
Simultaneous determination of methyltestosterone and its metabolite in fish by gas chromatography-mass spectrometry
Liufeng
Zhang†
ab,
Juan
Sun†
d,
Yinmeng
Ding
ab,
Li
Li
 *c and
Jing
Liu
*ab
*c and
Jing
Liu
*ab
aKey Laboratory of Drug Quality Control and Pharmacovigilance, Ministry of Education, China Pharmaceutical University, 24 Tongjiaxiang, Nanjing 210009, China
bDepartment of Pharmaceutical Analysis, China Pharmaceutical University, 24 Tongjiaxiang, Nanjing 210009, China
cJiangsu Academy of Agricultural Sciences, Nanjing 210014, China
dJiangsu Nanjing Environmental Monitoring Center, Nanjing 210041, China
First published on 6th December 2023
Abstract
Methyltestosterone is one of the banned drugs in aquaculture, and it should be monitored in food-producing animals. 17α-Methyl-5β-androstane-3α,17β-diol, as the main metabolite of methyltestosterone in vertebrates, could be used as another marker for controlling the administration of methyltestosterone, due to its high residual concentration and slow elimination rate. In this study, an analytical method based on gas chromatography-mass spectrometry (GC-MS) was developed and validated for the simultaneous determination of methyltestosterone and its main metabolite in fish. After pretreatment by liquid–liquid extraction with n-hexane and solid phase extraction with C18 and NH2 columns, the target analytes in the muscle tissues were extracted and concentrated, and the influence of the sample matrix was eliminated. Then, the prepared samples were separated and detected with GC-MS in the selected ion monitoring (SIM) mode. Methyltestosterone-D3 was chosen as the internal standard for quantitation. After optimization, the limits of detection for methyltestosterone and 17α-methyl-5β-androstane-3α,17β-diol were 20 μg kg−1 and 15 μg kg−1, respectively. The limits of quantitation were both 50 μg kg−1. The calibration curves showed good linearity in the concentration range from 50.0 ng mL−1 to 500.0 ng mL−1. The correlation coefficients of methyltestosterone and 17α-methyl-5β-androstane-3α,17β-diol were more than 0.9990. The recoveries of the analytes in real samples were in the range of 99.7–116.6% with the relative standard deviation of 5.2–8.3%. The established method could meet the demand for simultaneous detection of methyltestosterone and its major metabolite, and it could be used to provide more information on the abuse of methyltestosterone in food-producing animals.
Introduction
Sex determination and differentiation of fish is a matter of great interest in aquaculture as sex control brings great commercial value.1 For example, female Atlantic salmons always exhibit deterioration in flesh quality due to the ability of diversify energy towards the reproductive process,2 and male Nile tilapias show better growth with a lower feed conversion rate than females.3 Researchers have found that fish sex is determined by the male-promoting dmrt1/amh/gsdf/androgen pathway and the female-promoting foxl2/cyp19a1a/estrogen pathway. These two pathways work together and antagonize each other.4,5 When genes like cyp19a1a and foxl2 mutate and forfeit the original physiological function, the level of estrogen will decrease and the level of androgen will increase, inducing female-to-male sex reversal.6,7Methyltestosterone (MT), classified as an androgen and anabolic steroid (AAS), was initially identified in 1935 and subsequently introduced for medical applications in 1936.8 MT has also been employed in animal administration to induce changes in sex differentiation or stimulate growth.9,10 For example, studies have indicated that MT possesses the capability to inhibit the expression of a crucial steroidogenic enzyme (P450arom), thereby resulting in the masculinization of Nile tilapia.11 The levels of dmrt1 and foxl2 in the Orange-spotted grouper, a marine ray-finned fish species, exhibited significant alterations, resulting in evident masculinization traits upon administration of MT.12 Nonetheless, it is imperative to acknowledge the potential adverse consequences associated with the misuse of MT in aquaculture.13 Firstly, the potential consequences of MT include the induction of deformities in food-producing animals and the difficulty for farmers to effectively manage sexual differentiation.14,15 Secondly, the leakage and migration of MT from breeding sites can lead to its sorption onto soils and sediments.16,17 Finally, the consumption of residual MT by food-producing animals poses significant human health risks, including the potential for thrombotic and carcinogenic effects.18–20 Therefore, numerous nations have implemented restrictions on the utilization of MT in the domains of animal husbandry and aquaculture, except for its application for therapeutic intentions. In China, MT is classified as one of the proscribed substances for all food-producing animals during the breeding process, aiming to guarantee the safety of consumable animal-derived food items.
Up to now, not only the chemical additives themselves but also their metabolites are getting more and more attention due to their potential to prove the contamination or safety of food. For instance, both methylated and hydroxylated metabolites of phenylarsenic chemicals were considered as the promising target chemicals for detecting the contamination of some chemical warfare agents in fish.21 For robenidine hydrochloride (ROBH) which was an authorized feed additive for the prevention of coccidiosis in animal species, 4-chlorobenzoic acid (PBCA) was its predominant metabolite in the grass carp, and the elimination half-life of PBCA was 8 times that of ROBH. Thus, the detection results of PBCA could serve as reference data for the safety and reasonable use of ROBH in fish.22 Victoria Pure Blue BO (VPBO), a prohibited drug in aquaculture, was metabolized into deethyl-leuco-VPBO with longer residue time in rainbow trout, which could be used as a new residue marker for predicting the VPBO drug treatment of aquacultured species.23
For MT, seven metabolites have been found in vertebrates, arising from hydroxylation, reduction, isomerization, and rearrangement.24–27 17α-Methyl-5β-androstane-3α,17β-diol (M2) is one of its most prominent metabolites, owning high concentration and long half-life.28 In the urine of the heifer, the concentration of M2 is 19 times that of MT even after 7 days of administration.29 According to the pharmacokinetic research of MT and M2 in Nile tilapia, the elimination of M2 is extremely slow, and its residue concentration is high. The peak concentration (Cmax) and elimination half-life (T1/2b) of M2 were 5.604 μg mL−1 and 362.7 h, while Cmax and T1/2b of MT were 1.032 μg mL−1 and 18.4 h, respectively.30 Therefore, in order to strengthen the supervision and inspection of the use of MT in food-producing animals, it is very necessary to simultaneously detect MT and its metabolites.
In the field of food safety, more attention has been paid to the detection of methyl-testosterone monomers in meat products and dairy products. Several methods have been developed to determine MT in food-producing animals.31,32 Among them, mass spectrometry (MS) is the most effective and efficient detection technique because of its high sensitivity and resolution. In addition, the sample pretreatment and separation technology are also needed for the qualitative and quantitative analysis due to the complexity of the biological sample matrix. Regal et al. extracted MT from cow hair with acetonitrile, and then the determination was carried out by high-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) in multiple reaction monitoring (MRM) mode after the hydroxylamine derivatization.33 MT in muscle tissues could be directly determined by HPLC-MS/MS after the sample pretreatment of liquid–liquid extraction (LLE) and solid phase extraction (SPE).34 In terms of MT metabolites, two main compounds including M2 were measured by stable isotope analysis based on gas chromatography-mass spectrometry (GC-MS), in which M2 was derivatized with N-methyl-N-trimethylsilyl-trifluoroacetamide/dithioerythriol/ammonium iodide first.35
To the best of our knowledge, there was no method available for simultaneous qualitation and quantitation of MT and its main metabolites in food-producing animals. In this present research, a GC-MS method without any derivatization was established and validated for the simultaneous analysis of residual MT and M2 in fish muscle. LLE with n-hexane and SPE with C18 and NH2 columns were used to eliminate the influence of the sample matrix and concentrate the target compounds. The established method would provide more information to aid in further monitoring the administration of MT in food-producing animals.
Experimental
Reagents and materials
Methanol and acetonitrile were acquired from TEDIA (HPLC, Fairfield, USA). Acetone and n-hexane were purchased from Aladdin (HPLC, Shanghai, China). Methyltestosterone-D3 (D-MT, 100 μg mL−1), M2 (100 μg mL−1), and β-glucuronidase/arylsulfatase (≥85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 units per mL, ≤7500 units per mL) were purchased from Sigma-Aldrich (St. Louis, MO. USA). Methyltestosterone (MT, 98.3%) was purchased from Beijing Manhage Bio-tech Co., Ltd (Beijing, China). Diatomite (98%) was obtained from Macklin Biochemical Co., Ltd (Shanghai, China). C18 solid-phase extraction columns and NH2 solid-phase extraction columns were obtained from Agela Technologies (Tianjin, China).
000 units per mL, ≤7500 units per mL) were purchased from Sigma-Aldrich (St. Louis, MO. USA). Methyltestosterone (MT, 98.3%) was purchased from Beijing Manhage Bio-tech Co., Ltd (Beijing, China). Diatomite (98%) was obtained from Macklin Biochemical Co., Ltd (Shanghai, China). C18 solid-phase extraction columns and NH2 solid-phase extraction columns were obtained from Agela Technologies (Tianjin, China).
Preparation of standard solutions
The stock solution of MT was prepared at the concentration of 2.00 mg mL−1 with methanol. Then, it was further diluted by methanol to the concentration of 10.0 μg mL−1. The stock solutions of internal standard and M2 were prepared by diluting D-MT and M2 with methanol to 10.0 μg mL−1, respectively. For plotting the standard curve, the standard solutions were prepared at the concentrations of 50.0, 100.0, 200.0, 400.0, and 500.0 ng mL−1 by diluting the appropriate volume of the stock solutions with methanol. These solutions were all stored at −20 °C.Pretreatment of samples
The homogenized fish muscle (1.0 ± 0.1 g) was weighed into the mortar. Each sample was added with the stock solution of D-MT (5 μL) and diatomite (1.0 g), then the mixture was thoroughly ground to disperse the tissue efficiently. The sample was mixed with acetonitrile (5 mL) and β-glucuronidase/arylsulfatase (60 μL) by vortex and then incubated at 37 °C for 8 h.39 After that, the sample was centrifuged at 4000 rpm for 5 min at 4 °C, and the supernatant was collected. The above extraction step with acetonitrile was repeated twice. The supernatant was gathered and distilled to remove the solvent by rotary evaporator. The sample was redissolved with acetonitrile-saturated n-hexane (1 mL) and then centrifuged at 10000 rpm for 5 min at 4 °C. The underlying solution was transferred into a centrifuge tube with 10 mL of water.
For clean-up by SPE, C18 columns were successively activated with methanol and water, and NH2 columns were activated with acetonitrile. The sample was loaded on the C18 column first, and rinsed with water, acetone-aqueous solution (1:4, v/v), and n-hexane, respectively. After that, the C18 SPE column was connected in series with the NH2 column, and the solid-phase extraction system was eluted with acetonitrile (6 mL). In this process, the flow rate was controlled within 3 mL min−1. The eluate was collected and dried by a rotary evaporator. Then, methanol (1 mL) and anhydrous sodium sulphate were sequentially added and mixed. Finally, the supernatant was filtered by a membrane filter (0.22 μm) and applied for GC-MS analysis.
GC-MS instrument and conditions
The experiment was carried out on a QP2020 gas chromatography-mass spectrometer (Shimadzu, Kyoto, Japan). A fused-silica capillary column Shimadzu SH-Rxi-5MS (30 m × 0.25 mm × 0.25 μm) was used for separation. The injection volume was 1 μL, and the injection mode was splitless. The injector temperature was 280 °C. Helium gas (99.999%) was used as the carrier gas at a constant flow rate of 1.0 mL min−1. After optimization of separation, the initial temperature of the column was set at 170 °C for 2 min. It was increased at 16 °C min−1 to 250 °C, then increased at 5 °C min−1 to 280 °C, and maintained for 2 min. For MS detection, the electron impact ion source was operated with an ionization energy of 70 eV at 230 °C. Selected ion monitoring (SIM) mode was performed for the quantitation of analytes.Results and discussion
Selection of the targets for analysis
We used the software, named XenoSites and ADMET predictor, to predict all the potential metabolites and their toxicity of MT. As shown in Table 1, the reduction reaction products may be the main metabolites. The metabolite M2, as the primary reduction metabolite, has the most significant cardiotoxicity and acute toxicity. Then, we used the ADMET predictor and Chemprop to predict the half-life in vivo and liver microsomal clearance of MT and M2, respectively. The half-life period of MT is 7.4 h, and its microsomal clearance is 161.2 μL min−1 × 106 hepatocytes. The half-life period of M2 is 8.2 h, and its microsomal clearance is 49.8 μL min−1 × 106 hepatocytes. It means that M2 has a longer retention time and lower clearance rate. Thus, M2 was chosen as another marker to monitor the use of MT in this work.| Metabolites | Probability | Molecule | hERG toxicity (%) | Acute toxicity (LD50 mg kg−1) | Mutagenicity toxicity (%) |
|---|---|---|---|---|---|
| Reduction metabolite | High | CC12CCC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)CC1CCC3C2CCC4(C3CCC4(C)O)C (MT) O)CC1CCC3C2CCC4(C3CCC4(C)O)C (MT) |
0.03 | 2363 | 0.03 |
| CC12C(C)(CCC1C(CCC3C4(C)CCC(O)C3)C4CC2)O (M2) | 0.25 | 2386 | 0.03 | ||
| CC12C(C)(CCC1C(CCC(C3)C4(C)CCC3O)C4CC2)O |
0.25 | 1625 | 0.03 | ||
| CC12C(C)(CCC1C(CCC3CC(CCC43C)O)C4CC2)O |
0.21 | 686 | 0.08 | ||
| Dehydrogenation metabolite | Medium | CC12C(C)(CCC1C(CCC3CC(CCC43C)O)C4CC2)[O–] |
0.42 | 1031 | 0.03 |
| CC12C(C)(CCC1C([CH–]CC3CC(CCC43C)O)C4CC2)O |
0.45 | 1480 | 0.03 | ||
| CC12C(C)(CCC1C(C[CH–]C3CC(CCC43C)O)C4CC2)O |
0.42 | 1369 | 0.03 | ||
| CC12C(C)(CCC1C(CCC3CC(C[CH–]C43C)O)C4CC2)O |
0.40 | 1625 | 0.03 | ||
| CC12C(C)(CCC1C(CCC3CC([CH–]CC43C)O)C4CC2)O |
0.42 | 472 | 0.03 | ||
| CC12C(C)(CCC1C([CH–][CH–]C3CC([CH–][CH–]C43C)O)C4CC2)[O–] |
0.40 | 1625 | 0.03 | ||
| Oxygenation metabolite | Medium | CC12C(O)(CCC1C(CCC3CC(CCC43C)O)C4CC2)O |
0.03 | 1813 | 0.03 |
| CC12C(C)(C(O)CC1C(CCC3CC(CCC43C)O)C4CC2)O |
0.03 | 2281 | 0.03 | ||
| CC12C(C)(CC(O)C1C(CCC3CC(CCC43C)O)C4CC2)O |
0.03 | 1464 | 0.03 | ||
| CC12C(C)(CCC1C(C(O)CC3CC(CCC43C)O)C4CC2)O |
0.03 | 2262 | 0.03 | ||
| CC12C(C)(CCC1C(CC(O)C3CC(CCC43C)O)C4CC2)O |
0.03 | 1031 | 0.03 | ||
| CC12C(C)(CCC1C(CCC3CC(CC(O)C43C)O)C4CC2)O |
0.03 | 2262 | 0.03 | ||
| CC12C(C)(CCC1C(CCC3CC(C(O)CC43C)O)C4CC2)O |
0.03 | 1369 | 0.03 | ||
| OC12C(O)(C(O)C(O)C1C(C(O)C(O)C3CC(C(O)C(O)C43O)O)C4C(O)C2O)O |
0.03 | 1542 | 0.03 |
Selection of the sample pretreatment method
As is well known, fish is a complex biological sample. Thus, the determination of residues in fish always requires sample pretreatment to meet the analysis requirements, such as extraction, purification, and concentration. Meanwhile, the internal standard also needs to be added before pretreatment to improve the accuracy of quantification. Here, D-MT was chosen as the internal standard. As shown in Fig. 1, there is only a slight difference in the chemical structure of MT, M2, and D-MT. Thus, they possess similar physicochemical properties. We referred to the sample pretreatment procedure in the national standard method which specifies the methods of sample preparation, determination and confirmation by LC-MS/MS of 7 androgen residues in foodstuffs of animal origin and validated it.36 | ||
| Fig. 1 Chemical structures of MT, M2, and D-MT. | ||
The procedure of sample pretreatment is divided into two steps, including LLE and SPE. In the first step, the sample was digested by β-glucuronidase/arylsulfatase due to their ability to degrade extracellular matrix and reduce the sulfuric acid conjugates of MT into natural androgens. Then, the analytes were extracted by acetonitrile three times. Of course, some impurities are inevitably co-extracted. It should be noted that it is necessary to remove some of the lipid impurities before the clean-up of the sample through SPE, as the excess lipid will bind to the alkyl chain in the C18 SPE column, preventing the retention of MT, D-MT, and M2 on it. As we know, n-hexane is always used for removing lipids. However, n-hexane may not have the ability to eliminate sufficient lipids from the sample when the sample is rich in lipids. Repeated use of n-hexane may result in loss of the analytes. Therefore, after being extracted by acetonitrile and concentrated by rotary evaporation, the sample was re-dissolved with acetonitrile-saturated n-hexane, and stored at 4 °C for at least 6 h. It will coagulate the fat in the solution into a white solid, which is easy to be removed. In the second step of SPE, the C18 column was combined with the NH2 column to remove the liposoluble impurities with weaker polarity than MT and M2, and the strong polarity impurities. To evaluate the efficiency of the SPE procedure, the standard solutions of MT, M2, and D-MT dissolved in acetonitrile and blank matrix were directly loaded on SPE columns for test, and their recovery rates were analyzed in triplicate. The obtained data were all in the range of 93.2–112.6%, which met the requirement of sample pretreatment. It means that the proposed SPE procedure will not be affected by the presence of solvent and impurities in the sample.
GC-MS analysis programs
Temperature-programmed GC was employed for the separation due to the complicated matrix of fish muscle. Meanwhile, the full scan mode of MS was used at first for the identification of the targets and obtaining their specific mass-to-charge ratio (m/z) for SIM mode. After the optimization of GC-MS conditions, the retention time, qualitative and quantitative ions of the analytes were summarized in Table 2.| Analyte | Retention time (min) | Quantitative ion (m/z) | Qualitative ion 1 (m/z) | Qualitative ion 2 (m/z) |
|---|---|---|---|---|
| M2 | 11.11 | 230 | 235 | 135 |
| D-MT | 12.63 | 305 | 306 | 287 |
| MT | 12.66 | 302 | 303 | 284 |
Specificity
Specificity refers to the ability to unambiguously evaluate an analyte in the presence of possible components, such as impurity and matrix. As shown in Fig. 2, M2 was first detected with GC-MS analysis in SIM mode. D-MT and MT were nearly co-eluted. However, it does not affect their qualitative and quantitative analysis because of their different m/z. No other peaks with adjacent retention times of the targets were observed in the fish samples. All of the results indicated good specificity of this method.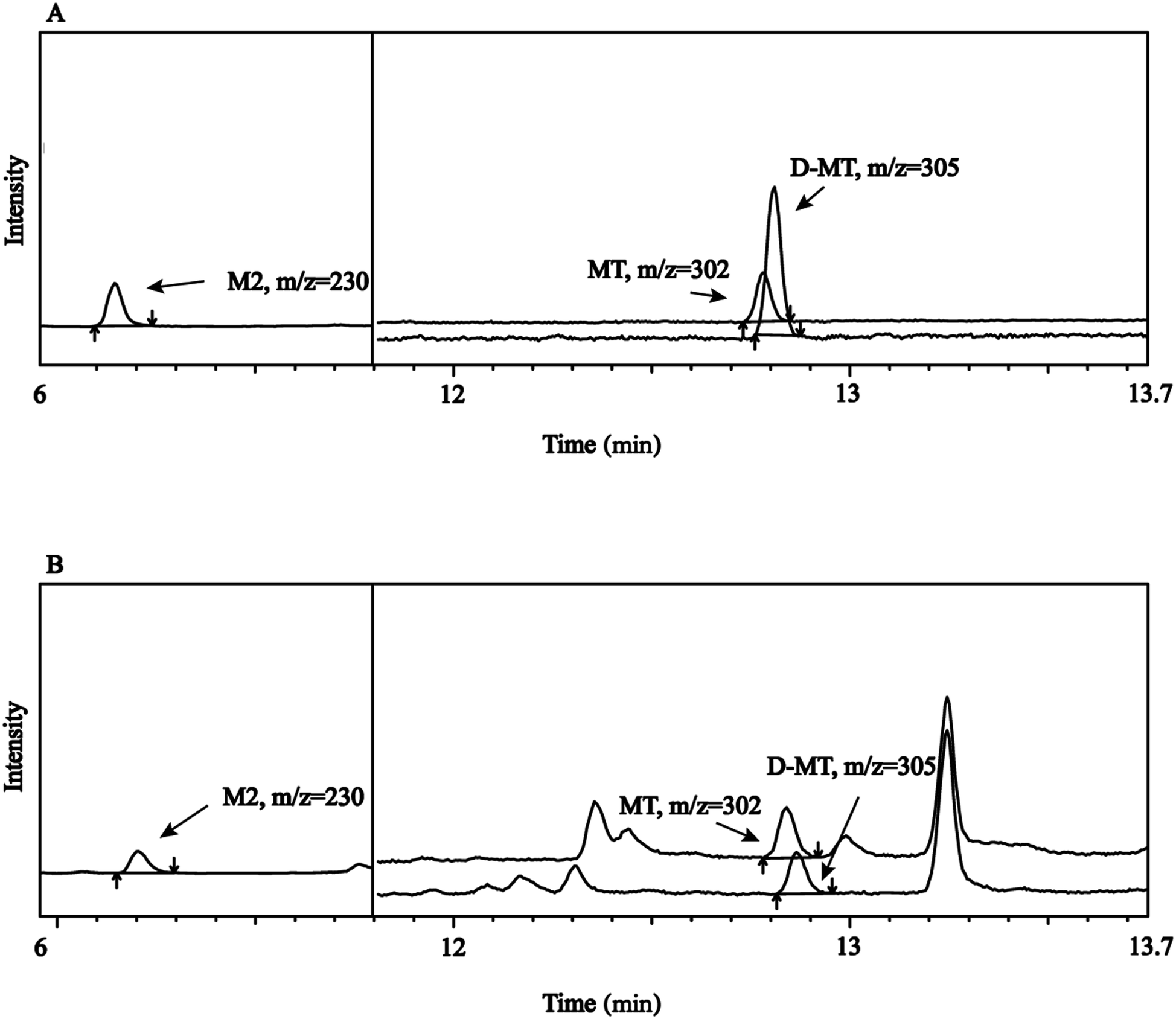 | ||
| Fig. 2 SIM profiles of (A) standard solution, (B) fish sample spiked with MT, M2, and D-MT. | ||
Limit of detection (LOD) and limit of quantitation (LOQ)
LOD and LOQ of each analyte were calculated based on the signal-to-noise ratio (S/N) of 3:1 and 10:1, respectively. Given that the real sample might exist the matrix effect, LOQ was also validated with the sample matrix. LODs of MT and M2 were determined to be 20 μg kg−1 and 15 μg kg−1, respectively. LOQs of MT and M2 were both 50 μg kg−1.
Linearity
The linearity of an analytical method refers to its ability to obtain test results proportional to the concentration or amount of analyte in the sample. In this work, D-MT was used as the internal standard to eliminate the potential interference and provide the most robust compensation for the impacts from the matrix effect, sample pretreatment, and measuring instruments. The ratio of the peak area of MT and M2 to D-MT was used for the calibration and calculation of samples. The calibration curves of MT and M2 were exhibited in Fig. 3. Within the concentration range of 50.0 to 500.0 ng mL−1, the correlation coefficients of MT and M2 were 0.9992 and 0.9996, respectively, indicating good linear relationships.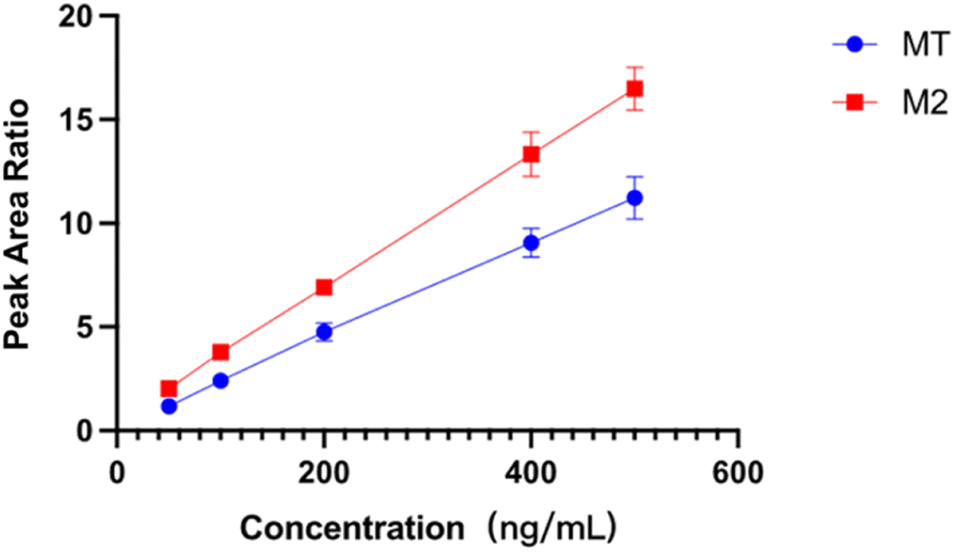 | ||
| Fig. 3 Calibration curve of MT and M2 (n = 3). | ||
Accuracy and precision
To further demonstrate the feasibility of the proposed analytical methodology, the recoveries of MT and M2 in fish muscle were measured at three spiked levels. The average recovery and relative standard deviation (RSD) were used to evaluate the accuracy and precision. The results in Table 3 showed the recoveries were in the range of 99.7–116.0% for MT, and 105.3–116.6% for M2. The RSD values were all below 8.3%.| Spiked concentration (μg kg−1) | Average recovery (%) | RSD (%) | ||
|---|---|---|---|---|
| MT | M2 | MT | M2 | |
| 50.0 | 104.4 | 105.3 | 5.2 | 6.8 |
| 150.0 | 99.7 | 116.6 | 8.2 | 8.3 |
| 400.0 | 116.0 | 115.7 | 6.3 | 5.6 |
Matrix effect
The matrix effect of MS may severely impair sensitivity and accuracy of the method. In this study, we evaluated the potential matrix effect by comparing the MS response with and without the presence of blank sample matrix. Six different fish species (Basa, Mackerel, Capelin, Salmon, Cod, and Flounder) were chosen for the purpose of examining. The analytes were spiked at two levels of concentration. To investigate the potential ion-suppression or ion-enhancement effect, matrix factors (MF) of each target were calculated. The results in Table 4 showed that the average MF of MT and M2 was in the range of 0.95–1.01, and the RSD values were all below 6.0%, suggesting little matrix effect.| Spiked concentration (μg kg−1) | Matrix factor (MF) | RSD (%) | ||
|---|---|---|---|---|
| MT | M2 | MT | M2 | |
| 100.0 | 0.99 | 1.01 | 6.0 | 3.5 |
| 300.0 | 1.00 | 0.95 | 3.4 | 4.1 |
Comparison with other methods
In Table 5, we summarized the instrument, pretreatment, sensitivity, repeatability, and accuracy in this work together with those obtained in other literatures. Recent research findings indicate that LLE exhibits superior recovery rates compared to SPE, refrigeration, and dispersed solid phase methods for the extraction of methyltestosterone from biological samples.37,39 Nevertheless, it is worth noting that LLE being used alone tends to introduce more impurities and result in a heightened interference signal.38 Consequently, we propose the integration of LLE with SPE in this work to enhance recovery rate and eliminate the background interference. In addition, MT and its main metabolite are simultaneously determined by GC-MS rather than LC-MS/MS. It means we offer another robust method for their qualitative and quantitative analysis in complex biological samples. Although the sensitivity of the proposed method is not good enough, it is mainly due to the scanning mode of MS being SIM. We believe that it could be improved by using GC-MS/MS for the determination.| Method | Pretreatment | Sample | Target | Recovery (%) | LOD (μg kg−1) | RSD (%) | Ref. |
|---|---|---|---|---|---|---|---|
| LC-MS/MS | LLE | Fish, shrimp, crab, bullfrog and mussel | MT | 86.4–104.6 | 0.4 | 0.45–7.31 | 37 |
| LC-MS/MS | LLE | Fish and shrimp | MT | 73–115 | 2 | 2.1–7.0 | 39 |
| LC-MS/MS | LLE | Fish and shrimp | MT | 96–105 | 0.4 | 96–105 | 32 |
| LC-MS/MS | QuEChERS | Pork, chicken and beef | MT | 80–96 | 0.4 | 1–5 | 40 |
| GC-MS | LLE, SPE | Fish | MT, M2 | 99.7–116.6 | MT: 20, MT: 15 | 5.2–8.3 | Present method |
Application to real samples
The developed method was applied to test MT and M2 in crucian carp and silver carp bought from the supermarket. Although there were no residues observed in the real samples, the method could still have significant application for the determination of MT and M2 in food of animal origin.Conclusions
MT, as a banned drug for food-producing animals, is needed to be strictly monitored and supervised. Although more and more methods for the detection of methyltestosterone have been developed, the research and application of its metabolites in the field of food safety have not been paid much attention. In this research, the main metabolite of MT was chosen as another marker for controlling the abuse of MT in the field of food analysis. A simple GC-MS method coupled with traditional sample pretreatment was established and validated for the determination of MT and M2 in fish. Compared with previous work, MT and its metabolite were simultaneously detected for the first time, and there was no need for any derivatization before the analysis of GC-MS. The established method was sensitive, accurate, and precise. The LOQs of MT and M2 were both 50 μg kg−1. The recoveries of real samples were all in the range of 99.7–116.6% with the RSDs between 5.2% and 8.3%. Obviously, the analysis of methyltestosterone and its metabolites in food has some feasibility, which is something we want to emphasize and present repeatedly in this work. It will help to provide more information on the safety of food of animal origin.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Key Research and Development Program of China (Grant No. 2019YFC1605400), and the Natural Science Foundation of Jiangsu Province (Grant No. BK20200567).References
- R. H. Devlin and Y. Nagahama, Aquaculture, 2002, 208, 191–364 CrossRef CAS.
- F. Piferrer, A. Beaumont, J. C. Falguiere, M. Flajshans, P. Haffray and L. Colombo, Aquaculture, 2009, 293, 125–156 CrossRef.
- M. T. Ridha, International Aquatic Research, Islamic Azad University, Tonekabon Branch, 2011, vol. 3 Search PubMed.
- X. Zhang, M. Li, H. Ma, X. Liu, H. Shi, M. Li and D. Wang, Endocrinology, 2017, 158, 2634–2647 CAS.
- M. H. Li, H. H. Yang, M. R. Li, Y. L. Sun, X. L. Jiang, Q. P. Xie, T. R. Wang, H. J. Shi, L. N. Sun, L. Y. Zhou and D. S. Wang, Endocrinology, 2013, 154, 4814–4825 CrossRef CAS PubMed.
- Q. Zhang, D. Ye, H. Wang, Y. Wang, W. Hu and Y. Sun, Endocrinology, 2020, 161 Search PubMed.
- D. N. Jiang, H. H. Yang, M. H. Li, H. J. Shi, X. B. Zhang and D. S. Wang, Mol. Reprod. Dev., 2016, 83, 497–508 CrossRef CAS PubMed.
- N. T. Shahidi, Clin. Therapeut., 2001, 23, 1355–1390 CrossRef CAS PubMed.
- R. T. Sparks, B. S. Shepherd, B. Ron, N. H. Richman III, L. G. Riley, G. K. Iwama, T. Hirano and E. G. Grau, Comp. Biochem. Physiol., B, 2003, 136, 657–665 CrossRef PubMed.
- M. Nagaraju and G. S. Devi, Ann. Romanian Soc. Cell Biol., 2021, 25, 20608–20616 Search PubMed.
- R. K. Bhandari, M. Nakamura, T. Kobayashi and Y. Nagahama, Gen. Comp. Endocrinol., 2006, 145, 20–24 CrossRef CAS PubMed.
- Q. Lyu, J. Hu, X. Yang, X. Liu, Y. Chen, L. Xiao, Y. Liu, Q. Wang, J. Chen, M. Huang, Z. Yu, H. Yang, H. Shi, Y. Zhang and H. Zhao, Gen. Comp. Endocrinol., 2019, 274, 26–36 CrossRef CAS PubMed.
- N. Mlalila, C. Mahika, L. Kalombo, H. Swai and A. Hilonga, Environ. Sci. Pollut. Res., 2015, 22, 4922–4931 CrossRef CAS PubMed.
- A. M. Budd, Q. Q. Banh, J. A. Domingos and D. R. Jerry, J. Mar. Sci. Eng., 2015, 3, 329–355 CrossRef.
- J. A. Beardmore, G. C. Mair and R. Lewis, Reproductive Biotechnology in Finfish Aquaculture, 2001, 283–301 Search PubMed.
- S. K. Ong, P. Chotisukarn and T. Limpiyakorn, Water, Air, Soil Pollut., 2012, 223, 3869–3875 CrossRef CAS.
- S. Biswas, C. Shapiro, W. Kranz, T. Mader, D. Shelton, D. D. Snow, S. L. Bartelt-Hunt, D. Tarkalson, S. Van Donk and T. C. Zhang, J. Soil Water Conserv., 2013, 68, 325–336 CrossRef.
- I. Thiblin, H. Garmo, M. Garle, L. Holmberg, L. Byberg, K. Michaelsson and R. Gedeborg, Drug Alcohol Depend., 2015, 152, 87–92 CrossRef CAS PubMed.
- S. Chang, A. B. Munster, J. Gram and J. J. Sidelmann, Semin. Thromb. Hemost., 2018, 44, 734–746 CrossRef CAS PubMed.
- C. L. G. Rivero-Wendt, A. L. Miranda-Vilela, I. Domingues, R. Oliveira, M. S. Monteiro, M. A. M. Moura-Mello, R. Matias, A. Soares and C. K. Grisolia, J. Environ. Sci. Health, Part A, 2020, 55, 1321–1332 CrossRef CAS PubMed.
- H. Niemikoski, D. Koske, U. Kammann, T. Lang and P. Vanninen, J. Hazard. Mater., 2020, 391, 122221 CrossRef CAS PubMed.
- Y. Liu, Y. Song, B. Cheng, J. Dong, N. Xu, S. Zhou, Q. Yang and X. Ai, Food Anal. Methods, 2020, 13, 516–529 CrossRef.
- E. Dubreil, M. Laurentie, J. M. Delmas, M. Danion, T. Morin, D. Hurtaud-Pessel, A. Viel, P. Sanders and E. Verdon, Chemosphere, 2021, 262, 127636 CrossRef CAS PubMed.
- W. Schänzer, G. Opfermann and M. Donike, Steroids, 1992, 57, 537–550 CrossRef PubMed.
- O. J. Pozo, P. Van Eenoo, K. Deventer, L. Lootens, W. Van Thuyne, M. K. Parr, W. Schanzer, J. V. Sancho, F. Hernandez, P. Meuleman, G. Leroux-Roels and F. T. Delbeke, Drug Metab. Dispos., 2009, 37, 2153–2162 CrossRef CAS PubMed.
- W. Schanzer and M. Donike, Anal. Chim. Acta, 1993, 275, 23–48 CrossRef.
- D. Martinez-Brito, X. de la Torre, M. K. Parr and F. Botre, Rapid Commun. Mass Spectrom., 2020, 34, e8834 CrossRef CAS PubMed.
- D. Martinez-Brito, M. Iannone, M. A. Tatangelo, F. Molaioni, X. de la Torre and F. Botre, Rapid Commun. Mass Spectrom., 2020, 34, e8870 CrossRef CAS PubMed.
- M. Blokland, H. Van Rossum, H. Herbold, S. Sterk, R. Stephany and L. Van Ginkel, Anal. Chim. Acta, 2005, 529, 317–323 CrossRef CAS.
- E. Hong, G. Zheng, D. Meng, S. Wu, B. Ma, L. Ma and Y. Song, J. Anhui Agric. Univ., 2011, 38, 911–915 CAS.
- X. Li, T. Yuan, T. Zhao, X. Wu and Y. Yang, J. Chromatogr. Sci., 2020, 58, 880–886 CAS.
- J. M. Storey, S. B. Clark, A. S. Johnson, W. C. Andersen, S. B. Turnipseed, J. J. Lohne, R. J. Burger, P. R. Ayres, J. R. Carr and M. R. Madson, J. Chromatogr. B, 2014, 972, 38–47 CrossRef CAS PubMed.
- P. Regal, C. Nebot, B. I. Vazquez, A. Cepeda and C. A. Fente, Meat Sci., 2010, 84, 196–201 CrossRef CAS PubMed.
- P. S. Chu, M. Lopez, S. Serfling, C. Gieseker and R. Reimschuessel, J. Agric. Food Chem., 2006, 54, 3193–3198 CrossRef CAS PubMed.
- Y. Shinohara, K. Isurugi and T. Hashimoto, J. Chromatogr. B Biomed. Sci. Appl., 2000, 741, 271–278 CrossRef CAS PubMed.
- L. M. Lin, L. Z. Chen, M. Du, Q. H. Meng, Z. F. Yue, T. Peng, S. P. Pan and D. Y. Ba, Chinese National Standard, 2010, SN/T 2677-2010 Search PubMed.
- X. Zhang, C. Fang, X. Lou, G. Yang, C. Kong, Y. Shi and D. Huang, Microchem. J., 2022, 183, 108119 CrossRef CAS.
- M. P. Juhascik and A. J. Jenkins, J. Chromatogr. Sci., 2009, 47, 553–557 CAS.
- W. Zheng, K. H. Yoo, J. M. Choi, D. H. Park, S. K. Kim, Y. S. Kang, A. M. Abd El-Aty, A. Hacimuftuoglu, J. Wang, J. H. Shim and H. C. Shin, Biomed. Chromatogr., 2019, 33, e4396 CrossRef PubMed.
- M. Lopez-Garcia, R. Romero-Gonzalez and A. G. Frenich, J. Chromatogr. A, 2018, 1540, 21–30 CrossRef CAS PubMed.
Footnote |
| † The first two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |