
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCobalt-catalyzed decarboxylative difluoroalkylation of nitrophenylacetic acid salts†
Ebbin
Joseph
,
Ian
Smith
and
Jon A.
Tunge
 *
*
Department of Chemistry, The University of Kansas, 1567 Irving Rd, Lawrence, KS 66045, USA. E-mail: tunge@ku.edu
First published on 16th November 2023
Abstract
The selective installation of fluorine-containing groups into biologically relevant molecules has been used as a common strategy for the development of pharmaceutically active molecules. However, the selective incorporation of gem-difluoromethylene groups next to sterically demanding secondary and tertiary alkyl groups remains a challenge. Herein, we report the first cobalt-catalyzed regioselective difluoroalkylation of carboxylic acid salts. The reaction allows for the facile construction of various difluoroalkylated products in good yields tolerating a wide range of functionalities on either reaction partner. The potential of the method is illustrated by the late-stage functionalization of molecules of biological relevance. Mechanistic studies support the in situ formation of a cobalt(I) species and the intermediacy of difluoroalkyl radicals, thus suggesting a Co(I)/Co(II)/Co(III) catalytic cycle.
Introduction
The incorporation of fluorine into organic substructures is one of the most widely studied areas of synthetic organic chemistry due to the numerous applications that fluorinated compounds possess.1 The gem-difluoromethylene unit is an important therapeutic moiety because of its ability to increase metabolic stability2 or improve the pharmacokinetic properties of molecules.3 The significance of this structural motif in drug discovery is further illustrated by the large variety of difluoroalkane-containing pharmaceutical compounds such as HIV-1 therapeutic agents,4 and chemotherapy drugs (Fig. 1).5 However, the incorporation of these fluorinated linkages remains a significant synthetic challenge and still relies on traditional methods such as the use of nucleophilic or electrophilic fluoride sources (e.g., DAST or Selectfluor).6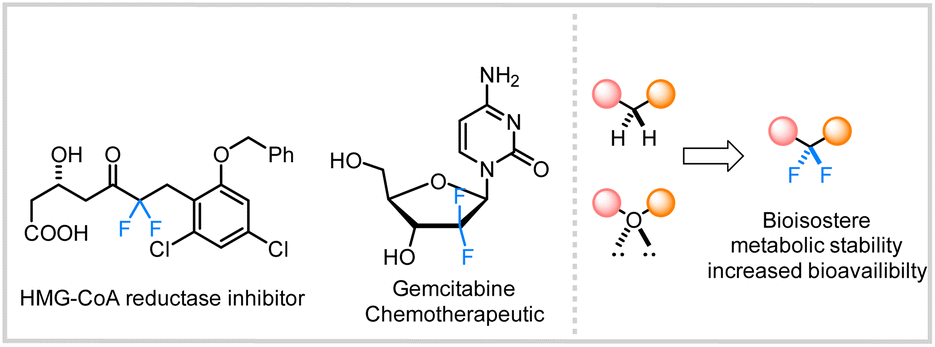 | ||
| Fig. 1 Select fluorine-containing therapeutics. | ||
In recent years, transition metal-catalyzed cross-couplings have emerged as convenient strategies for the construction of fluorine-containing organic compounds.6h,7–12 Recent efforts have led to the construction of various C(sp2 or sp)–CF2R bonds where the fluorinated alkane is often connected to the aryl,13 vinylic,12f,14 or propargylic9f,11b,15 positions. In contrast, the selective installation of the difluoromethylene group adjacent to aliphatic all carbon quaternary C(sp3)-centers remains innately challenging and sparsely reported (Scheme 1).11c,12b,16,17
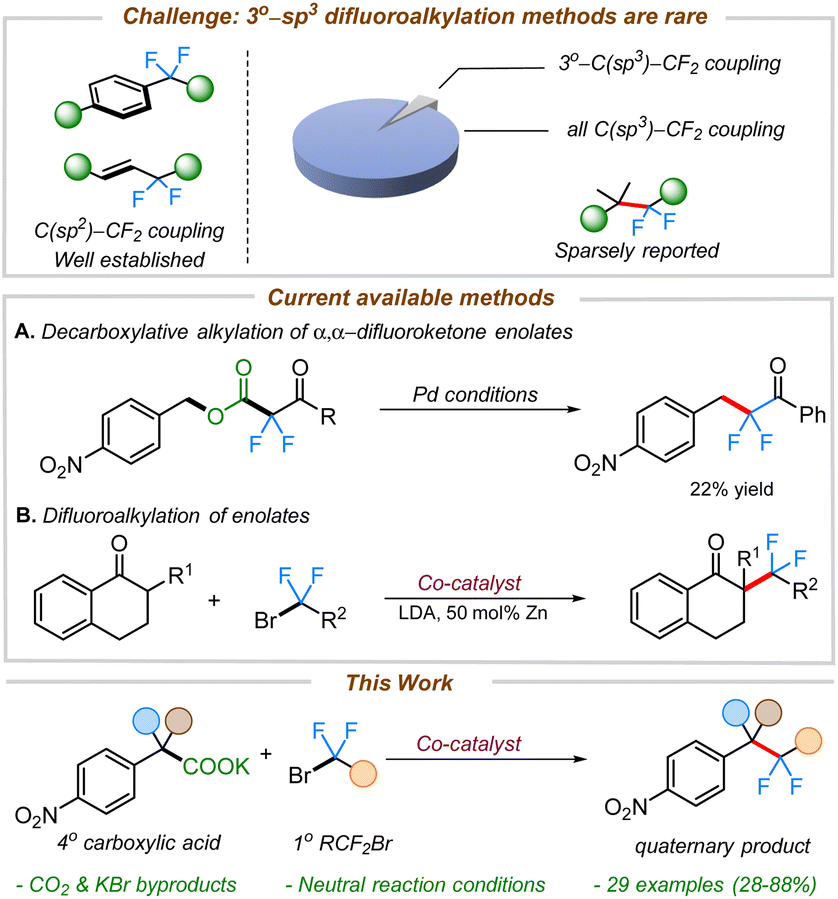 | ||
| Scheme 1 Decarboxylative difluoroalkylations. | ||
Nonetheless, integrating quaternary carbon centers has the potential to impart conformational rigidity and metabolic stability, leading to improved pharmacokinetic properties of molecules.18 With this in mind, we set out to develop difluoroalkylation of quaternary benzyl nucleophiles for the facile construction of all carbon quaternary C(sp3)–CF2 bonds.
We envisioned leveraging decarboxylation as an efficient strategy for the generation of benzylic nucleophiles from organic acids.19 There are a few reports on decarboxylative difluoroalkylations known in the literature.20,20d,21,22 Altman and co-workers have previously developed decarboxylative electrophilic benzylations of difluoroenolate nucleophiles (Scheme 1A).23 Although the chemistry proved highly effective for the difluoroalkylation of primary electron-rich benzyl electrophiles, the outcomes were substantially worse with electron-deficient benzyl electrophiles. Furthermore, coupling of 2° or 3° benzyl electrophiles was not possible. To address the challenge of difluoroalkylation of sterically-demanding benzyl moieties, we posited an alternate strategy involving umpolung of the reactive intermediates (i.e. using benzyl nucleophiles with α,α-difluorocarbonyl electrophiles).
Results and discussion
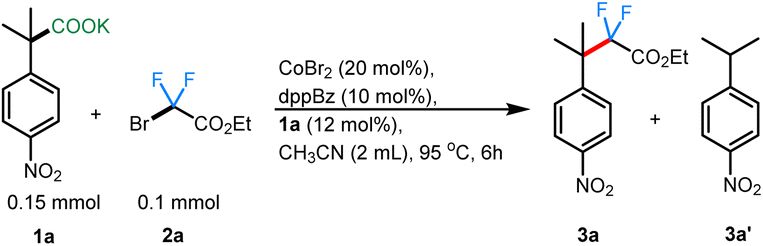
To begin, we took inspiration from Wang's cobalt-catalyzed gem-difluoroalkylation of α-tertiary aryl ketones (Scheme 1B).12b While that chemistry required the use of stoichiometric LDA and 50 mol% Zn reductant, it was anticipated that a decarboxylative coupling strategy would allow additive-free synthesis under more neutral conditions.24We initiated our studies by optimizing the conditions for the cobalt-catalyzed difluoroalkylation of 2-methyl-2-(4-nitrophenyl)propanoic acid potassium salt (1a) with bromodifluoroacetate (2a) using the conditions adapted from a related allylation study.24 Interestingly, with 10 mol% CoBr2 and 10 mol% of dppBz, we observed the corresponding difluoroalkylated (3a) in reasonable yields along with 30% of the protonated product 3a′ (Table 1, entry 4). Gratifyingly, when the cobalt loading was increased to 20 mol%, we observed the highest yield (81%) for the difluoroalkylated product 3a and decreased amount of the protonated byproduct 3a′ (Table 1, entry 1). Control studies confirmed the necessity of both cobalt and the ligand for efficient reactivity (Table 1, entries 2 & 3). Replacing CoBr2 with other cobalt sources such as Co(BF4)2 or CoI2 gave decreased yields of 3a (Table 1, entries 6 & 7). The initial solvent of choice, MeCN, was found to be the best for the reaction (Table 1, entry 8). Various bis-phosphine and diamine-containing ligands were screened; however, all of them failed to give an improvement in yield compared to that of dppBz (Table 1, entries 10–12). After additional screenings (see ESI† for more details), it was determined that CoBr2 (20 mol%), dppBz (10 mol%), and 1a (12 mol%) in CH3CN at 95 °C were optimal for this reaction, producing the desired product 3a in 77% isolated yield.
|
|
||
|---|---|---|
| Entry | Variations in conditions | Yielda3a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3a′ [%] :3a′ [%] |
| a Yields determined by quantitative 1H NMR analysis. Numbers in parentheses are isolated yields. | ||
| 1 | — | 84(77):16 |
| 2 | No cobalt | — |
| 3 | No ligand | — |
| 4 | 10 mol% of CoBr2 | 15:30 |
| 5 | 15 mol% of CoBr2 | 24:36 |
| 6 | Co(BF4)2 instead of CoBr2 | 40:80 |
| 7 | CoI2 instead of CoBr2 | 54:19 |
| 8 | DMF, DMSO, THF instead of MeCN | <25 |
| 9 | 60 °C instead of 95 °C | 40:18 |
| 10 | dppe instead of dppBz | 31:52 |
| 11 | dppf instead of dppBz | 45:50 |
| 12 | dtbbpy instead of dppBz | 42:28 |
With the optimized conditions in hand, we sought to expand the protocol to accommodate other fluoroalkylating reagents and carboxylate salts, enabling the construction of a unique range difluoroalkyl groups. Remarkably, in all cases, the product formation was regiospecific, with the C–CF2 bond formation occurring at the site where decarboxylation had occurred (3h & 3j–k). A wide range of potassium salts of various substituted 4-nitrophenyl acetic acids were found to be tolerant to the reaction conditions, providing the coupled products in moderate to good yields (Scheme 2). In addition to a simple methyl substituent (3a; 77%), the alkyl chain was extended to accommodate other longer alkyl chains (3b; 66% and 3c; 36%), albeit with lower yields. Both benzylic- and homobenzylic-substituted carboxylate salts gave reasonable yields for the corresponding fluoroalkylated product (3d; 45% and 3e; 56%). A carboxylate salt containing a cyclopentyl group at the alpha position gave the subsequent fluoroalkyated product in 60% yield (3g). Carboxylate salts bearing other important functional groups such as ester (3h; 55%), ether (3i; 49%), nitrile (3j; 61%), and ketone groups (3k; 71%) were all tolerated under the reaction conditions. Owing to the biological importance of heterocyclic compounds, the pyridine-containing carboxylate salt 1l was tested under our reaction conditions. We were delighted to find that 1l also underwent the transformation to deliver the corresponding difluoroalkylated product 3l in 82% yield. While many couplings occurred to provide products in moderate to good yield, it was noted that, as the steric hindrance around the quaternary carbon increased, the yields of coupling were adversely affected. This was especially clear with the naphthyl-substituted salt giving only 28% of the corresponding difluoroalkylated product (3m). Similar results were obtained with the α-phenyl carboxylate salt, giving only 28% of the corresponding difluoroalkylated product 3o. In instances with lower yields for the product, the mass balance was always accounted for by the amount of the protonated byproduct isolated.
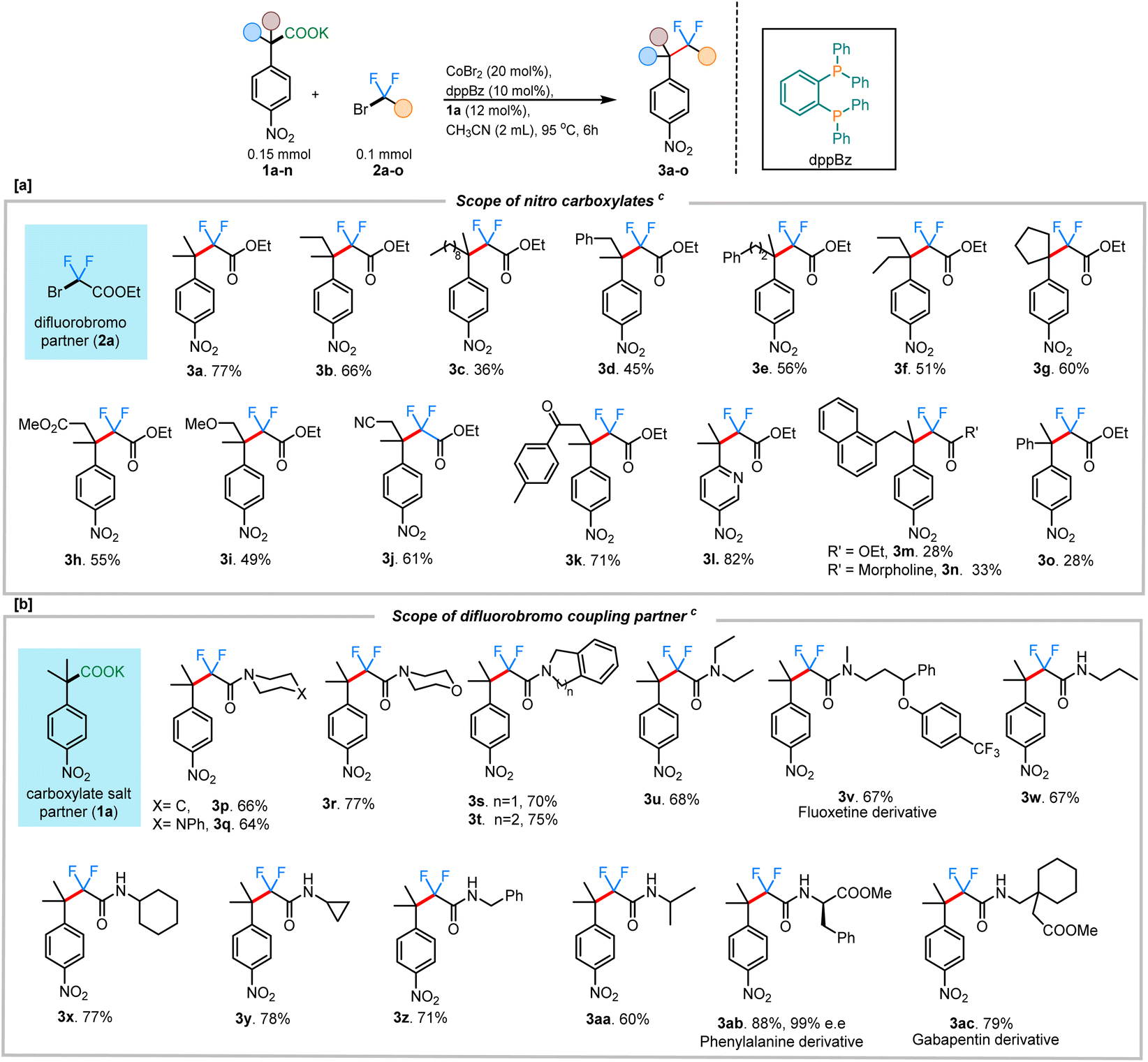 | ||
| Scheme 2 Scope of decarboxylative difluoroalkylations. a Scope of nitro carboxylates. b Scope of difluorobromo alkanes. c All reactions were run on a 0.1 mmol scale. Yields reported are isolated yields. Reaction conditions: CoBr2 (20 mol%), dppBz (10 mol%), 1a (12 mol%), CH3CN (2 mL), 95 °C. | ||
Additionally, the scope of different difluorobromo coupling partners was explored. Various acetamides, both cyclic and acyclic, were found to be well-tolerated during this transformation. Cyclic piperidine (3p; 66%), piperazine (3q; 64%), morpholine (3r; 77%), indoline (3s; 70%), and tetrahydroisoquinoline (3t; 75%) derived acetamides gave the corresponding fluoroalkylated product in good yields. The reaction was even successful with a fluoxetine-derived difluorobromoacetamide providing the corresponding cross-coupled product in 67% yield (3v). Simple alkyl substituted difluoroacetamides such as N-propyl (3w), N-cyclohexyl (3x), N-cyclopropyl (3y), N-benzyl (3z), and N-isopropyl (3aa) were also found to undergo the transformation efficiently, with the cyclopropyl ring staying intact under the reaction conditions. Importantly, a difluorobromoacetamide derived from L-phenylalanine also gave the cross-coupled product 3ab in 88% yield, without any observable racemization of the existing stereocenter (see ESI† for more details). This highlights the utility of decarboxylative couplings that obviate strong-base additives.12b The reaction with a gabapentin-derived difluorobromoacetamide likewise proceeded in good yield, and could be scaled up to a 1 mmol scale without large reduction in the yield (Scheme 3a).
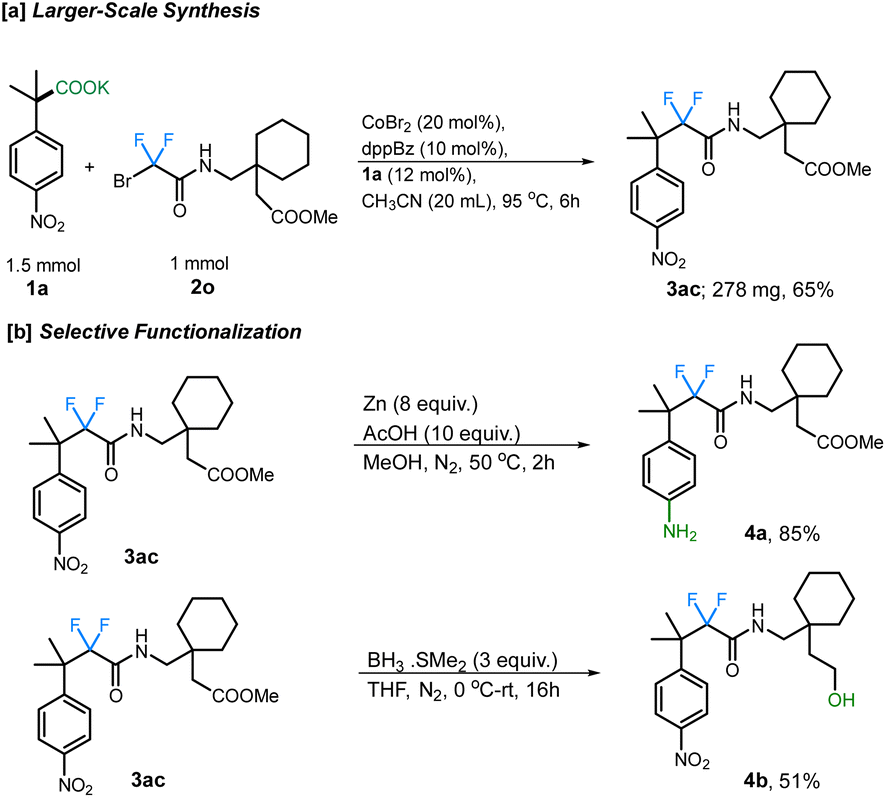 | ||
| Scheme 3 (a) Larger-scale reaction. (b) Synthetic utility. | ||
Finally, we further demonstrated the synthetic potential of this cobalt-catalyzed decarboxylative difluoroalkylation method through the synthetic modification of the difluoroalkylated products. For example, the resulting gabapentin-derived product 2m can be selectively reduced under Zn/AcOH conditions to the aniline derivative 4a. Moreover, the reduction of the ester group using BH3·SMe2 provides the corresponding alcohol 4b which can undergo further derivatizations (Scheme 3b).
To gain more insight into the mechanism of this cobalt-catalyzed decarboxylative difluoroalkylation reaction, a series of different experiments was performed. A competition experiment between bromodifluoroacetate (2a) and bromodifluoroacetamide (2d) showed that 2a reacted 10× faster than the related amide (2d) (Fig. 2a). This rate difference could result either from the more favorable oxidative addition of the bromodifluoroacetate to Co(I) or preferential single electron transfer from Co(I). Expectedly, the more electron deficient bromodifluoroacetates are easier to reduce than bromodifluoroacetamides.25 Importantly, concerted oxidative addition vs. single electron transfer pathways are distinguished by the intermediacy of a difluoroalkyl radical in the latter pathway.
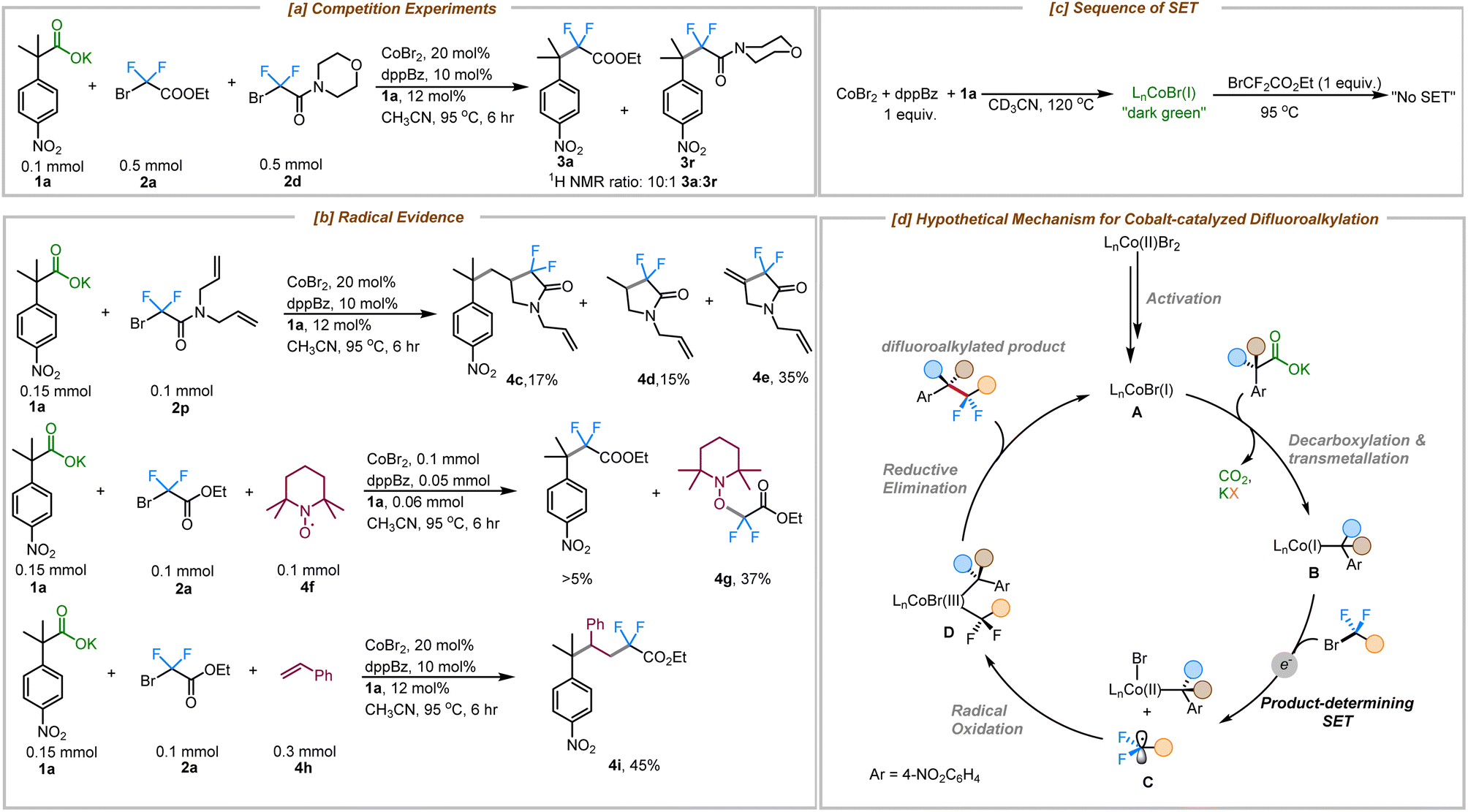 | ||
| Fig. 2 Mechanistic insights. (a) Competition experiment (b) radical evidence (c) sequence of SET (d) hypothetical difluoroalkylation mechanism. | ||
With this in mind, a radical clock experiment was performed with substrate 2p, which delivered the cyclized product 4c in 17% yield along with protocyclized product 4d and the dehydrocyclized product 4e in 15% and 35% respectively. Beyond that, a TEMPO trapping experiment showed the formation of adduct 4g in 37% yield and produced less than 5% of the coupled product (3a). Furthermore, the use of an external radical trap such as styrene delivered the corresponding three-component coupled product via a regioselective radical trapping pathway that furnished the product 4i exclusively (Fig. 2b). Based on our previous mechanistic studies for the cobalt-catalyzed decarboxylative allylation reaction, we have proposed the formation of an L1Co(I) species as the active catalytic species under these reaction conditions (see ESI for details; Fig. S10†).24 Furthermore, since the addition of the fluoroalkylating agent (2a) to the active Co–Br catalyst didn't show any evidence for irreversible bond scission products while being monitored using 19F NMR, we propose that decarboxylative metalation to form the more electron-rich alkyl-Co species might occur prior to SET (Fig. 2c, see ESI† for details).
Taken together, we propose the following mechanism for the cobalt-catalyzed decarboxylative difluoroalkylation reaction (Fig. 2d). The reduction of the CoBr2/dppBz complex by the carboxylate 1a generates the catalytically active Co(I) species (A).24 Decarboxylative metalation generates species B,19 which in turn reduces the difluoro alkyl bromide via SET, generating the difluoroalkyl radical C. Radical C undergoes subsequent radical oxidation and trapping by the cobalt complex to form species D. Reductive elimination from complex D delivers the difluroroalkyated product and regenerates the active cobalt(I) species.
Conclusions
In summary, we have developed a simple and efficient method for the regioselective difluoroalkylation of potassium salts of carboxylic acids. This cobalt-catalyzed decarboxylative approach allows for the facile construction of quaternary C(sp3)–CF2 bonds in a fully regio- and chemoselective fashion in moderate to good yields. The reaction proceeds with moderate to good efficacy, modest functional group tolerance, as well as a broad substrate scope, producing molecular CO2 and KBr as the only waste by-products. Mechanistic studies demonstrated a single electron transfer to the difluoroalkyl halides from a Co(I) species leading to the formation of a discrete difluoroalkyl radical.Data availability
The data underlying this study are available in the published article and its ESI.†Author contributions
J. T. and E. J. conceptualized and initiated the project. E. J. and I. S. performed the experiments and analyzed the data. J. T. and E. J. co-wrote the paper.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Science Foundation (CHE-2247708 and CHE-2155003). I. S. was supported through the NSF REU program (CHE-1950293). Support for the NMR instrumentation was provided by NSF Academic Research Infrastructure Grant No. 9512331, NIH Shared Instrumentation Grant No. S10RR024664, and NSF Major Research Instrumentation Grant No. 0320648.Notes and references
- (a) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef; (b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC; (c) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS; (d) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS; (e) N. A. Meanwell, J. Med. Chem., 2018, 61, 5822–5880 CrossRef CAS; (f) B. M. Johnson, Y.-Z. Shu, X. Zhuo and N. A. Meanwell, J. Med. Chem., 2020, 63, 6315–6386 CrossRef CAS.
- (a) W. B. Motherwell, M. J. Tozer and B. C. Ross, J. Chem. Soc., Chem. Commun., 1989, 1437–1439, 10.1039/C39890001437; (b) Q. Zhou, A. Ruffoni, R. Gianatassio, Y. Fujiwara, E. Sella, D. Shabat and P. S. Baran, Angew. Chem., Int. Ed., 2013, 52, 3949–3952 CrossRef CAS.
- (a) Y.-L. Liu, J.-S. Yu and J. Zhou, Asian J. Org. Chem., 2013, 2, 194–206 CrossRef CAS; (b) S. Holovach, K. P. Melnykov, A. Skreminskiy, M. Herasymchuk, O. Tavlui, D. Aloshyn, P. Borysko, A. B. Rozhenko, S. V. Ryabukhin, D. M. Volochnyuk and O. O. Grygorenko, Chem.–Eur. J., 2022, 28, e202200331 CrossRef CAS.
- (a) S. M. Woollard and G. D. Kanmogne, Drug Des., Dev. Ther., 2015, 9, 5447–5468 CAS; (b) X. Xie, Y.-G. Zheng, H. Chen, J. Li, R.-H. Luo, L. Chen, C.-B. Zheng, S. Zhang, P. Peng, D. Ma, L.-M. Yang, Y.-T. Zheng, H. Liu and J. Wang, J. Med. Chem., 2022, 65, 16526–16540 CrossRef CAS.
- (a) G. M. Blackburn, D. E. Kent and F. Kolkmann, J. Chem. Soc., Perkin Trans. 1, 1984, 1119–1125, 10.1039/P19840001119; (b) K. Holmsten, N. V. Jensen, L. S. Mouritsen, E. Jonsson, C. Mellnert, M. Agerbæk, C. Nilsson, M. Moe, A. Carus, E. Öfverholm, O. Lahdenperä, Y. Brandberg, H. Johansson, M. Hellström, H. v. d. Maase, H. Pappot and A. Ullén, Eur. J. Cancer, 2020, 127, 173–182 CrossRef CAS; (c) S. Ghosh, Z.-W. Qu, S. Roy, S. Grimme and I. Chatterjee, Chem.–Eur. J., 2023, e202203428 CrossRef CAS; (d) L. W. Hertel, G. B. Boder, J. S. Kroin, S. M. Rinzel, G. A. Poore, G. C. Todd and G. B. Grindey, Cancer Res., 1990, 50, 4417–4422 CAS; (e) F. Eckel, G. Schneider and R. M. Schmid, Expert Opin. Invest. Drugs, 2006, 15, 1395–1410 CrossRef CAS.
- (a) G. S. Lal, G. P. Pez, R. J. Pesaresi, F. M. Prozonic and H. Cheng, J. Org. Chem., 1999, 64, 7048–7054 CrossRef CAS; (b) J. Hu, W. Zhang and F. Wang, Chem. Commun., 2009, 7465–7478, 10.1039/B916463D; (c) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS; (d) M. G. Campbell and T. Ritter, Chem. Rev., 2015, 115, 612–633 CrossRef CAS; (e) C. Ni, M. Hu and J. Hu, Chem. Rev., 2015, 115, 765–825 CrossRef CAS; (f) D. E. Yerien, S. Barata-Vallejo and A. Postigo, Chem.–Eur. J., 2017, 23, 14676–14701 CrossRef CAS; (g) P. R. Melvin, D. M. Ferguson, S. D. Schimler, D. C. Bland and M. S. Sanford, Org. Lett., 2019, 21, 1350–1353 CrossRef CAS; (h) D. R. Carvalho and A. H. Christian, Org. Biomol. Chem., 2021, 19, 947–964 RSC.
- (a) B. Chen and D. A. Vicic, in Organometallic Fluorine Chemistry, ed. T. Braun and R. P. Hughes, Springer International Publishing, Cham, 2015, pp. 113–141, DOI:10.1007/3418_2014_87; (b) J. Rong, C. Ni and J. Hu, Asian J. Org. Chem., 2017, 6, 139–152 CrossRef CAS; (c) Z. Feng, Y.-L. Xiao and X. Zhang, Acc. Chem. Res., 2018, 51, 2264–2278 CrossRef CAS; (d) X. Li, X. Shi, X. Li and D. Shi, Beilstein J. Org. Chem., 2019, 15, 2213–2270 CrossRef CAS; (e) T. W. Butcher, W. M. Amberg and J. F. Hartwig, Angew. Chem., Int. Ed., 2022, 61, e202112251 CrossRef CAS; (f) H. Sindhe, B. Chaudhary, N. Chowdhury, A. Kamble, V. Kumar, A. Lad and S. Sharma, Org. Chem. Front., 2022, 9, 1742–1775 RSC.
- (a) J. Yang, H.-W. Zhao, J. He and C.-P. Zhang, Catalysts, 2018, 23, 1–35 CAS; (b) B. M. Trost, H. Gholami and D. Zell, J. Am. Chem. Soc., 2019, 141, 11446–11451 CrossRef CAS; (c) L. Li, Z. Zhao, J. Xu, H. Luo, Y. Li, X. Ma, L. Tang, B. Ren, X. Cao and Y.-N. Ma, Chem. Commun., 2020, 56, 9384–9387 RSC; (d) Y. He, Z. Huang, J. Ma, F. Huang, J. Lin, H. Wang, B.-H. Xu, Y.-G. Zhou and Z. Yu, Org. Lett., 2021, 23, 6110–6114 CrossRef CAS; (e) N. Lalloo, C. A. Malapit, S. M. Taimoory, C. E. Brigham and M. S. Sanford, J. Am. Chem. Soc., 2021, 143, 18617–18625 CrossRef CAS; (f) P. V. Saranya, T. Aneeja and G. Anilkumar, Appl. Organomet. Chem., 2022, 36, e6503 CrossRef CAS; (g) T. Xiang, Y. Liu, Q. Xu, K. He and F. Pan, J. Org. Chem., 2022, 87, 3135–3144 CrossRef CAS.
- (a) W. Kong, C. Yu, H. An and Q. Song, Org. Lett., 2018, 20, 4975–4978 CrossRef CAS; (b) F. Yuan, S. Zhou, Y. Yang, M. Guo, X. Tang and G. Wang, Org. Chem. Front., 2018, 5, 3306–3309 RSC; (c) X. Zeng, W. Yan, M. Paeth, S. B. Zacate, P.-H. Hong, Y. Wang, D. Yang, K. Yang, T. Yan, C. Song, Z. Cao, M.-J. Cheng and W. Liu, J. Am. Chem. Soc., 2019, 141, 19941–19949 CrossRef CAS; (d) Y. Yang, F. Yuan, X. Ren, G. Wang, W. Zhao, X. Tang and M. Guo, J. Org. Chem., 2019, 84, 4507–4516 CrossRef CAS; (e) D.-Q. Dong, S.-H. Yang, P. Wu, J.-Z. Wang, L.-H. Min, H. Yang, M.-Y. Zhou, Z.-H. Wei, C.-Z. Ding, Y.-L. Wang, J.-H. Gao, S.-J. Wang and Z.-L. Wang, Molecules, 2022, 27, 8461 CrossRef CAS; (f) G.-Q. Hu, L.-W. Yao, S.-S. Gui, C. Geng, W.-Y. Zhang, J.-H. Liu and B. Zhao, Synlett, 2022, 33, 594–598 CrossRef CAS.
- (a) L. An, Y.-L. Xiao, S. Zhang and X. Zhang, Angew. Chem., Int. Ed., 2018, 57, 6921–6925 CrossRef CAS; (b) Z. Wei, W. Miao, C. Ni and J. Hu, Angew. Chem., Int. Ed., 2021, 60, 13597–13602 CrossRef CAS; (c) S. Bertho, R. Maazaoui, D. Torun, I. Dondasse, R. Abderrahim, C. Nicolas and I. Gillaizeau, New J. Chem., 2021, 45, 17475–17482 RSC.
- (a) Y.-L. Xiao, W.-H. Guo, G.-Z. He, Q. Pan and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 9909–9913 CrossRef CAS; (b) L. An, C. Xu and X. Zhang, Nat. Commun., 2017, 8, 1460 CrossRef; (c) C. Li, Y.-X. Cao, R.-X. Jin, K.-J. Bian, Z.-Y. Qin, Q. Lan and X.-S. Wang, Chem. Sci., 2019, 10, 9285–9291 RSC; (d) R.-X. Jin, B.-B. Wu, K.-J. Bian, J.-L. Yu, J.-C. Dai, Y.-W. Zuo, Y.-F. Zhang and X.-S. Wang, Nat. Commun., 2022, 13, 7035 CrossRef CAS.
- (a) L. M. Kreis, S. Krautwald, N. Pfeiffer, R. E. Martin and E. M. Carreira, Org. Lett., 2013, 15, 1634–1637 CrossRef CAS; (b) C. Li, Y.-X. Cao, R. Wang, Y.-N. Wang, Q. Lan and X.-S. Wang, Nat. Commun., 2018, 9, 4951 CrossRef; (c) S. Han, S. Liu, L. Liu, L. Ackermann and J. Li, Org. Lett., 2019, 21, 5387–5391 CrossRef CAS; (d) X. Cheng, X. Liu, S. Wang, Y. Hu, B. Hu, A. Lei and J. Li, Nat. Commun., 2021, 12, 4366 CrossRef CAS; (e) Z. Hu, J. Ren, Z. Chen, S. Yin, F. Wu, C. Liu, Z. Wu and J. Wu, ChemistrySelect, 2021, 6, 12276–12279 CrossRef CAS; (f) J. Pan, H. Li, J. Wu and F. Wu, J. Organomet. Chem., 2021, 944, 121818 CrossRef CAS; (g) Y. Yamamoto, H. Suzuki, E. Kuroyanagi, K. Yamada and T. Yasui, Org. Biomol. Chem., 2022, 20, 2867–2872 RSC.
- (a) C. Guo, R.-W. Wang, Y. Guo and F.-L. Qing, J. Fluorine Chem., 2012, 133, 86–96 CrossRef CAS; (b) Z. Feng, Q.-Q. Min, Y.-L. Xiao, B. Zhang and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 1669–1673 CrossRef CAS.
- Z. Feng, Q.-Q. Min, H.-Y. Zhao, J.-W. Gu and X. Zhang, Angew. Chem., Int. Ed., 2015, 54, 1270–1274 CrossRef CAS.
- C. Xu, Z.-F. Yang, L. An and X. Zhang, ACS Catal., 2019, 9, 8224–8229 CrossRef CAS.
- (a) L. Dai, Y.-Y. Xu, Z.-H. Xia and S. Ye, Org. Lett., 2020, 22, 8173–8177 CrossRef CAS; (b) J. Liu, W. Ding, Q.-Q. Zhou, D. Liu, L.-Q. Lu and W.-J. Xiao, Org. Lett., 2018, 20, 461–464 CrossRef CAS; (c) E. Miller, S. Kim, K. Gibson, J. S. Derrick and F. D. Toste, J. Am. Chem. Soc., 2020, 142, 8946–8952 CrossRef CAS; (d) H. Song, R. Cheng, Q.-Q. Min and X. Zhang, Org. Lett., 2020, 22, 7747–7751 CrossRef CAS; (e) K. Li, J. Chen, C. Yang, K. Zhang, C. Pan and B. Fan, Org. Lett., 2020, 22, 4261–4265 CrossRef CAS.
- This Pie-chart was generated from a pool of 616 references obtained from a search in Sci-finder using the keyword “difluoroalkylation”. Only 7 reports involved difluoroalkylation of all carbon tertiary coupling partners.
- (a) T. T. Talele, J. Med. Chem., 2018, 61, 2166–2210 CrossRef CAS; (b) T. T. Talele, J. Med. Chem., 2020, 63, 13291–13315 CrossRef CAS.
- (a) P. J. Moon and R. J. Lundgren, ACS Catal., 2020, 10, 1742–1753 CrossRef CAS; (b) D. Kong, P. J. Moon, W. Qian and R. J. Lundgren, Chem. Commun., 2018, 54, 6835–6838 RSC; (c) P. J. Moon, A. Fahandej-Sadi, W. Qian and R. J. Lundgren, Angew. Chem., Int. Ed., 2018, 57, 4612–4616 CrossRef CAS.
- (a) Z. He, M. Hu, T. Luo, L. Li and J. Hu, Angew. Chem., Int. Ed., 2012, 51, 11545–11547 CrossRef CAS; (b) B. R. Ambler, M.-H. Yang and R. A. Altman, Synlett, 2016, 27, 2747–2755 CrossRef CAS; (c) D. L. Orsi and R. A. Altman, Chem. Commun., 2017, 53, 7168–7181 RSC; (d) W. Mei, Y. Kong and G. Yan, Org. Chem. Front., 2021, 8, 5516–5530 RSC.
- X. Li, R. Zhang, X. Zhang, P. Zhu and T. Yao, Chem.–Asian J., 2020, 15, 1175–1179 CrossRef CAS.
- (a) Z. Li, Z. Cui and Z.-Q. Liu, Org. Lett., 2013, 15, 406–409 CrossRef CAS; (b) G. Li, T. Wang, F. Fei, Y.-M. Su, Y. Li, Q. Lan and X.-S. Wang, Angew. Chem., Int. Ed., 2016, 55, 3491–3495 CrossRef CAS; (c) Y.-L. Lai, D.-Z. Lin and J.-M. Huang, J. Org. Chem., 2017, 82, 597–605 CrossRef CAS.
- M.-H. Yang, J. R. Hunt, N. Sharifi and R. A. Altman, Angew. Chem., Int. Ed., 2016, 55, 9080–9083 CrossRef CAS.
- E. Joseph, R. D. Hernandez and J. A. Tunge, Chem.–Eur. J., 2023, 29, e202302174 CrossRef CAS.
- H. Liang, G.-Q. Xu, Z.-T. Feng, Z.-Y. Wang and P.-F. Xu, J. Org. Chem., 2019, 84, 60–72 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc05583c |
| This journal is © The Royal Society of Chemistry 2023 |