbed on the CeO2 surface with a small barrier of 21 kJ mol−1 (CO2* → CO2(m)), forming a monodentate structure CO2(m). Then, the surface monomethyl carbonate species (MMCS) is generated with an activation energy of 77 kJ mol−1 (CO2(m) → MMCS). Before reaching to this transition state configurations, we find that a proton is transferred from a neighbouring methanol molecule to the surface carbonate species, resulting in the bicarbonate species. This bicarbonate species is attacked by methoxy moiety to form MMCS structure (see the inset of CO2(m) → MMCS in Fig. 5 and the snapshot near the transition state structure is shown in ESI, Fig. S13†). The resulting MMCS structure is less stable, and the carbomethoxy species (INT_1.1) is formed with a small barrier of 13 kJ mol−1 by eliminating a water molecule (snapshot near the transition state structure is also provided in ESI, Fig. S13†). This carbomethoxy configuration is meta-stable and it readily changes its configuration with a more stable monomethyl carbonate state accompanied by the creation of an oxygen vacancy during the MD simulation (shown as INT_1.2, and snapshot is provided in ESI, Fig. S13†). This intermediate is further attacked by another methoxy moiety, leading to the formation of INT_2 with an activation barrier of 49 kJ mol−1. During this attack of methoxy species, the surface vacancy is refilled by the oxygen atom. The final step is a bond cleavage between the carbon and surface oxygen atom, and this step requires an activation energy of 52 kJ mol−1. The free energy difference between INT_1 and the transition state between INT_2 and DMC* is estimated to be 87 kJ mol−1, which is comparable to the experimental value of 73 kJ mol−1 reported in ref. 16.
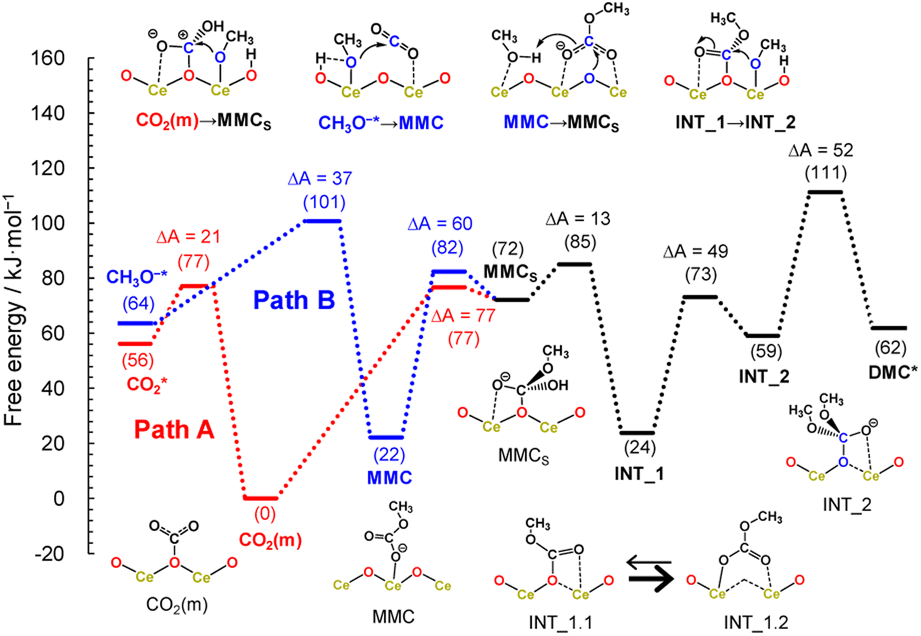 | ||
| Fig. 5 Free energy profiles along the two reaction pathways. The free energy of the monodentate structure CO2(m) is set to zero, and relative energy is shown in parenthesis. Activation energy (ΔA in kJ mol−1) in each step is also included. The inset on the bottom (INT_1 = INT_1.1 → INT_1.2) evidences the key step in the stabilization of the reaction intermediate and formation of an O-vacancy in the catalytic cycle over CeO2. | ||
Next, the reaction mechanism of Path B is analysed in detail. In the first step, the monomethyl carbonate species is formed by the reaction between a dissociated methanol and CO2 with an activation barrier of 37 kJ mol−1 (CH3O−* → MMC). The conformation of MMC species during the MD simulation is close to the adsorption structure of DM1 or DM2 shown in ESI, Fig. S12,† where in the MD simulations the hydrogen bond of the carbonyl group is observed with hydrogen in the methanol molecule (snapshot is provided in ESI, Fig. S13†). The next step is a nucleophilic attack of the surface oxygen atom to the carbon atom accompanying a proton transfer to the carboxylate group (MMC → MMCS). This process requires activation energy of 82 kJ mol−1.
The gaseous product (DMC) profile (Fig. 1) is in good agreement with the mechanistic insights obtained by the DFT calculations. Upon switching from CO2 to methanol, there is an induction period before reaching a stable DMC production. This clearly indicates a strong CO2 adsorption to the surface sites; it takes some time to replace the strongly adsorbed CO2 molecules by methanol. This competitive adsorption and replacement by methoxy retards the DMC formation and also implies that the methoxy and Ce3+ generation are crucial for the reactivity towards DMC formation. Also, the spectral features of bicarbonates in the region 1200–1800 cm−1 in the methanol atmosphere (Fig. 2, black) correspond to the bicarbonate species before the formation of MMCS structure. On the other hand, upon switching from methanol to CO2 there is an immediate formation of DMC with a boost in productivity, demonstrating that for the CO2 molecules it is much easier to kick out the previously adsorbed methanol from the surface through MMC intermediate. After that, DMC production ceases quite fast suggesting a very strong CO2 absorption to CeO2 surface and the lack of any methoxy species in the proximity. Based on the discussion above, we can also conclude that the stability of the intermediate (INT_1.2) and the creation of a defective surface is the key to make CeO2 an efficient catalyst for the direct synthesis of DMC while the reaction invokes the image of a Langmuir–Hinshelwood type of mechanism with methanol and CO2 molecules adsorbed in the immediate vicinity of a Ce–O pair.
Conclusions
In summary, the present study describes the in situ creation of a defective CeO2 surface and its involvement in the gas phase DMC synthesis from CO2 and methanol by using a combination of experimental and theoretical tools. While methanol seems to increase the electron density around Ce sites, CO2 behaves as an oxidizing agent leading to fully oxidized Ce4+. Furthermore, a boost in DMC production is observed when CO2 is passed over methoxy covered surface. The surface intermediate species directly correlated with DMC formation was elucidated by operando DRIFTS with the aid of the multivariate spectral analysis. It shows the feature of both methoxy and (bi)carbonates, in good agreement with the reported structure of monodentate methyl carbonate. We demonstrate, for the first time ever, the reconfiguration of this species on the CeO2 surface via a vacancy-assisted mechanism, observation which correlates well with the operando Raman results.Ultimately, direct DMC synthesis from methanol and CO2 is un-equivocally driven forward by the presence of neighbouring acid–base pairs over CeO2 surface. Nevertheless, the influence of O-vacancies in the reaction mechanism and the extraordinary redox (surface) properties of this oxide cannot be neglected anymore. It has already been shown that an increased surface reducibility which can ease the formation of surface defect sites – responsible for the intermediate stabilization – plays critical roles in the overall reaction mechanism involving the in situ water removal by 2-CP molecules (while the DMC formation mechanism is unchanged by the dopants and mainly catalysed by CeO2 surface).50 Hence, this study establishes new directions for designing better and more stable catalysts for this newly implemented and highly active route to DMC synthesis by direct methanol carboxylation reaction.
Data availability
Data will be made available on request.Author contributions
D. S. performed all experiments and T. S. performed the DFT-based calculations. A. B. and W. v. B. supported synchrotron X-ray-based experiments. F. M., J. H., A. N. and A. U. conceived and supervised this work. D. S., T. S., A. N. and A. U. analysed the data. The manuscript was drafted with the support and contribution from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Generalitat de Catalunya for financial support through CERCA Programme and recognition (2014 SGR 893) and thank MINECO (CTQ2016-75499-R (FEDER-UE)) for financial support through Severo Ochoa Excellence Accreditation 2014–2018 (SEV-2013-0319). The authors acknowledge the financial support by PRESTO from Japan Science and Technology Agency (JST) (Grant Number JPMJPR16S8). A part of this work is based on results obtained from a project commissioned by the New Energy and Industrial Technology Development Organization (NEDO). A part of the calculations was performed on supercomputers at RCCS (Okazaki, project no. 23-IMS-C172), RIIT (Kyushu Univ.), and CCMS, IMR (Tohoku Univ., proposal no. 202212-SCKXX-0002).Notes and references
- A.-A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951–976 CrossRef CAS PubMed.
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- Y. Ono, Appl. Catal. Gen., 1997, 155, 133–166 CrossRef CAS.
- M. Aresta and E. Quaranta, ChemTech, 1997, 27, 32–40 CAS.
- N. Keller, G. Rebmann and V. Keller, J. Mol. Catal. Chem., 2010, 317, 1–18 CrossRef CAS.
- D. Delledonne, F. Rivetti and U. Romano, Appl. Catal. Gen., 2001, 221, 241–251 CrossRef CAS.
- M. A. Pacheco and C. L. Marshall, Energy Fuels, 1997, 11, 2–29 CrossRef CAS.
- I. Garcia-Herrero, R. M. Cuéllar-Franca, V. M. Enríquez-Gutiérrez, M. Alvarez-Guerra, A. Irabien and A. Azapagic, ACS Sustain. Chem. Eng., 2016, 4, 2088–2097 CrossRef CAS.
- K. Tomishige, T. Sakaihori, Y. Ikeda and K. Fujimoto, Catal. Lett., 1999, 58, 225–229 CrossRef CAS.
- Y. Yoshida, Y. Arai, S. Kado, K. Kunimori and K. Tomishige, Catal. Today, 2006, 115, 95–101 CrossRef CAS.
- K. Tomishige, Y. Furusawa, Y. Ikeda, M. Asadullah and K. Fujimoto, Catal. Lett., 2001, 76, 71–74 CrossRef CAS.
- K. Tomishige, Curr. Top. Catal., 2002, 3, 81–101 CAS.
- M. G. Cutrufello, I. Ferino, V. Solinas, A. Primavera, A. Trovarelli, A. Auroux and C. Picciau, Phys. Chem. Chem. Phys., 1999, 1, 3369–3375 RSC.
- A. Bansode and A. Urakawa, ACS Catal., 2014, 4, 3877–3880 CrossRef CAS.
- M. Honda, M. Tamura, Y. Nakagawa, S. Sonehara, K. Suzuki, K. Fujimoto and K. Tomishige, ChemSusChem, 2013, 6, 1341–1344 CrossRef CAS PubMed.
- M. Honda, M. Tamura, Y. Nakagawa, K. Nakao, K. Suzuki and K. Tomishige, J. Catal., 2014, 318, 95–107 CrossRef CAS.
- D. Stoian, A. Bansode, F. Medina and A. Urakawa, Catal. Today, 2017, 283, 2–10 CrossRef CAS.
- S. Wada, K. Oka, K. Watanabe and Y. Izumi, Front. Chem., 2013, 1, 1–8 CAS.
- M. Aresta, A. Dibenedetto, C. Pastore, C. Cuocci, B. Aresta, S. Cometa and E. De Giglio, Catal. Today, 2008, 137, 125–131 CrossRef CAS.
- M. Aresta, A. Dibenedetto, C. Pastore, A. Angelini, B. Aresta and P. Imre, J. Catal., 2010, 269, 44–52 CrossRef CAS.
- B. Liu, C. Li, G. Zhang, X. Yao, S. S. C. Chuang and Z. Li, ACS Catal., 2018, 8, 10446–10456 CrossRef CAS.
- A. Trovarelli, Comments Inorg. Chem., 1999, 20, 263–284 CrossRef CAS.
- S. Kumar Meher and G. Ranga Rao, ACS Catal., 2012, 2, 2795–2809 CrossRef.
- H. C. Yao and Y. F. Yu, J. Catal., 1984, 86, 254–265 CrossRef CAS.
- A. Malik, A. de Juan and R. Tauler, in 40 Years of Chemometrics – From Bruce Kowalski to the Future, American Chemical Society, 2015, vol. 1199, pp. 95–128 Search PubMed.
- A. Voronov, A. Urakawa, W. van Beek, N. E. Tsakoumis, H. Emerich and M. Rønning, Anal. Chim. Acta, 2014, 840, 20–27 CrossRef CAS PubMed.
- T. Hyakutake, W. van Beek and A. Urakawa, J. Mater. Chem. A, 2016, 4, 6878–6885 RSC.
- L. Chen, S. Wang, J. Zhou, Y. Shen, Y. Zhao and X. Ma, RSC Adv., 2014, 4, 30968–30975 RSC.
- S.-Y. Zhao, S.-P. Wang, Yu-J. Zhao and X.-B. Ma, Chin. Chem. Lett., 2017, 28, 65–69 CrossRef CAS.
- G. N. Vayssilov, M. Mihaylov, P. S. Petkov, K. I. Hadjiivanov and K. M. Neyman, J. Phys. Chem. C, 2011, 115, 23435–23454 CrossRef CAS.
- O. Pozdnyakova, D. Teschner, A. Wootsch, J. Kröhnert, B. Steinhauer, H. Sauer, L. Toth, F. C. Jentoft, A. Knop-Gericke, Z. Paál and R. Schlögl, J. Catal., 2006, 237, 17–28 CrossRef CAS.
- P. Müller and I. Hermans, Ind. Eng. Chem. Res., 2017, 56, 1123–1136 CrossRef.
- D. Ferri, M. A. Newton and M. Nachtegaal, Top. Catal., 2011, 54, 1070 CrossRef CAS.
- A. Urakawa, Curr. Opin. Chem. Eng., 2016, 12, 31–36 CrossRef.
- K. Tomishige, Y. Ikeda, T. Sakaihori and K. Fujimoto, J. Catal., 2000, 192, 355–362 CrossRef CAS.
- S. Xie and A. T. Bell, Catal. Lett., 2000, 70, 137–143 CrossRef CAS.
- K. T. Jung and A. T. Bell, J. Catal., 2001, 204, 339–347 CrossRef CAS.
- J. Lamotte, V. Morávek, M. Bensitel and J. C. Lavalley, React. Kinet. Catal. Lett., 1988, 36, 113–118 CrossRef CAS.
- M. Bensitel, V. Morávek, J. Lamotte, O. Saur and J.-C. Lavalley, Spectrochim. Acta Part A Mol. Spectrosc., 1987, 43, 1487–1491 CrossRef.
- W. Y. Hernández, O. H. Laguna, M. A. Centeno and J. A. Odriozola, J. Solid State Chem., 2011, 184, 3014–3020 CrossRef.
- M. Machida, T. Kawada, H. Fujii and S. Hinokuma, J. Phys. Chem. C, 2015, 119, 24932–24941 CrossRef CAS.
- L. E. Gómez, J. F. Múnera, B. M. Sollier, E. E. Miró and A. V. Boix, Int. J. Hydrogen Energy, 2016, 41, 4993–5002 CrossRef.
- S. Lorentzou, G. Karagiannakis, D. Dimitrakis, C. Pagkoura, A. Zygogianni and A. G. Konstandopoulos, Energy Procedia, 2015, 69, 1800–1809 CrossRef CAS.
- T. Taniguchi, T. Watanabe, N. Sugiyama, A. K. Subramani, H. Wagata, N. Matsushita and M. Yoshimura, J. Phys. Chem. C, 2009, 113, 19789–19793 CrossRef CAS.
- M. Tamura and K. Tomishige, Angew. Chem., Int. Ed., 2015, 54, 864–867 CrossRef CAS PubMed.
- Z. Wu, M. Li, D. R. Mullins and S. H. Overbury, ACS Catal., 2012, 2, 2224–2234 CrossRef CAS.
- D. R. Mullins, M. D. Robbins and J. Zhou, Surf. Sci., 2006, 600, 1547–1558 CrossRef CAS.
- V. Matolín, J. Libra, M. Škoda, N. Tsud, K. C. Prince and T. Skála, Surf. Sci., 2009, 603, 1087–1092 CrossRef.
- J.-D. Cafun, K. O. Kvashnina, E. Casals, V. F. Puntes and P. Glatzel, ACS Nano, 2013, 7, 10726–10732 CrossRef CAS PubMed.
- D. Stoian, F. Medina and A. Urakawa, ACS Catal., 2018, 8, 3181–3193 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: A complete description of the reagents and lab-based/synchrotron setup is given. Multivariate curve resolution technique is briefly presented together with more results on its application over XAS/Raman data. Computational details for the DFT calculations and MD simulations, adsorption structures for the reagents and reaction intermediates and a comprehensive discussion are also included. See DOI: https://doi.org/10.1039/d3sc04466a |
| This journal is © The Royal Society of Chemistry 2023 |