
SERS-based immunoassay on a plasmonic syringe filter for improved sampling and labeling efficiency of biomarkers†
Eunice
Ebbah
,
Anthony
Amissah
,
Jun-Hyun
Kim
 * and
Jeremy D.
Driskell
*
* and
Jeremy D.
Driskell
*
Department of Chemistry, Illinois State University, Normal, IL 61790, USA. E-mail: jkim5@ilstu.edu; jdriske@ilstu.edu
First published on 21st November 2023
Abstract
Rapid, sensitive, and quantitative detection of biomarkers is needed for early diagnosis of disease and surveillance of infectious outbreaks. Here, we exploit a plasmonic syringe filter and surface-enhanced Raman spectroscopy (SERS) in the development of a rapid detection system, using human IgG as a model diagnostic biomarker. The novel assay design facilitates multiple passages of the sample and labeling solution through the detection zone enabling us to investigate and maximize sampling efficiency to the capture substrate. The vertical flow immunoassay process in this study involves the utilization of filter paper embedded with gold nanoparticles (AuNPs) to form a plasmonic substrate. Capture antibody (anti-human IgG) is then immobilized onto the prepared plasmonic paper and inserted into a vertical flow device (syringe filter holder). Sample solution is passed through the filter paper and the target antigen (human IgG) is selectively captured by the immobilized antibody to form an antibody–antigen complex. Next, functionalized AuNPs as extrinsic Raman labels (ERLs) are passed through the filter paper to label the captured biomarker molecules forming a layered structure. This sandwiched geometry enhances plasmonic coupling and SERS signal to provide highly sensitive detection of biomolecules. Systematic studies to investigate the impact of multiple infuse/withdraw cycles of the sample and labeling solutions reveal that antigen and ERL binding are maximized with 10 and 20 cycles, respectively. The optimized assay achieves a detection limit of ∼0.2 ng mL−1 for human IgG with a total assay time of less than 5 minutes, meeting the demands for rapid point of care diagnostics. Additionally, the optimized platform was implemented in the quantitative analysis of the SARS-CoV-2 nucleocapsid protein, the typical target in commercial, FDA-approved rapid antigen tests for COVID-19.
Introduction
The recent SARS-CoV-2 pandemic highlighted the importance of rapid and accurate diagnostic testing that can be widely deployed for large population screening and surveillance.1 Reverse transcriptase polymerase chain reaction (rtPCR) and lateral flow assay (LFA) rapid diagnostic tests were primarily employed to surveil patient infection and direct patient isolation; however, the slow processing time of rtPCR and poor sensitivity of rapid antigen tests, particularly for asymptomatic patients, exposed the need for better point of care (POC) diagnostics.2–4 Moreover, rtPCR and lateral flow rapid antigen tests provide limited capacity for multiplexed detection. Yet, multiplexed POC tests are highly desirable to identify the causative agent and differentiate patients with similar symptoms, such as fever, headache, and congestion and to predict disease severity via quantitation of multiple biomarkers.5Surface-enhance Raman spectroscopy (SERS)-based assays exhibit many of the desirable attributes for developing next-generation POC tests.6–10 Rationally designed SERS-promoting plasmonic architectures have been developed to achieve the requisite sensitivity for clinically accurate POC testing. Moreover, SERS spectra are spectroscopically narrow, enabling the differentiation of many Raman reporter labels for multiplexed detection, as evidenced in a recent report that successfully achieved multiplexed detection of 26 labels using SERS.11 Lastly, SERS measurements are readily acquired under ambient conditions with commercially available portable instruments. Given these known benefits, SERS-based assays have been intensively explored. These assays are typically designed in a sandwich format in which the analyte is captured by an antibody bound to a solid phase sensing surface. In a second step, a Raman reporter functionalized plasmonic particle then labels the bound antigen to facilitate detection and quantitation. Early works capitalizing on SERS for readout utilized solid, flat capture substrates that relied on diffusion for mass transport, mimicking the sample processing steps of an ELISA.7,8,12 The resulting assays required long incubation times and multiple labor-intensive washing steps, limiting utility for rapid diagnostics. Recently, SERS-based assays have been re-imagined to address the special requirements of POC testing, using magnetic beads13,14 and LFA formats.15–21
LFAs are particularly attractive because the method is well established with validated materials, fabrication methods, testing, and precedent for regulatory approval. Extension of LFAs to incorporate SERS-labels is straightforward and can be as simple as co-immobilizing a Raman reporter molecule on the antibody–gold nanoparticle conjugate extensively used in commercial LFAs for visual detection.16,20 Advances in SERS-based LFAs include optimization of the plasmonic particle used for labeling to maximize SERS sensitivity or the use of several unique labels for multiplexed detection.15,17–19,21–23 To this end, Zhang et al., developed a highly SERS-active core–shell plasmonic nanoparticle for use in a triplexed LFA to detect cardiac biomarkers achieving sub-picogram per milliliter LODs.21 However, even the advancements in sensitivity and multiplexing capacity does not address the inherent limitations in LFA designs, such as low sample volume, false negative incurred by the hook effect associated with excess antibodies and/or antigens, low multiplexing capacity, and moderate assay times of 5–30 minutes.24–26
Ongoing efforts to address some of the limitations of LFAs include the development of vertical flow assays (VFAs).24,27–29 VFAs are emerging as an alternative to LFAs that provide faster results, allow for larger volumes, afford greater multiplexing potential, and circumvent the hook effect.24 Much like LFAs, early VFAs relied on visual readout for detection that limited LOD and clinical accuracy. SERS-detection has been effectively coupled with VFAs to capitalize on the strengths of SERS detection and the VFA format.30–34 The concept of a SERS-based VFAs was first introduced and demonstrated by Clarke et al., using a commercially available nitrocellulose vertical flow device and functionalized spherical gold nanoparticles as the SERS tag.31 In an effort to improve assay sensitivity, novel plasmonic particles, such as core–shell nanostructures, have been incorporated into VFAs, achieving LODs less than 1 pg mL−1.30
Recently, our group reported on plasmonic membranes to facilitate plasmonic coupling for improved LODs, as an alternative approach to anisotropic or core–shell SERS tags.34,35 In that work, we demonstrated that a VFA designed with filter paper embedded with gold nanoparticles rather than the commonly used nitrocellulose yields substantial improvement in SERS signal.34 However, much like other LFA protocols, the sample and label made a single pass through the capture membrane. Moreover, the absorbent pad, limited sample, label, and wash volumes to a combined maximum volume of ∼350 μL. Here, we use a syringe to facilitate sample and label solutions passage through the capture membrane. The syringe allows for larger sample volumes to be analyzed, as well as multiple cycling steps of the sample and label solutions through the capture substrate to maximize binding efficiency. The capability of the designed filtration device as a SERS-based VFA is established using human IgG as a model antigen. The improvement in sensitivity with multiple sample passages through the capture substrate is clearly demonstrated. After optimizing and assessing the analytical figures of merit, the platform was configured with appropriate matched pair antibodies for the detection of SARS-CoV-2 nucleocapsid protein.
Experimental
Materials and reagents
Gold nanoparticles (AuNPs) were synthesized using tetrachloroauric(III) acid (HAuCl4·3H2O) and citric acid trisodium salt dihydrate from Acros Organics. Extrinsic Raman labels (ERLs) were prepared using unconjugated, citrate-capped AuNP (60 nm) purchased from Ted Pella Inc. Bovine serum albumin lyophilized powder (BSA) and purified IgG from human serum (I4506) were obtained from Sigma-Aldrich. Whatman grade 4 filter paper, phosphate buffered saline, 4-nitrobenzenethiol (NBT), Tween 20, trehalose anhydrous, polyclonal goat anti-human IgG antibody (31119), normal rabbit serum, and normal mouse serum were purchased from Thermo Scientific. Normal bovine serum, humanized monoclonal anti-SARS CoV-2 NP antibody (MBS355887), humanized monoclonal anti-SARS CoV-2 NP antibody (MBS355888), and recombinant SARS CoV-2 nucleocapsid protein (NP) (MBS355894) were purchased from MyBioSource. Normal human serum was purchased from Millipore. The syringe apparatus for the assay consisted of three parts purchased from Analytics Shop: male luer adapter to 1/4-28, 1.5 mm bore, PEEK (JR-CMlAPK), female luer adapter to 1/4-28, 1.5 mm bore, PEEK (JR-CFLAPK), and union, PEEK, LP 1.3 mm bore, body only, 1/4-28 (JR-065).AuNP synthesis and plasmonic paper capture substrate preparation
Highly concentrated spherical AuNPs (60 nm) were synthesized to prepare plasmonic papers. The synthesis of AuNP was achieved using a slightly modified thermal reduction method.36–38 Nanopure water (98 mL) was added to a 2.0 mL aliquot of gold solution (1 wt% HAuCl4·3H2O) in a 250 mL Erlenmeyer flask with a magnetic stirring bar. The solution was vigorously stirred for 30 min, and the flask was heated to boiling. Trisodium citrate (1.5 mL at 1 wt%) was quickly added to the boiling solution and immediately removed from heat. The resulting solution was then allowed to stir at room temperature for 40 min, resulting in the formation of spherical AuNPs. The final solution was then stored at room temperature without further purification prior to use. It is noted that the synthetic conditions readily resulted in the formation of a high concentration of AuNPs, allowing for effective fabrication with a filter paper to prepare plasmonic substrates in a cost-effective way.Whatman grade 4 filter papers with 20–25 μm pore sizes were utilized in the preparation of the plasmonic capture substrate. Employing a previously developed dip coating method,39 the papers were first dried at 40 °C in an oven overnight and then submerged in 10 mL of the synthesized AuNP suspension in a plastic Petri dish (60 mm × 15 mm) for 24 h to allow for AuNP adsorption. The filter papers were removed from the AuNP suspension, treated with 95% EtOH to remove excess AuNPs and dried in the oven (∼40 °C) for 30 min. The resulting plasmonic paper was then cut into circles of 3 mm diameter using a single-hole punch. To prepare the plasmonic capture substrate, 2 μL of 1 mg mL−1 goat anti-human IgG in a solution of 1% (wt/v) trehalose in PBS was applied onto the 3 mm prepared plasmonic paper circles and oven dried for 30 min at 37 °C. Previously, it has been established that passively adsorbed antibodies bind to AuNPs through cysteine residues (e.g., Au–S interactions) to form a robust, irreversibly bound capture antibody layer.40–45 The oven dried SERS capture substrates were then blocked with 100 μL, 1% (wt/v) BSA solution for 1 h to minimize nonspecific binding before use.
Extrinsic Raman label (ERL) preparation
Following a previously established procedure, a Raman reporter molecule (4-nitrobenzenethiol, NBT) together with a detection antibody were both adsorbed onto commercially available 60 nm AuNPs to prepare extrinsic Raman labels (ERLs). Commercially sourced AuNPs were used due to high uniformity and stability34,46 and the 60 nm diameter was selected because it has been reported as the ideal size of Au-based plasmonic NPs for SERS application.47,48 Briefly, the pH of a 1.0 mL aliquot of 60 nm AuNPs (Ted Pella, Inc.) was adjusted to pH 8.0 with the addition of 50 mM phosphate buffer (40 μL). A detection antibody (30 μg) and 1 mM NBT (10 μL) were simultaneously added to the AuNP suspension. The suspension was vortexed and incubated at room temperature for 90 min to allow maximum adsorption of the antibody and NBT onto the AuNPs surface, forming the ERLs. As noted above, antibodies passively bind to form a robust layer.40–45 Excess, unbound detection antibody and NBT were removed from the ERL suspension via centrifugation at 5000g for 5 min to pellet the ERL conjugate, removal of the clear supernatant, and resuspension in fresh 2 mM phosphate buffer (pH 8.0). Two additional centrifugation purification cycles were performed to thoroughly remove unbound antibody and reporter molecules. The concentration of ERLs was increased by resuspending the pelleted conjugates in half the original AuNP volume of buffer during the final resuspension step of the washing cycles for a final ERL concentration of ∼5.2 × 1010 ERLs per mL. To further block any exposed surface of the AuNPs and mimic physiological ionic strength to prevent protein unfolding, 10 μL of 10% (wt/v) BSA and 10 μL of 10% (wt/v) NaCl were added to the purified ERLs, respectively.Syringe filter immunoassay protocol
The plasmonic capture substrate with pre-immobilized goat anti-human IgG and blocked with BSA was placed in a syringe filter apparatus and the fittings screwed to keep the capture substrate in place. Sample solutions of antigen ranging in concentration from 0.1–200 ng mL−1 human IgG were prepared by dilution of 1 mg mL−1 human IgG stock solution in PBS. PBS was used as a negative control to determine nonspecific binding of ERLs. A 1 mL syringe was loaded with 100 μL of sample solution containing antigen and passed through the plasmonic capture substrate. Importantly, the sample delivery by the syringe and porous capture substrate allowed for the sample to be passed through the capture substrate by flowing in a forward and reverse direction multiple times to optimize antigen capture efficiency. Next, a 1 mL syringe was loaded with 100 μL of ERLs (2× concentrated relative to stock concentration unless noted otherwise) and passed through the plasmonic capture substrate, again allowing for optimization of the number of passages through the filter to optimize labeling of captured antigen. Lastly, a 100 μL wash buffer (1% wt/v BSA, 5% Tween 20 in PBS) was passed through the filter followed by two additional rinses to remove excess unbound antigens and ERLs, leaving only the specifically bound components on the plasmonic paper. The paper was removed from the filter holder and allowed to dry in a desiccator before SERS analysis. This entire method of analysis, i.e., sampling, labeling, washing, and spectral acquisition, required approximately 5 minutes.Enzyme-linked immunosorbent assay (ELISA)
An ELISA was performed as a gold standard to compare the analytical performance of the SERS filter assay. To this end, 100 μL of 5 μg mL−1 goat anti-human IgG antibody was added to each well of a 96-well Immulon 2HB microtiter plate and incubated overnight at 4 °C to allow for antibody adsorption. The capture antibody solution was removed, each well was rinsed three times with 300 μL of PBS, and the wells were blocked by three sequential additions of 300 μL SuperBlock. Following the third addition/removal of SuperBlock, 100 μL of standard solutions of human IgG prepared in PBS with 1% BSA (0–1000 ng mL−1) were added to the wells in triplicate and incubated for 2 h. The sample solutions were then removed, each well rinsed three times with 300 μL of PBS containing 1% BSA, and 100 μL of horseradish peroxidase (HRP)-labelled goat anti-human IgG was added. The HRP antibody was allowed to react for 2 h before removal and each well rinsed three times with 300 μL of PBS containing 1% BSA. Finally, 100 μL of 1-step ABTS solution was added to each well and the absorbance at 410 nm was recorded on a Varioskan plate reader (Thermo Scientific, Inc.) after allowing 20 min for color development of the enzymatic reaction.Instrumentation
Results and discussion
Design and principle of filter assay
Fig. 1A shows the overview of filtration-based immunoassay, where an antibody is pre-immobilized onto a plasmonic paper to form a robust SERS capture substrate. The plasmonic capture substrate is inserted into the filtration device and flow of sample solution through the porous substrate is achieved using a syringe. The highly specific nature of antibodies and antigens allows targeted analytes in sample solutions to be selectively captured and concentrated by the immobilized antibody on the plasmonic substrate. Next, the addition of ERLs, delivered via syringe, specifically labels the bound antigen, and allows for SERS detection. In our previous work, we established that the AuNPs reliably and uniformly embedded in the capture substrate,35,49i.e., filter paper, are essential for irreversible adsorption of the capture antibody to the filter support40–45 and yields a sandwiched geometry when ERLs bind to support plasmonic coupling between the AuNPs allowing hotspots to be generated to significantly enhance the SERS signal.12,35,48,50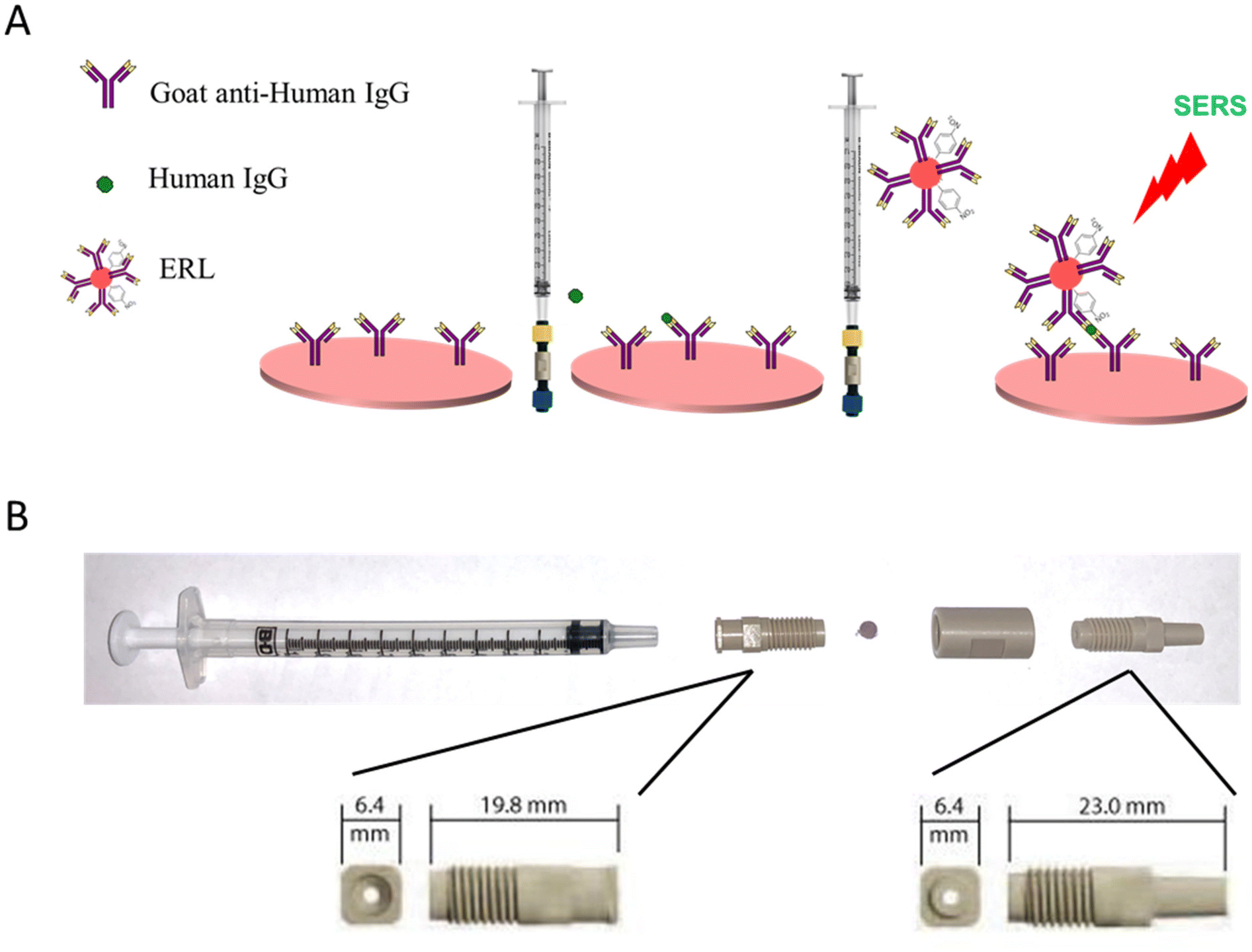 | ||
| Fig. 1 Overview of SERS-based VFA (A). Photograph of syringe filter apparatus (B). | ||
Previously, a SERS-based vertical flow immunoassay (VFA) was developed in which sample and ERL solutions were sequentially drawn through the plasmonic capture using an absorbent pad.34 The SERS VFA provided rapid and sensitive detection with a non-technical protocol. However, the combined volume of the sample, ERL solution, and rinse buffer was limited to ∼350 μL by the absorbance capacity of the VFA plug. Moreover, the sample and ERLs solutions made a single passage through the capture substrate for binding. The syringe filter assay explored in this work has the advantage of using the syringe filter system to potentially improve the binding efficiency of the antigen and ERL and detect much lower concentrations by incorporating multiple forward and reverse passages of sample and label through the capture filter. Additionally, volume of sample, label, and wash solutions that can be passed through the filter paper is theoretically unlimited, unlike VFA and LFAs, to potentially improve sensitivity and further minimize non-specific binding.
A holder for the plasmonic filter paper was assembled using commercially available components. The filtration device was carefully selected to meet the following criteria: readily assemble/disassemble to accommodate user-selected filter paper, form a tight seal, in-line flow channel above and below the filter to maintain column flow through the filter in both directions, and restrict solution flow to a small, defined area of the filter paper. Plasmonic paper is sandwiched between a male and female threaded luer adapter and held together with a union (Fig. 1B). Solution is confined to a 1.5 mm diameter flow channel defined by the luer adapters. The threaded fittings provide a tight seal around the plasmonic filter paper and enable easy access to load/unload the filter paper.
Feasibility of SERS filtration assay
Whatman grade 4 filter paper was selected as the capture substrate support to which AuNPs were embedded.34 The UV-vis extinction spectra of the AuNP suspension before and after the filter paper was submersed for AuNP adsorption are shown in Fig. S1A.† The decrease in color intensity of the AuNP suspension in the cuvette from purple to pink as seen in Fig. S1A† indicates that there has been an effective deposition of AuNPs onto the filter paper to form a plasmonic paper. Additionally, the UV-vis spectra provided semi-quantitative data where the initial concentration of AuNP solution before loading showed a 2.4 extinction at 540 nm while the AuNP solution after loading showed a 0.6 extinction indicating the deposition of AuNP to form the plasmonic paper. Likewise, the SEM image (Fig. S1B†) also confirms the presence of AuNPs on the filter paper which suggests a successful adsorption of AuNPs on the filter paper. In addition, the surface absorption band was compared to the extinction spectrum of colloidal AuNP solution (Fig. S2†). Unlike well-dispersed colloidal AuNPs in aqueous solution, the surface UV-Vis-near IR spectrum displayed a very broad peak with a gradually declining shoulder line in the entire visible range. This phenomenon has been explained by the presence of small gaps between adjacent plasmonic particles on the substrate to induce interparticle coupling. As randomly aggregated AuNPs are abundantly distributed across filter paper, numerous spacings/gaps between AuNPs readily govern the overall wavelength shift and overlap (i.e., broad band). Although the absorption measurement on a 2-D substrate is challenging due to the measurement conditions involving a strong absorber paper material,51,52 the absorption pattern of the plasmonic paper clearly indicated the presence of a certain degree of locally aggregated AuNPs to induce plasmonic coupling.53–55Dynamic light scattering (DLS) and UV-vis extinction spectrophotometry were used to characterize the synthesized ERLs. Fig. S3A† shows DLS data where the hydrodynamic diameter of unmodified AuNPs is ∼60 nm and that of the ERLs is ∼80 nm. The increase in hydrodynamic diameter with the immobilization of the antibody is consistent with the size of an IgG molecule and indicates monolayer formation with no detectable aggregation. UV-vis extinction was also used as a complementary technique to confirm successful formation of ERLs. The unconjugated AuNPs exhibited a maximum extinction at 538 nm (Fig. S3B†). The UV-vis data shows a ∼4 nm red shift for the prepared ERLs indicating a change in the refractive index as a result of antibody conjugation to form stable bioconjugates.
Following the successful characterization of ERLs and plasmonic paper, the feasibility of the assay was assessed by performing positive and negative control experiments using the filtration device (Fig. 1). The plasmonic paper was fabricated with 55 mm diameter filter paper; thus, after forming the plasmonic paper it was cut into 3 mm diameter pieces prior to functionalizing with capture antibody, blocking with BSA, and loading into the filter holder device (Fig. 1B). As a positive control, 100 μL of a 100 ng mL−1 human IgG sample in PBS was loaded into a 1 mL syringe and passed through the plasmonic capture substrate. The sample solution was infused/withdrawn 10 times to evaluate the integrity of the filter paper and confirm a tight seal around the capture substrate while confining liquid flow through a defined area of the filter. Following the sampling cycles, 100 μL of ERLs prepared with goat anti-human IgG antibodies were passed through the capture substrate using 10 cycles to label the captured antigen. Lastly, 300 μL of wash buffer was passed through the capture substrate using a syringe to remove any non-specifically bound or excess ERLs. The wash buffer was added by cycling 100 μL of solution through the capture filter ten times (infuse/withdraw cycles) using the syringe and repeating two additional times (with fresh 100 μL of wash buffer solution) to thoroughly remove excess unbound ERLs. Upon removal of the capture substrate from the filtration device, it was evident that the solutions were confined to a small area in the center of the filter paper equivalent to the inner diameter of the flow channel of the filter holder. Moreover, visual inspection of the plasmonic paper confirmed that the paper was not damaged as a result of multiple infuse/withdraw cycles of sample, label, and wash solutions. The average of six SERS spectra collected from two independent plasmonic papers used to analyze the positive control is presented in Fig. 2. As anticipated an intense SERS spectrum was observed with spectral features characteristic of NBT, the Raman reporter molecule used to prepare the ERL. A negative control PBS sample was then analyzed by flowing through a capture substrate for 10 cycles, followed by 10 ERL passages, and the same rinsing cycle as the 100 ng mL−1 hIgG positive control sample. In contrast to the positive control sample, a much smaller intensity was recorded for the negative control. These results establish feasibility of the device to effectively deliver antigen and label to the capture substrate, confirm antigen and ERL are specifically captured and concentrated on the filtration device as a result of antibody–antigen interactions, and non-specific binding of ERLs is minimal.
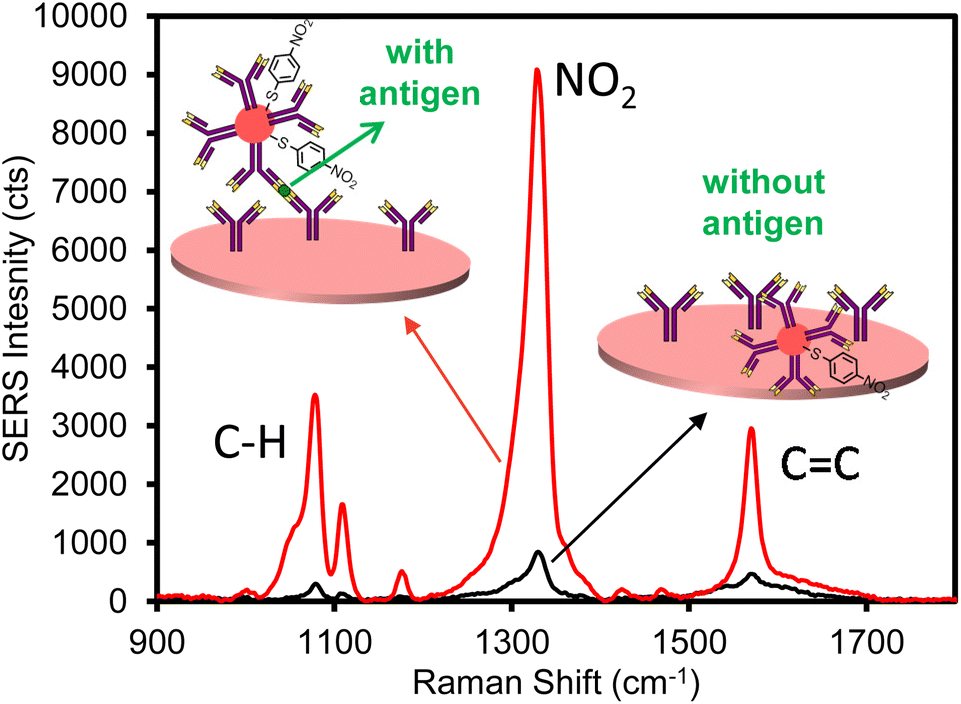 | ||
| Fig. 2 Average SERS spectra for 100 ng mL−1 human IgG positive control (red) and PBS negative control (black) samples. | ||
Preliminary optimization studies also investigated the use of nitrocellulose as the supporting filter paper. The previous rapid VFA developed by our group used Whatman grade 4 filter paper as an affordable and ideal substrate for making the plasmonic paper.34 However, most commercially available LFIAs uses nitrocellulose as a membrane to immobilize capture antibodies because the slightly hydrophobic nature of nitrocellulose allows proteins to bind through hydrophobic interactions.56,57 Therefore, we deposited AuNPs onto nitrocellulose membrane (Fig. S4†), and functionalized with goat anti-hIgG as a SERS capture substrates in the VFA. A 100 ng mL−1 hIgG positive control sample and PBS negative control sample were analyze following the sampling, labeling, and washing procedure used to establish feasibility as described above. The intensity of positive control for the plasmonic capture substrate fabricated with nitrocellulose paper was greater than that of the Whatman grade 4 paper (Fig. S5†). However, the use of nitrocellulose paper resulted in substantially higher non-specific binding of the ERLs in the negative control assay leading to a high SERS intensity as compared to that of the Whatman grade 4 paper (20–25 μm), possibly due to the much smaller pore size distribution (∼0.45 μm).
Effect of filtration cycles on binding efficiency
The primary motivation for developing a filtration-based immunoassay was to enable multiple sampling and labeling cycles to improve binding efficiency. It is well established that actively transporting antigen to an antibody-functionalized capture substrate can reduce the time required for antigen–antibody binding by overcoming binding kinetics limited by diffusional mass transport.58–61 However, if mass transport is sufficiently increased, the binding efficiency decreases as the antigen is delivered to the antibody capture substrate at a faster rate than antibody–antigen recognition occurs. For example, the signal due to antigen binding in a filter-based immunoassay decreased as the flow rate increased.62 Thus, in previous works, a balance was required to maximize binding, while minimizing assay time, i.e., faster flow rates. In addition, binding is highly flow rate-dependent; thus, flow must be carefully controlled using a syringe pump to achieve acceptable precision and afford quantitative results. Here, multiple passages of sample and labeling solution through the filter using a syringe to infuse/withdraw solutions provide multiple opportunities for interaction to increase the cumulative effective binding efficiency, independent of flow rate, such that binding becomes concentration dependent.The use of the syringe filter apparatus served as an ideal tool to enable multiple passages of sample and/or labeling solutions back and forth through the plasmonic capture substrate. The number of infuse/withdraw cycles of a 100 μL positive control sample consisting of 50 ng mL−1 hIgG in PBS was varied to investigate the effect of multiple sampling cycles on antigen binding. The ERL labeling step was held constant for each paper at 10 infuse/withdraw cycles and each capture substrate was rinsed with 300 μL of wash buffer. SERS analysis reveals that the antigen binding increases with increasing sampling cycles up to 10 cycles (Fig. 3A). The binding reaches a maximum at 10 cycles and no additional antigen binding is observed for 15, 20, or 30 sample passages through the capture substrate. Moreover, the plasmonic paper substrate maintained visual integrity with the increased number of sampling cycles (Fig. S6†). A negative control sample was analyzed to determine if multiple passages impacted non-specific binding. The signal for the negative control samples were statistically equivalent up to 20 cycles; however, a slight increase in non-specific binding was consistently observed after 30 sampling cycles.
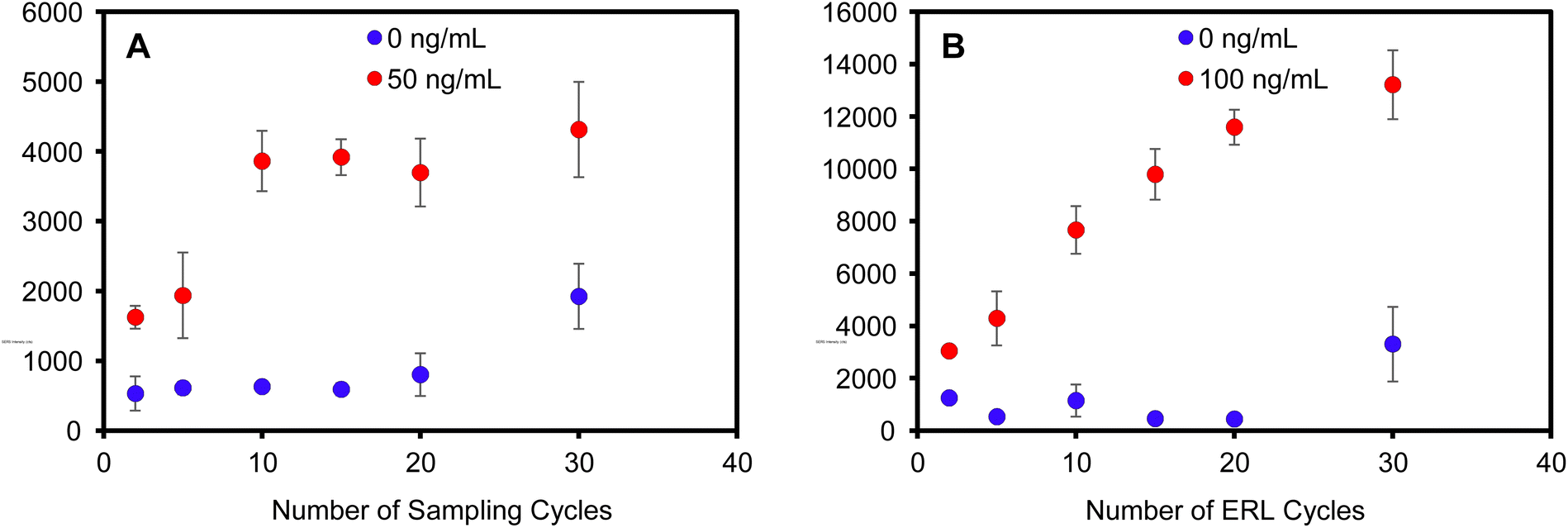 | ||
| Fig. 3 Average SERS intensity of the 1338 cm−1 peak as a function of the number of (A) sampling cycles (ERL labeling step was held constant at 10 infuse/withdraw cycles) and (B) ERL cycles (sampling step was held constant at 10 infuse/withdraw cycles). | ||
In a similar experiment, positive control hIgG samples (100 ng mL−1) and negative control samples (PBS) were cycled through the capture substrate 10 times. The number of ERL solution passages were then varied to investigate multiple labeling cycles on specific and non-specific binding. Fig. 3B shows that the SERS intensity increased with increased ERL cycles. Unlike the antigen binding step, the signal did not saturate after cycling the ERLs through the capture substrate 30 times. Analysis of the negative samples also show equivalent non-specific binding for less than 20 ERL cycles, but signal for non-specific binding substantially increased with 30 ERL passages. Collectively, these data establish that the optimized assay requires 10 passages of the sample and 20 passages of the ERLs to maximize binding efficiency while minimizing non-specific binding. It is reasonable to expect that more passages of the ERLs are required to maximize binding efficiency compared to antigen because the number of antigen molecules are significantly greater than the number of ERLs in the 100 μL volumes – 100 ng mL−1 antigen is equivalent to 4.0 × 1011 hIgG per mL (670 pM) compared to 5.2 × 1010 ERL per mL (86 pM). Thus, the probability of ERL collision with a target is less than that of the more concentrated antigen, requiring more passages for complete labeling of bound antigen.
Analytical performance of optimized filter assay
Standard solutions of human IgG in PBS were analyzed to establish the dose-dependent response and define the analytical figures of merit of the optimized assay. Two independently prepared plasmonic capture substrates and two sets of calibration standards ranging from 0 to 200 ng mL−1 hIgG were analyzed to incorporate inter-assay variability. Fig. 4A shows the average SERS spectra acquired for each standard concentration. As is evident, the intensity correlates with antigen concentration up to approximately 50 ng mL−1, where the signal approaches a maximum value. The concentration-dependent response is plotted in Fig. 4B. The data clearly demonstrated that the optimized assay provides quantitative results and the inter-assay variability in measured signal is ∼10–15% for concentrations greater than 1 ng mL−1. In addition, the calibration curve shape without any sign of decreasing SERS intensity at high antigen concentrations indicates that this two-step sandwich assay is not limited by the common hook effect.63–66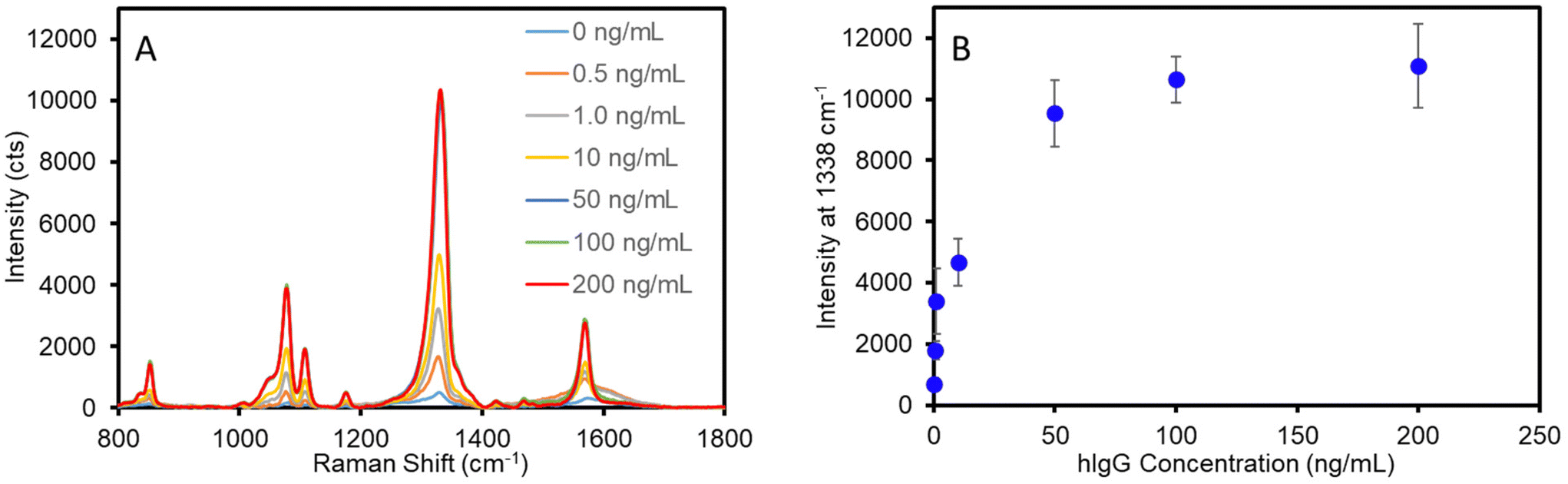 | ||
| Fig. 4 Average SERS spectra (n = 6) for hIgG calibration standards (A). Calibration curve for optimized assay (B). Error bars represent the standard deviation for 6 measurements across two independent substrates for each concentration. | ||
The detection limit of the assay was determined from the linear best-fit regression analysis of the data for the low concentration range (0–2 ng mL−1) (Fig. S7†). Using the best-fit equation for the data in Fig. S7,† the detection limit was calculated as the lowest antigen concentration that would produce a signal equal to that of the mean blank signal plus three times the standard deviation of the blank signal. The optimized assay afforded a detection limit of 0.2 ng mL−1 (1.3 pM) for hIgG. A previously developed SERS-based vertical flow immunoassay for IgG detection achieved a detection limit of 3–8 ng mL−1 (20–53 pM);34 however, as discussed, the VFA only allows the sample and ERL solution to pass through the plasmonic capture substrate once. Thus, these results suggest that improved sampling efficiency afforded by multiple sampling/labeling passages through the sensing substrate improves the detection limit approximately 20-fold. This detection limit is comparable to those achieved by recently developed SERS-based LFAs for IgG detection that report LODs ranging from 0.1 to 5.0 ng mL−1 using optimized plasmonic tags.22,23 However, it is important to consider that the antibody binding affinity impacts the assay detection limit and prevents the accurate comparison of inter-laboratory/inter-method LODs in which different antibody–antigen systems are used. To better evaluate the analytical performance of this SERS filtration assay, a sandwich ELISA was conducted using the same antibody–antigen system to allow for appropriate comparison of LODs (Fig. S8†). The ELISA resulted in a detection limit of 1.1 ng mL−1, while requiring ∼8 h to complete the analysis. This is in contrast to the SERS filter assay developed in this work that results in a 5-fold improvement in LOD, while significantly reducing the assay time.
The specificity of the assay was assessed by analyzing relatively simple negative control samples, e.g., PBS and 10% BSA, and a series of serum samples from various species that were diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100, which represent more complex biological samples. Fig. 5 demonstrates that each of the human IgG-negative samples, e.g., PBS, 10% BSA, rabbit serum, bovine serum, and mouse serum, resulted in minimal signal. Importantly, the more complex non-human sera samples yielded similar signals, i.e., non-specific binding, to the PBS control. In contrast, the human serum sample resulted in saturating signal, which is expected considering the concentration of IgG is 5–18 mg mL−1 in undiluted normal human serum corresponding to 50–180 μg mL−1 in the 100× diluted samples analyzed here. Collectively, these results confirm the specificity of the SERS filter assay.
:100, which represent more complex biological samples. Fig. 5 demonstrates that each of the human IgG-negative samples, e.g., PBS, 10% BSA, rabbit serum, bovine serum, and mouse serum, resulted in minimal signal. Importantly, the more complex non-human sera samples yielded similar signals, i.e., non-specific binding, to the PBS control. In contrast, the human serum sample resulted in saturating signal, which is expected considering the concentration of IgG is 5–18 mg mL−1 in undiluted normal human serum corresponding to 50–180 μg mL−1 in the 100× diluted samples analyzed here. Collectively, these results confirm the specificity of the SERS filter assay.
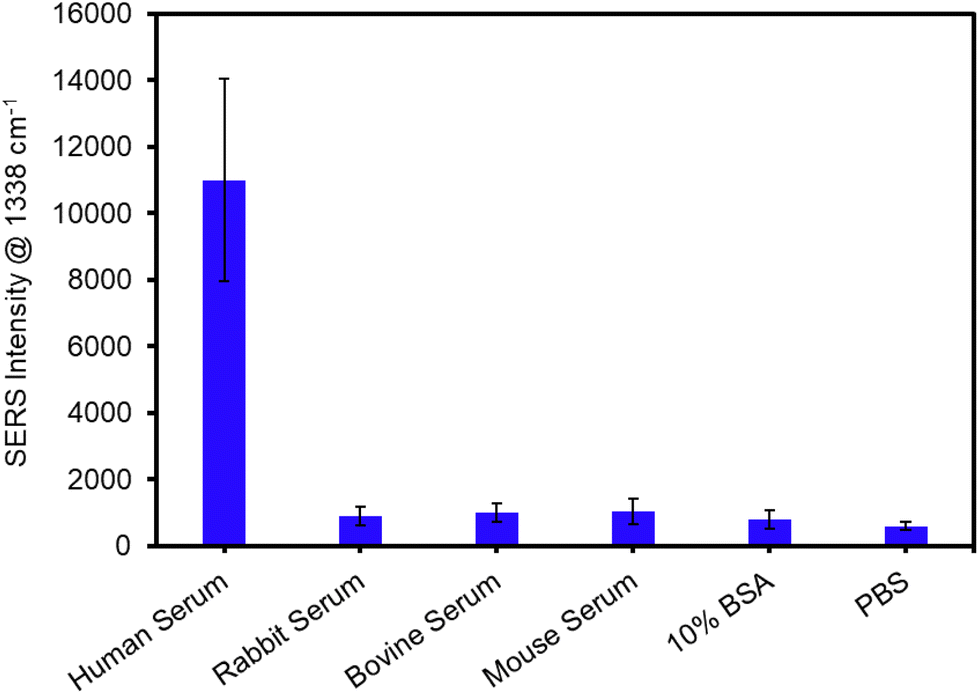 | ||
| Fig. 5 Average SERS intensities for the analysis of sera samples diluted 100-fold with PBS. PBS and 10% BSA were also analyzed as negative control samples. Error bars represent the standard deviation for 6 measurements across two independent substrates for each concentration. | ||
Detection of SARS-CoV-2 antigen
The assay was re-configured for the detection of SARS-CoV-2 nucleocapsid (N) protein. The N protein is the most common biomarker target of commercial COVID-19 rapid antigen tests. The N protein has a low mutation rate and is antigenically stable among SARS-CoV-2 viral strains for antibody binding,1,67 as evidenced by clinical studies demonstrating that commercial rapid antigen tests (LFAs) targeting the N protein provide equivalent sensitivities for the detection of the Omicron, Delta, and Wuhan-Hu-1 G614 variants.67,68 To this end, the plasmonic paper was functionalized with a humanized monoclonal antibody against N protein and the SERS label was functionalized with a second humanized monoclonal antibody against N protein. The matched pair antibodies were designed to function in a sandwich immunoassay format. Standard solutions of N protein were prepared in PBS and analyzed using the optimized sampling and labeling parameters determined in previous sections. A calibration curve is presented in Fig. 6 and shows a similar concentration-dependent trend observed for the detection of hIgG. A slight increase in non-specific binding for this system, relative to the anti-hIgG model, resulted in an LOD of 1.0 ng mL−1 (21 pM) for N protein.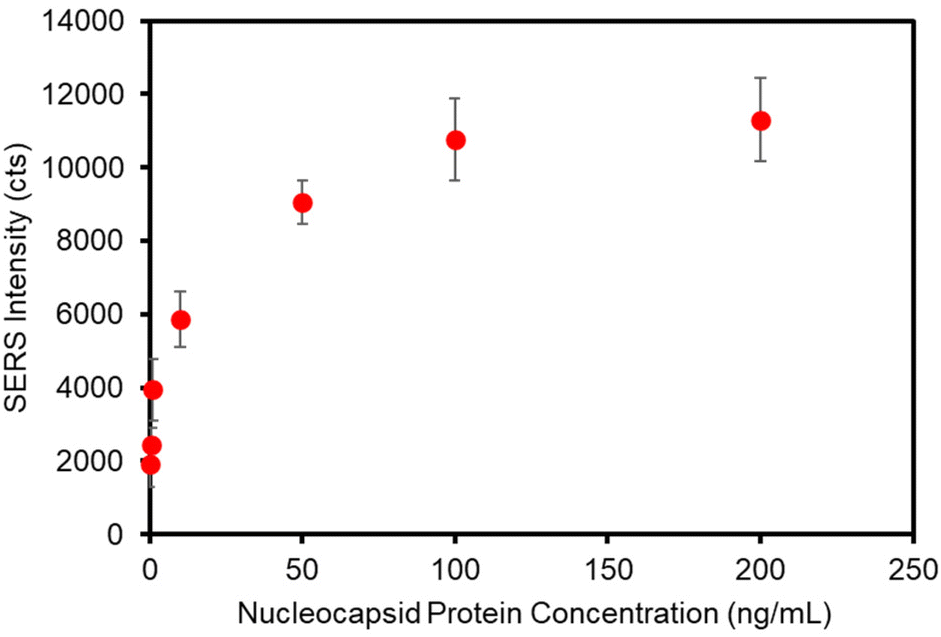 | ||
| Fig. 6 Calibration curve for the detection of SARS-CoV-2 nucleocapsid protein. Each data point represents the average intensity of the 1338 cm−1 band collected from 6 different locations on two independently prepared substrates. The standard deviation is represented by the error bars. | ||
Conclusions
With the aim of developing a novel POC test that is easy to use, rapid, and sensitive, we have designed an optimized SERS-based vertical flow immunoassay that incorporates a syringe filter system to flow the sample and labeling reagent through the detection membrane. This work builds on a previous platform developed in our lab that exploits plasmonic coupling between a nanoparticle-embedded membrane and nanoparticle label to generate exceptionally large SERS enhancements for detection. Here, we explored the use of a syringe filtration device to drive solution through the vertical flow membrane to investigate antigen and label binding efficiency as a function of infuse/withdraw cycles. These results reveal that multiple passages of the sample and ERL suspension through the capture membrane substantially increase antigen–antibody binding efficiency to improve the assay sensitivity with minimal increase in assay time. Current rapid diagnostic platforms such as LFAs and VFAs rely on capillary action to actively transport sample and expedite antigen–antigen binding at the detection zone; however, these platforms limit the number of interactions between the sample and detection zone to a single passage. The data presented here highlights an opportunity to improve antibody–antigen binding efficiency by implementing multiple, rapid, successive interactions during sampling, and can readily be adapted to non-SERS-based vertical flow assays without restraint. Furthermore, successful demonstration of specificity for the analysis of a series of serum samples establishes that this platform is capable of translation to clinical sample analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the National Science Foundation through the Macromolecular, Supramolecular and Nanochemistry Program, Award # CHE-2203740. Partial support was also provided by Illinois State University. The FESEM multi-user facility was acquired with the support from the Division of Material Research (DMR), National Science Foundation (NSF) (Award # 2116612).References
- O. Vandenberg, D. Martiny, O. Rochas, A. van Belkum and Z. Kozlakidis, Nat. Rev. Microbiol., 2021, 19, 171–183 CrossRef CAS
.
- S. Lambert-Niclot, A. Cuffel, S. L. Pape, C. Vauloup-Fellous, L. Morand-Joubert, A.-M. Roque-Afonso, J. L. Goff and C. Delaugerre, J. Clin. Microbiol., 2020, 58, e00977–e00920 CrossRef CAS PubMed
- G. C. K. Mak, P. K. C. Cheng, S. S. Y. Lau, K. K. Y. Wong, C. S. Lau, E. T. K. Lam, R. C. W. Chan and D. N. C. Tsang, J. Clin. Virol., 2020, 129, 104500 CrossRef CAS PubMed
- M. Nagura-Ikeda, K. Imai, S. Tabata, K. Miyoshi, N. Murahara, T. Mizuno, M. Horiuchi, K. Kato, Y. Imoto, M. Iwata, S. Mimura, T. Ito, K. Tamura and Y. Kato, J. Clin. Microbiol., 2020, 58, e01438–20 CrossRef CAS PubMed
- R. Lei and C. Mohan, Crit. Rev. Immunol., 2020, 40, 497–512 CrossRef PubMed
- Z. Cheng, R. Wang, Y. Xing, L. Zhao, J. Choo and F. Yu, Analyst, 2019, 144, 6533–6540 RSC
- D. S. Grubisha, R. J. Lipert, H.-Y. Park, J. Driskell and M. D. Porter, Anal. Chem., 2003, 75, 5936–5943 CrossRef CAS
- N. Guarrotxena and G. C. Bazan, Chem. Commun., 2011, 47, 8784–8786 RSC
- M. D. Porter, R. J. Lipert, L. M. Siperko, G. Wang and R. Narayanana, Chem. Soc. Rev., 2008, 37, 1001–1011 RSC
- J. Langer, D. Jimenez de Aberasturi, J. Aizpurua, R. A. Alvarez-Puebla, B. Auguié, J. J. Baumberg, G. C. Bazan, S. E. J. Bell, A. Boisen, A. G. Brolo, J. Choo, D. Cialla-May, V. Deckert, L. Fabris, K. Faulds, F. J. García de Abajo, R. Goodacre, D. Graham, A. J. Haes, C. L. Haynes, C. Huck, T. Itoh, M. Käll, J. Kneipp, N. A. Kotov, H. Kuang, E. C. Le Ru, H. K. Lee, J.-F. Li, X. Y. Ling, S. A. Maier, T. Mayerhöfer, M. Moskovits, K. Murakoshi, J.-M. Nam, S. Nie, Y. Ozaki, I. Pastoriza-Santos, J. Perez-Juste, J. Popp, A. Pucci, S. Reich, B. Ren, G. C. Schatz, T. Shegai, S. Schlücker, L.-L. Tay, K. G. Thomas, Z.-Q. Tian, R. P. Van Duyne, T. Vo-Dinh, Y. Wang, K. A. Willets, C. Xu, H. Xu, Y. Xu, Y. S. Yamamoto, B. Zhao and L. M. Liz-Marzán, ACS Nano, 2020, 14, 28–117 CrossRef CAS PubMed
- O. E. Eremina, A. T. Czaja, A. Fernando, A. Aron, D. B. Eremin and C. Zavaleta, ACS Nano, 2022, 16, 10341–10353 CrossRef CAS PubMed
- J. D. Driskell, R. J. Lipert and M. D. Porter, J. Phys. Chem. B, 2006, 110, 17444–17451 CrossRef CAS PubMed
- D. Antoine, M. Mohammadi, M. Vitt, J. M. Dickie, S. S. Jyoti, M. A. Tilbury, P. A. Johnson, K. E. Wawrousek and J. G. Wall, ACS Sens., 2022, 7, 866–873 CrossRef CAS
- Y. Li, C. Lin, Y. Peng, J. He and Y. Yang, Sens. Actuators, B, 2022, 365, 131974 CrossRef CAS
- L. Blanco-Covián, V. Montes-García, A. Girard, M. T. Fernández-Abedul, J. Pérez-Juste, I. Pastoriza-Santos, K. Faulds, D. Graham and M. C. Blanco-López, Nanoscale, 2017, 9, 2051–2058 RSC
- J. Hwang, S. Lee and J. Choo, Nanoscale, 2016, 8, 11418–11425 RSC
- Z. Rong, R. Xiao, S. Xing, G. Xiong, Z. Yu, L. Wang, X. Jia, K. Wang, Y. Cong and S. Wang, Analyst, 2018, 143, 2115–2121 RSC
- V. Tran, B. Walkenfort, M. Konig, M. Salehi and S. Schlucker, Angew. Chem., Int. Ed., 2019, 58, 442–446 CrossRef CAS
- R. Xiao, L. Lu, Z. Rong, C. Wang, Y. Peng, F. Wang, J. Wang, M. Sun, J. Dong, D. Wang, L. Wang, N. Sun and S. Wang, Biosens. Bioelectron., 2020, 168, 112524 CrossRef CAS PubMed
- S. Yadav, M. A. Sadique, P. Ranjan, N. Kumar, A. Singhal, A. K. Srivastava and R. Khan, ACS Appl. Bio Mater., 2021, 4, 2974–2995 CrossRef CAS PubMed
- D. Zhang, L. Huang, B. Liu, H. Ni, L. Sun, E. Su, H. Chen, Z. Gu and X. Zhao, Biosens. Bioelectron., 2018, 106, 204–211 CrossRef CAS
- S. Chen, L. Meng, L. Wang, X. Huang, S. Ali, X. Chen, M. Yu, M. Yi, L. Li, L. Yuan, W. Shi and G. Huang, Sens. Actuators, B, 2021, 348, 130706 CrossRef CAS PubMed
- H. Liu, E. Dai, R. Xiao, Z. Zhou, M. Zhang, Z. Bai, Y. Shao, K. Qi, J. Tu, C. Wang and S. Wang, Sens. Actuators, B, 2021, 329, 129196 CrossRef CAS PubMed
- R. Lei, D. Wang, H. Arain and C. Mohan, Diagnostics, 2022, 12, 1107 CrossRef CAS
- Y. Liu, L. Zhan, Z. Qin, J. Sackrison and J. C. Bischof, ACS Nano, 2021, 15, 3593–3611 CrossRef CAS
- K. M. Koczula and A. Gallotta, Essays Biochem., 2016, 60, 111–120 CrossRef PubMed
- P. Chen, M. Gates-Hollingsworth, S. Pandit, A. Park, D. Montgomery, D. AuCoin, J. Gu and F. Zenhausern, Talanta, 2019, 191, 81–88 CrossRef CAS PubMed
- Y. K. Oh, H.-A. Joung, S. Kim and M.-G. Kim, Lab Chip, 2013, 13, 768–772 RSC
- G. M. S. Ross, G. I. Salentijn and M. W. F. Nielen, Biosensors, 2019, 9, 143 CrossRef CAS PubMed
- R. Chen, B. Liu, H. Ni, N. Chang, C. Luan, Q. Ge, J. Dong and X. Zhao, Analyst, 2019, 144, 4051–4059 RSC
- O. J. R. Clarke, B. L. Goodall, H. P. Hui, N. Vats and C. L. Brosseau, Anal. Chem., 2017, 89, 1405–1410 CrossRef CAS PubMed
- J. H. Granger, A. Skuratovsky, M. D. Porter, C. L. Scaife, J. E. Shea, Q. Li and S. Wang, Anal. Methods, 2017, 9, 4641–4646 RSC
- D. Zhang, L. Huang, B. Liu, Q. Ge, J. Dong and X. Zhao, Microchim. Acta, 2019, 186, 699 CrossRef PubMed
- R. Frimpong, W. Jang, J.-H. Kim and J. D. Driskell, Talanta, 2021, 223, 121739 CrossRef CAS PubMed
- J. A. Lartey, J. P. Harms, R. Frimpong, C. C. Mulligan, J. D. Driskell and J.-H. Kim, RSC Adv., 2019, 9, 32535–32543 RSC
- J. Kimling, M. Maier, B. Okenve, V. Kotaidis, H. Ballot and A. Plech, J. Phys. Chem. B, 2006, 110, 15700–15707 CrossRef CAS PubMed
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55–75 RSC
- M. Wuithschick, A. Birnbaum, S. Witte, M. Sztucki, U. Vainio, N. Pinna, K. Rademann, F. Emmerling, R. Kraehnert and J. Polte, ACS Nano, 2015, 9, 7052–7071 CrossRef CAS PubMed
- J. Magura, A. Zeleňáková, V. Zeleňák and M. Kaňuchová, Appl. Surf. Sci., 2014, 315, 392–399 CrossRef CAS
- O. Awotunde, S. Okyem, R. Chikoti and J. D. Driskell, Langmuir, 2020, 36, 9241–9249 CrossRef CAS PubMed
- H. de Puig, I. Bosch, M. Carré-Camps and K. Hamad-Schifferli, Bioconjugate Chem., 2017, 28, 230–238 CrossRef CAS PubMed
- S. L. Filbrun and J. D. Driskell, Analyst, 2016, 141, 3851–3857 RSC
- G. Ruiz, N. Ryan, K. Rutschke, O. Awotunde and J. D. Driskell, Langmuir, 2019, 35, 10601–10609 CrossRef CAS PubMed
- K. Siriwardana, A. Wang, K. Vangala, N. Fitzkee and D. Zhang, Langmuir, 2013, 29, 10990–10996 CrossRef CAS
- A. Wang, K. Vangala, T. Vo, D. Zhang and N. C. Fitzkee, J. Phys. Chem. C, 2014, 118, 8134–8142 CrossRef CAS
- A. Lopez, F. Lovato, S. Hwan Oh, Y. H. Lai, S. Filbrun, E. A. Driskell and J. D. Driskell, Talanta, 2016, 146, 388–393 CrossRef CAS PubMed
- S. Hong and X. Li, J. Nanomater., 2013, 2013, 1–9 Search PubMed
- J. K. Yoon, K. Kim and K. S. Shin, J. Phys. Chem. C, 2009, 113, 1769–1774 CrossRef CAS
- W. Jang, H. Byun and J.-H. Kim, Mater. Chem. Phys., 2020, 240, 122124 CrossRef CAS
- M. Schütz and S. Schlücker, Phys. Chem. Chem. Phys., 2015, 17, 24356–24360 RSC
- T. Horibe, K. Ishii, D. Fukutomi and K. Awazu, Laser Ther., 2015, 24, 303–310 CrossRef
- R. G. J. Strens and B. J. Wood, Mineral. Mag., 1979, 43, 347–354 CrossRef CAS
- D. Bartkowiak, V. Merk, V. Reiter-Scherer, U. Gernert, J. P. Rabe, J. Kneipp and E. Kemnitz, RSC Adv., 2016, 6, 71557–71566 RSC
- M. Iatalese, M. L. Coluccio, V. Onesto, F. Amabo, E. D. Fabrizio and F. Gentile, Nanoscale Adv., 2019, 1, 228–240 RSC
- L. Litti and M. Meneghetti, Phys. Chem. Chem. Phys., 2019, 21, 15515–15522 RSC
- B. L. Petros, P. M. Procell, G. H. Campbell and F. H. Collins, Bull. W. H. O., 1989, 67, 525–533 CAS
- E. R. Tovey and B. A. Baldo, J. Biochem. Biophys. Methods, 1989, 19, 169–183 CrossRef CAS PubMed
- D. G. Myszka, T. A. Morton, M. L. Doyle and I. M. Chaiken, Biophys. Chem., 1997, 64, 127–137 CrossRef CAS PubMed
- H. Nygren, M. Werthen and M. Stenberg, J. Immunol. Methods, 1987, 101, 63–71 CrossRef CAS
- P. E. Sheehan and L. J. Whitman, Nano Lett., 2005, 5, 803–807 CrossRef CAS
- M. Stenberg and H. Nygren, J. Theor. Biol., 1985, 113, 589–597 CrossRef CAS PubMed
- M. A. Penn, D. M. Drake and J. D. Driskell, Anal. Chem., 2013, 85, 8609–8617 CrossRef CAS PubMed
- C. Parolo, A. Sena-Torralba, J. F. Bergua, E. Calucho, C. Fuentes-Chust, L. Hu, L. Rivas, R. Álvarez-Diduk, E. P. Nguyen, S. Cinti, D. Quesada-González and A. Merkoçi, Nat. Protoc., 2020, 15, 3788–3816 CrossRef CAS PubMed
- S. Dodig, Biochem. Med., 2009, 19, 50–62 CrossRef CAS
- X. Liu and Q. Huo, J. Immunol. Methods, 2009, 349, 38–44 CrossRef CAS
- A. Lopez, F. Lovato, S. H. Oh, Y. H. Lai, S. Filbrun, E. A. Driskell and J. D. Driskell, Talanta, 2016, 146, 388–393 CrossRef CAS PubMed
- J. Deerain, J. Druce, T. Tran, M. Batty, Y. Yoga, M. Fennell, D. E. Dwyer, J. Kok and D. A. Williamson, J. Clin. Microbiol., 2022, 60, e02479–e02421 CrossRef CAS PubMed
- P. de Michelena, I. Torres, Á. Ramos-García, V. Gozalbes, N. Ruiz, A. Sanmartín, P. Botija, S. Poujois, D. Huntley, E. Albert and D. Navarro, J. Infect., 2022, 84, e64–e66 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3an01899g |
| This journal is © The Royal Society of Chemistry 2024 |