
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Targeted recovery of metals from thermoelectric generators (TEGs) using chloride brines and ultrasound†
Guillaume
Zante
 *a,
Evangelia
Daskalopoulou
a,
Christopher E.
Elgar
a,
Rodolfo Marin
Rivera
a,
Jennifer M.
Hartley
a,
Kevin
Simpson
b,
Richard
Tuley
b,
Jeff
Kettle
c and
Andrew P.
Abbott
a
*a,
Evangelia
Daskalopoulou
a,
Christopher E.
Elgar
a,
Rodolfo Marin
Rivera
a,
Jennifer M.
Hartley
a,
Kevin
Simpson
b,
Richard
Tuley
b,
Jeff
Kettle
c and
Andrew P.
Abbott
a
aSchool of Chemistry, University of Leicester, Leicester, LE1 7RH, UK. E-mail: gz45@leicester.ac.uk
bEuropean Thermodynamics Ltd, Kibworth, LE8 0RX, UK
cJames Watt School of Engineering, University of Glasgow, Glasgow, G12 8QQ, UK
First published on 30th May 2023
Abstract
Recovery of elemental copper, bismuth, tellurium, antimony and tin from thermoelectric generators (TEGs) is vital to recover the high content of critical metals and potential risk of environmental pollution as a result of incorrect disposal of TEGs and to enable the circular economy. In this work, aqueous choline chloride and calcium chloride hexahydrate brines were characterised and used in combination with copper(II) as an oxidising agent to leach copper and tin from TEGs. This permitted the Bi2−xSbxTe3 legs to be readily separated from the ceramic substrates by filtration. It was shown that at low chloride content, surface passivation and solubility of the oxidised species were the limiting factors towards oxidation, whereas solvent viscosity (mass transport) was the limiting factor at high chloride content. The copper(II) species formed in the different brines were determined via UV-vis spectroscopy. The redox potentials of the oxidising species were found to be significantly altered by choline chloride content, but not so much by calcium chloride hexahydrate content, suggesting variation in chloride activity within the different brines. The developed approach has been shown to be a viable and scalable method to recover high value critical metals from e-waste containing TEGs.
Sustainability spotlightThermoelectric generators are sustainable and cost-effective devices that can convert waste heat into additional electrical power. The global demand for bismuth and tellurium is forecast to increase, hence the recycling of old devices is becoming more important. The sustainable advancement of the present work is the selective recovery of bismuth tellurides from scrap thermoelectric devices via the use of copper(II) as a mild oxidising agent in aqueous chloride brines. This work falls under Goal 12 “Responsible consumption and production”, as it describes a method for enabling quick and clean recovery of the critical semiconductor elements, which in turn could reduce the current reliance on natural resources to supply demand for these devices. |
1 Introduction
Energy efficiency is a vital topic and it has been estimated that about 75% of the energy produced worldwide dissipates as heat into the environment.1 Thermoelectric generators (TEGs, also called Seebeck generators) are sustainable and cost-effective, although inefficient, devices that have demonstrated promising results to convert waste heat into additional electrical power.2 TEGs are widely used in many applications, such as power generators – especially in automotives, thermal energy sensors, for cooling in electronics and photonics, and for medical treatment, among others,3 resulting in increased global demand for the key elements.4–7 Interest in TEGs has grown rapidly in recent years in order to harvest energy for IoT devices such as sensors and actuators.8 TEGs have a simple design, no moving parts, a long lifespan and require low maintenance enabling their application in numerous situations. The most common modules are based of bismuth telluride (Bi2Te3), doped with antimony to produce either a p-type ((Sb0.8Bi0.2)2Te3) or n-type (Bi2(Te0.8Se0.2)3) materials.9 The scarcity of these elements has limited the development of Bi2Te3 based TEGs.10,11 Alternative materials, such as skutterudites, Ca/Mg oxides, and Mg2Si, along with the discovery of new materials such as Heusler compounds (magnetic intermetallic compounds), has allowed the development of potentially more cost-effective TEGs,12 although such devices have not yet proven their commercial success.Bi is listed as Critical Raw Material by the EU in 2020 owing to its scarcity and supply chain risks, whilst Te and Se are amongst the least abundant elements on the Earth's crust.13 All three tend to be sourced as by-products of other metal processing,5,14,15 and are therefore vulnerable to supply chain risks. Hence, improved recovery of both bismuth, tellurium and antimony from end-of-life TEG devices has an increasing importance.
Current recycling processes for TEGs are relatively under studied. Mechanical cutting and milling of the thermoelectric materials has been used to liberate different fractions, which can then be size separated.16 However, processes reported so far result in dissolution of the TEGs or reduction of their size, therefore re-processing is required to remanufacture the TEG from the obtained recycled materials. Some hydrometallurgical methods for recycling TEGs have been reported in the literature, such as non-selective leaching with hydrochloric, nitric, or sulphuric acids, followed by the use of hydroxide and hydrazine to precipitate the bismuth and tellurium powders, or directly regenerate new forms of bismuth telluride.17–19 The bacterial recovery of tellurium in both solid and gaseous forms has also been attempted.20 These approaches require further processing of the leaching solution to recover the metals.
An alternative method for recovery of the thermoelectric legs from waste TEGs would be to selectively leach out the base metal layers that adhere the thermoelectric materials to the ceramic substrates. Solvents such as deep eutectic solvents (DESs) and concentrated chloride brines have the advantage of high chloride contents, which leads to different speciation and redox behaviour, hence allowing to selectively dissolve target metals. This approach has been applied successfully to the selective leaching of metals from minerals, dissolution of metal oxides from end-of-life batteries, leaching of silver and aluminium from solar cells and copper from circuit boards, among others.21–24 In addition to DESs, chloride brines have previously been used as solvents for the leaching of metals from minerals,25–28 and the leaching of silver and aluminium from solar panels,24 using a number of different oxidising agents, including CuII and FeIII.
In this work, choline chloride (ChCl) and calcium chloride hexahydrate (CaCl2·6H2O) brines have been applied for selective dissolution of copper from thermoelectric devices using copper(II) chloride as the oxidising agent to liberate the Bi2−xSbxTe3 legs from the ceramic substrate. Copper(II) was selected as the oxidising agent to minimise impurity contamination because the CuII/I redox potential is less anodic relative to the target metals, resulting in more noble metals remaining unaffected. This allows for easy recovery of the individual thermoelectric legs via simple filtration. Both materials used in this process are abundantly available; CaCl2·6H2O is a waste by-product from the Solvay process,29 and ChCl is a bulk commodity chemical, which has no additional REACH requirements. The use of these materials enables a rapidly scalable recycling process. The copper(II) species present in solution are characterised along with their redox properties as a way to understand the relative oxidising ability towards the target elements. Chloride concentration (and activity) will likely be different between the organic and inorganic chloride sources, affecting the soluble metal species formed. This method was compared with high-intensity ultrasonication, which allows efficient removal of the brittle Bi2−xSbxTe3 legs from the ceramic substrate.
2 Materials and methods
2.1 Reagents and solvent preparation
The solvents used in this study are prepared by dissolving choline chloride (ChCl, Sigma Aldrich, >98%) or calcium chloride hexahydrate (CaCl2·6H2O, Honeywell, >97%) in deionised water (Elga Purelab Option apparatus) at varying molar ratios of salt-to-water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, 1:4, 1:10, and 1:50). The mixtures were stirred at 50 °C until a clear and homogeneous liquid was obtained.
:3, 1:4, 1:10, and 1:50). The mixtures were stirred at 50 °C until a clear and homogeneous liquid was obtained.
The leaching solutions were prepared by dissolving 0.1 mol dm−3 anhydrous copper(II) chloride (Acros Organics, 99%) in the appropriate solvent. The solutions for voltammetric analysis were prepared by dissolving 0.02 mol dm−3 of anhydrous copper(II) chloride (Alfa Aesar, 99.9%), tin(II) chloride (Alfa Aesar, 98%), tellurium(IV) chloride (Sigma Aldrich, 99%), antimony(III) chloride (Sigma Aldrich, 99%) and bismuth(III) chloride (Alfa Aesar, 98%) in the different brines. The UV-vis spectra were recorded using the same solutions. The elements investigated have a concentration higher than 1 wt% in the thermoelectric materials.
2.2 Leaching studies
Metal leaching was carried out by placing ca. 30 mg of a Cu foil, or Sn wire, Sb lump, Te or Bi chunk in 5 mL of the appropriate solvent containing the oxidising agent (0.1 mol dm−3). Without an oxidising agent, the solvent alone is unable to dissolve the metals (no leaching was observed for any of the metals). The system was stirred (200 rpm) at 50 °C for 1 hour, before the remaining solid metal was removed, rinsed with deionised water, air-dried, and weighed to determine the mass of metal dissolved. Table 1 shows the salt-to-water ratio of the brines selected together with their theoretical water and chloride concentration. Variation in the water content of brines (due to purity of the chemical sources, losses during heating…) is unlikely to affect the chemistry of the solutes since brines already contain a large amount of water. The values are compared with DESs using ethylene glycol as an HBD, which provide a similar chloride content.| Brine/molar ratio | Chloride conc./mol kg−1 | Water conc./mol kg−1 | Viscosity/mPa s |
|---|---|---|---|
| a Data taken from D'Agostino et al.30 b Data taken from Hartley et al.31 | |||
| CaCl 2 ·6H 2 O brines | |||
| CaCl2·6H2O:3H2O |
7.3 | 33.0 | 7.72(8) |
| CaCl2·6H2O:4H2O |
6.9 | 34.4 | 6.11(5) |
| CaCl2·6H2O:10H2O |
5.0 | 40.1 | 2.58(3) |
| CaCl2·6H2O:50H2O |
1.8 | 50.0 | 1.26(7) |
|
|||
| ChCl brines | |||
| ChCl:3H2O |
5.2 | 15.5 | 14.08(6) |
| ChCl:4H2O |
4.7 | 18.9 | 8.0(1) |
| ChCl:10H2O |
3.1 | 31.3 | 4.56(2) |
| ChCl:50H2O |
1.0 | 48.1 | 1.327(5) |
|
|||
| Deep eutectic solvents | |||
| ChCl:2EGa |
3.8 | 7.6 (EG) | 37 |
| CaCl2·6H2O:2EGb |
5.8 | 23.3 (EG + H2O) | 59.48(8) |
Thermoelectric materials were provided by European Thermodynamics Ltd, with a schematic of their structure shown in Fig. S1.† They are composed of two layers of Al2O3 ceramic wafer coated with a Cu conductor strip of 8 mm2 size with 380 μm thickness. Between the Cu strip and the ceramic, an 8 mm2 sized and 3 μm thick W interface layer is used to complete the thermal junction and attach to the underlying solder. A 300 μm thick layer of solder containing mostly Sn, but also Zn coats the Cu strips. Finally, two 1 mm3 thermoelectric legs of bismuth–antimony telluride or bismuth telluride doped with selenium (hereafter referred to as Bi2−xSbxTe3) are sandwiched between these solder layers to complete the device. Overall, the ceramic plate represents around 40 wt% of the thermoelectric materials, while soldering and junction elements other than Sn represent less than 1 wt% of the materials. The composition of the thermoelectric materials, obtained after aqua regia (a 3:1 volume ratio of hydrochloric and nitric acid) digestion of 100 mg of materials in 10 mL and analysis of the solution with ICP-MS is displayed in Table 2. The composition reported is in accordance with the composition of European thermodynamics' products.
| Metal | Composition, wt% |
|---|---|
| Cu | 21.3 |
| Te | 18.6 |
| Bi | 13.9 |
| Sb | 3.5 |
| Sn | 3.5 |
| Ni | 0.9 |
| W | 0.8 |
| Se | 0.5 |
| Others (ceramic plate, Zn) | 37.0 |
During leaching experiments, a sample of thermoelectric material (around 1 × 0.5 cm) was immersed in different leaching solutions containing 0.1 mol dm−3 of Cu(II) chloride as an oxidising agent (the liquid to solid ratio is maintained at 20 mL g−1). The solution was then ultrasonicated at room temperature for 30 minutes with a Fisherbrand FB15055 ultrasonic bath delivering an ultrasonic frequency of 37 kHz, a power supply of 150 W, and a power density of around 0.05 W cm−3. The sample was then removed from the solution, rinsed with deionised water, air-dried, and weighed. The metal content in the solution is estimated by ICP-MS after filtration with 0.2 μm PET syringe filters. The Bi2−xSbxTe3 legs are filtered from the solution with filter paper, rinsed with deionised water, air-dried, and weighed.
High intensity ultrasound experiments were carried out using a commercial ultrasonic system (Branson Sonics, 1.25DCXa20-V), using a 20 mm diameter cylinder sonotrode. The system operates at 20 kHz, with power variable up to 1250 W, and a maximum power intensity of 398 W cm2. The two ceramic plates were manually separated first, allowing for a closer distance between the sonotrode and the thermoelectric material. During high intensity ultrasound experiments, a sample of thermoelectric material (around 1 × 1.5 cm) was submerged under deionised water (20 mL) and placed under the sonotrode at a distance of 1 mm. Ultrasound was applied to the surface at 50% power intensity (198 W cm−2) for 15 seconds. The resultant solids were collected via filtration, and rinsed with deionised water.
2.5 Instrumentation
The viscosity values of the different brines were measured at 25 °C using a Seiko EG&G QCM922A Quartz Crystal Microbalance (QCM), according to previously described procedures (Hartley et al., 2022). Solvent densities were estimated by weighing 100 mL of the solvent heated at 25 °C in a 100 mL volumetric flask, using a 4 d.p. balance (Mettler Toledo). Conductivity values were recorded using a commercial chemical-resistant type conductivity probe (SC72SN-31-AA; Yokogawa) and metre (SC72 Personal Conductivity Meter; Yokogawa), with an integrated temperature sensor (accuracy within ± 0.7 °C). The cell constant for the commercial conductivity probe was 5.2 ± 0.5 cm−1, and conductivity standards (certified traceable to NIST; VWR) of 44479, 11419, 8863, 1249, 885, 442, and 74 μS cm−1 (at 19 °C) were used as received to perform calibrations. The reported conductivity values are an average of at least three repeat measurements.
The metal content and Bi:Sb ratio of the thermoelectric devices was obtained via ICP-MS with a Thermo Scientific iCAP-Qc apparatus. Digestion of a sample containing two of the Cu strip-BiTe legs was done in aqua regia. These solutions were filtered with 0.2 μm PET syringe filters before being diluted (1000 fold) with a 2% trace metal grade nitric acid (Fisher Scientific, 70%) solution. Lanthanum and rhenium are used as internal standard (10 μg dm−3 of each). Concentrations were obtained from calibration curves between 0.1 and 1 μg dm−3 for Te, 10 and 500 μg dm−3 for Sb and Bi, and 25 and 5000 μg dm−3 for Cu.
Absorption spectra were recorded with a Mettler Toledo UV5 Bio spectrometer, using quartz cuvettes with a path length of 10 mm or 0.1 mm, depending on the strength of the absorptions. To determine the composition of the materials, Scanning Electron Microscopy (SEM) and Energy Dispersive X-ray (EDX) images were obtained on a Field Emission Gun Scanning Electron Microscopy (Quanta 650 FEG-SEM apparatus) with an accelerating voltage of 20 kV.
The electrochemical behaviour of the target metals was studied by cyclic voltammetry (CV), using an IVIUMnSTAT potentiostat. A 0.5 mm diameter platinum disk was used as the working electrode, with a platinum flag counter electrode, and a 3 mol dm−3 KCl Ag/AgCl reference electrode. Prior to each experiment, the working electrode was polished with a 0.3 μm and 0.05 μm alumina slurry, rinsed with deionised water, and air dried. The reference electrode potential was calibrated to the [Fe(CN)6]3−/4− redox couple, which is used as an internal standard to ensure comparability between the different solvents. The scan rate was 20 mV s−1.
3 Results and discussion
The first section of this study investigates the electrochemical properties of the constituent elements in a TEG and compared these with the leaching solutions used in two brines, one with a high charge density (Ca2+) and one with a lower charge density (choline). The second half of the study investigated the application of ultrasound to recycling of TEG devices.3.1 Speciation of Cu(II) and the thermoelectric leg elements in the brines
In order to understand the ability of the selected oxidising agent to oxidise other metallic species, identification of metal speciation in the brines is essential. The speciation of CuII in the different brines is discussed in the ESI.† The absorption spectra of Cu(II) chloride in ChCl and CaCl2·6H2O brines with salt-to-water ratios of 1:50; 1:10; 1:4 and 1:3 are shown in Fig. S2(a) and (b),† and photographs of these systems are shown in Fig. S2(c).† The absorbance maxima for these spectra and predicted species are presented in Table S1,† while the plot of the molar extinction coefficient at 402 nm as a function of water content in the brines is displayed in Fig. S3.† These spectra indicate the presence of [CuCl4]2− in the most concentrated choline chloride brines ([Cl−] ≥ 4.7 mol kg−1), mixed chloride–water species at lower chloride concentrations ([CuCl(H2O)n]+ and [CuCl3(H2O)n]−) while no chloride species are present at the lowest chloride concentration ([CuCl(H2O)n]+ and [Cu(H2O)n]2+). In the CaCl2·6H2O brines, mixed chloride–aqua complexes are found at different chloride concentrations.
Absorption spectroscopy of the Bi(III), Te(IV), Sb(III) and Sn(II) chloride salts dissolved in different brines was used to determine the species present (Fig. S4,† absorbance maxima and proposed species are displayed in Table 3). In all eight of the different brines, BiIII ions were present as chloride species, as shown by absorbance maxima present at ca. 210, 225, and 330 nm.32–34 There is a minor solvatochromic shift between the ChCl and CaCl2·6H2O brines (ca. 3 nm), which is much smaller than was seen for Cu(II) ions. Literature suggests the presence of [BiCl6]3− in ChCl:2EG, although multiple species were not ruled out.35 In concentrated acidic brines (11 mol per kg chloride), EXAFS spectroscopy indicated an average coordination of 5–6 chloride ligands, whereas at low chloride content (1 mol kg−1) an average of ca. 3–4 chloride ligands were coordinated, probably in the form of a mixture36 of [BiCl2]+ and [BiCl5]2−. Therefore, the most likely species present in these ChCl and CaCl2·6H2O brines will be a mixture of 5–6 chloride coordination, except for the ChCl brine with a chloride content of 1 mol kg−1, where a lower coordination number is expected.
| Solvent/chloride content | Absorbances/nm | Predicted species |
|---|---|---|
| Bi | ||
| CaCl2·6H2O brines | ||
| [Cl−] ≥ 1.8 mol kg−1 | 210, 222, 328 | [BiCl5]2−/[BiCl6]3− |
| ChCl brines | ||
| [Cl−] ≥ 3.1 mol kg−1 | 211, 224, 329 | [BiCl5]2−/[BiCl6]3− |
| [Cl−] = 1.0 mol kg−1 | 209, 222, 324 | [BiCl2]+ and [BiCl5]2− |
|
||
| Sb | ||
| CaCl2·6H2O brines | ||
| [Cl−] ≥ 6.9 mol kg−1 | 229, 264, 289 | [SbCl4]− |
| [Cl−] = 5.0 mol kg−1 | 219, 263, 280 | [SbCl2]+ and [SbCl3]0 |
| ChCl brines | ||
| [Cl−] ≥ 4.7 mol kg−1 | 226, 241, 266, 285 | [SbCl4]− |
|
||
| Sn | ||
| CaCl2·6H2O brines | ||
| [Cl−] ≥ 1.8 mol kg−1 | 220 | [SnCl4]2− |
| ChCl brines | ||
| [Cl−] ≥ 1.0 mol kg−1 | 222 | [SnCl4]2− |
The spectra for TeCl4 dissolved in the different brines show a broad absorbance maximum below 200 nm, hence it is not possible to determine the exact species present via this technique. However, it is known that Te(IV) ions can form mixed ligand complexes, depending on chloride content. For example, in highly concentrated aqueous brines (13–14 mol per kg chloride) there is the possibility for [TeCl4]− or [TeO0.3Cl3.6]n to form, where the oxygen may be from oxide, hydroxide, or water. However, in low chloride systems (<5 mol kg−1) the species are fully oxygen-coordinated. In ChCl:2EG a solution of TeCl4 displays absorbance maxima at 248 and 289 nm,32 which is potentially related to [TeCl6]2−. Hence, the most likely species present in the ChCl and CaCl2·6H2O brines will be mixed ligand complexes.
The spectra for Sb(III) are more complex, as multiple absorbance maxima are present. In the ChCl brines there are four maxima present in the region from 200 to 350 nm. These are less distinct in the CaCl2·6H2O brines. UV-vis spectra of [SbCl4]− in acetonitrile show two main absorbance regions at ca. 230–260 nm and ca. 280 nm, whereas the spectrum of [SbCl6]3− displays absorbance maxima at ca. 240–270 nm and ca. 310 nm.34 It is therefore most likely that in the brines with a ChCl concentration of 4.7 mol kg−1 and higher presented here, that the most likely species is [SbCl4]−. In the CaCl2·6H2O brine with a chloride content of 5.0 mol kg−1, another species is present, perhaps relating to a mixture of [SbCl2]+ and [SbCl3]0, as observed in acidic chloride brines containing ca. 3.5 mol kg per chloride.37 For the CaCl2·6H2O and ChCl brines with the lowest chloride content, Sb was found to precipitate instantaneously, possibly as insoluble antimony oxychloride species (SbOCl), since antimony is known to hydrolyse quickly when diluted in water.38 Therefore, Sb behaviour in ChCl brine having a chloride content lower than 4.7 mol kg−1 as well as CaCl2·6H2O brine having a chloride content of 1.8 mol kg−1 was not investigated further.
The absorbance maxima for the Sn(II) complexes vary from 222 nm in the highest chloride concentration systems, down to 209 nm in the least concentrated systems. Sn(II) is known to form [SnCl3]− and [SnCl4]2− complexes in aqueous media, even at relatively low chloride contents (0.35 mol dm−3 HCl39). In DES media, [SnCl4]2− complexes are formed.40 Therefore, it is likely that the majority species in these chloride brines will be [SnCl4]2−, with the shift in absorbance maxima values being related to the change in ionic strength and solvation environment of the different solvents. There is minimal solvatochromic shift between the ChCl and CaCl2·6H2O brines.
3.2 Electrochemistry and leaching
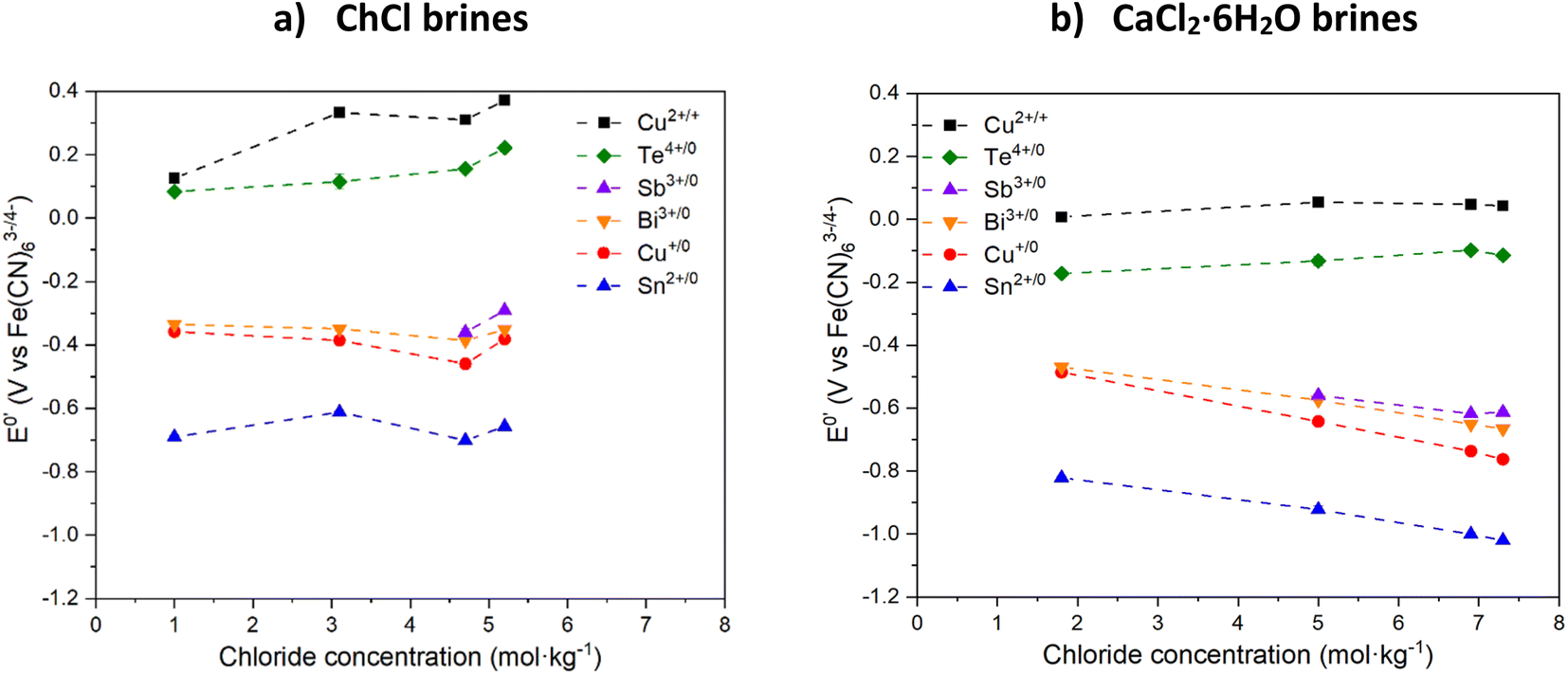 | ||
| Fig. 1 Formal electrode potentials of the different redox couples in (a) ChCl, and (b) CaCl2·6H2O brines, as a function of solvent chloride content. Values corrected for small changes in concentration and temperature using the Nernst equation, and referenced to the [Fe(CN)6]3−/4− couple. | ||
The formal electrode potential of the CuII/I couple was measured in the different brines and it was found that the presence of different Cu species resulted in a cathodic shift between most and least concentrated chloride systems of up to 0.3 V in the ChCl brines, and of up to 0.05 V in the CaCl2·6H2O brines. Increasing the chloride content causes the formal electrode potential to increase. This means that the CuII species present in the systems with the highest water contents are less oxidising relative to the [Fe(CN)6]3−/4− couple compared to those in the highest chloride content systems. This effect can be used to tune the oxidising strength of CuII towards other metals, assuming that the formal electrode potentials of the target metals are not altered adversely by the changing solvent composition. Interestingly, the redox potential of the CuII/I couple is consistently more than 250 mV more cathodic in CaCl2·6H2O brines compared to ChCl brines having the same salt-to-water ratios, regardless of the actual chloride content. It should be noted that the redox potential of the CuII/I couple in ChCl:2EG is of a similar value to that of the ChCl brines containing >3 mol per kg chloride.
The CVs of Bi chloride in the various brines show two redox events, the first being related to the Bi3+/0 couple (at around −0.6 V vs. [Fe(CN)6]3−/4− in CaCl2·6H2O brines and around −0.4 V vs. [Fe(CN)6]3−/4− in ChCl brines) while the other is related to the formation of platinum–bismuth alloys,41 and the subsequent oxidation of Bi from the alloy at more anodic potentials.42 With increasing chloride concentration, the Bi3+/0 redox couple becomes more cathodic, i.e. the Bi3+ ion is stabilised relative to elemental Bi, meaning that Bi metal should be easier to oxidise. This effect is more significant in the CaCl2·6H2O brines, probably because of the higher chloride concentration. The Sb3+/0 couple is found on Sb CVs at around −0.6 V vs. [Fe(CN)6]3−/4− in CaCl2·6H2O brines and around −0.4 V vs. [Fe(CN)6]3−/4− in ChCl brines, while the Sb5+/3+ couple is visible in some CVs for potentials higher than +0.4 V43vs. [Fe(CN)6]3−/4−. Similarly to the Bi3+/0 couple, the Sb3+/0 couple seems to be more cathodic when the CaCl2·6H2O content of the brine is increased. However, due to the low solubility of Sb in the least concentrated chloride brines, the lack of data makes the trend difficult to analyse.
The CV of Te in the brines show one couple at around −0.2 V vs. [Fe(CN)6]3−/4− in CaCl2·6H2O brines and around +0.1 V vs. [Fe(CN)6]3−/4− in ChCl brines which could be attributed to the TeIV/0 couple, while the second couple appearing on some CVs may be related to the presence of the Te0/2− couple.44 Contrarily to Bi and Sb behaviour, the addition of chloride in the brines causes an anodic shift in the TeIV/0 couple, meaning that increasing chloride content increases the formal redox potential and that elemental Te becomes harder to oxidise in the brines with the highest chloride content.
The SnII/0 and CuI/0 couples are visible in ChCl brines at around −0.5 V and −0.4 V, respectively, all vs. [Fe(CN)6]3−/4−. In CaCl2·6H2O brines, the same redox couples are present at around −0.9 V, and −0.6 V, respectively. For these three couples, increasing the CaCl2·6H2O content causes a cathodic shift of the redox potential and makes their oxidation easier. The effect of increasing chloride content is much higher when using CaCl2·6H2O as compared to ChCl, probably due to the higher chloride content and a higher impact on speciation.
In all cases, the Ecell values for oxidation of the target elements by CuII are positive, i.e. ΔG = −nFEcell is negative, indicating that the reaction will be thermodynamically viable regardless of the brine selected. For all the metals investigated except Te, Ecell is increasing with increasing CaCl2·6H2O content since it causes a slight anodic shift of the redox potential of the CuI/0 couple while a cathodic shift is observed for the other metals, except Te. Increasing ChCl content has a much lower effect, with Ecell values remaining approximately constant along the series for all elements, except Te.
| nCuII + M →nCuI + Mn+ | (1) |
Metallic Cu is seen to be dissolved in all systems, but the leaching efficiency does was not shown to improve as a function of chloride concentration (Fig. 2a and b). The greatest amounts of metal leached were obtained in the solutions containing the highest water contents (ca. 0.45 mmol of Cu dissolved over the course of an hour), rather than the highest chloride contents (ca. 0.15 to 0.25 mmol of Cu dissolved). In the brines with lower chloride content, white Cu(I) chloride precipitates were observed that can hinder further leaching. This indicates that the stability of the CuI species in solution is less important than the increase in solvent viscosity and corresponding decrease in mass transport. The solder metal, Sn, is dissolved at a similar rate to Cu, as it is highly reactive and therefore easily forms stable solution species.
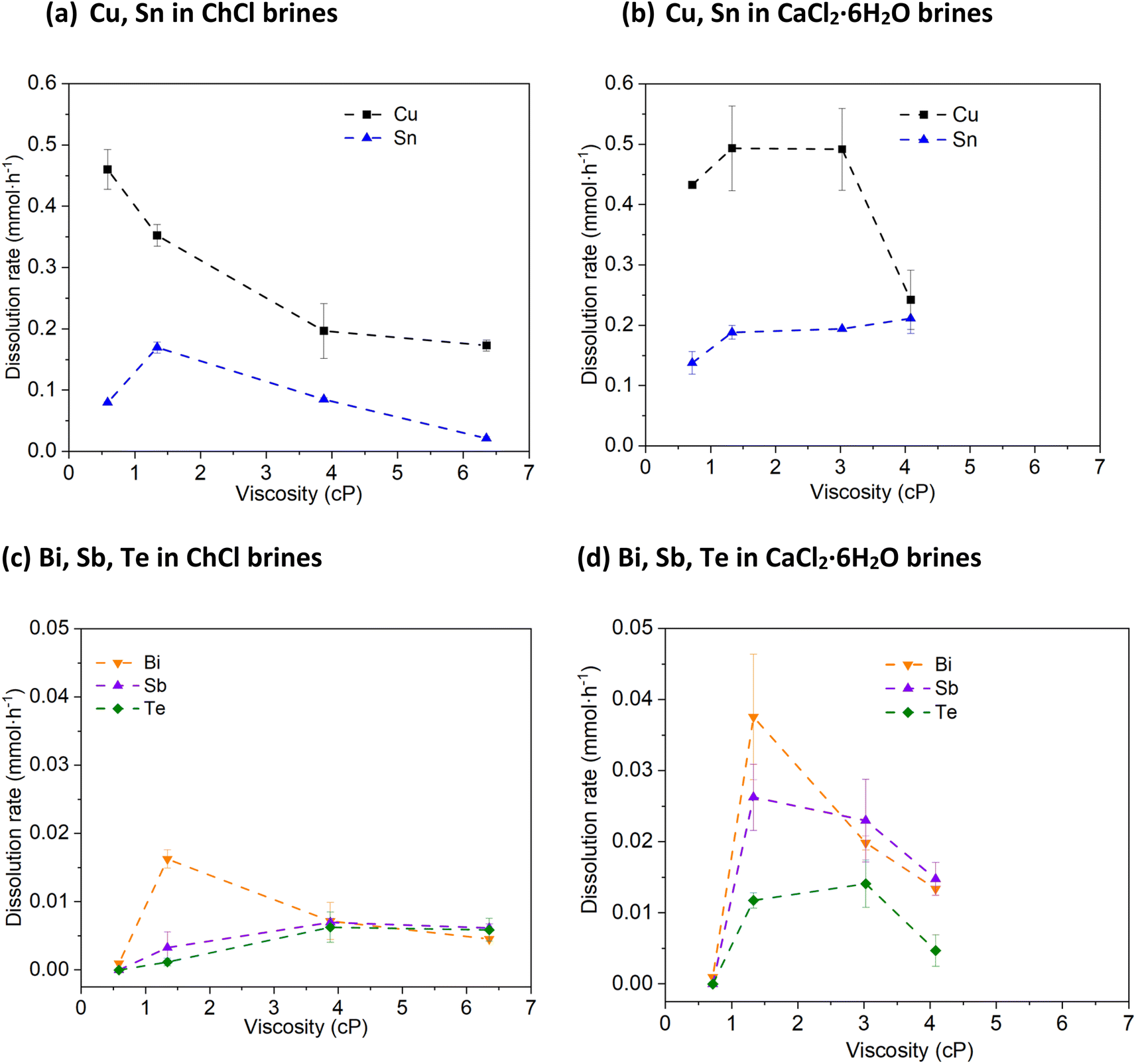 | ||
| Fig. 2 Rate of metal dissolution using 0.1 mol dm−3 of Cu(II) chloride as an oxidising agent for: (a) Cu, Sn in ChCl brines, (b) Cu, Sn in CaCl2·6H2O brines, (c) Bi, Sb, Te in ChCl brines, and (d) Bi, Sb, Te in CaCl2·6H2O brines as a function of solvent viscosity at 50 °C. Temperature = 50 °C, stirring rate = 200 rpm. | ||
Bi, however, is much more resistant to being leached by CuII, despite the relative redox potentials indicating that this process is thermodynamically favourable. Fig. 2c and d shows that when the chloride content is less than 3 mol kg−1, no dissolution occurs, and similarly to Cu, a decrease in dissolution rate is seen in the systems with the highest chloride contents, also being the most viscous liquids. However, even in the system which worked most favourably towards Bi, only ca. 0.04 mmol is dissolved – a factor of 10 smaller than the amount for Cu. The oxidation of Sb follows a similar profile to Bi with respect to chloride content, with maximum leaching of ca. 0.03 mmol. The leaching of Te with CuII is poor in all systems investigated here, with a maximum of only ca. 0.015 mmol Te oxidised within one hour. Overall, the trend of higher solubility relative to chloride content is maintained, and can be explained by the oxidised species formed; at low chloride content, Bi, Sb and Te ions can form oxide and oxychloride species, which may be passivating. Given that the oxidation of these elements is a 3- or 4-electron process, mass transport is likely to be more critical than for a 1-electron oxidation process. While these poor leaching efficiencies towards the semiconductor elements may appear less than desirable in a recycling process, thermoelectric devices such as the one described in the Experimental section have a layer of Cu that attaches the Bi2−xSbxTe3 to the ceramic substrates.
Hypothetically, by selectively leaching the Cu away, the Bi2−xSbxTe3 legs are released from the ceramic substrate and can be recovered by size grading (as the ceramic substrates are much larger). The solvent will then only contain a mixture of Cu(I) and Cu(II) ions, which can then be regenerated to all being Cu(II) via reaction with atmospheric oxygen, or by electrolytic means. The Bi2−xSbxTe3 legs can then be simply rinsed with deionised water to remove any solvent and Cu ions, before further processing. Therefore, an optimal process would ensure maximum Cu and solder leaching efficiency, while minimising solubility of the other elements is the ChCl and CaCl2·6H2O brines with a chloride concentration of 1.0 and 1.8 mol kg−1, respectively.
3.3 Recovery of Bi2−xSbxTe3 from thermoelectric devices
To minimise the number of processing steps required to separate the individual elements from each other, it is advantageous to selectively remove the Bi2−xSbxTe3 legs by leaching the Cu tabs that ensure the structural integrity of the thermoelectric materials. It was determined that the most optimal solvent to ensure selectivity towards Cu was the ChCl and CaCl2·6H2O brines with the lowest chloride content (1.0 and 1.8 mol kg−1, respectively). The two ceramic plates were manually separated from each other to ensure a more efficient removal process, and immersed in the solvents containing 0.1 mol dm−3 CuCl2 as an oxidising agent. Ultrasonication was preferred over mechanical stirring, as it allowed removal of the Bi2−xSbxTe3 legs within 30 minutes (with mechanical stirring at 200 rpm and room temperature, it took more than an hour to obtain the same result). After 20 minutes of ultrasonication at room temperature, the Bi2−xSbxTe3 legs became separated from the substrate, revealing the underlying Cu layer, partially contaminated with a thin layer of Bi2−xSbxTe3 particles (Fig. 3b). The legs remain undissolved and can be easily recovered from the solution via filtration, as shown in Fig. 3c, then re-used for the manufacture of new thermoelectric materials, assuming their quality has not been affected by the leaching process. The Cu plates can also be detached from the ceramic substrate and recovered by filtration, as evidenced in Fig. 3c, despite some selective leaching. This could be related to the removal of any solder between the Cu plates and the ceramic substrate, or that the Cu layer has been undercut. Overall, all of the Bi2−xSbxTe3 legs are removed from the Cu plate in about 30 minutes. After three repeated contacts with fresh Cu chloride brines, Cu is completely dissolved or removed from the ceramic substrate, as evidenced in Fig. S7.† | ||
| Fig. 3 Photographs of thermoelectrics samples before and after 20 minutes of leaching with 0.1 mol dm−3 Cu(II) chloride in (a) ChCl brine (1.0 mol per kg chloride); (b) CaCl2·6H2O brine (1.8 mol per kg chloride); (c) photograph of the Bi2−xSbxTe3 legs and Cu plates recovered from the solution and washed. | ||
Whilst the ultrasonication at room temperature successfully remove the Bi2−xSbxTe3 legs, the time taken was 20 min. To increase the separation, alternative approaches were sought to speed up the Bi2−xSbxTe3 using mechanical separation of both the ceramic substrate and Cu plates without the need for additional chemicals via the use of a high intensity ultrasound, as opposed to an ultrasonic bath. As a result of the higher and better directed power density of the ultrasound (198 W cm−2) the Bi2−xSbxTe3 legs rapidly broke apart as the sample passed under the tip of the sonotrode. After processing, the solution was filtered to collect the fragmented Bi2−xSbxTe3. The recovered ceramic plate retained a thin layer of Bi2−xSbxTe3 attached to the solder on the surface of the Cu tabs (Fig. 4a), indicating that the recovery percentage of the semiconductor materials (>80%) was less than obtained after chemical leaching (>95%). Bi2-xSbxTe3 was recovered as a powder with a wide range of particle sizes (from 1 mm3 to 10 μm3), as seen in Fig. 4b. As no material was dissolved during this process, the different products will be easier to separate from one another, while also minimising the amount of waste solvent requiring remediation.
 | ||
| Fig. 4 (a) Photographs of thermoelectric samples before and after 15 seconds of ultrasonication in water. (b) Photograph of the recovered Bi2−xSbxTe3 powder. | ||
Overall, it is clear that the lower volumes of chemicals, types of chemicals and processing time provides a clear economic benefit over the use of chemical oxidising agents in chloride brines. Further work is clearly needed to establish whether it is preferential to recover material in this manner; whereby TEG material that is recovered in a variety of sizes and needs to be re-processed into thermoelectric legs, or whether the recovery of fully intact thermoelectric legs is a better approach to create a recycled TEG. This would be highly dependent upon the condition and especially, the degradation, of TEG devices when recovered at end-of-life. There are likely to be multiple regulatory and quality control issues in order to directly reuse thermoelectric legs with both manners. With both techniques, the thermoelectric legs are removed, thus mixing the n-type (which contain Bi, Se and Te) and the p-type (which contain Bi, Sb and Te) legs. Due to the different composition of the legs, further refining is likely needed. Both processes are therefore useful to separate the legs from the initial thermoelectric materials, but require additional steps for either purifying Bi, Sb, Te and Se or remanufacturing p- and n-type thermoelectric legs. High intensity ultrasonication does not involve chemical reactions and is probably more appropriate to avoid chemical modification of the legs (formation of passivation layers…) which could change their properties.
3.4 Additional concerns
Dissolution of the solder is feasible using different chemicals such as hydrochloric acid (Fig. S8†). However, thermoelectric legs are still attached to the copper electrode after removal of the solder. To achieve thermoelectric leg removal, (at least partial) dissolution of the copper electrode is needed. An alternative method to achieve copper dissolution, strong oxidising agents like nitric acid solutions can be used, but it also achieves complete dissolution of the thermoelectric leg, which would require further separation and precipitation of Bi, Sb and Te.When using copper chloride as a mild oxidising agent, the low amount of nickel (10 mg kg−1), Te (<2 mg kg−1), Bi (<1 mg kg−1), selenium (<1 mg kg−1), Sb (<0.1 mg kg−1), Sn (ca. 160 mg kg−1) and Zn (ca. 10 mg kg−1) present in the leaching solution after use makes the recovery of the dissolved elements economically unviable. However, after a currently unknown number of cycles, the leaching solution will become enriched in these elements or precipitation of insoluble species may occur. Recovery methods could include selective electrowinning or precipitation, as seen for Zn in DESs by the addition of ammonia solution.47
A potential solution to make the process semi-continuous would be to use a leaching vessel equipped with a sieve, as depicted in Fig. S9.† Here, the thermoelectric materials are placed in a leaching solution containing a sieve that has apertures of no more than 2 mm diameter. This sieve will retain the large ceramic parts (average length 1 cm) while letting the Bi2−xSbxTe3 legs (average size 1 mm3) pass through, enabling simple gravimetric separation of the components. The used leaching solution could be tapped-off, and regenerated by either using oxygen from air or via electrolytic means. After prolonged use of thermoelectric materials, a thin layer of copper oxide could be formed at the surface of the copper electrode, which can partially protect the bulk copper from further oxidation and also reduce the efficiency of the dissolution process. However, copper oxides could be dissolved with acetic acid48 or DESs based on organic acids.49
Both processes allow the removal of thermoelectric legs without using strong acids and facilitate further processing by separating in one step the legs from the ceramics. Clearly the removal of such acids, with a benign non-acid based method is desirable as it removed the need to handle and dispose of hazardous materials.
With low power ultrasounds, copper oxidation is needed, which requires the use of an oxidising agent, which will require wastewater treatment (although the oxidising agent can be regenerated and the dissolution of elements outside of Cu, Zn and Sn is minimal). In our conditions, the process takes around 20 minutes and requires an electricity consumption of 0.05 kW h per piece of thermoelectric material. With high power ultrasounds, water is used as a propagation media for ultrasound waves. The physical effect of ultrasounds is sufficient to pulverise the brittle thermoelectric legs. This process does not produce wastewater as no elements are dissolved in water. The process is fast (15 seconds) and requires an electricity consumption of around 0.003 kW h. The high-power intensity ultrasonication process looks a more promising processing route considering that it is faster, consumes less electricity and does not produce wastewater. However, it does reduce the thermoelectric legs into pieces of different sizes which may be harder to post-process. Further studies are needed to determine how to remanufacture thermoelectric materials from (relatively) intact pieces or pulverised materials. This would also require further optimisation of the application of ultrasonication.
The two techniques proposed in this manuscript target the weaknesses at the interface between different material layers. The first method allows the dissolution of base metals such as Cu and Sn which have a low redox potential and are relatively easy to oxidise. This technique can potentially be applied to other electronic waste for the selective, acid-free, and low-cost dissolution of soldering elements. It has successfully been demonstrated for the recovery of gold flakes from waste printed circuit boards.22 The second technique takes advantage of the brittle nature of the thermoelectric legs to controllably “blast” the thermoelectric materials with high-power ultrasound. When compared to conventional crushing, it has significant advantages as the approach allows separation of two materials with different brittleness (thermoelectric legs and ceramic) by controllably reducing the size of the most brittle material. This technique is especially important for symbiosis in recycling e-waste when characteristically different brittle materials are present.
4 Conclusions
The electrochemical properties and speciation behaviour of Cu(II) chloride was investigated in ChCl and CaCl2·6H2O brines containing varying salt-to-water molar ratios. Increasing chloride content in the brines allows forming anionic chloride complexes which impact the electrochemical behaviour of Cu. Additionally, the speciation and electrochemical of the elements present in thermoelectric materials- (Sb, Bi, Te, Sn) relative to Cu was studied. It was found that chloride species were present for all the elements, except in the most aqueous systems. For all elements to be dissolved, the redox potentials were more cathodic than the oxidising agent, resulting in thermodynamically preferential reactions.These brines were used for the leaching of pure metals from TEGs, and it was found that in the least concentrated chloride solutions (1.0 mol kg−1 and 1.8 mol kg−1 chloride for ChCl and CaCl2·6H2O brines, respectively) showed the greatest selectivity towards Cu and the solder elements. This was determined to be due to passivation of the thermoelectric elements (Bi, Sb, and Te), most likely through the formation of insoluble precipitates. At high chloride contents, the relatively high viscosity of these systems hindered mass transport and decreased leaching rates. This means that for optimal selectivity, there is a balance to be maintained between chloride concentration and solvent properties.
Thermoelectric devices containing Bi2−xSbxTe3 were treated with the 1:50 CaCl2·6H2O and ChCl brines. It was found that the Cu and solder elements could be leached away, resulting in pure output streams of clean ceramics and liberated Bi2-xSbxTe3 legs which could easily be separated by sieving. When a junction layer of W between the Cu and ceramic was present, the Cu strips were liberated, rather than fully oxidised. The recovery of metallic Cu rather than a dissolved species will improve the viability of the recycling process from an economic point of view, and also result in less change to the leaching bath composition. While the application of high-powered ultrasound to the thermoelectric devices in water resulted in separation of the semiconductor materials from the ceramics and Cu tabs, a powdered material of uneven grain sizes was recovered which would require further processing steps to be useful. Therefore, further work is needed to establish whether fully intact thermoelectric legs are needed for a circular economy for TEGs. Or ultimately TEGS will be treated in the same manner as conventional e-waste, with further size reduction and homogenisation before material recovery.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation program under the Marie Sklodowska-Curie grant agreement number 101026159. The authors would also like to thank the Faraday Institution (Faraday Institution grant code FIRG027, project website https://relib.org.uk), and the UKRI Interdisciplinary Circular Economy Centre for Technology Metals, Met4Tech project (EP/V011855/1) for funding this work. Authors would like to thank Mr G. Clark and Mr A. Cox for their help with SEM and ICP-MS experiments. Authors are very grateful to European Thermodynamics Limited for the thermoelectric samples.References
- C. Forman, I. K. Muritala, R. Pardemann and B. Meyer, Renewable Sustainable Energy Rev., 2016, 57, 1568–1579 CrossRef.
- M. A. Zoui, S. Bentouba, J. G. Stocholm and M. Bourouis, Energies, 2020, 13, 3606 CrossRef CAS.
- S. B. Riffat and X. Ma, Appl. Therm. Eng., 2003, 23, 913–935 CrossRef.
- A. Bahrami, G. Schierning and K. Nielsch, Adv. Energy Mater., 2020, 10, 1904159 CrossRef CAS.
- N. T. Nassar, H. Kim, M. Frenzel, M. S. Moats and S. M. Hayes, Resour., Conserv. Recycl., 2022, 184, 106434 CrossRef CAS.
- E. Rombach and B. Friedrich, Handbook of Recycling, Elsevier, 2014, pp. 125–150 Search PubMed.
- M.-A. Shahbazi, L. Faghfouri, M. P. A. Ferreira, P. Figueiredo, H. Maleki, F. Sefat, J. Hirvonen and H. A. Santos, Chem. Soc. Rev., 2020, 49, 1253–1321 RSC.
- A. Chatterjee, C. N. Lobato, H. Zhang, A. Bergne, V. Esposito, S. Yun, A. R. Insinga, D. V. Christensen, C. Imbaquingo, R. Bjørk, H. Ahmed, M. Ahmad, C. Y. Ho, M. Madsen, J. Chen, P. Norby, F. M. Chiabrera, F. Gunkel, Z. Ouyang and N. Pryds, JPhys Energy, 2023, 5, 022001 CrossRef.
- N. Jaziri, A. Boughamoura, J. Müller, B. Mezghani, F. Tounsi and M. Ismail, Energy Rep., 2020, 6, 264–287 CrossRef.
- G. Calvo, G. Mudd, A. Valero and A. Valero, Resources, 2016, 5, 36 CrossRef.
- N. Rötzer and M. Schmidt, Resources, 2018, 7, 88 CrossRef.
- D. Champier, Energy Convers. Manage., 2017, 140, 167–181 CrossRef.
- Selenium and Tellurium Chemistry: from Small Molecules to Biomolecules and Materials, ed. J. D. Woollins and R. S. Laitinen, Springer, Heidelberg; New York, 2011 Search PubMed.
- E. Deady, C. Moon, K. Moore, K. M. Goodenough and R. K. Shail, Ore Geol. Rev., 2022, 143, 104722 CrossRef.
- V. A. Krenev, N. F. Drobot and S. V. Fomichev, Theor. Found. Chem. Eng., 2015, 49, 540–544 CrossRef CAS.
- O. Velázquez-Martinez, A. Kontomichalou, A. Santasalo-Aarnio, M. Reuter, A. J. Karttunen, M. Karppinen and R. Serna-Guerrero, Resour., Conserv. Recycl., 2020, 159, 104843 CrossRef.
- W.-B. Kim, Bull. Korean Chem. Soc., 2013, 34, 2167–2170 CrossRef CAS.
- H. Y. Lee and J. K. Lee, Sep. Sci. Technol., 2015, 50, 1665–1670 CrossRef CAS.
- R. Sasai, T. Fujimura and T. Sano, J. Ceram. Soc. Jpn., 2021, 129, 118–121 CrossRef CAS.
- W. D. Bonificio and D. R. Clarke, J. Appl. Microbiol., 2014, 117, 1293–1304 CrossRef CAS PubMed.
- G. R. T. Jenkin, A. Z. M. Al-Bassam, R. C. Harris, A. P. Abbott, D. J. Smith, D. A. Holwell, R. J. Chapman and C. J. Stanley, Miner. Eng., 2016, 87, 18–24 CrossRef CAS.
- R. Marin Rivera, G. Zante, J. Hartley, K. S. Ryder and A. P. Abbott, Green Chem., 2022, 24, 3023–3034 RSC.
- D. L. Thompson, I. M. Pateli, C. Lei, A. Jarvis, A. P. Abbott and J. M. Hartley, Green Chem., 2022, 24, 4877–4886 RSC.
- G. Zante, R. Marin Rivera, J. M. Hartley and A. P. Abbott, J. Cleaner Prod., 2022, 370, 133552 CrossRef CAS.
- M. F. C. Carneiro and V. A. Leão, Hydrometallurgy, 2007, 87, 73–82 CrossRef CAS.
- J. E. Dutrizac, Hydrometallurgy, 1992, 29, 1–45 CrossRef CAS.
- P. R. Holmes and F. K. Crundwell, Geochim. Cosmochim. Acta, 2000, 64, 263–274 CrossRef CAS.
- J. Liddicoat and D. Dreisinger, Hydrometallurgy, 2007, 89, 323–331 CrossRef CAS.
- G. Steinhauser, J. Cleaner Prod., 2008, 16, 833–841 CrossRef.
- C. D'Agostino, R. C. Harris, A. P. Abbott, L. F. Gladden and M. D. Mantle, Phys. Chem. Chem. Phys., 2011, 13, 21383 RSC.
- J. M. Hartley, J. Allen, J. Meierl, A. Schmidt, I. Krossing and A. P. Abbott, Electrochim. Acta, 2022, 402, 139560 CrossRef CAS.
- F. Bevan, H. Galeb, A. Black, I. M. Pateli, J. Allen, M. Perez, J. Feldmann, R. Harris, G. Jenkin, A. Abbott and J. Hartley, ACS Sustainable Chem. Eng., 2021, 9, 2929–2936 CrossRef CAS.
- C. Merritt, H. M. Hershenson and L. B. Rogers, Anal. Chem., 1953, 25, 572–577 CrossRef CAS.
- H. Nikol and A. Vogler, J. Am. Chem. Soc., 1991, 113, 8988–8990 CrossRef CAS.
- L. Vieira, J. Burt, P. W. Richardson, D. Schloffer, D. Fuchs, A. Moser, P. N. Bartlett, G. Reid and B. Gollas, ChemistryOpen, 2017, 6, 393–401 CrossRef CAS PubMed.
- B. E. Etschmann, W. Liu, A. Pring, P. V. Grundler, B. Tooth, S. Borg, D. Testemale, D. Brewe and J. Brugger, Chem. Geol., 2016, 425, 37–51 CrossRef CAS.
- G. S. Pokrovski, A. Yu. Borisova, J. Roux, J.-L. Hazemann, A. Petdang, M. Tella and D. Testemale, Geochim. Cosmochim. Acta, 2006, 70, 4196–4214 CrossRef CAS.
- M. Filella, N. Belzile and Y.-W. Chen, Earth-Sci. Rev., 2002, 59, 265–285 CrossRef CAS.
- D. M. Sherman, K. V. Ragnarsdottir, E. H. Oelkers and C. R. Collins, Chem. Geol., 2000, 167, 169–176 CrossRef CAS.
- J. M. Hartley, C.-M. Ip, G. C. H. Forrest, K. Singh, S. J. Gurman, K. S. Ryder, A. P. Abbott and G. Frisch, Inorg. Chem., 2014, 53, 6280–6288 CrossRef CAS PubMed.
- N. Shekhovtsova, T. Glyzina, S. Romanenko and N. Kolpakova, J. Solid State Electrochem., 2012, 16, 2419–2423 CrossRef CAS.
- L.-Y. Hsieh, J.-D. Fong, Y.-Y. Hsieh, S.-P. Wang and I.-W. Sun, J. Electrochem. Soc., 2018, 165, D331–D338 CrossRef CAS.
- J. A. Barragan, C. Ponce de León, J. R. Alemán Castro, A. Peregrina-Lucano, F. Gómez-Zamudio and E. R. Larios-Durán, ACS Omega, 2020, 5, 12355–12363 CrossRef CAS PubMed.
- A. Sorgho, M. Bougouma, G. De Leener, J. Vander Steen and T. Doneux, Electrochem. Commun., 2022, 140, 107327 CrossRef CAS.
- O. Cakir, J. Mater. Process. Technol., 2006, 175, 63–68 CrossRef CAS.
- T. Keskitalo, J. Tanskanen and T. Kuokkanen, Resour., Conserv. Recycl., 2007, 49, 217–243 CrossRef.
- A. P. Abbott, J. Collins, I. Dalrymple, R. C. Harris, R. Mistry, F. Qiu, J. Scheirer and W. R. Wise, Aust. J. Chem., 2009, 62, 341 CrossRef CAS.
- K. L. Chavez and D. W. Hess, J. Electrochem. Soc., 2001, 148, G640 CrossRef CAS.
- A. P. Abbott, G. Capper, D. L. Davies, K. J. McKenzie and S. U. Obi, J. Chem. Eng. Data, 2006, 51, 1280–1282 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00087g |
| This journal is © The Royal Society of Chemistry 2023 |