
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Both sides matter: anode configurations alter the activity of electrolyzers for organic hydrogenations†
Kevinjeorjios
Pellumbi‡
 ab,
Jonas
Wolf‡
ab,
Sangita Conjeevaram
Viswanathan
ab,
Leon
Wickert
ab,
Mena-Alexander
Kräenbring
c,
Julian T.
Kleinhaus
b,
Kai
junge Puring
a,
Fatih
Özcan
c,
Doris
Segets
cd,
Ulf-Peter
Apfel
*ab and
Daniel
Siegmund
*ab
ab,
Jonas
Wolf‡
ab,
Sangita Conjeevaram
Viswanathan
ab,
Leon
Wickert
ab,
Mena-Alexander
Kräenbring
c,
Julian T.
Kleinhaus
b,
Kai
junge Puring
a,
Fatih
Özcan
c,
Doris
Segets
cd,
Ulf-Peter
Apfel
*ab and
Daniel
Siegmund
*ab
aFraunhofer Institute for Environmental, Safety and Energy Technology, UMSICHT, Oberhausen, Germany. E-mail: daniel.siegmund@umsicht.fraunhofer.de
bInorganic Chemistry, Faculty of Chemistry and Biochemistry, Ruhr University Bochum, Bochum, Germany. E-mail: ulf.apfel@rub.defor
cInstitute for Energy and Materials Processes – Particle Science and Technology (EMPI-PST), University of Duisburg-Essen, Duisburg, Germany
dCenter for Nanointegration Duisburg-Essen (CENIDE), University of Duisburg-Essen, Duisburg, Germany
First published on 29th March 2023
Abstract
Electrifying the chemical industry is a key step towards the generation of green and sustainable chemical products. Electrochemical hydrogenations have gained a central role in this effort, generating important synthons in compact and modular zero-gap electrolyzers. Despite the significant achievements in recent years, clear guidelines to improve the efficiency and selectivity of zero-gap electrolyzers are still missing. Herein, by means of the hydrogenation of 2-methyl-3-butyn-2-ol (MBY), we present a holistic investigation for understanding the role of the anode porous transport layer and employed membrane on the efficiency of a zero-gap electrolyzer. Alongside, we provide a series of optimization steps, demonstrating that ECH electrolyzers can efficiently operate electrochemical hydrogenations at IrO2 loadings below 1 mg cm−2. We believe our results provide a crucial step towards accelerating the establishment of electrochemical methods in the chemical industry.
Sustainability spotlightElectrification of the chemical industry requires electrochemical cells tailored to the desired reactions. Despite decades of work in fuel-cell and electrolytic systems, clear guidelines regarding industrially relevant electrosynthetic reactors are still missing. Aiming to establish such protocols and to accelerate the adoption of electrosynthesis, we set out to develop optimal cell assemblies for electrochemical hydrogenations. We herein pivot towards revealing the role of the anode materials, their processing and configuration on the cathodic electrochemical hydrogenation. We show that electrosynthesis can be performed using only one-tenth of the required noble-metal loading compared to H2-producing cells. Our work clearly aligns with the Sustainable Development Goals 7 (affordable and clean energy) and 12 (responsible consumption and production) set by the UN. |
Introduction
The increasingly obvious consequences of climate change triggered a great political and societal push towards the development of more sustainable and greener technologies to fuel the modern world. Such sustainable alternatives include e.g., the establishment of a hydrogen-economy as well as CO2-recycling technologies. Going beyond small synthons, recent focus has also been laid upon electro-organic syntheses, aiming to create a more sustainable chemical industry powered directly by renewables.1,2Among the different areas that electro-organic reactions encompass, the field of electrochemical hydrogenation (ECH) is particularly attractive due to significantly milder and safer reaction conditions and avoidance of gaseous hydrogen compared to the current thermocatalytic state-of-the-art solutions. Inspired by previous protocols as well as reactor designs in the fields of fuel cells, hydrogen generation and CO2 reduction, the field of ECH has demonstrated significant potential for the chemical transformation of a palette of functional groups in scalable cell designs and elevated current densities.3–8
Despite the obvious potential, significant hurdles decelerating the broader application of ECH reactions at the industrial level still remain.9 The mentioned inspiration sources primarily originate from reactor designs involving gaseous/liquid reactions and interfaces, not fully addressing the challenging environment of ECH reactions. Notably, although ECH reactions involve electrochemical transformation at interfaces between organics and aqueous solvents, the material and cell designs applied are usually not tailored to such conditions. Specifically, since most industrially relevant substrates are non-water soluble, the required use of organic solvents often augments failure-modes, such as swelling and cracking of membranes. These effects thus not only decrease the energy efficiency of the electrolyzer but can lead to an increased crossover of organic material and ultimately cell failure.10
Although significant work continues to advance the ECH field in terms of catalyst and electrode design at an accelerated rate, evident knowledge gaps regarding the transition of efficient electrolyzers from the lab-scale to an industrially relevant alternative, able to reliably replace the current thermocatalytic state-of-the-art, still persist.2,11,12 Focusing on zero-gap electrolyzers, in which the ion-exchange membrane is pressed between the two electrodes to form the so-called membrane electrode assembly (MEA), as a highly energy-efficient and scalable cell design, we identified two key points playing a fundamental role in ECH optimization. These are the choice of the anode porous transport layer and the configuration of the MEA.13–15
Regarding the former, in the case of zero-gap electrolyzers and oxygen evolution (OER) as anodic reaction, the anode porous transport layer (PTL) takes multiple roles. It must ensure the conduction of current and the adequate transport of water to the catalytic layer, simultaneously removing generated gaseous products.14,16 The porous structure, electrical conductivity, and compressibility of the anode PTL furthermore influence the performance of the overall electrolyzer. Further factors to be considered for the choice of the material for the anode PTL are costs and stability under demanding reaction conditions and elevated current densities. Hence, titanium PTLs are among the most employed materials in the case of industrially relevant electrolyzers.17 As an important note, while some of the most active ECH electrolyzers often employ anion-exchange membranes and cheap and robust anodes made from Ni-foams, such systems are not easily transferable to a larger pool of organic reactants, which often require acidic conditions within the electrolyzer.18 This limitation is often either associated with the employed product not being able to withstand alkaline conditions, or with the decreased stability of the current anion-exchange membranes against organic media.19
Regarding the MEA configuration, in principle, two main approaches for the application of the catalyst are known, namely the catalyst-coated substrate (CCS) with the catalytic layer being deposited directly on the electrode and the catalyst-coated membrane (CCM), in which the catalyst is deposited on the ion-exchange membrane.20,21 The effect of this configuration for different membrane types, such as cation- and anion-exchange membranes (CEMs and AEMs) in the case of zero-gap cells has been a topic of decades-long research in the fuel cell and H2-electrolysis communities. However, only recently such differences have also been explored in the case of CO2 electroreduction.22 Although the CCS approach is characterized by a higher degree of robustness, able to stabilize the catalytic layer more efficiently,20,21 overall, CCM is the preferred approach, since it allows for a more efficient catalyst utilization and improved ionic conductivity to the catalytic layer.
Overall, this short overview of key points discussed in neighbouring fields of research underlines our main target question: are current state-of-the-art electrolyzer concepts and materials transferable to other electro-synthetic applications, in which the electrolyte environments between the two half-cells can be vastly different?
Aiming to fill this important knowledge gap in the current literature, we herein set out to investigate both the anode PTL for acidic and alkaline environments and the effect of the MEA configuration. Thereby we employed a series of anode PTLs with different pore structures and compressibility factors and tested both cation- and anion-exchange membranes (CEMs and AEMs). Herein, we demonstrate the significant effect of the anode configuration on the efficiency of cathodic organic reactions in electrolyzers. Employing the hydrogenation of the vitamin synthon 2-methyl-3-butyn-2-ol, MBY, as our ground-laying reaction, we present a series of optimized cell assemblies and approaches, reaching FE values of up to 50% at some of the lowest cell voltages reported for electro-organic transformations. Furthermore, our investigation demonstrates that cathodic electro-organic reactions can efficiently be operated with an anodic IrO2 loading below 1 mg cm−2. This is important as it enables dramatically lower acquisition costs of the overall electrolyzer, hence moving closer towards industrial applicability.
Results and discussion
All experiments were performed in our previously reported in-house built zero-gap electrolyzer. As a catalyst for the hydrogenation of 2-methyl-3-butyn-2-ol (MBY), we selected Fe3Ni6S8, a robust and cheap pentlandite catalyst that possesses high selectivity towards the generation of 2-methyl-3-buten-2-ol (MBE) at high current densities.23 For the performance of the oxygen evolution reaction (OER) at the anode, we selected IrO2, a state-of-the-art catalyst, at a loading of 1.5 mg cm−2, aiming to minimize possible bottlenecks due to catalytic limitations.10,24 A Nafion 115 and Fumasep FAA 3 PK 130 were used as cation-exchange (CEM) and anion-exchange (AEM) membranes, respectively, while electrolysis was performed at 80 mA cm−2 for 1 h. The generated products MBE and 2-methyl-3-butan-2-ol (MBA) were analysed as previously reported.23To understand the role of the anode PTL, we selected a broad spectrum of materials with different degrees of compressibility ranging from Ti sintered fibre felts with either 1 mm (Ti1 mm) or 0.15 mm (Ti0.15 mm) thickness, stainless steel mesh (SSM) and an H23 carbon paper (H23).15 The most promising cathode PTLs from our previous investigation were used as cathode supports, namely the carbon-fleece SGL-GFD (SGL) and the carbon paper H23.23 As a general convention, we refer to the different assemblies according to the following scheme: zero-gap electrolyzer (ZGE) type Anode MEA configuration-Membrane_Anode|Cathode. For example, a ZGE separated by an IrO2-loaded AEM membrane, employing H23 as the anode and SGL at the cathode support respectively, will be referred to as ZGE type CCM–AEM_H23|SGL.
Analyzing the different Faraday Efficiency (FE) values for the ECH (FEECH) between the two types of membranes, it is evident that AEMs clearly outperform CEM-separated cells in all of the tested configurations (Fig. 1A). We attribute this difference to the altering pH-environments governed by the two membrane types. The acidic environment that CEMs create via the conduction of protons limits the ECH, favouring the parasitic hydrogen evolution reaction (HER). In comparison, the conduction of generated hydroxides through an AEM appears to favour the hydrogenation of MBY. This finding is in accordance with literature results, in which the use of AEMs overall promoted the ECH.4,6 Furthermore, in the case of AEM-based cells, the selected materials and chosen MEA configuration appear to have only a small influence on the observed performance. Specifically, in the case of AEMs, the FEECH values for CCM-anodes lie in the range of 40–54%, while CCS-based anodes possess FEECH values between 48–60%. In contrast, CEM-separated cells demonstrate a significant response to both the MEA configuration and the support material. Notably, in the case of our CCM–CEM-separated electrolyzer, SGL-based cathodes employing anode materials with a higher degree of compressibility such as H23 and SSM anodes, leads to FEECH values of 35% and 40%, respectively. In contrast, the use of Ti-sintered fibre PTLs favours the ECH mostly in CCM configurations with CCM–CEM_Ti1 mm|SGL reaching a value of 40% for the ECH of MBY. Interestingly, the acidic environment associated with the use of CEMs does not lead to a significant over-hydrogenation of MBY to MBA, with the corresponding FE values being in the range of 2–10% depending on the chosen cell configuration. This finding indicates that the undesired over-hydrogenation is not limited by proton availability and suggests that the product desorption from the catalyst occurs instantly, i.e., not allowing for the second hydrogenation step. Then, the overall reduced hydrogenation efficiency at lower pH affects both hydrogenation steps, however does not influence the observed ratio between MBE and MBA.
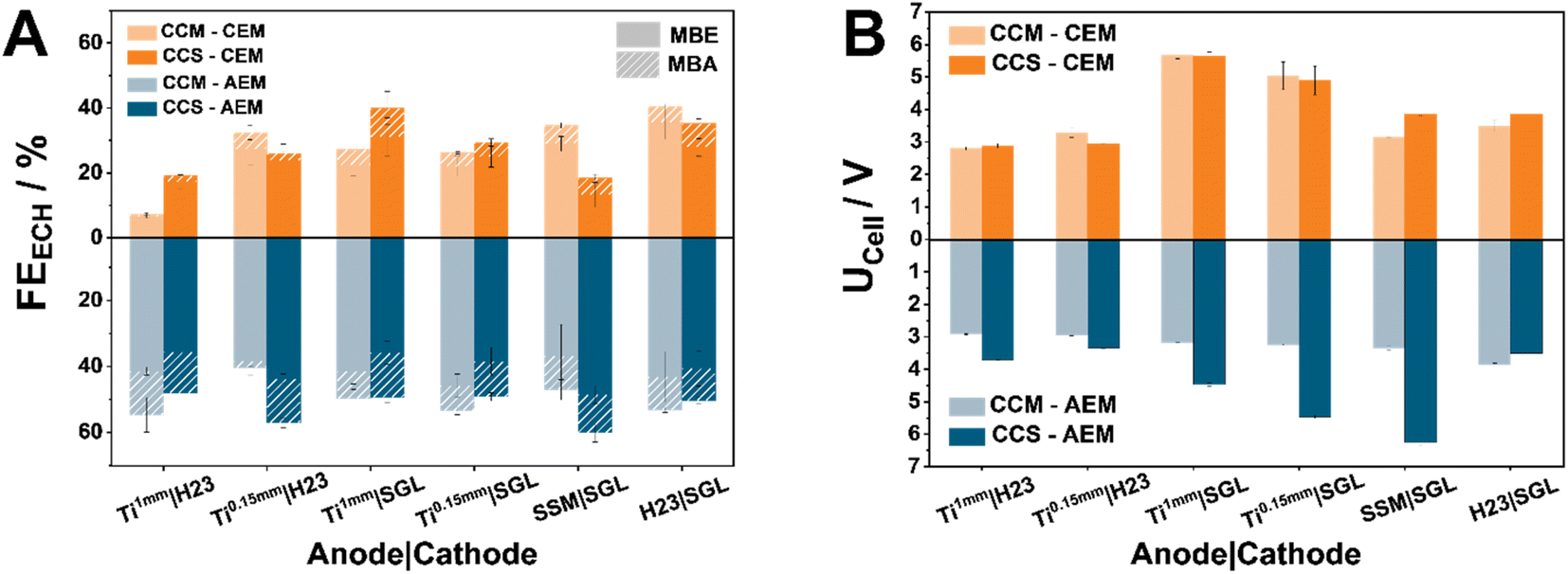 | ||
| Fig. 1 Investigation of the effect of the cell assembly and the deposition method on the MBY-ECH at 80 mA cm−2 (catholyte: 1 M MBY, 0.3 M KOH in H2O, anolyte: H2O). Comparison of the obtained FE (A) with and without hot-pressing and the respective cell voltages (B) after 1 h of electrolysis. Here the filled FE values denote the detected quantified FE for MBE, while the striped one the FE for MBA, detected in the catholyte (all results are also presented in tabular form in Table S1† to provide a detailed overview). | ||
Despite different cell assemblies demonstrating similar FEECH values, the observed trends are not mirrored in the cell voltage (Fig. 1B). Compared to their CCS-counterparts, apart from the CCM–AEM_H23|SGL assembly, CCM–AEMs demonstrate significantly lower cell voltages (UCell) ranging from 2.9 V in the case of Ti|H23 combinations to more than 3 V when SGL-based cathodes are used. Here the CCM–AEM_Ti0.15 mm|H23 demonstrates the lowest cell voltage of 2.9 V at 80 mA cm−2, while the CCS-AEM_Ti0.15 mm|SGL possesses the highest UCell value of 6.2 V. In the case of CEMs, the contrast between CCM and CCS-based MEAs is not as pronounced as for AEMs, which indicates that the contact between the anode catalyst layer and the membrane is the main origin for voltage losses in the latter case. The importance of the contact between anode and AEM could be explained by the higher local pH at the AEM surface which reduces the kinetic limitations of the water oxidation. In addition, the contact between AEM and cathode appears to be either less important or optimal independent of the used PTLs. Accordingly, the cell voltages for the CCM–AEM setups are comparable throughout all PTL combinations, while for CEMs it is mainly governed by the used PTLs, where apparently an optimized cell compression is crucial for low cell voltages. In the case of CEMs, the Ti|H23 combinations show the lowest cell voltages at 2.8 V and 3.2 V for Ti1 mm and Ti0.15 mm, respectively. Transitioning to SGL-cathodes, for both SSM and H23 as the anode material, CCM appears to be the more efficient MEA configuration with the respective UCell values lying at 3.1 V and 3.5 V, respectively. The most interesting exception in the described trends are clearly the Ti|SGL cell assemblies. Independently of the MEA configuration and Ti-thickness, both demonstrate the highest and similar UCell values at 5.6 V and 4.9 V for Ti1 mm and Ti0.15 mm, respectively.
While we so far demonstrated the stability of pentlandite for the ECH via experiments for 100 h at 160 mA cm−2, each newly developed MEA system must always undergo long-term electrolytic testing to gain important insights into the overall system stability. Therefore, we investigated the long-term performance of two exemplary MEAs; CCM–CEM_Ti1 mm|H23 and its AEM-based counterpart. While these cell assemblies show similar UCell values, their corresponding FEECH values are significantly different. Over 10 h of electrolysis at 80 mA cm−2, we can attribute the unaccounted FE to hydrogen formation as evidenced by online GC-quantification (Fig. S1†).
Prior and post electrolysis EIS analysis allows to better understand changes in the above-mentioned systems. Both membranes undergo electrochemical conditioning, with the high-frequency resistance (HFR), mainly attributed to resistances caused by the employed membrane, significantly decreasing after 10 h of electrolysis. Here, in both EIS measurement-points, the AEM-separated electrolyzer shows a higher HFR, lying at 2.25 Ω cm−2 against the respective value of the CEM-based system of 1.41 Ω cm2 after electrolysis. This difference could be the result of the added PEEK support within the polymer matrix since both membranes in their dry form possess similar thicknesses.
Regarding the cathodic reaction, the charge transfer resistance of the cathode is strongly dependent on the employed membrane and governing reaction. For the case of the CCM–CEM, for which the kinetically fast HER is largely the primary reduction reaction, the charge transfer resistance is almost half (1.1 Ω cm2) of the respective value (2 Ω cm2), obtained for the MBE-generating CCM–AEM. Despite the observed electrochemical decrease in HFR/membrane resistance, both tested electrolyzers showed an increasing cell voltage during electrolysis. Notably, the cell voltage of the Nafion 115 separated electrolyzer shows a significant voltage increase of 14.8 mV h−1, while its AEM-counterpart demonstrates a significantly lower increase of 3.2 mV h−1. This observed performance loss is possibly caused by swelling of the Nafion-containing components, i.e. the membrane itself as well as the Nafion-bound IrO2 catalytic layer. Similar behaviours have also been observed by Chen et al. during the ECH of biomass derivatives.25 Nevertheless, changes in the chemical environment of the electrolyte cannot be excluded, possibly suggesting that continuously operated single-pass systems could be more promising for ECH operations under elevated current densities. Furthermore, from an industrial standpoint both the obtained voltage decay and observed resistance values from our EIS analysis demonstrate the need of multi-componential improvements to reach competitive values with industrial zero-gap electrolyzers. Here, understanding, developing, and testing the role of different membranes, binders as well as electrolyte compositions under long-term experiments, coupled with tailored EIS protocols, will certainly facilitate this transition.26
Visualisation of the cell compression to understand cell voltages
Evidently, a variety of factors can influence the overall cell efficiency towards the ECH, starting from the assembly parameters to the intrinsic electrochemical processes at the different interfaces. Therefore, to decipher the ongoing mechanisms, we followed a three-pathway approach involving physical, electrochemical, and microscopic methods. Specifically, we employed pressure-sensitive paper to visualize the compression between the electrodes at the membrane. With the help of atomic force microscopy (AFM), we furthermore investigated the morphology of the IrO2 catalytic layer for CCM-membranes. Finally, we modified our electrolyzer set-up to measure the half-cell potential of the cathode during electrolysis (Fig. 2B) and thus quantified the influence of the anode on the electrochemical performance of the cathode.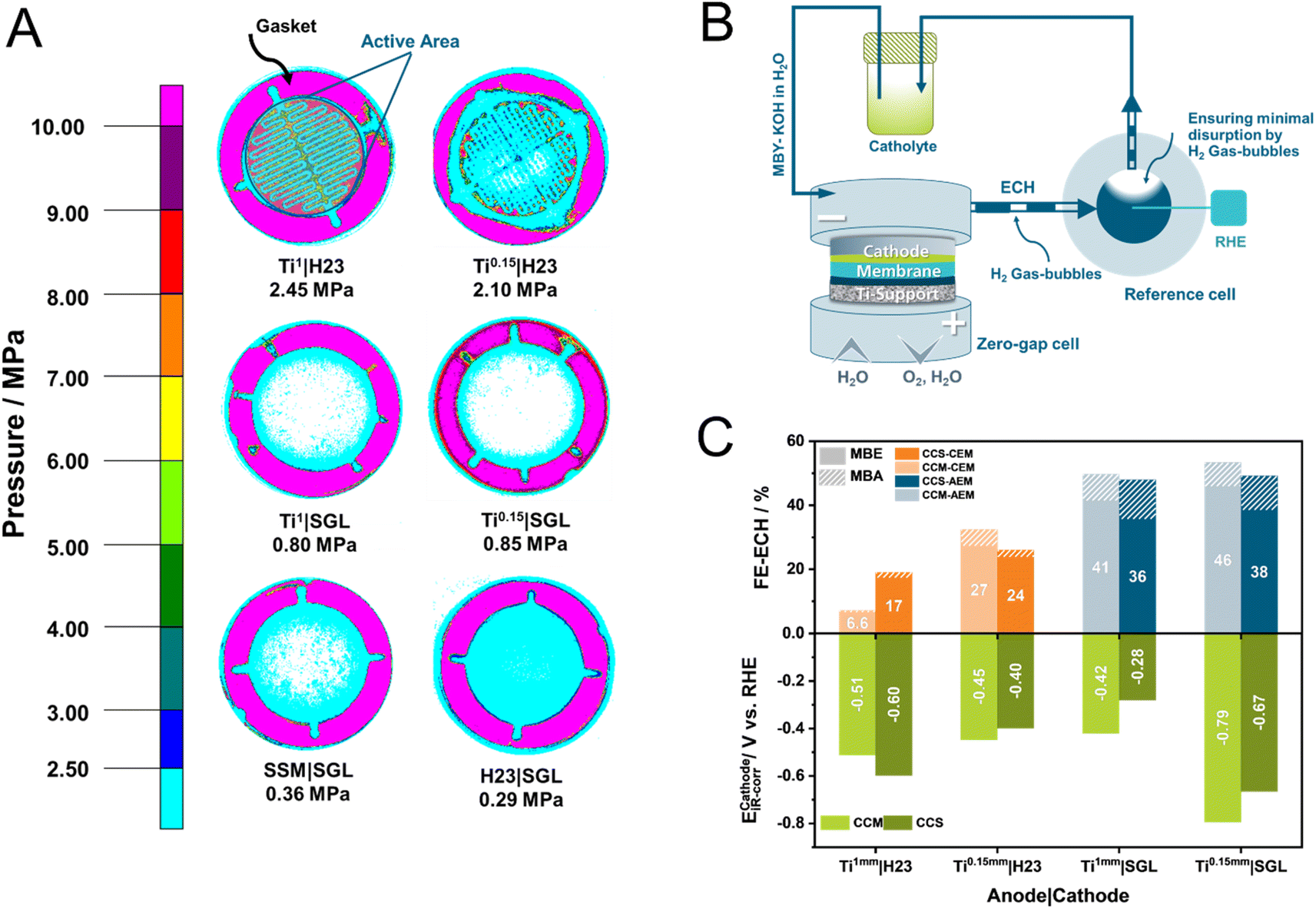 | ||
| Fig. 2 Visualization of the applied pressure within the employed 12.57 cm2 electrolyzer (A). Schematic representation of the modified set-up used to determine the half-cell voltages at the cathode (B). Investigation of the interplay between cathode half-cell potential and FE-ECH at 80 mA cm−2 (catholyte: 1 MBY, 0.3 M KOH in H2O, anolyte: H2O) (C). | ||
In a previous investigation, we showed that the use of pressure sensitive paper can be a powerful tool for every electrochemist working with zero-gap electrolyzers, allowing them to visualize and understand the pressure distribution within a cell in a highly cost-effective manner.31 Comparing the herein presented combinations of cathode and anode support (Fig. 2A), interesting trends become apparent. Notably, Ti|H23 combinations present overall the highest compression in the active area of the electrolyzer. The average applied pressure in our 40 mm active area increases with the thickness of the Ti-supports from 2.1 to 2.4 MPa for Ti0.15 mm to Ti1 mm, respectively. Surprisingly, switching from the H23 cathode to the SGL-one is accompanied by a major implication for the applied pressure. Visually, it is directly evident that in the case of the SGL-supports the applied pressure is significantly declined, lying below the previously reported optimal compression values for H2 and CO2 electrolyzers between 2–4 MPa.27,28 As an interesting side note, the compression between the two electrodes appears to become more homogeneous as the mechanical compressibility of the anode support increases from Ti1 to the carbon paper H23.
Connecting these results with the observed UCell trends, we can conclude that in the case of Ti|H23-based cells and for both tested cation and anion-exchange membranes, overall, some of the lowest cell voltages, could be demonstrated. This is attributed to the overall elevated average pressure between the electrodes. In the case of the SGL-cathodes, the correlation between the homogeneity of the applied pressure and the obtained UCell values is not directly evident. Although in the case of CEMs, the homogeneity of the applied pressure appears to play a significant role, with the UCell value decreasing from 5.8 V to 3.2 V when more compressible anode supports, such as SSM and H23 are employed, this is not mirrored for AEMs. Specifically for AEMs, the MEA configuration appears to heavily influence the obtained cell performance. We attribute this behaviour to the existence of the PEEK support for the used Fumasep membrane, decreasing the influence of the cell compression in the case of the AEM. To further understand this interplay between the MEA configuration and the applied pressure, we employed AFM. Interestingly, CCM–AEMs possess a significantly lower roughness factor compared to their CCM–CEM counterparts of 175 nm to 306 nm, respectively (Fig. S1†). Possibly, alongside the PEEK support, the more homogeneous IrO2 catalytic layer of CCM–AEMs allows for better electrical conductivity and ion transport within the anode compartment.
In contrast, for CCM–CEMs, a homogeneous compression appears to be necessary to optimize the current transport within the rougher catalytic layer, demonstrating how, via a combination of techniques, trends in ECH electrolyzers can not only be quickly understood but also rapidly improved.
Half-cell monitoring towards rapid catalytic screening
Evidently, the different components of a zero-gap ECH electrolyzer contribute in different manners to the observed cell selectivity and activity. Aiming to further explore the interesting trends beyond the overall cell assembly, we set out to measure the half-cell potential of the cathode and how this is influenced by the overall cell assembly (Fig. 2C). Here, we focused our investigation on cells consisting of Ti-supports, which demonstrated high FEECH values and cell voltages. Specifically, to study the behaviour of AEMs, we used SGL as the reference cathode and H23 for the CEMs under our standard conditions.Although half-cell potentials have been studied for ECH reactions, such studies have mainly focused on the electrochemical hydrogenation of furfural, performed in H-type electrolyzers at low current densities.11,29 By coupling a reference-hydrogen electrode to the outlet of our electrolyzer, we were – to the best of our knowledge for the first time – able to quantify the half-cell in the field of ECH within a zero-gap electrolyzer, notably for rather unexplored reactants such as alkynols (Fig. S2†). Here, the role of the anode-composition on the cathode becomes surprisingly evident. In the case of SGL-cathodes in AEM electrolyzers, our iR-corrected half-cell results demonstrate the significant effect the anode support can have on the performance of the cathode. Although both cell assemblies possess similar FEECH values, the use of Ti1 mm anode supports leads to the lowest half-cell potential at 280 mV for the CCS approach. In contrast, the use of the thinner Ti supports (0.15 mm) is associated with the highest ECathode values within the tested series with −790 mV and −670 mV vs. RHE for the CCM and CCS-AEM configurations, respectively. Interestingly, the inverse trend is obtained for CEMs, with thicker support leading to higher half-cell potentials at the cathode.
Moreover, by comparing the overall cell resistance against our half-cell measurements, we can show that for ECH electrolyzers the cathode plays a minimal role towards the total cell potential (Table S2†). Specifically, the lower cell compression associated with the use of a carbon-felt cathode limits the performance of the anode in CCS configurations. Such trends are in line with previous reports where the optimization of the anode architecture and the performance of the OER played a critical role not only on the UCell, but also on the obtained selectivity, and similar to studies reported for the electroreduction of CO2.28,30 Finally, by carefully observing the obtained half-cell values, a further trend arises. Comparing the different MEA configurations, more negative half-cell potentials are accompanied by higher FEECH and FEMBE values, for the same membranes and PTLs. We assume that changes in the local environment, e.g. the pH at the cathode, suppress the competing HER and with it shift the half-cell potential to more cathodic values, hence favoring the ECH. Specifically, to holistically understand the interplay of different components, operando analysis techniques as well as sophisticated simulation models involving not only the generation of organic products but also the generation of gaseous ones via the HER, alongside ion-conduction and crossover of organic material through the membrane are needed. This is however a challenging issue we aim to tackle in future investigations as more tailored and sophisticated systems are produced by us.
Improving the efficiency of zero-gap ECH electrolyzers
Our investigations aim not only at providing the community with helpful insights on the effect of the cell composition on the ECH, but also with possible improvement pathways. In the toolbox of every electrochemical engineer working with zero-gap electrolyzers, hot-pressing has been shown to be a useful technique to improve the overall cell efficiency. Herein, we envisioned that dimensional changes of the membrane could be minimized through hot-pressing, leading to better cell efficiency (Fig. 3). For this comparison, we selected Ti0.15 mm-based assemblies for both H23 and SGL. These showed some of the highest FEECH values, with optimization potential regarding the cell voltage.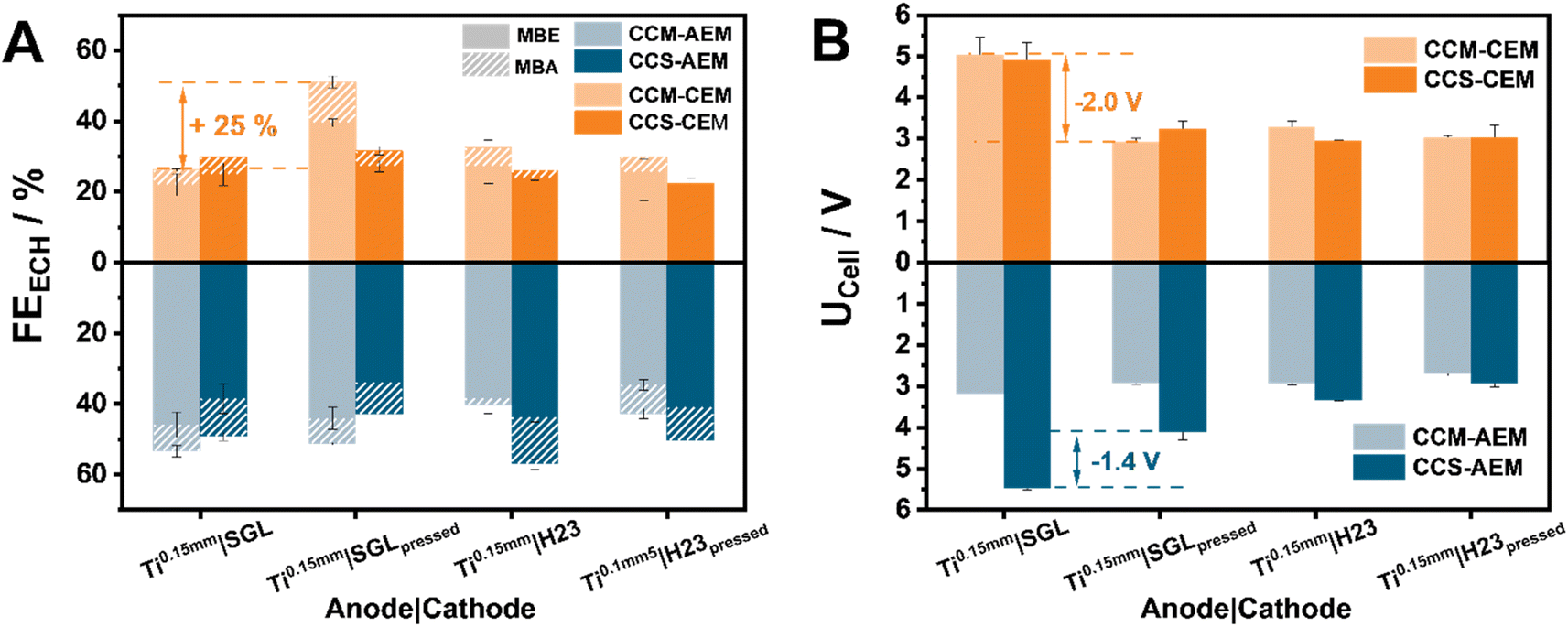 | ||
| Fig. 3 Investigation of the effect of hot-pressing the anode support on the ion-exchange membrane prior to electrolysis, MBY-ECH at 80 mA cm−2 (catholyte: 1 MBY, 0.3 M KOH in H2O, anolyte: H2O). Comparison of the obtained FE (A) with and without hot-pressing and the respective cell voltages (B) after 1 h of electrolysis. The filled FE values denote the detected quantified FE for MBE, while the striped one denotes the FE for MBA, detected in the catholyte. | ||
In the case of AEMs, hot-pressing appears to minimally influence the observed ECH behaviour, with the same observation being valid also for their CEM counterparts. The most striking change can be observed in the case of the CCM–CEM_Ti0.15 mm|SGL assembly. Effectively in this case, the FE towards the hydrogenation of MBY is doubled, from 21% and 4% for MBE and MBA to 39% and 11% for MBE and MBA, respectively, after hot-pressing. In addition to this improvement, the assembly demonstrates a significant cell voltage decrease of 2.0 V to a UCell of 2.9 V after hot-pressing. A similar improvement is also observed in the case of AEMs, with a decrease of 1.4 V being observed for the CCS-AEM_Ti0.15 mm|SGL cell assembly, however, without being accompanied by an increased current efficiency. Evidently, by pressing the membrane onto the titanium prior to cell assembly, the necessary compression and electrical conductivity between the Ti-PTL and the IrO2 layer can be guaranteed. Furthermore, this improvement could also be attributed to the minimization of dimensional changes in the tested membranes. Interestingly, in the case of the H23-based cathodes, the effect of the followed strategy is less pronounced, decreasing the obtained cell voltage by approximately 200 mV across the different membranes and anode supports. We attribute this behaviour to the degree of compression in this assembly which may already lie close to the maximal point that can be reached within our cell with hot-pressing playing only a minimal role in this case. Regarding the different effects of the hot pressing on the FE we conclude that the origin of the voltage decrease is different for AEM and CEM, similar to the different effects of the MEA configuration. The effect of hot-pressing in the case of AEMs is the same as using CCM instead of CCS, namely it ensures an optimized contact between the membrane and the catalyst layer. As described above, this effect is significantly less pronounced in the case of CEMs. In this case, the voltage losses are related to the PTLs and the cell compression, that can also influence the FE on the cathode side as described previously.
Furthermore, via the addition of conductive salts in the anolyte in these hot-pressed variants, we were able to decrease the required cell voltage to 2.2 V and 3.0 V for the assemblies CCM–AEM_Ti0.15 mm|H23 and CCM–CEM_Ti1 mm|SGL, respectively. Effectively, these demonstrate some of the lowest cell voltage values reported for electro-organic reactions, while maintaining FEECH values of at least 50% at 80 mA cm−2 (Fig. S3 & Table S3†).3,5,7,31
Decreasing IrO2-loadings, increasing the sustainability of ECH electrolyzers
Increasing the energy efficiency of an ECH electrolyzer is possibly only part of the equation towards reliably establishing electrochemical hydrogenation on the industrial scale. Another important factor is the cost of the electrolyzer. Similarly to the case of H2-producing and CO2-reducing electrolyzers, the use of IrO2 as the anode catalyst has been shown to be the major cost factor, with current research heavily focusing on lowering the required catalytic loading below 1 mg cm−2. Therefore, to address this critical issue, we performed a loading variation of IrO2 at 80, 160, and 240 mA cm−2, to understand the advantages and limitations of electro-synthetic electrolyzers against types of electrolyzers (Fig. 4). Here, we employed the most active cell assembly of CCM–AEM_Ti0.15 mm|H23 combined with a supporting electrolyte of 2 M KOH flowing through the anode.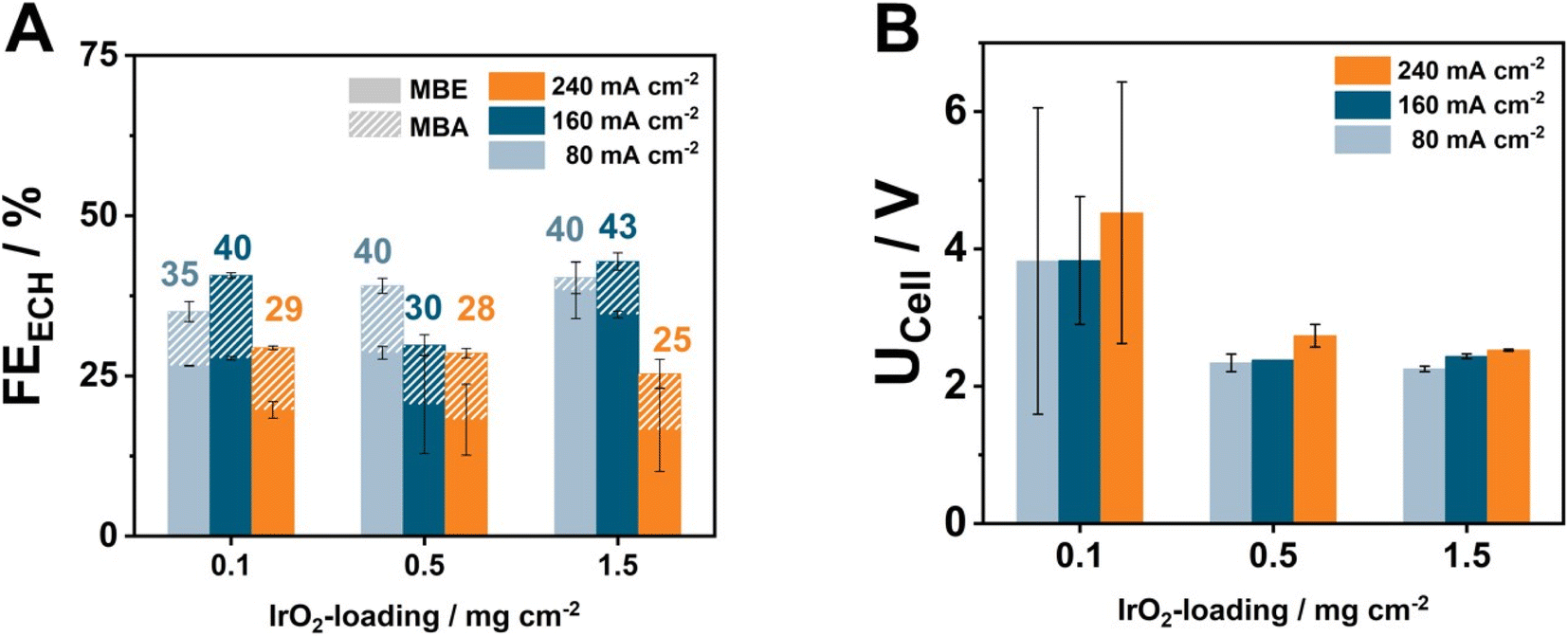 | ||
| Fig. 4 Investigation of the effect of IrO2 loading on the CCM–AEM_Ti0.15 mm|H23 on the MBY-ECH at three different current densities (catholyte: 1 MBY, 0.3 M KOH in H2O, anolyte: 2 M KOH). Comparison of the obtained FE (A) and the respective cell voltages (B) after 1 h of electrolysis. Here the filled FE values denote the detected quantified FE for MBE, while the striped one represents the FE for MBA, detected in the catholyte. | ||
At the lowest tested current density of 80 mA cm−2, anodes containing 0.5 and 1.5 mg cm−2 IrO2 possess similar FEECH values of 40%, followed by the lowest loading of 0.1 mg cm−2 at 35%. Simultaneously, at the highest current density of 240 mA cm−2, the FEECH values decrease below 30% for all the tested IrO2 loadings. This observation possibly hints at mass-transport limitations hindering the performance of the ECH under these current densities.32 Surprisingly, the IrO2 loading appears to also have a direct effect on the MBE![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MBA ratio, with lower loading leading to higher FEMBA increasing from 2% to 8% and 10% at 80 mA cm−2.
:MBA ratio, with lower loading leading to higher FEMBA increasing from 2% to 8% and 10% at 80 mA cm−2.
Regarding the observed cell voltage, minimal differences of 90–200 mV exist between IrO2 loadings of 0.5 and 1.5 mg cm−2, with the UCell values at 240 mA cm−2 being 2.7 V and 2.6 V, respectively. This difference could be attributed to a slightly lower electrical conductivity between the catalytic particles, with our AFM investigations showing that AEMs with 0.5 mg cm−2 IrO2 possess a significantly higher roughness factor of 317 nm against 175 nm in the case of loadings of 1.5 mg cm−2. Notably, cells containing only 0.1 mg cm−2 evidently break this trend. Here, the obtained UCell values lie in the range of 3.5–4.5 V, and increase with current density. Moreover, despite our best efforts, the large error bars reflect the difficulty of reaching optimal current conduction to and between the catalytic particles at such low loadings.33 Although our results already address the necessity of lowering the loading with noble metal at the anode, the future use of automated coating systems as well as of conductive filler particles could be two promising approaches to further lower the required loadings, accompanied by improved cell voltages.24,34
As an interesting note, since the generated products of ECH-electrolyzers have a higher market value compared to target products from water and CO2 electrolysis, it may also be viable to operate such electrolyzers under lower electrolytic loads, possibly allowing for the use of more cost-effective and sustainable anode catalysts.35 Nevertheless, we believe that the value of such techno-economic models would be further augmented by the development of continuously operating ECH-electrolyzers, as well as protocols for accelerated stress tests. These are currently the topic of future investigations.36
Conclusions
We herein demonstrate the significant influence of the anode, i.e., MEA configuration and anode support, not only on the overall efficiency of an ECH zero-gap electrolyzer but also on its selectivity towards the hydrogenation of alkynols. Furthermore, by providing a series of important tools to deconvolute the observed trends, such as the visualization of the pressure distribution and roughness of the catalytic layer, we illustrate a stepwise improvement of the electrolyzer efficiency. Via this multi-angle approach, we were able to reach FE values of 50% for the electrochemical hydrogenation at cell voltages of 2.2 V, demonstrating that ECH electrolyzers can reliably operate under significantly lower noble-metal loadings at the anode compared to the state of the art. In addition, through the use of our operando half-cell investigation, we are able to provide another electrochemical tool towards further understanding the mechanism of alkynol hydrogenation under different pH conditions by systematic studies.29,37 At the same time, through such an approach and a larger reactant pool, this technique could provide insights of predicting catalytic trends for the FEECH from the half-cell potential of the cathode, prior to quantification, ultimately opening the pathway towards lab-scale automation for ECH investigations under standardized cell assemblies, generated by coherent workflows. Evidently, the anode matters, though since component interplay characterizes electrochemical systems, the employed binder, membrane and electrode and catalyst architecture must be carefully and simultaneously tailored to the target substrate. These results further underline the economic and sustainable character of the herein presented concept. We believe that our investigation not only provides researchers with significant optimization options for their individual cells and substrates but also lays the groundwork for a broader series of investigations ranging from operando measurements to automation and large-scale techno-economic considerations.Author contributions
Conceptualization: K. P., J. W., K. J. P., D. Se., U.-P. A., and D. Si.; methodology: K. P., J. W., J. T. K., K. J. P., U.-P. A., and D. Si.; data curation: K. P., J. W., S. C. V., L. W., and M.-A. K.; formal analysis: K. P., L. W., J. W., M.-A. K., and J. T. K.; funding acquisition: U.-P. A., D. Se, and D. Si.; supervision: F. Ö., K. J. P., D. Se., U.-P. A., and D. Si.; writing-original draft preparation: K. P., J. W., L. W., J. T. K., M.-A. K., F. Ö. D. Se., U.-P. A., and D. Si. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
K. P. acknowledges the Fonds of the Chemical Industry for a PhD Fellowship, while the work of J. W. has been supported by a PhD Fellowship by the Studienstiftung des deutschen Volkes. The work of J. T. K. and U.-P. A. was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2033 – 390677874 – RESOLV, the Fraunhofer Internal Programs under Grant no. Attract 097-602175 and the Fraunhofer Lighthouse project ShaPID. D. Si. is grateful for the financial support by the BMBF within the NanoMatFutur Project ‘‘H2Organic’’ (No. 03XP0421). L. W., M-A. K., F. Ö., D. Se, and U.-P. A. are also thankful for support by the Mercator Research Center Ruhr (MERCUR.Exzellenz, ‘DIMENSION’ Ex-2021-0034 and ‘KataSign’ Ko-2021-0016).References
- (a) D. Pletcher, R. A. Green and R. C. D. Brown, Chem. Rev., 2018, 118, 4573 CrossRef CAS PubMed; (b) M. Garedew, F. Lin, B. Song, T. M. DeWinter, J. E. Jackson, C. M. Saffron, C. H. Lam and P. T. Anastas, ChemSusChem, 2020, 13, 4214 CrossRef CAS PubMed.
- C. H. Lam, W. Deng, L. Lang, X. Jin, X. Hu and Y. Wang, Energy Fuels, 2020, 34, 7915 CrossRef CAS.
- H. Liu, T.-H. Lee, Y. Chen, E. W. Cochran and W. Li, ChemElectroChem, 2021, 8, 2817 CrossRef CAS.
- L. A. Diaz, T. E. Lister, C. Rae and N. D. Wood, ACS Sustainable Chem. Eng., 2018, 6, 8458 CrossRef CAS.
- M. N. Dell'Anna, M. Laureano, H. Bateni, J. E. Matthiesen, L. Zaza, M. P. Zembrzuski, T. J. Paskach and J.-P. Tessonnier, Green Chem., 2021, 23, 6456 RSC.
- R. S. Delima, M. D. Stankovic, B. P. MacLeod, A. G. Fink, M. B. Rooney, A. Huang, R. P. Jansonius, D. J. Dvorak and C. P. Berlinguette, Energy Environ. Sci., 2022, 15, 215 RSC.
- T. Imada, M. Chiku, E. Higuchi and H. Inoue, ACS Catal., 2020, 10, 13718 CrossRef CAS.
- R. S. Sherbo, A. Kurimoto, C. M. Brown and C. P. Berlinguette, JACS, 2019, 141, 7815 CrossRef CAS.
- D. Siegmund, S. Metz, V. Peinecke, T. E. Warner, C. Cremers, A. Grevé, T. Smolinka, D. Segets and U.-P. Apfel, JACS Au, 2021, 1, 527 CrossRef CAS PubMed.
- H. Zhang and P. K. Shen, Chem. Rev., 2012, 112, 2780 CrossRef CAS PubMed.
- S. A. Akhade, N. Singh, O. Y. Gutiérrez, J. Lopez-Ruiz, H. Wang, J. D. Holladay, Y. Liu, A. Karkamkar, R. S. Weber, A. B. Padmaperuma, M.-S. Lee, G. A. Whyatt, M. Elliott, J. E. Holladay, J. L. Male, J. A. Lercher, R. Rousseau and V.-A. Glezakou, Chem. Rev., 2020, 120, 11370 CrossRef CAS PubMed.
- L. Zhang, T. U. Rao, J. Wang, D. Ren, S. Sirisommboonchai, C. Choi, H. Machida, Z. Huo and K. Norinaga, Fuel Process. Technol., 2022, 226, 107097 CrossRef CAS.
- B. Chen, P. Mardle and S. Holdcroft, J. Power Sources, 2022, 550, 232134 CrossRef CAS.
- A. Fallisch, L. Schellhase, J. Fresko, M. Zechmeister, M. Zedda, J. Ohlmann, L. Zielke, N. Paust and T. Smolinka, Int. J. Hydrogen Energy, 2017, 42, 13544 CrossRef CAS.
- T. Lickert, M. L. Kiermaier, K. Bromberger, J. Ghinaiya, S. Metz, A. Fallisch and T. Smolinka, Int. J. Hydrogen Energy, 2020, 45, 6047 CrossRef CAS.
- (a) A. T. Mayyas, M. F. Ruth, B. S. Pivovar, G. Bender and K. B. Wipke, Manufacturing Cost Analysis for Proton Exchange Membrane Water Electrolyzers, National Renewable Energy Lab. (NREL), Golden, CO (United States), 2019, NREL/TP-6A20-72740 CrossRef; (b) E. T. Ojong, J. T. H. Kwan, A. Nouri-Khorasani, A. Bonakdarpour, D. P. Wilkinson and T. Smolinka, Int. J. Hydrogen Energy, 2017, 42, 25831 CrossRef CAS.
- (a) H. Teuku, I. Alshami, J. Goh, M. S. Masdar and K. S. Loh, Int. J. Energy Res., 2021, 45, 20583 CrossRef CAS; (b) O. Schmidt, A. Gambhir, I. Staffell, A. Hawkes, J. Nelson and S. Few, Int. J. Hydrogen Energy, 2017, 42, 30470 CrossRef CAS; (c) B. Mayerhöfer, D. McLaughlin, T. Böhm, M. Hegelheimer, D. Seeberger and S. Thiele, ACS Appl. Energy Mater., 2020, 3, 9635 CrossRef.
- Y. P. Wijaya, K. J. Smith, C. S. Kim and E. L. Gyenge, Green Chem., 2020, 22, 7233 RSC.
- (a) D. A. Salvatore, C. M. Gabardo, A. Reyes, C. P. O'Brien, S. Holdcroft, P. Pintauro, B. Bahar, M. Hickner, C. Bae, D. Sinton, E. H. Sargent and C. P. Berlinguette, Nat. Energy, 2021, 6, 339 CrossRef CAS; (b) Z. Sun, J. Pan, J. Guo and F. Yan, Adv. Sci., 2018, 5, 1800065 CrossRef PubMed.
- H. A. Miller, K. Bouzek, J. Hnat, S. Loos, C. I. Bernäcker, T. Weißgärber, L. Röntzsch and J. Meier-Haack, Sustainable Energy Fuels, 2020, 4, 2114 RSC.
- R. Phillips and C. W. Dunnill, RSC Adv., 2016, 6, 100643 RSC.
- K. U. Hansen, L. H. Cherniack and F. Jiao, ACS Energy Lett., 2022, 7, 4504 CrossRef CAS.
- K. Pellumbi, L. Wickert, J. T. Kleinhaus, J. Wolf, A. Leonard, D. Tetzlaff, R. Goy, J. A. Medlock, K. Junge Puring, R. Cao, D. Siegmund and U.-P. Apfel, Chem. Sci., 2022, 13, 12461 RSC.
- J. E. Park, S. Kim, O.-H. Kim, C.-Y. Ahn, M.-J. Kim, S. Y. Kang, T. I. Jeon, J.-G. Shim, D. W. Lee, J. H. Lee, Y.-H. Cho and Y.-E. Sung, Nano Energy, 2019, 58, 158 CrossRef CAS.
- W. Chen, G. He, F. Ge, W. Xiao, J. Benziger and X. Wu, ChemSusChem, 2015, 8, 288 CrossRef CAS PubMed.
- (a) D. E. Blanco, A. Z. Dookhith and M. A. Modestino, ACS Sustainable Chem. Eng., 2020, 8, 9027 CrossRef CAS; (b) D. E. Blanco, A. Z. Dookhith and M. A. Modestino, React. Chem. Eng., 2019, 4, 8 RSC; (c) K. Nagasawa, A. Kato, Y. Nishiki, Y. Matsumura, M. Atobe and S. Mitsushima, Electrochim. Acta, 2017, 246, 459 CrossRef CAS; (d) K. Takano, H. Tateno, Y. Matsumura, A. Fukazawa, T. Kashiwagi, K. Nakabayashi, K. Nagasawa, S. Mitsushima and M. Atobe, Bull. Chem. Soc. Jpn., 2016, 89, 1178 CrossRef CAS.
- S. H. Ahn, B.-S. Lee, I. Choi, S. J. Yoo, H.-J. Kim, E. Cho, D. Henkensmeier, S. W. Nam, S.-K. Kim and J. H. Jang, Appl. Catal., B, 2014, 154–155, 197 CrossRef CAS.
- L. Hoof, N. Thissen, K. Pellumbi, K. Junge Puring, D. Siegmund, A. K. Mechler and U.-P. Apfel, Cell Rep. Phys. Sci., 2022, 3, 100825 CrossRef CAS.
- L. Zhou, Y. Li, Y. Lu, S. Wang and Y. Zou, Chin. J. Catal., 2022, 43, 3142 CrossRef CAS.
- (a) T. Schuler, J. M. Ciccone, B. Krentscher, F. Marone, C. Peter, T. J. Schmidt and F. N. Büchi, Adv. Energy Mater., 2020, 10, 1903216 CrossRef CAS; (b) T. Schuler, T. Kimura, T. J. Schmidt and F. N. Büchi, Energy Environ. Sci., 2020, 13, 2153 RSC.
- J. Benziger and J. Nehlsen, Ind. Eng. Chem. Res., 2010, 49, 11052 CrossRef CAS.
- (a) C. Rozain, E. Mayousse, N. Guillet and P. Millet, Appl. Catal., B, 2016, 182, 123 CrossRef CAS; (b) T. L. Doan, H. E. Lee, M. Kim, W. C. Cho, H. S. Cho and T. Kim, J. Power Sources, 2022, 533, 231370 CrossRef CAS.
- U. Babic, E. Nilsson, A. Pătru, T. J. Schmidt and L. Gubler, J. Electrochem. Soc., 2019, 166, F214–F220 CrossRef CAS.
- (a) S. Siracusano, N. van Dijk, R. Backhouse, L. Merlo, V. Baglio and A. S. Aricò, Renewable Energy, 2018, 123, 52 CrossRef CAS; (b) V. K. Puthiyapura, S. Pasupathi, H. Su, X. Liu, B. Pollet and K. Scott, Int. J. Hydrogen Energy, 2014, 39, 1905 CrossRef CAS.
- (a) G. Li, H. Yu, W. Song, X. Wang, Y. Li, Z. Shao and B. Yi, Int. J. Hydrogen Energy, 2012, 37, 16786 CrossRef CAS; (b) T. Audichon, E. Mayousse, T. W. Napporn, C. Morais, C. Comminges and K. B. Kokoh, Electrochim. Acta, 2014, 132, 284 CrossRef CAS; (c) A. Loh, X. Li, O. O. Taiwo, F. Tariq, N. P. Brandon, P. Wang, K. Xu and B. Wang, Int. J. Hydrogen Energy, 2020, 45, 24232 CrossRef CAS.
- (a) M. J. Orella, Y. Román-Leshkov and F. R. Brushett, Curr. Opin. Chem. Eng., 2018, 20, 159 CrossRef; (b) M. J. Orella, S. M. Brown, M. E. Leonard, Y. Román-Leshkov and F. R. Brushett, Energy Technol., 2020, 8, 1900994 CrossRef.
- X. H. Chadderdon, D. J. Chadderdon, J. E. Matthiesen, Y. Qiu, J. M. Carraher, J.-P. Tessonnier and W. Li, JACS, 2017, 139, 14120 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00043e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |