
Visual detection of microplastics using Raman spectroscopic imaging†
Kaili
Liu
 ,
Xu
Pang
,
Huacai
Chen
* and
Li
Jiang
,
Xu
Pang
,
Huacai
Chen
* and
Li
Jiang
College of Optical and Electronic Technology, China Jiliang University, 310018 Hangzhou, China. E-mail: 544867537@qq.com
First published on 31st October 2023
Abstract
As a new type of pollutant in the marine environment and terrestrial ecosystems, microplastics have attracted widespread attention. Assessing the ecological risk of microplastics relies on accurately detecting small-sized particles in the environment. Microplastics exhibit unique “fingerprint” characteristics in Raman spectroscopy, making them suitable for rapid identification. In this study, we achieved visualization of microplastics through pseudo-color images generated by Raman spectroscopy imaging. Pseudo-color imaging maps were generated by selecting characteristic peaks and the classical least-squares fitting method was used to visually represent the distribution of different microplastics. The study explored the potential of Raman spectroscopy and its mapping mode in distinguishing various types of mixed microplastics and demonstrated that this approach can identify microplastics in complex environmental samples. Specifically, a cloud-point extraction followed by membrane filtration method was successfully applied to identifying mixed-component microplastics. In summary, the category, quantity, location, and differentiation of microplastics can be accurately analyzed by Raman spectroscopy, which provides a basis for assessing their ecological risk.
Introduction
Microplastics (MPs) are plastic fragments, particles, or fibers in the size range of 1 μm–5 mm.1,2 Plastics in the environment can be degraded and transformed into smaller plastic particles by a combination of UV radiation, physical wear and tear, chemical degradation, and biodegradation.3 As a new type of environmental pollutant, they are increasingly becoming a global hot issue due to their wide distribution and difficulty in degradation.4 Studies have shown that microplastics were detected in seawater,5,6 freshwater,7–9 drinking water,10 bottled water,11,12 airborne dust,13 tea bags,14 sediment,15 and soil.16 In addition, microplastics were detected in fish,17 shellfish,18 and human metabolites.19 Heather A. Leslie found microplastic particles as small as 700 nm in the blood of human volunteers for the first time, and the probability of detecting microplastics in the blood of 22 volunteers was as high as 77.2%.20 This study demonstrates that microplastics are diffused not only throughout the environment but also in our bodies. In a sense, plastic particles have already spread throughout various corners of the ecosystem, but the consequences of long-term deposition in the ecosystem are still not very clear. Therefore, accurate and quantitative detection of small-sized microplastics in the environment is the basis for assessing the ecological risk of microplastics.21 However, due to the limitations of sampling and detection methods, the size of environmental plastics detected in existing studies is generally large, and it is not possible to visualize and accurately identify small-sized microplastics, so their actual numbers may be significantly underestimated.22Currently, the analysis and identification of microplastics in environmental and biological samples are usually by visual, spectroscopic, and thermal analysis methods.23 The visual method mainly used auxiliary tools such as electron microscopy to achieve subjective identification based on the appearance characteristics of microplastics.15 Spectroscopic methods were mainly used to identify polymer composition by obtaining information on microplastic functional groups, and common methods include fast large-area Raman spectroscopy24 and micro-Fourier-transform infrared spectroscopy (micro-FT-IR).25 Micro-FT-IR exhibits a limited spatial recognition rate and struggles to identify particles smaller than 10 μm. Conversely, Raman spectroscopy boasts superior spatial resolution, enabling precise identification of microplastic particles, even those of extremely minute dimensions.26 Raman spectroscopy is widely used in the study of the molecular structure and chemical composition of substances. A mono-chromatic excitation laser beam is generally employed to interact with the target to excite Raman scattering and then form the fingerprint spectrum. Characterization of the chemical structure of the sample and comparison with the fingerprint spectrum of the reference sample enables qualitative analysis of unknown samples.27
The mapping mode of Raman spectroscopy enables signal acquisition on a point-by-point basis within the designated area. The utilization of bright spots in the pseudo-color map generated by Raman imaging facilitates the determination of the sample's type, shape, and size, which significantly enhances both the efficiency and accuracy of detection.28,29 A multivariate curve resolution-alternating least squares (MCR-ALS) analysis of Raman hyperspectral imaging data can achieve direct identification and visualisation of MPs in a complex serum background.30 In addition to spectroscopic identification and quantitative methods, thermogravimetric Fourier-transform infrared spectroscopy coupled with gas chromatography/mass spectrometry (TGA-FTIR-GC/MS) is feasible for the analysis of the type and total mass of microplastics and the additives in them in complex samples.31,32 However, when multiple microplastic particles are mixed, the morphology of the particles in the microscopic observation field is similar, and it is difficult to distinguish the components accurately.5
Thomas Maes and other researchers visualized the analysis of microplastics by staining them with Nile Red and by detecting fluorescence emission.33,34 This method can visually classify polymers into different categories such as hydrophilic or hydrophobic but cannot accurately identify polymer types.35 Raman mapping can automatically detect and analyze each suspected plastic particle point by point and perform mapping analysis, which not only can identify the polymer composition of microplastics and avoid false-positive results of non-plastic particles but also can provide quantitative information, size and distribution of the measured range of microplastics.36
In this study, the feasibility of Raman spectroscopy was tested, and the identification and visualization of microplastics (MPs) are accomplished using Raman spectroscopy and Raman imaging techniques. Whether it is a single component, binary components, multiple samples, or a mixed sample of microplastics with environmental impurities, they all can be effectively distinguished. In addition, taking into account that identifying multiple-component microplastics requires extensive scanning or multi-point detection, the cloud-point extraction followed by membrane filtration method had been applied for the identification of mixed-component microplastics.
Experimental
Samples
This study focused on five plastics commonly found in daily life. Polyethylene (PE), polypropylene (PP), polyvinyl chloride (PVC), polystyrene (PS), and polytetrafluoroethylene (PTFE) were selected to study microplastics discrimination. The samples mainly consisted of granular raw materials, all of which were purchased from Shanghai Fengtai Plastic Chemical Co.PE, PP, PVC, PS and PTFE are all irregularly sized powdered solid forms with particle sizes ranging from 5 μm to 500 μm, and can be directly used for Raman spectroscopy. Hexadecyl trimethyl ammonium bromide (CTAB) and Triton X-45 (TX-45) were purchased from Sigma-Aldrich.
Instrumentation
The Raman mapping model was used to map microplastic samples, respectively, and the bright spots in the Raman imaging pseudo-color map can be used to determine the location, shape and size of the microplastics in the scanned area. Mapping detection of a 500 μm level single component sample was performed with a scan range size of 200 μm × 200 μm and a step size of 10 μm, covering a total of 441 points. The scan range size of binary-component samples is 200 μm × 200 μm with a step size of 8 μm, covering a total of 676 points. The scan range size of 10 μm level binary-component samples is 24 μm × 24 μm with a step size of 2 μm, covering a total of 169 points. For environmental impurity mixtures, the scanning range was 400 μm × 400 μm with a step size of 20 μm, covering a total of 441 points. The scanning range of multi-component samples is 400 μm × 400 μm with a step size of 20 μm, covering a total of 441 points. Raman signals were automatically collected point by point during each scan, resulting in as many spectra as there were points. By testing the samples under different conditions, the ideal conditions for detecting microplastics at the 500 μm level with a 10× magnification objective were obtained. The accumulated integration time is 3 s and the Raman spectrum acquisition range is 200–1800 cm−1. However, the microplastic sample with a size of 10 μm is better detected under a 50× magnification objective. The Labspec6 software imaging analysis method was used to draw the pseudo-color map. The collected spectra are de-baselined and the background is deducted to select a range of characteristic peaks of different plastics, which can show the intensity imaging map of the peaks in the clamped range. The distribution and size of different kinds of microplastic samples can be seen from the clear light and dark distinction of bright spots. The CLS fitting method selects the definitive spectra of microplastics or experimentally collected instantaneous spectra as standard spectra, performs a least-squares fit to the scanned spectral data to determine the detection of sample components by the degree of fit, and forms a pseudo-color map to visualize the analysis of microplastic samples.37
A micro confocal Raman spectrometer (HORIBA LabRAM HR Evolution) consists of a confocal microscope, a laser, a spectrometer with a photodetector, a computer, and other components. It not only is able to obtain a high-resolution microscopic morphology of the sample but also can be selected according to the magnification image of samples for Raman spectroscopy of the specimen micro-region. A micro confocal Raman spectrometer is equipped with four common laser wavelengths, 325 nm, 532 nm, 633 nm, and 785 nm, which can be selected according to the characteristics of the detected substances. The laser source removes stray light and plasma lines from the surrounding area through an interference filter. The polarized light produced by the polaroid enters the microscope through a plane mirror and other optical devices, and the light irradiated on the sample is scattered by the action of the sample. The scattered light collected by the microscope is passed through a Rayleigh filter to remove the Rayleigh scattered light and obtain Raman scattered light. The Raman scattered light is spectroscopically separated by the grating to form the Raman signal in different wavelength bands, and finally the Raman spectrum is displayed on the computer terminal by a CCD detector.
Results and discussion
Raman spectroscopy
Fig. 4 shows the Raman spectra of the microplastic samples after pre-processing the spectral data. There are obvious differences between the Raman spectra of different types of plastics, with different characteristic peak positions. PE has typical peaks near 1067 cm−1, 1132 cm−1 and 1308 cm−1, which are generated due to the stretching vibration of the C–C bond of the functional group inside the PE and CH2 twisting.38 PP has typical peaks near 808 cm−1, which correspond to CH2 rocking and C–C stretching.39 The Raman spectrum of PVC is dominated by the C–Cl stretching band group ranging from 610 to 700 cm−1 and the CH2 deformation band near 1432 cm−1.40 PTFE has unique bands near 732 cm−1 and 1380 cm−1, which correspond to CF2 stretching and CF stretching.41 PS has unique bands near 1001 cm−1 and 1603 cm−1, which are associated with the CC stretches of the aromatic ring and the aromatic CCH quadrant stretch mode.42Raman analysis of single-component microplastics
To perform an accurate qualitative analysis of the microplastics in a region and reduce the false detection rate, a point-by-point scanning approach was adopted to detect their Raman spectra. 300–500 μm PE and PP particles were evenly spread on slides for Raman mapping, and then the microplastic samples were identified by classification on the mapping image as shown in Fig. 1.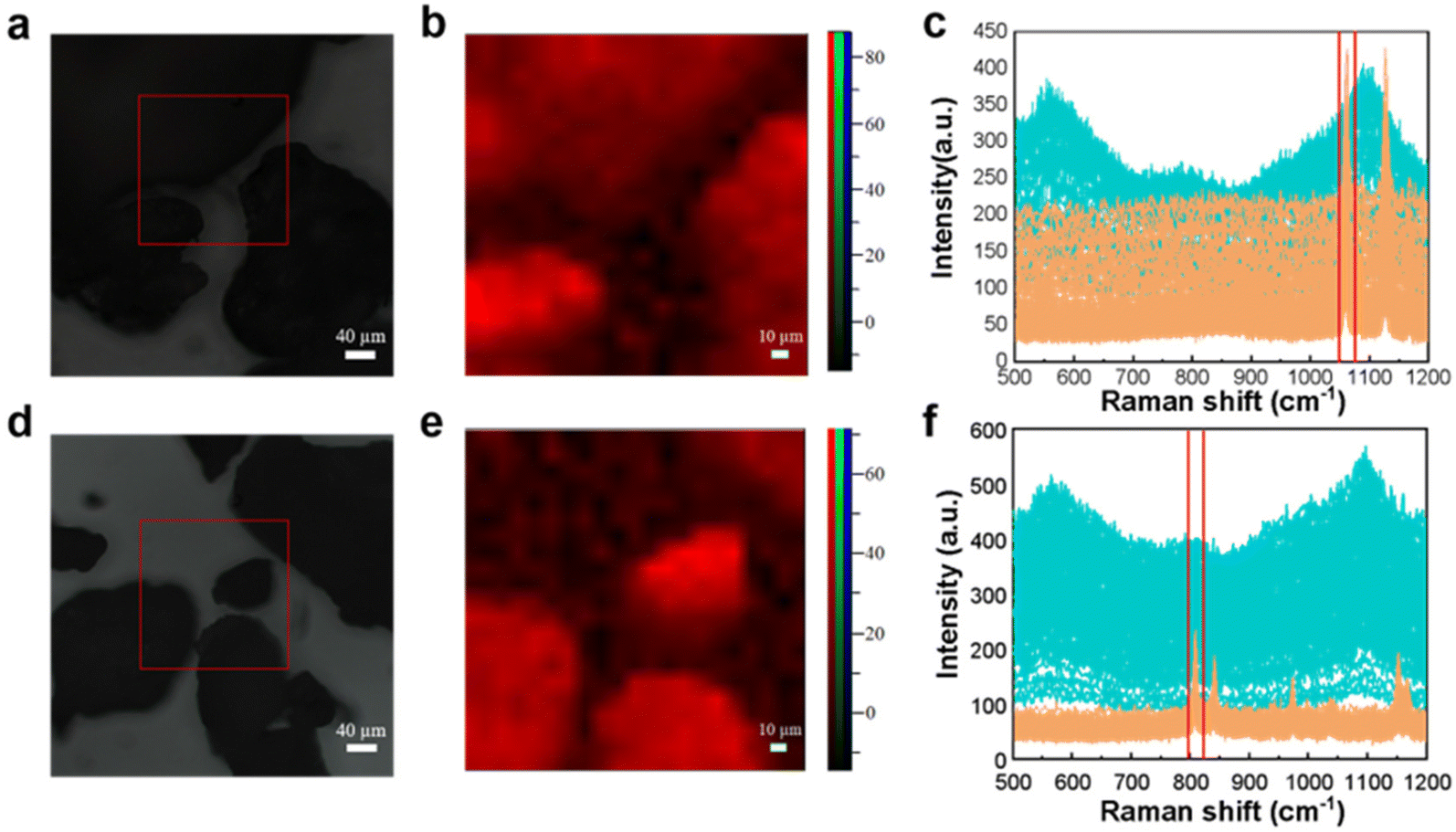 | ||
| Fig. 1 Microscopy images, mapping images, and full Raman spectra of PE (a–c) and PP (d–f). | ||
Fig. 1(a) shows that the PE sample is observed under the microscope as a fragmented inhomogeneous black solid. Raman mapping was performed sequentially for 441 points in the selected area, and the Raman spectrum of each point was plotted, as shown in Fig. 1(c). To enhance visualization, the sample spectrum is displayed in blue, while the blank background spectrum is presented in yellow. It can be seen that the background of the blank background spectrum curve is higher and there is no Raman signal. The Raman spectrum of the sample is shown in Fig. 1(c), which was extracted using software. It has been identified and confirmed to be PE because the peaks matched well with the Raman spectrum (fingerprint) of PE and were consistent with published studies.36 Under 532 nm laser excitation, the characteristic peak at 1067 cm−1 is generated due to the stretching vibration of the C–C stretching. With the cursor clamped to the beam range of 1055–1069 cm−1 where this characteristic peak is located, the peak intensity map of this region is shown in Fig. 1(b). The bright red area indicates that the characteristic peak intensity at this position is higher; more precisely the PE content is higher. The location of PE and its size can be identified by the light–dark distinction of the peak intensity map, and it can be seen that the sample field of view under the microscope corresponds well to the peak pseudo-color map in good agreement. This indicates that the PE samples can be accurately visualized and analyzed by Raman mapping.
Similarly, Fig. 1(d) shows the PP sample as a fragmented black solid under the microscope. Raman mapping of PP samples and plotting of point-to-point Raman spectral images are shown in Fig. 1(f). The PP characteristic peak at 808 cm−1 was selected in the beam range for analysis using the “peak-clamping method”, and the peak intensity map is shown in Fig. 1(e). The identification of PP can be achieved through the brightness of the pseudo-color map. This feature enables the analysis of PP particles using Raman mapping, allowing for accurate visualization and detection of microplastic particles, including their composition and distribution.
Raman analysis of binary-component microplastics
In order to distinguish different mixed microplastics in the same area, the Raman spectrum is also detected by point-by-point scanning. A homogeneous mixture of microplastics with a size of about 500 μm was mapped and plotted with Raman spectra.To obtain more accurate observation results, PE was chosen to be mixed in this experiment. Fig. 2 illustrates the utilization of Raman mapping for the identification and visualization of mixed microplastics with multiple components. The microscopy image of homogeneously mixed PE and PP particles is shown in Fig. 2(a), which shows a noticeable gap between the two black solids. Based on visual observation, there are three possible scenarios: first, both particles are composed of PE; second, both particles are composed of PP; third, one particle is PP while the other is PE. Fig. 2(c) demonstrates that the Raman mapping of selected regions can effectively provide qualitative information about the two particles and display their distribution clearly.
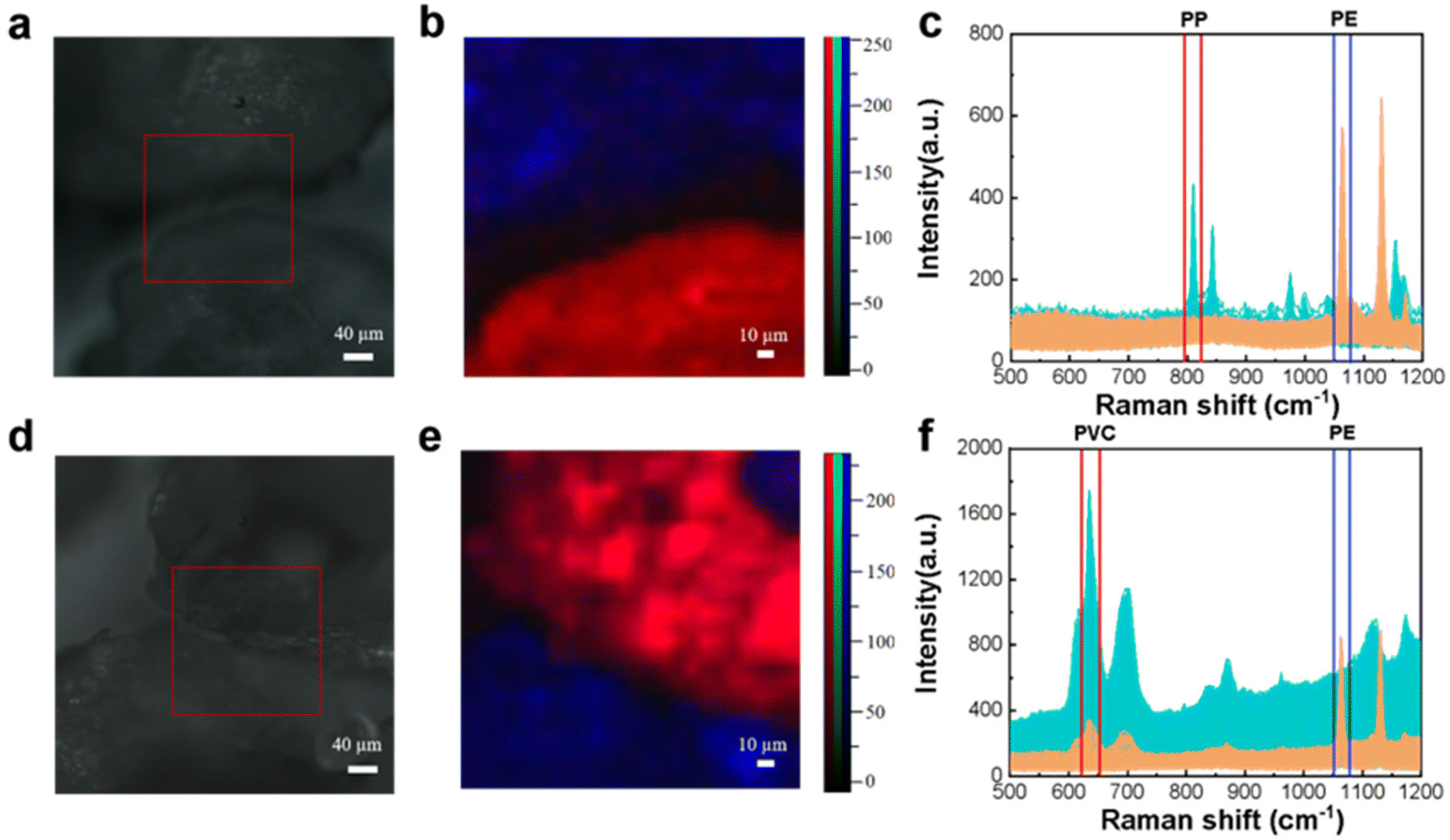 | ||
| Fig. 2 Microscopy images, mapping images, and full Raman spectra of microplastic mixtures (a–c: PP and PE; d–f: PVC and PE). | ||
The PE and PP Raman spectra of multiple scans are shown in yellow and blue, respectively. The C–C bond stretching vibration of PE samples under laser excitation produced a characteristic peak at 1067 cm−1. The PP sample interatomic stretching vibration and CH2 wobble vibration produced a characteristic peak at 808 cm−1.
The characteristic peaks of the two microplastics in the ranges of 1055–1069 cm−1 and 794–820 cm−1 were selected by clamping the peaks with blue and red cursors, respectively, and the peak intensities of the clamped ranges are shown in the corresponding colors in Fig. 2(b). When the characteristic peak at 1067 cm−1 is selected, a bright blue area appears in the upper left region, indicating a higher intensity and a larger presence of the PE component. Similarly, when the characteristic peak at 808 cm−1 is chosen, a bright red area appears below, also indicating a higher intensity and a larger presence of the PP component. The color of the pseudo-color map corresponds to the color of the cursor used to capture the peaks, and it aligns well with the samples observed under the microscope. This suggests that accurate visual analysis can be conducted.
The characteristic peaks at 1067 cm−1 and 636 cm−1 of PE and PVC selected, respectively, for feature identification are shown in Fig. 2(f). The Raman spectra of the PE sample show that some of the data follow the same trend as those of the PVC sample, with the characteristic peaks of both PE and PVC. The reason for this occurrence is that when scanning the junction of two samples, one plastic sample becomes the background information of the other sample, and the specific identification of the microplastic can be done using the intensity of the characteristic peak. If the peak intensity at 636 cm−1 is relatively low compared to the intensity at 1067 cm−1, the point is identified as PE; otherwise, it is identified as PVC. The composition was identified by the color of the bright spot of the pseudo-color image obtained by the pinch peak method, and it can be seen that the pseudo-color image of Raman mapping has good correspondence with the sample distribution image under the microscope.
It is worth noting that the image observed under the microscope in general is of two irregular black solids connected. It is likely to be incorrectly identified as plastic I on the lower left region and plastic II on the upper right region by visual inspection. However, the pseudo-color map of Raman mapping suggests that the characteristic peak information of PE is at the upper right region of the PVC particles.
Raman mapping draws spectral images by acquiring Raman signals point by point. Its inherently sensitive detection performance allows weak Raman signals to be presented. This performance can not only identify the type of microplastic sample but also display its location and size. Raman mapping greatly improves the detection efficiency of microplastic identification and reduces false detection rates.
Raman analysis of 10 μm sized samples
Experiments had also been carried out to identify microplastics of smaller sizes that are still present in actual environmental samples. When detecting mixed microplastic solids of 10 μm size, there is a significant overlap of numerous particles during Raman detection due to their small sizes, which prevent them from agglomerating and adsorbing onto each other. The particles cannot be completely separated for scanning. It is difficult to accurately distinguish the location and characterize the different particles by Raman mapping. To address this situation, a one-to-one homogeneous mixture of 10 μm mixed microplastic solids and 0.01 mol L−1 hexadecyl trimethyl ammonium bromide (CTAB) was prepared, ensuring complete dispersion and configuration into a solution form for testing. The comparison between Fig. 3(a) and (b) shows that the mapping effect is better and that accurate visualization is possible for small particle size microplastics.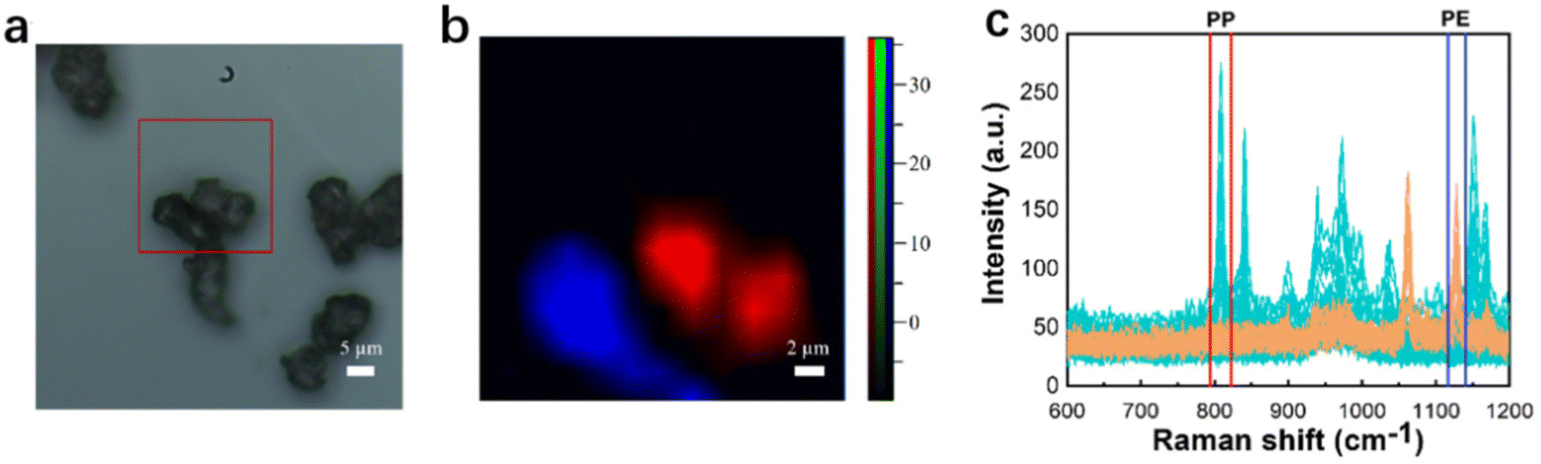 | ||
| Fig. 3 Microscopy image, mapping image, and full Raman spectra of 10 μm PE and PP (a–c). | ||
Raman analysis of multiple-component microplastics
However, due to the software's limitation of having only red, green, and blue channels, the peak-clamping method can only analyze a maximum of three samples simultaneously, making it unsuitable for identifying mixtures with more than three components. The classical leas-squares (CLS) fitting method was employed to address this issue allowing for the simultaneous identification of mixtures of three or more components. The definitive spectra of several components were selected as standard spectra and fitted by CLS to the collected mapping spectral data. Besides, cloud-point extraction as an eco-friendly procedure43 using TX-45 as the extracting agent was applied to pre-concentrate microplastics, and the samples are closely spaced. By selecting an appropriate scanning range or replacing a small amount of the collected area, qualitative analysis of mixed microplastic samples can be achieved.In particular, the samples were placed on a clean silicon wafer for Raman spectroscopy analysis to obtain the standard spectra applied to CLS, which are shown in Fig. 4. A mixed cellulose ester (MCE) filter membrane without obvious peaks was selected for microplastic filtration.37 A mixed solution of microplastic particles was filtered through the MCE filter membrane after cloud-point extraction, followed by drying and in situ Raman spectroscopy detection on the membrane.
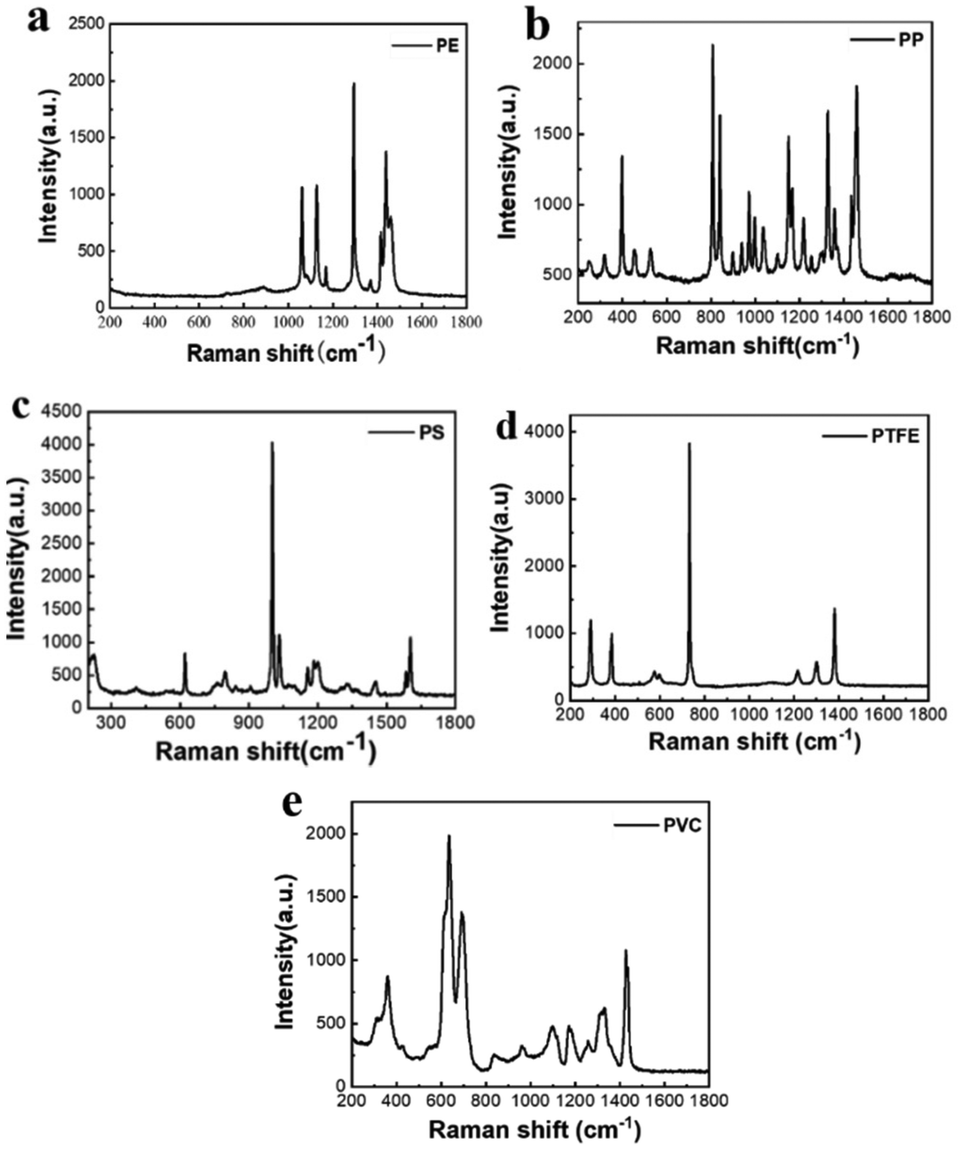 | ||
| Fig. 4 Raman spectra of (a) PE, (b) PP, (c) PS, (d) PTFE, and (e) PVC. | ||
The CLS determines the nature of each spectrum from the percentage probability of its fit and plots the pseudo-color map in different colors. In Fig. 5(c), it is apparent that the PS fraction corresponds to the red color, the PTFE fraction corresponds to the blue color, the PP fraction corresponds to the green color, and the PE fraction corresponds to the yellow color. The black color represents the Raman spectrum of MCE. The pseudo-color diagram shows the distribution and the size of the various components in a clear way. Fig. 5(d) shows all the spectral data collected under Raman mapping. The results of feature identification for these spectral data are displayed in the figure, where each characteristic peak of the microplastics is clear and distinct without any overlap. The experimental results demonstrate that Raman mapping can accurately distinguish microplastic samples of multiple components, which improves the efficiency of microplastic identification and reduces the false detection rate.
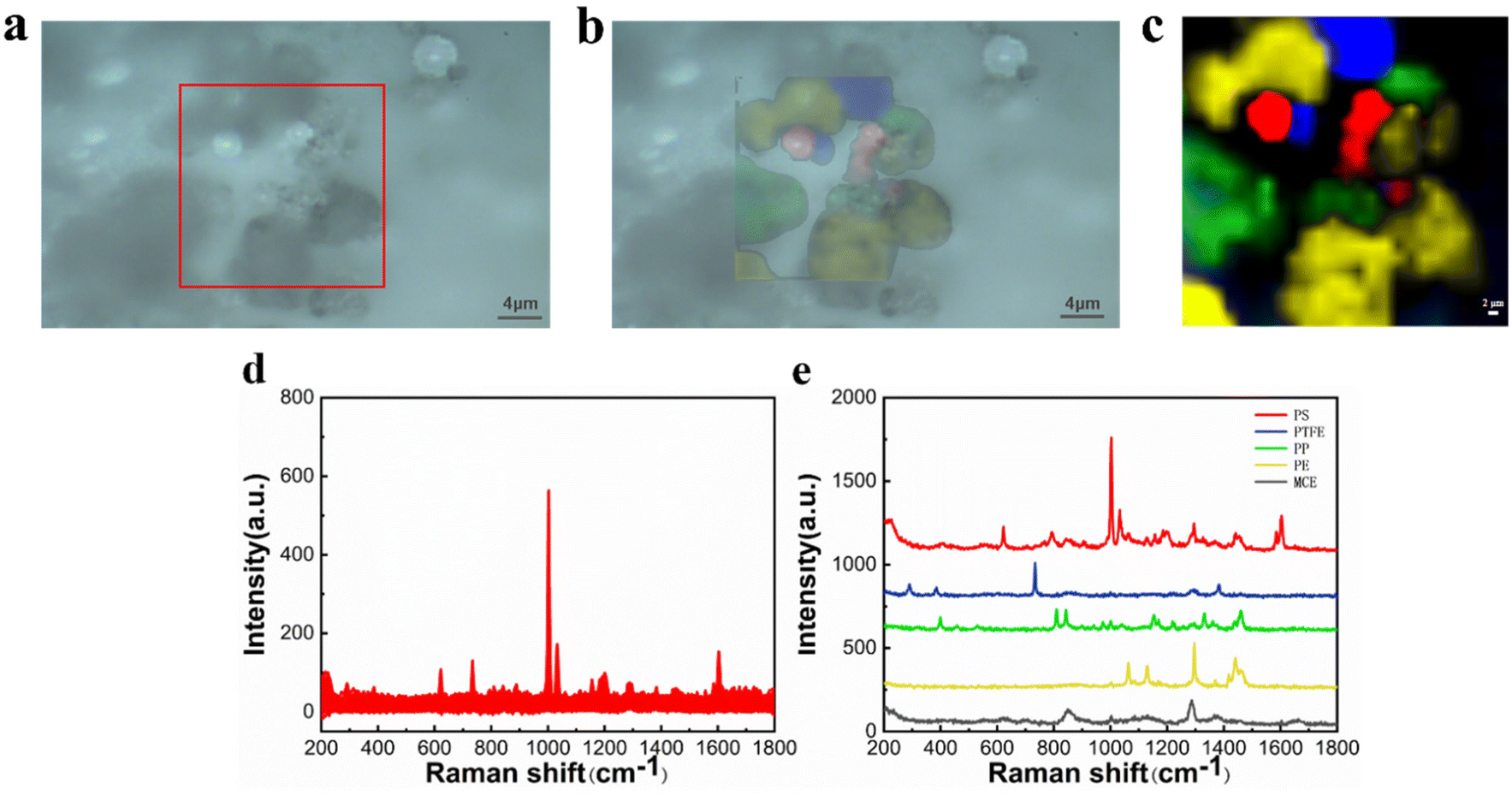 | ||
| Fig. 5 Microscopy image (a), fitting image (b), mapping image (c), original Raman spectra (d), and Raman spectra of PS, PTFE, PP, PE and MCE (e). | ||
Raman analysis of a simulated environmental sample
In order to study more challenging evidence, it was decided to test the Raman imaging and the mapping mode on real-world environmental samples. Fig. 6 shows the evidence analyzed by Raman imaging and the corresponding results obtained using the mapping mode. A sample of off-white perlite was selected to simulate impurities in the environment that might interfere with the detection of microplastics, and was mixed with two “unknown” microplastics by randomly selecting two samples from the existing samples and then mapped for scanning. The image observed under the microscope and the Raman spectra acquired by mapping are shown in Fig. 6(a and c), respectively. The perlite sample has no distinctive characteristic peaks, so it is not possible to use the peak-clamping method for component analysis of mixed samples. Therefore, the classical least-squares (CLS) fitting method was used. Experimentally collected instantaneous spectra of several components were selected as standard spectra and fitted by CLS to the collected mapping spectral data. The Raman spectrum of the sample is shown in Fig. 6(c), which was extracted using software. The yellow one has been identified and confirmed to be PE because the peaks matched well with the Raman spectrum (fingerprint) of PE, which involved the typical peaks near 1067 cm−1, 1132 cm−1 and 1308 cm−1. The blue one has been confirmed to be PP because of the unique peaks near 808 cm−1. In Fig. 6(b), it can be seen that the red color and the blue color correspond to the PP fraction and the PE fraction, respectively, and the green color corresponds to the simulated environmental sample impurities. The pseudo-color diagram shows the distribution and the size of the various component samples in a clear and visual way. The experimental results demonstrate that Raman mapping can accurately distinguish microplastic samples from environmental impurities in the presence of interference.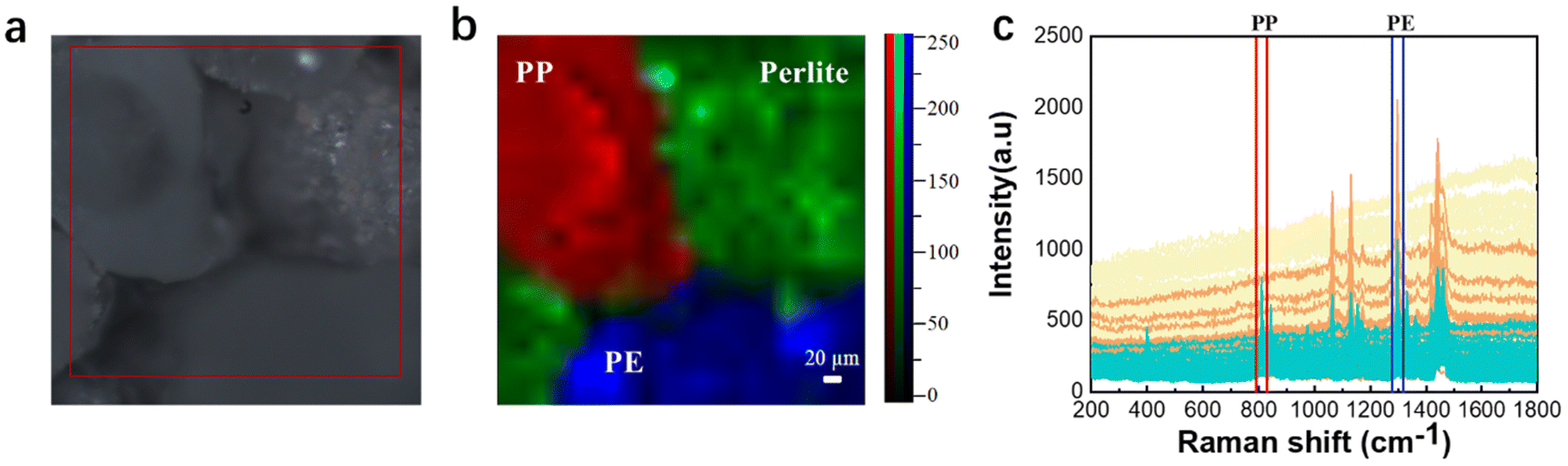 | ||
| Fig. 6 Microscopy image (a), mapping image (b), and full Raman spectra of PE, PP, and impurities (c). | ||
Conclusions
In summary, experimental identification of single- and multi-component mixed microplastics of different particle sizes had been accomplished by Raman mapping. The characteristic peaks of the point-to-point Raman spectra were analyzed by the peak-clamping method and the CLS fitting method and plotted in different pseudo-color images. This not only helps visualize the size and distribution of the different microplastics in the mixed samples but also allows visualization of the small components present in the sample when their spectral signals are weak, making Raman spectroscopy more accurate and comprehensive. Raman mapping has high detection efficiency for microplastic samples mixed with environmental impurities and can well distinguish environmental background impurities from microplastic samples.However, in the process of detecting microplastics, the success rate of identifying plastic particles with a particle size of less than 1 μm is not yet very high. And in the actual environmental samples, there are many organic substances attached to the surface of microplastics, which will interfere with Raman detection. The current steps to eliminate organic matter are still relatively cumbersome. How to remove the organic matter attached to the surface of microplastics by a simple method for more efficient visual detection of microplastics using Raman mapping – this is still a question that we need to study in the future.
Author contributions
Kaili Liu: data curation, software, formal analysis, validation, investigation, visualization, and original draft. Xu Pang: formal analysis, investigation, review and editing. Huacai Chen: conceptualisation, supervision, investigation, methodology, project administration, review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the team members for their assistance and contribution in sampling and experiments, and their valued opinions and advice, especially for helping with this study.References
- R. C. Thompson, Y. Olsen, R. P. Mitchell, A. Davis, S. J. Rowland, A. W. G. John, D. McGonigle and A. E. Russell, Science, 2004, 304, 838 CrossRef CAS PubMed.
- J. C. Anderson, B. J. Park and V. P. Palace, Environ. Pollut., 2016, 218, 269 CrossRef CAS PubMed.
- M. O. Rodrigues, N. Abrantes, F. J. M. Gonçalves, H. Nogueira, J. C. Marques and A. M. M. Gonçalves, Sci. Total Environ., 2018, 633, 1549 CrossRef CAS PubMed.
- A. Isobe, S. Iwasaki, K. Uchida and T. Tokai, Nat. Commun., 2019, 10, 1 CrossRef.
- V. Hidalgo-Ruz, L. Gutow, R. C. Thompson and M. Thiel, Environ. Sci. Technol., 2012, 46, 3060 CrossRef CAS.
- W. Zhang, S. Zhang, J. Wang, Y. Wang, J. Mu, P. Wang, X. Lin and D. Ma, Environ. Pollut., 2017, 231, 541 CrossRef CAS.
- D. Eerkes-Medrano, R. C. Thompson and D. C. Aldridge, Water Res., 2015, 75, 63 CrossRef CAS PubMed.
- M. C. M. Blettler, E. Abrial, F. R. Khan, N. Sivri and L. A. Espinola, Water Res., 2018, 143, 416 CrossRef CAS.
- J. Li, H. Liu and J. P. Chen, Water Res., 2018, 137, 362 CrossRef CAS PubMed.
- M. Pivokonsky, L. Cermakova, K. Novotna, P. Peer, T. Cajthaml and V. Janda, Sci. Total Environ., 2018, 643, 1644 CrossRef CAS PubMed.
- R. Lehner, C. Weder, A. Petri-Fink and B. Rothen-Rutishauser, Environ. Sci. Technol., 2019, 53, 1748 CrossRef CAS PubMed.
- D. Schymanski, C. Goldbeck, H.-U. Humpf and P. Fürst, Water Res., 2018, 129, 154 CrossRef CAS.
- M. Klein and E. K. Fischer, Sci. Total Environ., 2019, 685, 96 CrossRef CAS PubMed.
- L. M. Hernandez, E. G. Xu, H. C. E. Larsson, R. Tahara, V. B. Maisuria and N. Tufenkji, Environ. Sci. Technol., 2019, 53, 12300 CrossRef CAS PubMed.
- J. C. Prata, J. P. D. Costa, A. C. Duarte and T. Rocha-Santos, Trends Anal. Chem., 2018, 110, 150 CrossRef.
- D. He, Y. Luo, S. Lu, M. Liu, Y. Song and L. Lei, Trends Anal. Chem., 2018, 109, 163 CrossRef CAS.
- A. L. Lusher, N. A. Welden, P. Sobral and M. Cole, Anal. Methods, 2017, 9, 1346 RSC.
- C. M. Rochman, A. Tahir, S. L. Williams, D. V. Baxa, R. Lam, J. T. Miller, F.-C. Teh, S. Werorilangi and S. J. Teh, Sci. Rep., 2015, 5, 14340 CrossRef CAS.
- P. Schwabl, S. Köppel, P. Königshofer, T. Bucsics, M. Trauner, T. Reiberger and B. Liebmann, Ann. Intern. Med., 2019, 171, 453 CrossRef PubMed.
- H. A. Leslie, M. J. M. van Velzen, S. H. Brandsma, A. D. Vethaak, J. J. Garcia-Vallejo and M. H. Lamoree, Environ. Int., 2022, 163, 107199 CrossRef CAS.
- K. D. Cox, G. A. Covernton, H. L. Davies, J. F. Dower, F. Juanes and S. E. Dudas, Environ. Sci. Technol., 2019, 53, 7068 CrossRef CAS.
- E. L. Teuten, J. M. Saquing, D. R. U. Knappe, M. A. Barlaz, S. Jonsson, A. Björn, S. J. Rowland, R. C. Thompson, T. S. Galloway, R. Yamashita, D. Ochi, Y. Watanuki, C. Moore, P. H. Viet, T. S. Tana, M. Prudente, R. Boonyatumanond, M. P. Zakaria, K. Akkhavong, Y. Ogata, H. Hisashi, I. Satoru, M. Kaoruko, H. Yuki, I. Ayako, S. Mahua and T. Hideshige, Philos. Trans. R. Soc., B, 2009, 364, 2027 CrossRef CAS.
- E. Besseling, B. Wang, M. Lürling and A. A. Koelmans, Environ. Sci. Technol., 2014, 48, 12336 CrossRef CAS.
- R. Lenz, K. Enders, C. A. Stedmon, D. M. A. Mackenzie and T. G. Nielsen, Mar. Pollut. Bull., 2015, 100, 82 CrossRef CAS.
- S. Primpke, C. Lorenz, F. R. Rascher and G. Gerdts, Anal. Methods, 2017, 9, 1499 RSC.
- A. Käppler, D. Fischer, S. Oberbeckmann, G. Schernewski, M. Labrenz, K.-J. Eichhorn and B. Voit, Anal. Bioanal. Chem., 2016, 408, 8377 CrossRef PubMed.
- R. R. Jones, D. C. Hooper, L. Zhang, D. Wolverson and V. K. Valev, Nanoscale Res. Lett., 2019, 14, 1 CrossRef PubMed.
- J. Hutchings, C. Kendall, B. Smith, N. Shepherd, H. Barr and N. Stone, J. Biophotonics, 2009, 2, 91 CrossRef CAS PubMed.
- C. F. Araujo, M. M. Nolasco, A. M. P. Ribeiro and P. J. A. Ribeiro-Claro, Water Res., 2018, 142, 426 CrossRef CAS.
- M. Tian, C. L. M. Morais, H. Shen, W. Pang, L. Xu, Q. Huang and F. L. Martin, J. Hazard. Mater., 2022, 422, 126892 CrossRef CAS PubMed.
- Y. Liu, R. Li, J. Yu, F. Ni, Y. Sheng, A. Scircle, J. V. Cizdziel and Y. Zhou, Environ. Pollut., 2020, 272, 115946 CrossRef.
- R. Mansa and S. Zou, Environ. Adv., 2021, 5, 100117 CrossRef CAS.
- T. Maes, R. Jessop, N. Wellner, K. Haupt and A. G. Mayes, Sci. Rep., 2017, 7, 1 CrossRef.
- G. Erni-Cassola, M. I. Gibson, R. C. Thompson and J. A. Christie-Oleza, Environ. Sci. Technol., 2017, 51, 13641 CrossRef CAS PubMed.
- W. J. Shim, Y. K. Song, S. H. Hong and M. Jang, Mar. Pollut. Bull., 2016, 113, 469 CrossRef CAS.
- Z. Sobhani, M. A. Amin, R. Naidu, M. Megharaj and C. Fang, Anal. Chim. Acta, 2019, 1077, 191 CrossRef CAS PubMed.
- D. Liu, Y. Song, F. Li and L. Chen, China Environ. Sci., 2020, 40, 4429 CAS.
- H. Sato, M. Shimoyama, T. Kamiya, T. Amari, S. Šašic, T. Ninomiya, H. W. Siesler and Y. Ozaki, J. Appl. Polym. Sci., 2002, 86, 443 CrossRef CAS.
- A. S. Nielsen, D. N. Batchelder and R. Pyrz, Polymer, 2002, 43, 2671 CrossRef CAS.
- E. Rusen, B. Marculescu, L. Butac, N. Preda and L. Mihut, Fullerenes, Nanotubes Carbon Nanostruct., 2008, 16, 178 CrossRef CAS.
- J. Mihály, S. Sterkel, H. Ortner, L. Kocsis, L. Hajba, E. Furdyga and J. Mink, Croat. Chem. Acta, 2006, 79, 497 Search PubMed.
- D. B. Menezes, A. Reyer, A. Marletta and M. Musso, Mater. Res. Express, 2017, 4, 015303 CrossRef.
- W. I. Mortada, Microchem. J., 2020, 157, 105055 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3an01270k |
| This journal is © The Royal Society of Chemistry 2024 |