Structure regulation induced high capacity and ultra-stable cycling of conjugated organic cathodes for Li-ion batteries†
Received
14th October 2022
, Accepted 16th November 2022
First published on 17th November 2022
Abstract
A polymerization strategy to integrate conjugated structures with rich redox-active units for lithium ion batteries has been rapidly adopted to realize highly efficient polymer cathodes, because polymerization can solve the poor conductivity issue as well as solubility problem, existing in small organic molecules. However, the structural–property correlation of such polymers has not been systematically investigated, and the great impact of the conjugated structures on the overall performance has not been clearly revealed. In this work, we design and synthesize three novel pyrene-4,5,9,10-tetraone (PTO)-based polymers containing different thiophene derivatives as linking units, poly(2,7-thiophene pyrene-4,5,9,10-tetraone) (P(PTO-T1)), poly(2,7-(2,2′-bithiophene)pyrene-4,5,9,10-tetraone) (P(PTO-T2)) and poly(2,7-(thieno[3,2-b]thiophene)pyrene-4,5,9,10-tetraone) (P(PTO-TT)), to tune their electronic structures for high-performance lithium-ion batteries (LIBs). All these three materials deliver high specific capacity and long-cycle stability, which benefits from the high activity of the PTO units, the good conductivity of conjugated frameworks, and the insoluble properties of the polymers. Moreover, polymers with different thiophene moieties show distinct electrochemical performances, among which P(PTO-TT) exhibits the best rate capability (a reversible capacity of 129 mA h g−1 at a high current density of 2 A g−1 and a high capacity retention of 94% after 1200 cycles). Electrochemical and theoretical analyses reveal that an optimized electronic structure of P(PTO-TT) determined by using the thieno[3,2-b]thiophene (TT) linking units is crucial to realize the excellent battery performance. Our work unveils the structure–property correlation of PTO-based polymers as cathode materials, which provides inspiration for rational design of polymer cathodes for high-performance LIBs.
Introduction
Nowadays, it is urgent to develop high performance and sustainable energy storage devices to meet the energy and power demands of rapid social and economic development and release environmental stress.1–3 As one of the outstanding energy storage devices, lithium-ion batteries (LIBs) have been widely applied in electric vehicles and other portable consumer electronics, dominating the energy storage market.4–7 However, commercial cathode materials of LIBs are mostly inorganic materials, such as transition metal oxides, LiCoO2 and LiFePO4, which suffer from low specific capacities and environmental issues such as limited raw resources and difficult recycling.8–15 Thus, searching for new materials to address these issues is important and highly desirable. Organic materials, with structural diversity, reasonable sustainability, tunable properties, and possible high capacity/high energy density, have been demonstrated as potential candidates for next generation LIBs.8,9,16–20 Many types of organic materials containing different redox active centers (carbonyls, nitriles, radicals, imine compounds, etc.) have been extensively studied.21 Among them, conjugated carbonyl compounds are considered as the most promising candidates for the next generation organic electrode materials due to their high capacity and good electronic stability. Considering the importance of conductivity towards electrochemical performances, the design and synthesis of conjugated polymers with an optimized electronic structure are highly desirable. Consequently, investigation on structural regulation of conjugated polymers is essential for the rational design of highly efficient organic cathode materials.
Pyrene-4,5,9,10-tetraone (PTO), which can make full use of four carbonyl active sites and deliver an extraordinarily high theoretical capacity of 408 mA h g−1, has attracted extensive attention as a cathode material for high energy density LIBs.22 However, the serious dissolubility and low conductivity impede the application of PTO in metal-ion batteries. Many strategies, including polymerization23–25 or regulating the electrolyte,26 have been projected to suppress the dissolution problem to some extent. As an example, Chen et al. reported a polymer, polyphenyl-1,3,5-(pyrene-4,5,9,10-tetraone) (PPh-PTO), using 1 M LiClO4/tetraethylene glycol dimethyl ether (G4) as the electrolyte, delivering an excellent capacity of 235 mA h g−1 at 0.1 A g−1 with 95% capacity retention after 1400 cycles.26 On the other hand, introducing electron-withdrawing groups27–29 and conjugated structures, and loading into an appropriate matrix30 are of great benefit to improve the working potentials and electro-conductivity. It is noteworthy that rigid linear polymers possess high active center utilization and remain stable in the electrolyte, which is attributed to their non-twistable rigid structure. For instance, the Yao group introduced a π-conjugated redox linear polymer, poly{[N,N′-bis(2-octyldodecyl)-1,4,5,8-naphthalenedicarboximide-2,6-diyl]-alt-5,5′-(2,2′-bithiophene)} (P(NDI2OD-T2)), which delivered 95% of its theoretical capacity at 100C and maintained 96% capacity retention after 3000 cycles at 10C.31 By synthesizing poly(2,7-ethynylpyrene-4,5,9,10-tetraone) (PEPTO) containing a carbon carbon-triple bond, Zhang et al. confirmed that suitable linking units between two adjacent PTOs can improve the stability, capacity and energy density of the as-prepared polymers, which can be attributed to the enhanced conjugation and planarity.32 Therefore, it is an effective way to improve the electrochemical performance of PTO based polymers by introducing conductive linking units. However, there are few reports focusing on the tuning of the linking units between redox-active structures in comparison with those on exploiting new redox-active functional groups. In-depth study on the structure–property correlation of such polymers is of great importance for developing highly efficient polymer cathodes. Among various small molecules, thiophene and its derivatives as common monomers in conducting polymers stand out as promising linkers for redox-active structures, mainly due to their conjugation properties and facile copolymerization through a simple Stille coupling reaction, showing their great potential as efficient linkers to improve the conjugated structures of resulting polymers.31,33,34 In addition, the introduce of thiophene and its derivatives in π-conjugated polymers can lead to enhanced stability. For example, the Yao group introduced a π-conjugated polymer, poly{[N,N′-bis(2-octyldodecyl)-1,4,5,8-naphthalenedicarboximide-2,6-diyl]-alt-5,5′-(2,2′-bithiophene)} (P(NDI2OD-T2)) by using 2,2′-bithiophene as a linking unit, which exhibited excellent cycling stability (96% capacity retention after 3000 cycles).31 Moreover, Li et al. synthesized naphthalene dicarboximide (NDI)-based conjugated polymers with ion-conducting ethylene glycol (EG) side chains (PNDI-T2EG), which exhibited a more excellent cycling stability and rate performance.34
Herein, we synthesized three π-conjugated linear polymers (P(PTO-T1), P(PTO-T2), P(PTO-TT)) based on the carbonyl-rich monomer PTO as the cathodes of LIBs. Three different linking moieties, thiophene, 2,2′-bithiophene and thieno[3,2-b]thiophene are applied as the linking units to study their impact on the polymer structures and the effect of planarity and conjugation on the stability and conductivity of polymer cathodes. Electrochemical analysis based on the battery performance and theoretical calculations based on density functional theory (DFT) are performed to simulate the molecular structure, which enables the in-depth study of the structure–property correlation.
Results and discussion
Material syntheses and characterization
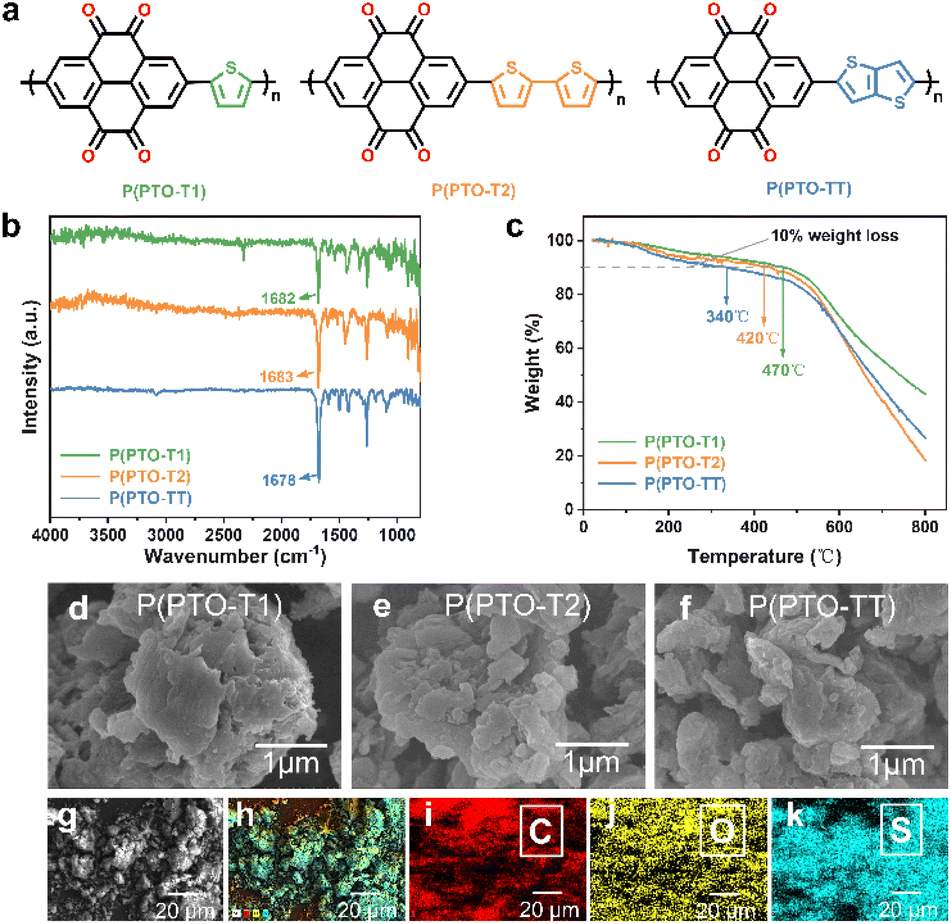
P(PTO-T1), P(PTO-T2) and P(PTO-TT) (Fig. 1a) were synthesized via Stille coupling reactions. The detailed synthetic routes (Schemes S1 and S2, ESI†) and the Experimental details are described in the ESI.† The obtained products P(PTO-T1), P(PTO-T2) and P(PTO-TT) have been characterized by Fourier transform infrared (FT-IR) spectroscopy, elemental analysis (EA), scanning electron microscopy (SEM) and thermogravimetric analysis (TGA). As shown in Fig. 1b, P(PTO-T1), P(PTO-T2) and P(PTO-TT) exhibit three distinct peaks at around 1682 cm−1, 1683 cm−1 and 1678 cm−1 respectively, which can be attributed to the vibrations of the carbonyl (CO) stretching. The presence of carbonyl groups indicates the successful recovery of CO functional groups after deprotection. The elemental analysis (EA) has been conducted on P(PTO-T1), P(PTO-T2) and P(PTO-TT), which is consistent with their elemental composition (refer to the ESI†). Thermal gravimetric analysis (TGA) (Fig. 1c) of P(PTO-T1), P(PTO-T2) and P(PTO-TT) shows that their decomposition temperatures with 10% weight loss are 470 °C, 420 °C and 340 °C, respectively, indicating their high thermal stability as active electrochemical materials in rechargeable batteries. As depicted in Fig. 1d–f and S3 (ESI),† the morphologies of these three polymers have been characterized by scanning electron microscopy (SEM), and they all exhibit microsized structures. Energy dispersive spectroscopy (EDS) analysis of the as-synthesized P(PTO-TT) confirms the homogeneous distribution of C, O, and S (Fig. 1g–k). After mixing P(PTO-TT) with Super P as a conducting additive and polyvinylidene fluoride (PVDF) as the binder, a composite electrode can be successfully fabricated (Fig. S4, ESI†).
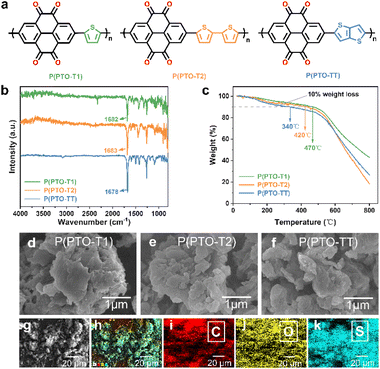 |
| | Fig. 1 (a) Scheme for the synthetic route of redox-active P(PTO-T1), P(PTO-T2) and P(PTO-TT) cathode materials. (b) FT-IR spectra and (c) TGA curves of P(PTO-T1), P(PTO-T2) and P(PTO-TT). (d–f) SEM images of P(PTO-T1) (d), P(PTO-T2) (e) and P(PTO-TT) (f). (g–k) SEM image of P(PTO-TT) and the corresponding element mapping. | |
Electrochemical performance
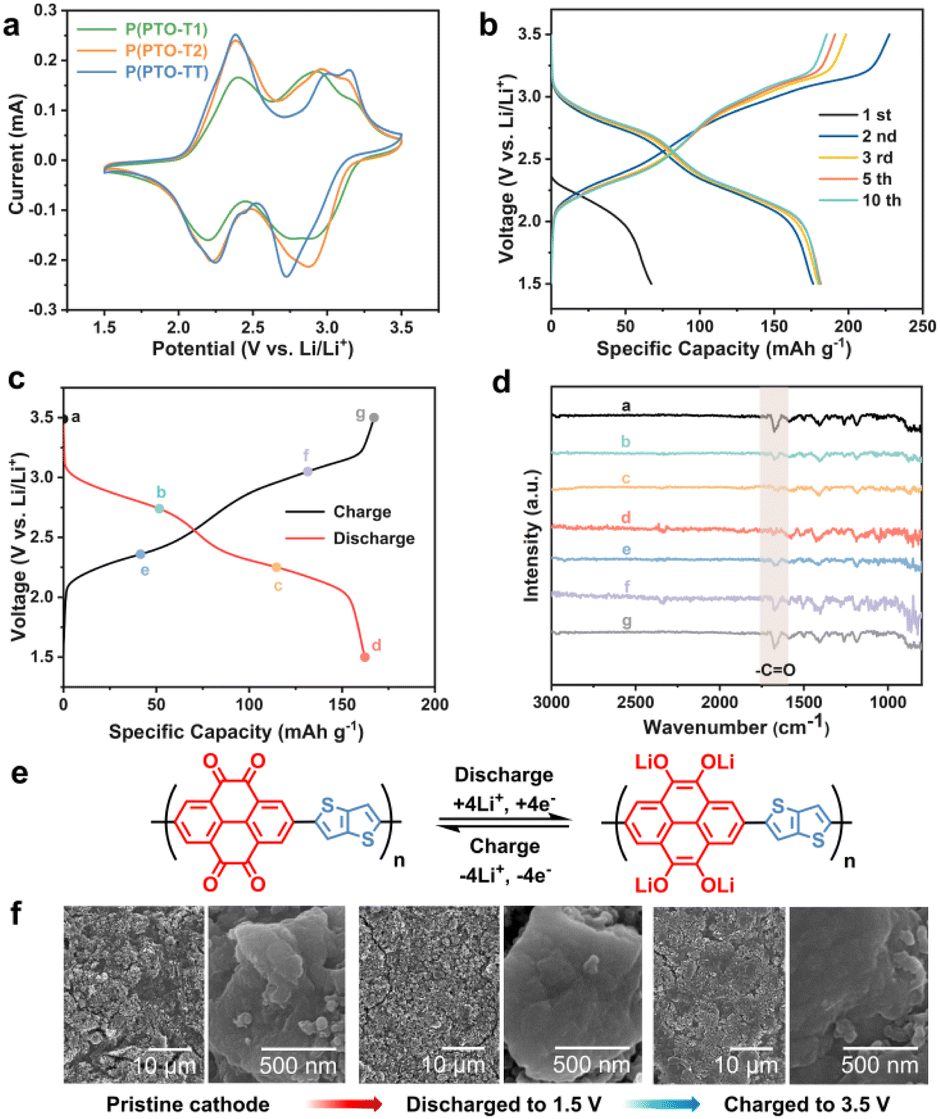
The theoretical specific capacities of P(PTO-T1), P(PTO-T2) and P(PTO-TT) as cathode materials are calculated to be 288, 236 and 250 mA h g−1. In order to investigate the electrochemical performance of P(PTO-T1), P(PTO-T2) and P(PTO-TT), the coin-type 2032 half-cell batteries with 1.0 M LiTFSI (DOL![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DME = 1:1 (v/v)) electrolyte were fabricated. The CV curves in the range of 1.5 to 3.5 V (vs. Li/Li+) at a scan rate of 0.2 mV s−1 are shown in Fig. 2a. There are two couples of enolation redox peaks of carbonyl groups for P(PTO-T1), P(PTO-T2) and P(PTO-TT) between 3.5 and 1.5 V (2.77/2.90 V and 2.21/2.36 V for P(PTO-T1), 2.88/2.96 V and 2.24/2.38 V for P(PTO-T2) and 2.74/3.05 V and 2.25/2.36 V for P(PTO-TT)), corresponding to two pairs of two-electron reaction of the four carbonyl groups, which is consistent with the plateaus between 3.5 V and 1.5 V shown in the charge–discharge profiles (Fig. 2b and S5, ESI†). No additional redox peaks in the CV curves or additional plateaus in the charge–discharge profiles can be found for P(PTO-T1), P(PTO-T2) and P(PTO-TT) except for those assigned to the carbonyl groups, indicating that thiophene, 2,2′-bithiophene and thieno[3,2-b]thiophene as linkers are electrochemically inactive in the polymers between 1.5 and 3.5 V (vs. Li/Li+). As shown in Fig. S6 (ESI),† the cathodic and anodic curves of the three cathodes remained similar in the 2nd–5th scans in terms of the shape, peak current and potential, indicating their good stability and reversibility. Additionally, during the first five cycles, the potential difference between cathodic and anodic peaks in the same redox couple decreases slightly, implying decreased polarization after the activation process. Ex situ FT-IR spectra after each charge and discharge step of P(PTO-TT) cathodes have been obtained to gain insight into the redox reaction. As shown in Fig. 2c and d, there is a strong vibration absorption peak at around 1676 cm−1, which is attributed to the carbonyl group. Upon discharging to 1.5 V, the peak of the CO group decreases gradually and disappears at the end of discharge, corresponding to the formation of Li–O. Subsequently, the CO group peak with sharp and strong absorption recovers gradually when the electrode is fully recharged to 3.5 V, indicating a good reversible redox activity of P(PTO-TT). This is coincident with the results of previous cathode investigations on PTO-based compounds (as shown in Fig. 2e).25,35,36Ex situ SEM measurement has been performed to investigate the morphology of the polymer electrodes at different charge/discharge states. As shown in Fig. 2f, after full charge to 3.5 V and discharge to 1.5 V, the morphologies of the P(PTO-TT) cathode show almost no change and the grain sizes are similar to those of the pristine materials, confirming the structural stability of the P(PTO-TT) cathode.
:DME = 1:1 (v/v)) electrolyte were fabricated. The CV curves in the range of 1.5 to 3.5 V (vs. Li/Li+) at a scan rate of 0.2 mV s−1 are shown in Fig. 2a. There are two couples of enolation redox peaks of carbonyl groups for P(PTO-T1), P(PTO-T2) and P(PTO-TT) between 3.5 and 1.5 V (2.77/2.90 V and 2.21/2.36 V for P(PTO-T1), 2.88/2.96 V and 2.24/2.38 V for P(PTO-T2) and 2.74/3.05 V and 2.25/2.36 V for P(PTO-TT)), corresponding to two pairs of two-electron reaction of the four carbonyl groups, which is consistent with the plateaus between 3.5 V and 1.5 V shown in the charge–discharge profiles (Fig. 2b and S5, ESI†). No additional redox peaks in the CV curves or additional plateaus in the charge–discharge profiles can be found for P(PTO-T1), P(PTO-T2) and P(PTO-TT) except for those assigned to the carbonyl groups, indicating that thiophene, 2,2′-bithiophene and thieno[3,2-b]thiophene as linkers are electrochemically inactive in the polymers between 1.5 and 3.5 V (vs. Li/Li+). As shown in Fig. S6 (ESI),† the cathodic and anodic curves of the three cathodes remained similar in the 2nd–5th scans in terms of the shape, peak current and potential, indicating their good stability and reversibility. Additionally, during the first five cycles, the potential difference between cathodic and anodic peaks in the same redox couple decreases slightly, implying decreased polarization after the activation process. Ex situ FT-IR spectra after each charge and discharge step of P(PTO-TT) cathodes have been obtained to gain insight into the redox reaction. As shown in Fig. 2c and d, there is a strong vibration absorption peak at around 1676 cm−1, which is attributed to the carbonyl group. Upon discharging to 1.5 V, the peak of the CO group decreases gradually and disappears at the end of discharge, corresponding to the formation of Li–O. Subsequently, the CO group peak with sharp and strong absorption recovers gradually when the electrode is fully recharged to 3.5 V, indicating a good reversible redox activity of P(PTO-TT). This is coincident with the results of previous cathode investigations on PTO-based compounds (as shown in Fig. 2e).25,35,36Ex situ SEM measurement has been performed to investigate the morphology of the polymer electrodes at different charge/discharge states. As shown in Fig. 2f, after full charge to 3.5 V and discharge to 1.5 V, the morphologies of the P(PTO-TT) cathode show almost no change and the grain sizes are similar to those of the pristine materials, confirming the structural stability of the P(PTO-TT) cathode.
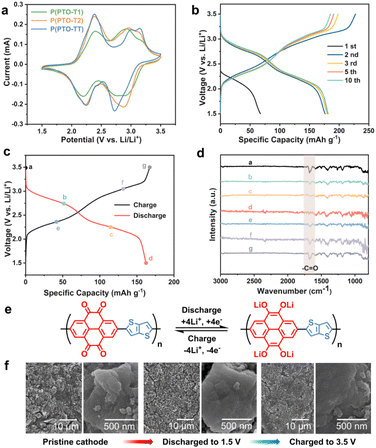 |
| | Fig. 2 Electrochemical properties of the P(PTO-T1), P(PTO-T2) and P(PTO-TT) cathodes between 3.5 and 1.5 V. (a) CV measurements at a scan rate of 0.2 mV s−1. (b) Galvanostatic charge–discharge profiles of P(PTO-TT) cathodes at 50 mA g−1. (c) Charge/discharge profiles of P(PTO-TT) electrodes with marked points at different discharge and charge states at 50 mA g−1. (d) Ex situ FT-IR spectra for the sample taken at different states as marked in (c). (e) Proposed reaction mechanisms for P(PTO-TT) cathodes. (f) SEM images of the as-prepared P(PTO-TT) electrodes at different states. | |
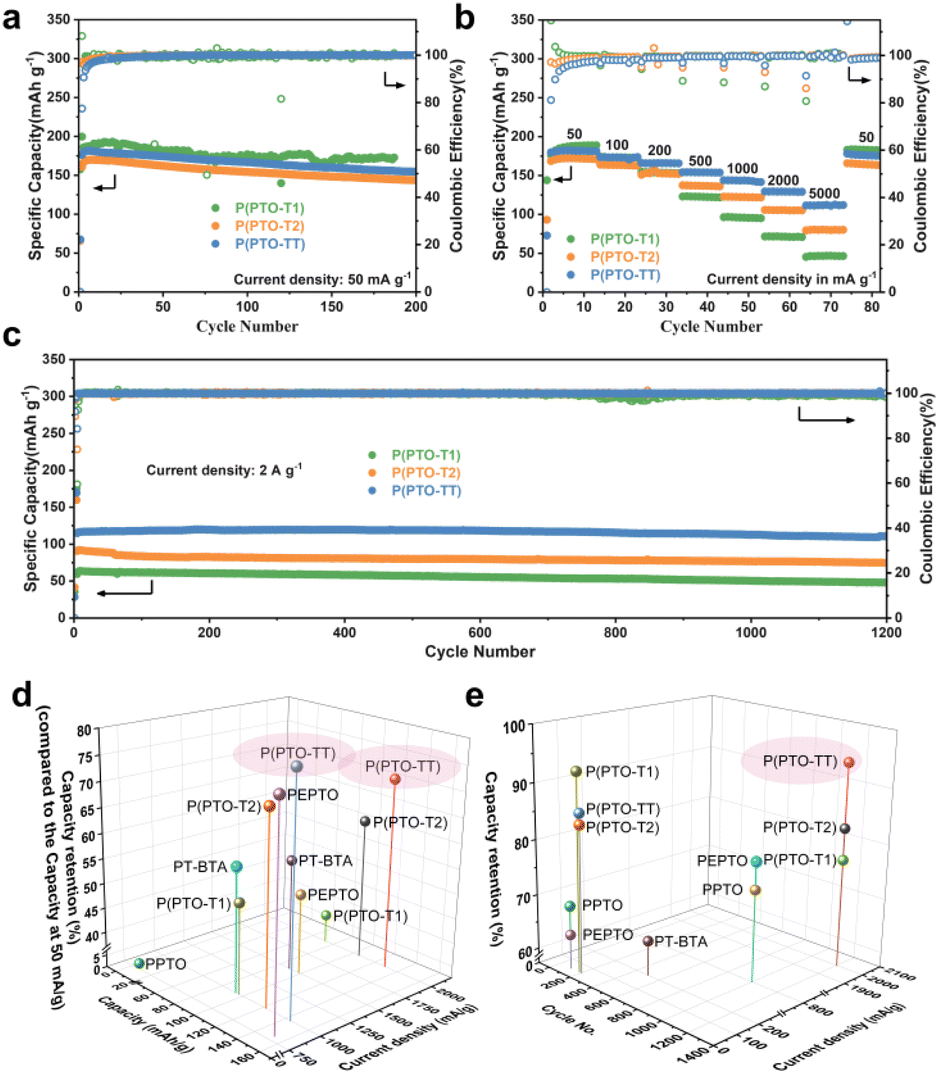
The cycling performance of the PTO-based cathodes is shown in Fig. 3a. P(PTO-T1), P(PTO-T2) and P(PTO-TT) exhibit reversible capacities of 186 mA h g−1, 167 mA h g−1 and 180 mA h g−1 at the 2nd discharge (50 mA g−1), respectively. The capacity contribution from the conductive agent (Super P) is neglectable in the same test voltage range (Fig. S7†). The relatively low capacities of the three materials at the first discharge can be attributed to the self-discharge of the coin-type cell. The slightly lower coulombic efficiency of the three cathodes between the 1st and the following cycles can be attributed to the irreversible oxidation of the electrolyte at a high voltage. Nevertheless, this side reaction can be inhibited by increasing the current rate. An obvious increase in capacity along with the Coulomb efficiency can be observed for the three cathode materials in the first few cycles, which can be ascribed to the gradual stabilization of the electrode interface and the utilization of more activated redox sites.37 After 200 cycles, the capacities of P(PTO-T2) and P(PTO-TT) remain as high as 144 and 155 mA h g−1, respectively, indicating the good cycling performance of both materials. For P(PTO-T1), higher specific capacities of 187 mA h g−1 and 173 mA h g−1 are obtained at the second and 186 cycles, respectively. The capacity is higher than that of P(PTO-T2) and P(PTO-TT), which can be attributed to the higher theoretical specific capacity due to the contribution of the lower molecular weight of the inactive thiophene structure. Importantly, the coulombic efficiency values of all three materials approach as high as 100% after the activation processes.
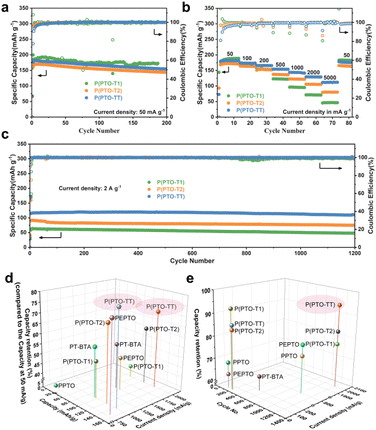 |
| | Fig. 3 (a) Cycling performance at 50 mA g−1 and (b) rate performance of P(PTO-T1), P(PTO-T2) and P(PTO-TT). (c) Long-term cycling stability of P(PTO-T1), P(PTO-T2) and P(PTO-TT) at 2 A g−1. (d and e) Comparison of the electrochemical performances of P(PTO-T1), P(PTO-T2), and P(PTO-TT) with that of other reported PTO-based polymers (PT-BTA,40 PPTO,32 and PEPTO32) using 1 M LiTFSI (DOL:DME = 1:1 (v/v)) as the electrolyte. The capacity retention in (d) is compared with the capacity at 50 mA g−1, and the capacity retention in (e) is compared with the capacity at 1st cycle at the same current density. See Table S1 (ESI)† for detailed information used in (d) and (e) for more comparisons using different electrolyte systems. | |
The rate performance and long-term cycling stability of P(PTO-T1), P(PTO-T2) and P(PTO-TT) have been further investigated. As shown in Fig. 3b, galvanostatic charge–discharge cycling has been performed at current densities ranging from 50 mA g−1 to 5 A g−1. When the current density increases from 50 mA h g−1 to 2 A g−1, the capacity of P(PTO-T1) decreases from 187 mA h g−1 to 72 mA h g−1 with a low capacity retention of 39%, while the capacity of P(PTO-T2) decreased from 172 to 105 mA h g−1 with a moderate capacity retention of 61%. In special, the capacity of P(PTO-TT) remains as high as 129 mA h g−1 when the current density increases to 2 A g−1, with a capacity retention of 71% compared to that at 50 mA g−1, i.e. 182 mA h g−1. More impressively, P(PTO-TT) delivers a capacity of 111 mA h g−1 at a high current density of 5 A g−1 with a retention of 61%. These results show that P(PTO-T1) delivers higher capacity at a slow rate (current densities lower than 200 mA g−1) compared to P(PTO-T2) and P(PTO-TT), which can be ascribed to the relatively higher theoretical capacity. In contrast, P(PTO-TT) exhibits much higher capacity than P(PTO-T1) and P(PTO-T2) at high rates (current densities higher than 200 mA g−1), which indicates much better rate performance of P(PTO-TT). Moreover, the long-term cycling performance of P(PTO-TT) at a high rate has also been studied. As shown in Fig. 3c, P(PTO-TT) exhibits excellent cycling stability at 2 A g−1. After 1200 cycles, P(PTO-TT) can still achieve a high capacity of 111 mA h g−1, corresponding to a capacity retention of 94% with respect to its initial capacity (117 mA h g−1). However, after 1200 cycles, the capacities of P(PTO-T1) and P(PTO-T2) are 48 mA h g−1 and 75 mA h g−1, respectively, and the corresponding capacity retentions were 76% and 82%, which are much lower than that of P(PTO-TT). More comparison with previous reports on PTO-based compounds has been summarized in Table S1 (ESI).† Higher specific capacity or better rate capability can be found in several recent reports such as 2,7-diethylnylpyrene-4,5,9,10-tetraone (DE-PTO) developed by Li et al.38 and a ketoenol COF exploited by Cooper et al.39 In these studies, the superior performances result from multiple functions of the reported materials such as anion storage ability of the electron-rich diyne linkages in DE-PTO, and the greatly improved carbonyl utilization benefitting from the well-defined porous structure of crystalline PTO-COFs. Additionally, some other representative studies using the standard electrolyte of 1 M LiTFSI (DOL:DME = 1:1 (v/v)) for Li-ion batteries are selected for a direct comparison (Fig. 3d and e). Note that compounds containing redox-active functional groups, which contribute to the specific capacity in addition to PTO have been excluded in this direct comparison but are shown in Table S1 (ESI)† instead. As shown in Fig. 3d and e, P(PTO-TT) in this work outperforms others in terms of long-term cycling stability, rate capability and capacity retention at over 1 A g−1 (with regard to the capacity at 50 mA g−1), indicating a significant improvement arising from the integration of thieno[3,2-b]thiophene linkers.32,40–42
Reaction kinetics
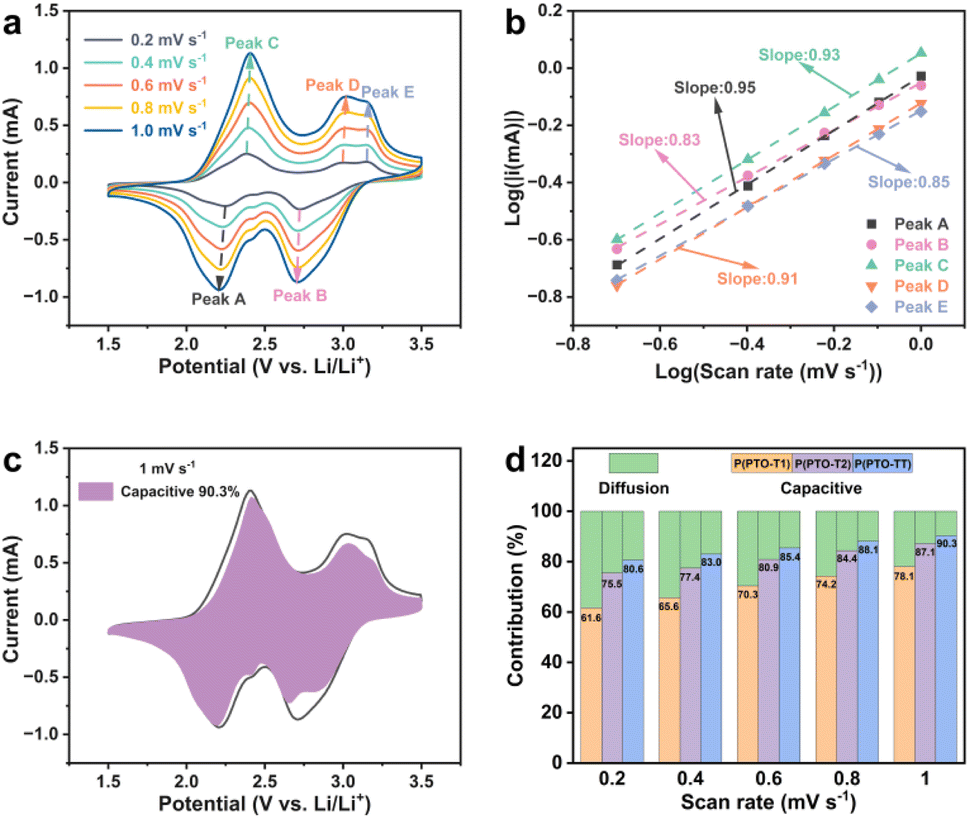
Superior rate stability of P(PTO-TT) (compared to P(PTO-T1) and P(PTO-T2)) can be observed from the specific capacity versus current density plots (Fig. S8, ESI†), implying a great impact of different thiophene moieties on the activity of PTO units. When the current density is higher than 200 mA h g−1, P(PTO-TT) provides significantly highest capacities compared to P(PTO-T1) and P(PTO-T2), suggesting the favourable kinetics of P(PTO-TT). To further elucidate this behaviour, cyclic voltammetry (CV) measurements at different scan rates have been conducted (Fig. 4a, S9 and S10, ESI†). As shown in Fig. 4a, in addition to slight shifts in the cathodic and anodic peaks, the CV curves of P(PTO-TT) are almost identical when increasing the scanning rate (0.2–1.0 mV s−1). Theoretically, the capacitive effect can be expressed through the relation of i = avb, where i is the peak current, v is the scan rate, and a and b are parameters.43 The b value can be obtained from the slope of the linear fit of a log(v) vs. log(i) plot. For P(PTO-TT) (Fig. 4b), the b values of peaks A, B, C, D and E are 0.95, 0.83, 0.93, 0.91 and 0.85 respectively, indicating that the charge storage of P(PTO-TT) is mainly controlled by capacitive processes. Similar results can be obtained for P(PTO-T1) and P(PTO-T2) (Fig. S9 and S10, ESI†). Furthermore, the equation of i = k1v1.0 + k2v0.5, where k1v corresponds to the pseudocapacitive contribution and k2v0.5 represents the diffusion-controlled contribution, is used to calculate the contribution of the capacitive component at a specific scan rate. For P(PTO-TT), the pseudocapacitive contribution increases from 80.6% to 90.3% with the increase of the scan rate from 0.2 mV s−1 to 1.0 mV s−1, which is much higher than those of P(PTO-T1) and P(PTO-T2) cathodes (61.6–78.1% for P(PTO-T1) and 75.5–87.1% for P(PTO-T2), respectively). Obviously, the fast pseudocapacitive behaviour contributes to good cycling and outstanding rate performance at high current densities.
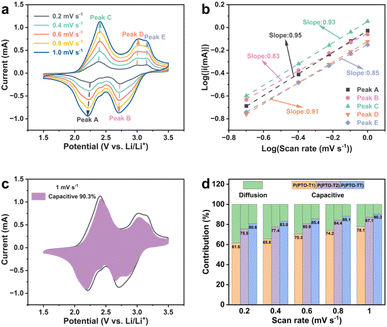 |
| | Fig. 4 (a) CV curves from a P(PTO-TT) electrode at different scan rates from 0.2 to 1.0 mV s−1 and (b) the relationship between logi and logv. (c) Capacitive and diffusion-controlled contributions at a scan rate of 1 mV s−1. (d) The pseudocapacitive charge storage contributions (in percentage) of P(PTO-T1), P(PTO-T2) and P(PTO-TT) at different scan rates. | |
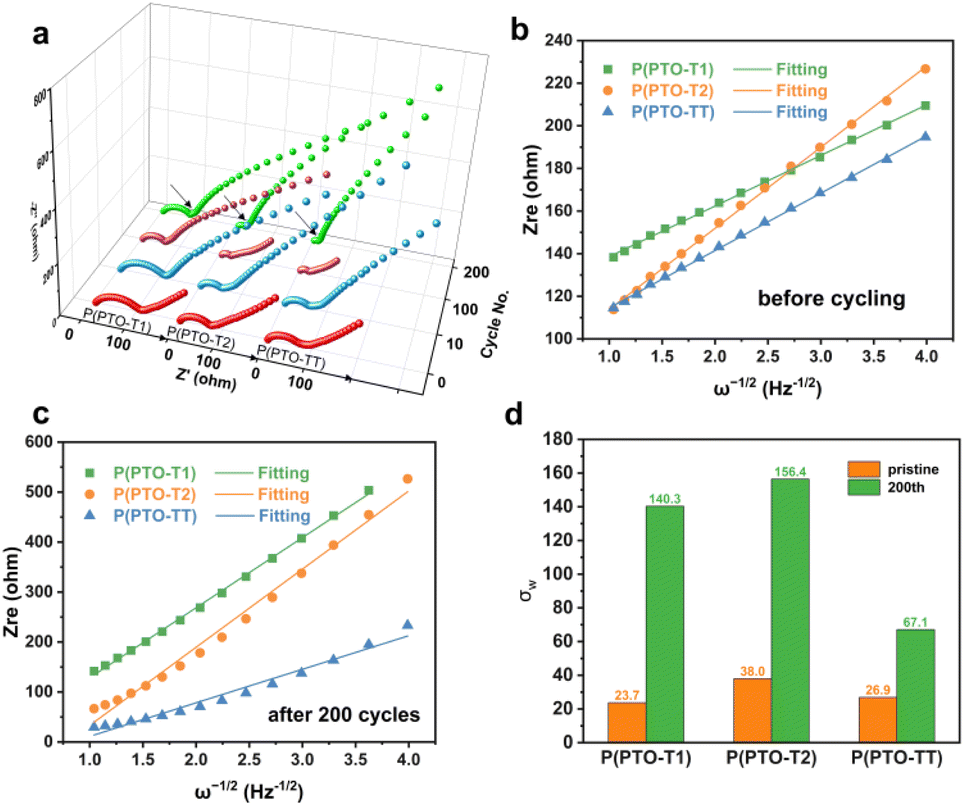
The electrochemical impedance spectroscopy (EIS) measurement has also been conducted to study the reaction kinetics of Li+ intercalation/deintercalation during cycling. From the Nyquist plots in Fig. 5a, the charge-transfer resistance (Rct) of P(PTO-TT) and P(PTO-T2) decreases obviously with the increase in the cycling number, which suggests that Li+ diffuses faster in P(PTO-TT) and P(PTO-T2) after cycling. However, the Rct of P(PTO-T1) decreases first and then increases, indicating the slightly sluggish Li+ diffusion. Notably, after 200 cycles, the lowest charge transfer resistance (Rct) of P(PTO-TT) is observed among the three polymers, indicating a fast charge transfer capability.
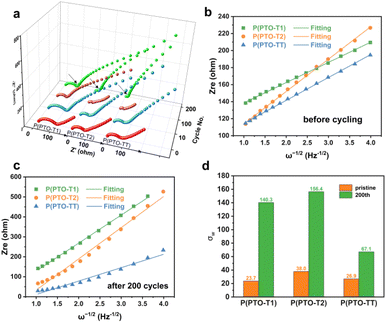 |
| | Fig. 5 (a) Nyquist plots for P(PTO-T1), P(PTO-T2) and P(PTO-TT) electrodes during cycling. The linear relationship between Zre and the inverse of the square root of the angular frequency (ω−1/2) in the low frequency region of P(PTO-T1), P(PTO-T2) and P(PTO-TT) before cycling (b) and after 200 cycles (c). (d) The Warburg impedance coefficient (σw) of P(PTO-T1), P(PTO-T2) and P(PTO-TT) at the pristine state and after 200 cycles. | |
In addition, by linearly fitting the relationship between Zre and the inverse of the square root of the angular frequency (ω−1/2),44 the Warburg impedance coefficient (σw, Fig. 5d and Table S2, ESI†) related to Li+ diffusion characteristics can be obtained from the inclined line in the low frequency region, as shown in Fig. 5b and c.45 By comparing the slopes of P(PTO-T1), P(PTO-T2) and P(PTO-TT), the σw values for pristine P(PTO-T1), P(PTO-T2) and P(PTO-TT) are similar at the pristine state. However, after 200 cycles, the slopes of P(PTO-T1) and P(PTO-T2) are much steeper than that of P(PTO-TT), suggesting that Li+ diffused much faster in P(PTO-TT) after cycling, contributing to the outstanding rate performance of P(PTO-TT).
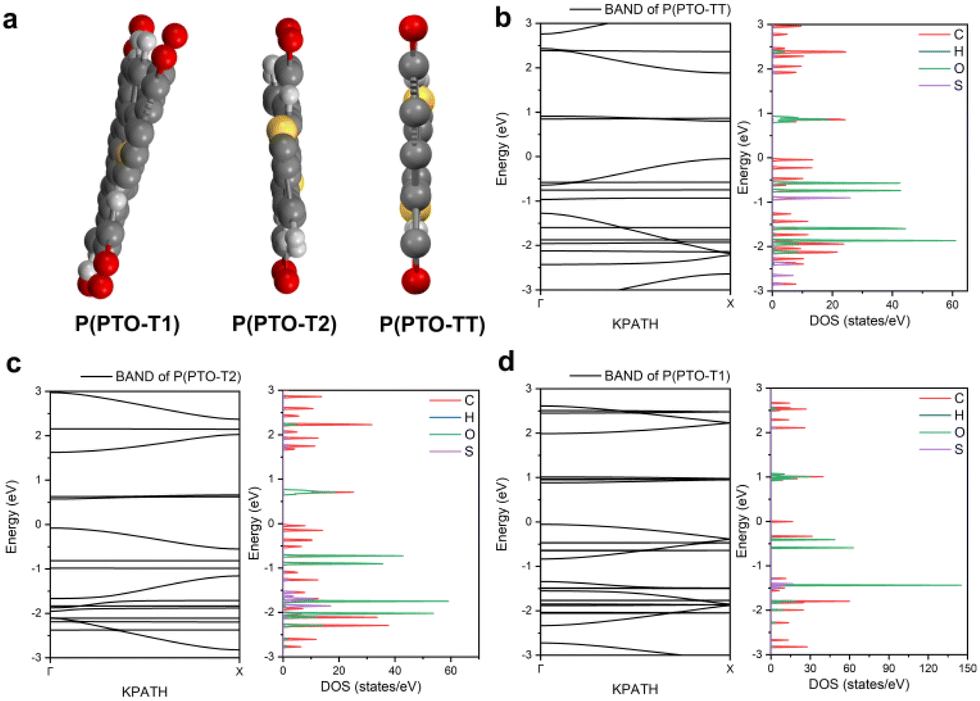
DFT calculations of the geometry and electron distributions
Apart from electrochemical analysis which demonstrates the fast kinetics of P(PTO-TT), investigation on the molecular structures is of great importance to reveal the structure–property correlation of the polymers. Theoretical calculations based on density functional theory (DFT) have been performed to simulate the polymer structures of P(PTO-T1), P(PTO-T2) and P(PTO-TT). The polymer monomers are arranged periodically along the a-axis of the crystal lattice and the polymers are far enough apart from each other. Compared with P(PTO-T1) and P(PTO-T2), the conjugation and planarity of P(PTO-TT) is enhanced as depicted in the side view in Fig. 6a. The band structures and the projected density of states (PDOS) of elements of P(PTO-T1), P(PTO-T2) and P(PTO-TT) polymers are depicted in Fig. 6b–d. Conversely, the electron and hole effective masses based on the second derivative of band structures 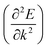 can be an indicator to compare the electronic conduction. The electron and hole effective masses of these polymers are calculated by using eqn (1) and (2),
can be an indicator to compare the electronic conduction. The electron and hole effective masses of these polymers are calculated by using eqn (1) and (2),| | 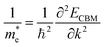 | (1) |
| | 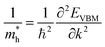 | (2) |
where ℏ is the reduced Planck constant, and 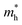 and
and 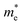 is the hole effective mass and the electron effective mass, respectively. Consistent with our assumption, P(PTO-T2) and P(PTO-TT) exhibit small effective masses both for an electron and hole with regard to those of P(PTO-T1), indicating enhanced electronic conduction compared with P(PTO-T1). In addition, as shown in Table S3 (ESI),† P(PTO-TT) has the smallest effective mass and therefore the best electronic conduction, which can be a reason why P(PTO-TT) shows much better rate performance than P(PTO-T2) and P(PTO-T1). Apparently, P(PTO-TT) gained the optimized conjugated structure due to the employment of the thieno[3,2-b]thiophene linker, which accounts for the fast kinetics during battery charge–discharge processes and leads to excellent cycling and rate performances.
is the hole effective mass and the electron effective mass, respectively. Consistent with our assumption, P(PTO-T2) and P(PTO-TT) exhibit small effective masses both for an electron and hole with regard to those of P(PTO-T1), indicating enhanced electronic conduction compared with P(PTO-T1). In addition, as shown in Table S3 (ESI),† P(PTO-TT) has the smallest effective mass and therefore the best electronic conduction, which can be a reason why P(PTO-TT) shows much better rate performance than P(PTO-T2) and P(PTO-T1). Apparently, P(PTO-TT) gained the optimized conjugated structure due to the employment of the thieno[3,2-b]thiophene linker, which accounts for the fast kinetics during battery charge–discharge processes and leads to excellent cycling and rate performances.
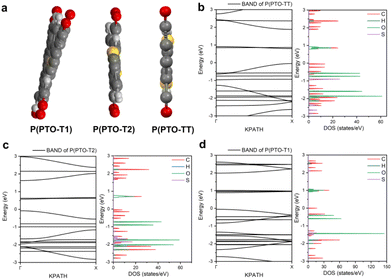 |
| | Fig. 6 (a) The side view of P(PTO-T1), P(PTO-T2) and P(PTO-TT). The band structure calculation and density of states (DOS) calculations of (b) P(PTO-TT), (c) P(PTO-T2) and (d) P(PTO-T1). | |
Conclusions
In summary, we have successfully synthesized three novel PTO-based polymers, P(PTO-T1), P(PTO-T2) and P(PTO-TT), and investigated them as redox-active cathodes for lithium organic batteries (LIBs). By tailoring different thiophene moieties between the PTO structures, we compared the electrochemical performance of the three as-prepared polymers. It is demonstrated that P(PTO-TT) exhibited the best rate performance and cycling stability, which can be attributed to the enhanced conjugation and planarity as confirmed by the theoretical analysis. Furthermore, the dominant pseudocapacitive behavior of P(PTO-TT) facilitated the electrochemical reactions. Therefore, carbonyl-rich P(PTO-TT) delivered a high capacity (182 mA h g−1 at 50 mA g−1), excellent rate performance (129 mA h g−1 at 2 A g−1) and improved cycling stability (more than 85% capacity retention after 200 cycles at 50 mA g−1 and a high capacity retention of 111 mA h g−1 after 1200 cycles at 2 A g−1). This work could inspire the structure tuning of rigid linear polymers for their use as cathode materials in high-stability and high-power lithium-ion batteries.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful to the National Natural Science Foundation of China (No. 22075027, 52003030, and 52103218), and for financial support from the State Key Laboratory of Explosion Science and Technology (YBKT21-06). Q. Z. is thankful for the funding support from City University of Hong Kong (9380117, 7005620 and 7020040) and Hong Kong Institute for Advanced Study, City University of Hong Kong, Hong Kong, P. R. China as well as from State Key Laboratory of Supramolecular Structure and Materials, Jilin University (sklssm202233).
References
- W. Guo and Y. Fu, Energy Environ. Mater., 2018, 1, 20–27 CrossRef CAS
 .
.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS .
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed .
- J. B. Goodenough and K. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS .
- K. Kang, Y. S. Meng, J. Bréger, C. P. Grey and G. Ceder, Science, 2006, 311, 977–980 CrossRef CAS PubMed .
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 CrossRef .
- S. Cao, H. Zhang, Y. Zhao and Y. Zhao, eScience, 2021, 1, 28–43 CrossRef .
- J. Xie, P. Gu and Q. Zhang, ACS Energy Lett., 2017, 2, 1985–1996 CrossRef CAS .
- Y. Lu and J. Chen, Nat. Rev. Chem., 2020, 4, 127–142 CrossRef CAS .
- T. Sun, J. Xie, W. Guo, D. S. Li and Q. Zhang, Adv. Energy Mater., 2020, 10, 1904199 CrossRef CAS .
- J. Xie, Z. Wang, Z. J. Xu and Q. Zhang, Adv. Energy Mater., 2018, 8, 1703509 CrossRef .
- M. T. Michael, W. Christopher and D. I. Eric, Energy Environ. Sci., 2012, 5, 7854–7863 RSC .
- L. Yuan, Z. Wang, W. Zhang, X. Hu, J. Chen, Y. Huang and J. B. Goodenough, Energy Environ. Sci., 2011, 4, 269–284 RSC .
- M. Winter, B. Barnett and K. Xu, Chem. Rev., 2018, 118, 11433–11456 CrossRef CAS PubMed .
- J. Zhu, X. Chen, A. Q. Thang, F. Li, D. Chen, H. Geng, X. Rui and Q. Yan, SmartMat, 2022, 3, 384–416 CrossRef CAS .
- Y. Liang, Z. Tao and J. Chen, Adv. Energy Mater., 2012, 2, 742–769 CrossRef CAS .
-
(a) J. Xie and Q. Zhang, J. Mater. Chem. A, 2016, 4, 7091–7106 RSC ;
(b) J. Xie and Q. Zhang, Mater. Today Energy, 2020, 18, 100547 CrossRef CAS ;
(c) X. Zhan, Z. Chen and Q. Zhang, J. Mater. Chem. A, 2017, 5, 14463–14479 RSC .
-
(a) H. Wang, C. Yao, H. Nie, K. Wang, Y. Zhong, P. Chen, S. Mei and Q. Zhang, J. Mater. Chem. A, 2020, 8, 11906–11922 RSC ;
(b) Y. Tong, Z. Sun, J. Wang, W. Huang and Q. Zhang, SmartMat, 2022 DOI:10.1002/smm2.1115 ;
(c) W. Zhou, M. Zhang, X. Kong, W. Huang and Q. Zhang, Adv. Sci., 2021, 2004490 CrossRef CAS PubMed .
- T. Sun, Q. Sun, Y. Yu and X. Zhang, eScience, 2021, 1, 186–193 CrossRef .
- S. Xu, H. Dai, S. Zhu, Y. Wu, M. Sun, Y. Chen, K. Fan, C. Zhang, C. Wang and W. Hu, eScience, 2021, 1, 60–68 CrossRef .
- S. Lee, G. Kwon, K. Ku, K. Yoon, S. K. Jung, H. D. Lim and K. Kang, Adv. Mater., 2018, 30, e1704682 CrossRef .
- Y. Liang, P. Zhang and J. Chen, Chem. Sci., 2013, 4, 1330 RSC .
- Y. Liang, Y. Jing, S. Gheytani, K. Y. Lee, P. Liu, A. Facchetti and Y. Yao, Nat. Mater., 2017, 16, 841–848 CrossRef CAS PubMed .
- A. Shimizu, H. Kuramoto, Y. Tsujii, T. Nokami, Y. Inatomi, N. Hojo, H. Suzuki and J. Yoshida, J. Power Sources, 2014, 260, 211–217 CrossRef CAS .
- T. Nokami, T. Matsuo, Y. Inatomi, N. Hojo, T. Tsukagoshi, H. Yoshizawa, A. Shimizu, H. Kuramoto, K. Komae, H. Tsuyama and J. Yoshida, J. Am. Chem. Soc., 2012, 134, 19694–19700 CrossRef CAS .
- S. Zheng, L. Miao, T. Sun, L. Li, T. Ma, J. Bao, Z. Tao and J. Chen, J. Mater. Chem. A, 2021, 9, 2700–2705 RSC .
- Y. Zhang, J. Wang and S. N. Riduan, J. Mater. Chem. A, 2016, 4, 14902–14914 RSC .
- X. Yin, S. Sarkar, S. Shi, Q. Huang, H. Zhao, L. Yan, Y. Zhao and J. Zhang, Adv. Funct. Mater., 2020, 30, 1908445 CrossRef CAS .
- Y. Chen and C. Wang, Acc. Chem. Res., 2020, 53, 2636–2647 CrossRef CAS .
- R. Shi, L. Liu, Y. Lu, Y. Li, S. Zheng, Z. Yan, K. Zhang and J. Chen, Adv. Energy Mater., 2021, 11, 2002917 CrossRef CAS .
- Y. Liang, Z. Chen, Y. Jing, Y. Rong, A. Facchetti and Y. Yao, J. Am. Chem. Soc., 2015, 137, 4956–4959 CrossRef CAS .
- J. Xie, W. Chen, G. Long, W. Gao, Z. J. Xu, M. Liu and Q. Zhang, J. Mater. Chem. A, 2018, 6, 12985–12991 RSC .
- L. Liu, F. Tian, X. Wang, Z. Yang, M. Zhou and X. Wang, React. Funct. Polym., 2012, 72, 45–49 CrossRef CAS .
- X. Li, Y. Li, K. Sarang, J. Lutkenhaus and R. Verduzco, Adv. Funct. Mater., 2021, 31, 2009263 CrossRef CAS .
- Y. Liang, Y. Jing, S. Gheytani, K. Lee, P. Liu, A. Facchetti and Y. Yao, Nat. Mater., 2017, 16, 841–848 CrossRef CAS PubMed .
- Y. Liang, P. Zhang and J. Chen, Chem. Sci., 2013, 4, 1330 RSC .
- Y. Cao, W. Sun, C. Guo, L. Zheng, M. Yao and Y. Wang, ACS Nano, 2022, 16, 9830–9842 CrossRef CAS PubMed .
- H. Pan, Z. Zuo, F. He and Y. Li, Energy Storage Mater., 2022, 52, 465–472 CrossRef .
- H. Gao, A. R. Neale, Q. Zhu, M. Bahri, X. Wang, H. Yang, Y. Xu, R. Clowes, N. D. Browning, M. A. Little, L. J. Hardwick and A. I. Cooper, J. Am. Chem. Soc., 2022, 144, 9434–9442 CrossRef CAS PubMed .
- K. Li, Q. Li, Y. Wang, H. Wang, Y. Li and Z. Si, Mater. Chem. Front., 2020, 4, 2697–2703 RSC .
- T. Nokami, T. Matsuo, Y. Inatomi, N. Hojo, T. Tsukagoshi, H. Yoshizawa, A. Shimizu, H. Kuramoto, K. Komae, H. Tsuyama and J. Yoshida, J. Am. Chem. Soc., 2012, 134, 19694–19700 CrossRef CAS PubMed .
- C. Yao, Z. Wu, J. Xie, F. Yu, W. Guo, Z. J. Xu, D. Li, S. Zhang and Q. Zhang, ChemSusChem, 2020, 13, 2457–2463 CrossRef CAS PubMed .
- J. Wang, J. Polleux, J. Lim and B. Dunn, J. Phys. Chem. C, 2007, 111, 14925–14931 CrossRef CAS .
- Y. Huang, J. Chen, J. Ni, H. Zhou and X. Zhang, J. Power Sources, 2009, 188, 538–545 CrossRef CAS .
- X. Shan, G. Zhou, L. Yin, W. Yu, F. Li and H. Cheng, J. Mater. Chem. A, 2014, 2, 17808–17814 RSC .
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 c,
Shilin
Mei
*a,
Guankui
Long
c,
Shilin
Mei
*a,
Guankui
Long
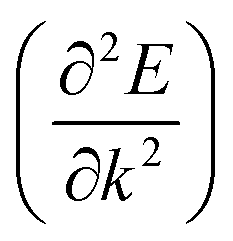 can be an indicator to compare the electronic conduction. The electron and hole effective masses of these polymers are calculated by using eqn (1) and (2),
can be an indicator to compare the electronic conduction. The electron and hole effective masses of these polymers are calculated by using eqn (1) and (2),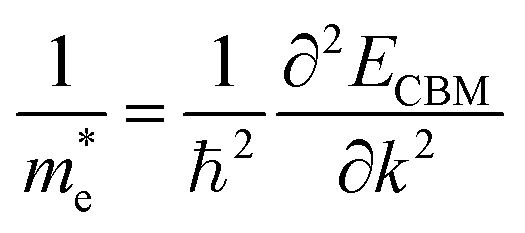
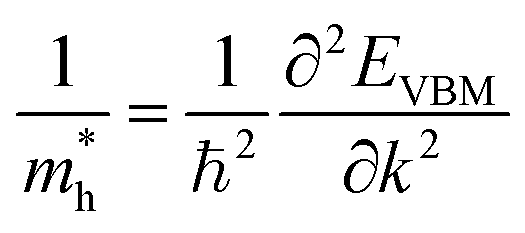
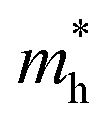 and
and 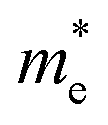 is the hole effective mass and the electron effective mass, respectively. Consistent with our assumption, P(PTO-T2) and P(PTO-TT) exhibit small effective masses both for an electron and hole with regard to those of P(PTO-T1), indicating enhanced electronic conduction compared with P(PTO-T1). In addition, as shown in Table S3 (ESI),† P(PTO-TT) has the smallest effective mass and therefore the best electronic conduction, which can be a reason why P(PTO-TT) shows much better rate performance than P(PTO-T2) and P(PTO-T1). Apparently, P(PTO-TT) gained the optimized conjugated structure due to the employment of the thieno[3,2-b]thiophene linker, which accounts for the fast kinetics during battery charge–discharge processes and leads to excellent cycling and rate performances.
is the hole effective mass and the electron effective mass, respectively. Consistent with our assumption, P(PTO-T2) and P(PTO-TT) exhibit small effective masses both for an electron and hole with regard to those of P(PTO-T1), indicating enhanced electronic conduction compared with P(PTO-T1). In addition, as shown in Table S3 (ESI),† P(PTO-TT) has the smallest effective mass and therefore the best electronic conduction, which can be a reason why P(PTO-TT) shows much better rate performance than P(PTO-T2) and P(PTO-T1). Apparently, P(PTO-TT) gained the optimized conjugated structure due to the employment of the thieno[3,2-b]thiophene linker, which accounts for the fast kinetics during battery charge–discharge processes and leads to excellent cycling and rate performances.