
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMonitoring host–pathogen interactions using chemical proteomics
Angela
Weigert Muñoz
 a,
Weining
Zhao
*b and
Stephan A.
Sieber
*ac
a,
Weining
Zhao
*b and
Stephan A.
Sieber
*ac
aCenter for Functional Protein Assemblies, Department of Bioscience, TUM School of Natural Sciences, Technical University of Munich, Ernst-Otto-Fischer-Straße 8, D-85748 Garching, Germany. E-mail: stephan.sieber@tum.de
bCollege of Pharmacy, Shenzhen Technology University, Shenzhen 518118, China. E-mail: zhaoweining@sztu.edu.cn
cHelmholtz Institute for Pharmaceutical Research Saarland (HIPS), Germany
First published on 10th November 2023
Abstract
With the rapid emergence and the dissemination of microbial resistance to conventional chemotherapy, the shortage of novel antimicrobial drugs has raised a global health threat. As molecular interactions between microbial pathogens and their mammalian hosts are crucial to establish virulence, pathogenicity, and infectivity, a detailed understanding of these interactions has the potential to reveal novel therapeutic targets and treatment strategies. Bidirectional molecular communication between microbes and eukaryotes is essential for both pathogenic and commensal organisms to colonise their host. In particular, several devastating pathogens exploit host signalling to adjust the expression of energetically costly virulent behaviours. Chemical proteomics has emerged as a powerful tool to interrogate the protein interaction partners of small molecules and has been successfully applied to advance host–pathogen communication studies. Here, we present recent significant progress made by this approach and provide a perspective for future studies.
 Angela Weigert Muñoz | Angela Weigert Muñoz is a postdoctoral researcher in the group of Stephan Sieber at the Technical University of Munich and has completed her PhD in the Sieber group this year. In her research, she uses chemical proteomics to unravel bacterial responses to eukaryotic hormones. Prior to that, she obtained her MSc in biochemistry at the Technical University of Munich. She has stayed abroad during her Master's and PhD studies at the Georgia Institute of Technology (USA) and the Emory University (USA). |
 Weining Zhao | Weining Zhao studied chemistry in Zhengzhou University (China) and obtained his PhD from Technical University of Munich in 2016 under the supervision of Prof. Stephan A. Sieber. He further conducted two years’ postdoctoral research in Sieber's group with a focus on target evaluation of bioactive small molecules in Gram-negative bacteria via chemical proteomics. After working as a senior mass spectrometry scientist in Beijing Genomics Institute (BGI) for two years, he has been an associate professor at the Department of Pharmacy in Shenzhen Technology University since July 2020 and started his independent research career. His main research interests are related to functional proteomics and synthetic biology. |
 Stephan A. Sieber | Stephan A. Sieber obtained his graduate degree from the University of Marburg in Germany. Following his studies, he conducted postdoctoral research at the Scripps Research Institute in La Jolla, USA, as part of the Cravatt group. In 2006, he started his independent research career at the University of Munich (LMU), Germany, with the support of a DFG Emmy-Noether grant. In 2009, he accepted the position of a full professor at the Technical University of Munich. Research in his lab focuses on chemical biology and chemical proteomics, with a special emphasis on investigating bioactive small molecules to discover new antibiotic targets in pathogenic bacteria as well as developing unprecedented strategies to interfere with bacterial virulence. |
1. Introduction
The evolution and spread of antimicrobial resistance have made drug-resistant bacterial infections one of the most serious global threats to human health in the 21st century.1 Classical antibiotics target essential processes in bacteria and thus exert selection pressure, which drives antimicrobial resistance formation.2 This is further aggravated by the dissemination and maintenance of resistance factors among pathogens, e.g. by widespread horizontal gene transfer. In contrast to this, the development of novel antibiotics is extremely sluggish3 and no new antimicrobial drug has been developed by traditional methods in the last 30 years.4 Therefore, the speed of microbial resistance development and the difficulties in the antibiotic discovery have culminated in a serious global threat which urgently necessitates the development of novel therapeutic strategies with unprecedented modes of action beyond the conventional mechanisms.2 Innovative novel approaches have already exhibited promising antibacterial effects in in vitro and/or in vivo experiments such as phage therapy,5 phage endolysins,6,7 CRISPR-Cas systems,8 efflux pump inhibitors,9 combinatorial therapy,10 antibacterial monoclonal antibodies,11 and virulence inhibition,12 among others.2 However, additional antimicrobial strategies are still urgently needed.While conventional antimicrobial research mainly focuses on bactericidal or bacteriostatic compounds, the inhibition of bacterial communication has been put forward as a novel therapeutic strategy.13–15 Although this approach may not kill bacteria, limiting their ability to propagate an infection is an attractive way to block pathogenicity (pathoblocker) and help the immune system to take over their elimination. Microbial pathogens have lived and evolved for millions of years in close association with their hosts and their mutual interaction has become important for their life cycle and infectivity.16 Pathogens have developed elaborate strategies to circumvent multiple eukaryotic defence lines and can thus pose severe threats to human health. Often, these pathogenicity mechanisms rely on the interactions with the host environment, therefore, deciphering the molecular processes at the interface of eukaryotic hosts and bacterial pathogens could help to conceive unprecedented antimicrobial therapeutic strategies and to reveal novel antibiotic targets against infectious diseases.17 Chemical proteomics is a valuable technique to interrogate this molecular communication since it can be applied to track the interaction of proteins with small molecules involved in interkingdom signalling. Here, we illustrate popular chemical proteomics techniques and summarise recent advances by this approach in the field of host–pathogen communication.18–20 This review focusses on studies employing chemical probes to elucidate biological processes involved in the interplay of animal hosts with pathogenic bacteria/mycobacteria and how this communication can contribute to virulence. Host interactions with the microbiome,21–25 viruses,26–28 or protozoans,29,30 have been reviewed elsewhere. Moreover, recent reviews have covered related topics such as chemical proteomics to elucidate bacterial biology,31 host biology during infection,32 or cell–cell communication.33
2. Chemical proteomics tools
Chemical proteomics is a multidisciplinary approach which utilises small molecule probes to label and detect specific proteins or subsets of the proteome. Probes may be used for the identification of protein targets of small molecules or to label proteins using chemical analogues of enzymatic substrates or of precursors of post-translational modifications (PTMs) via metabolic incorporation by endogenous cellular biosynthesis machineries.34 Activity-based protein profiling (ABPP)/affinity-based protein profiling (AfBPP) is a strategy to identify protein targets of chemical compounds and requires the derivatisation of the bioactive molecules with chemical handles.35,36 These probes can be used for target identification, searching for druggable targets as well as for functional studies of proteins.35,36 It is noteworthy that this technique is based on the activity or affinity between small molecule probes and their targets regardless of protein abundance.37–46 Central to ABPP and AfBPP is the design and synthesis of suitable probes closely mimicking the native bioactive small molecules. It is essential that the probes maintain all structural elements necessary to retain the bioactivity of the parent compound, often requiring an assessment of the structure activity relationship. The activity-based probes (ABPs) typically consist of functionally different features: (1) a bioactive small molecule-based binding group to direct the engagement of the probe to its targets. In the case of affinity-based reversible binding, photoreactive groups such as diazirines, benzophenones, or aryl azides are incorporated into the probe. (2) A reporter tag which enables subsequent visualisation or enrichment after bioorthogonal ligation. (3) Optionally, a linker can connect the ligand and the reporter tag to provide spatial separation as well as to fine-tune physicochemical properties such as solubility.Metabolic labelling is commonly used to tag biomolecules by incorporating a reporter group via the endogenous enzymatic machinery of a cell. This is achieved by feeding a chemically modified building block (e.g., lipid, amino acid, nucleotide, carbohydrate) to a biological system such as cells or animals.47–49 As many components are distinct in microbial pathogens from eukaryotic cells, substantial progress in host–pathogen interaction studies has been made using metabolic labelling.50–58
In both approaches, chemical probes may directly be modified with a reporter group such as a fluorophore or an affinity handle (e.g., biotin) to enable the analysis of target proteins. Often, however, it is more convenient to use smaller, latent tags which perturb biological functions to a lesser extent, and which can be chemically derivatised at a later point. This two-step approach typically applies azides or terminal alkynes as tags which can be derivatised by bioorthogonal chemistries such as copper(I)-catalysed azide–alkyne cycloaddition (CuAAC),59,60 the strain-promoted azide–alkyne cycloaddition (SPAAC)61–63 or the Staudinger ligation.61,64,65 A typical chemoproteomics workflow includes the following steps: the probes are incubated with live cells to allow the ligand bind to its targets or facilitate metabolic incorporation. In case of labelling with reversible binders, the affinity-based probe should be irradiated with UV light after incubation to trigger covalent bond formation between its photoreactive group and its binding partners. This covalent bond assures a robust complex that remains stable in the following steps. Next, the reporter tag is appended most typically by “click chemistry”. This can be a fluorophore for the visualisation of target proteins of the bioactive small molecules, or a biotin tag which allows the purification of the covalently bound targets on avidin beads followed by tryptic digestion and liquid chromatography tandem mass spectrometry analysis (LC-MS/MS). Current mass spectrometry technologies combined with label-free or isotopic labelling enable global and comprehensive profiling of protein targets of small molecules by the comparison of probe-treated proteome to control samples (Fig. 1(a) and (b)).66 Alternative chemoproteomic methods have been developed which circumvent the need for chemically modified probes. Proteomic strategies such as drug affinity responsive target stability (DARTS),67 cellular thermal shift assay (CETSA),68 thermal proteome profiling (TPP),69,70 or limited proteolysis coupled with MS (LiP-MS)71–73 employ native bioactive molecules to elucidate shifts in protein stability upon small molecule binding.
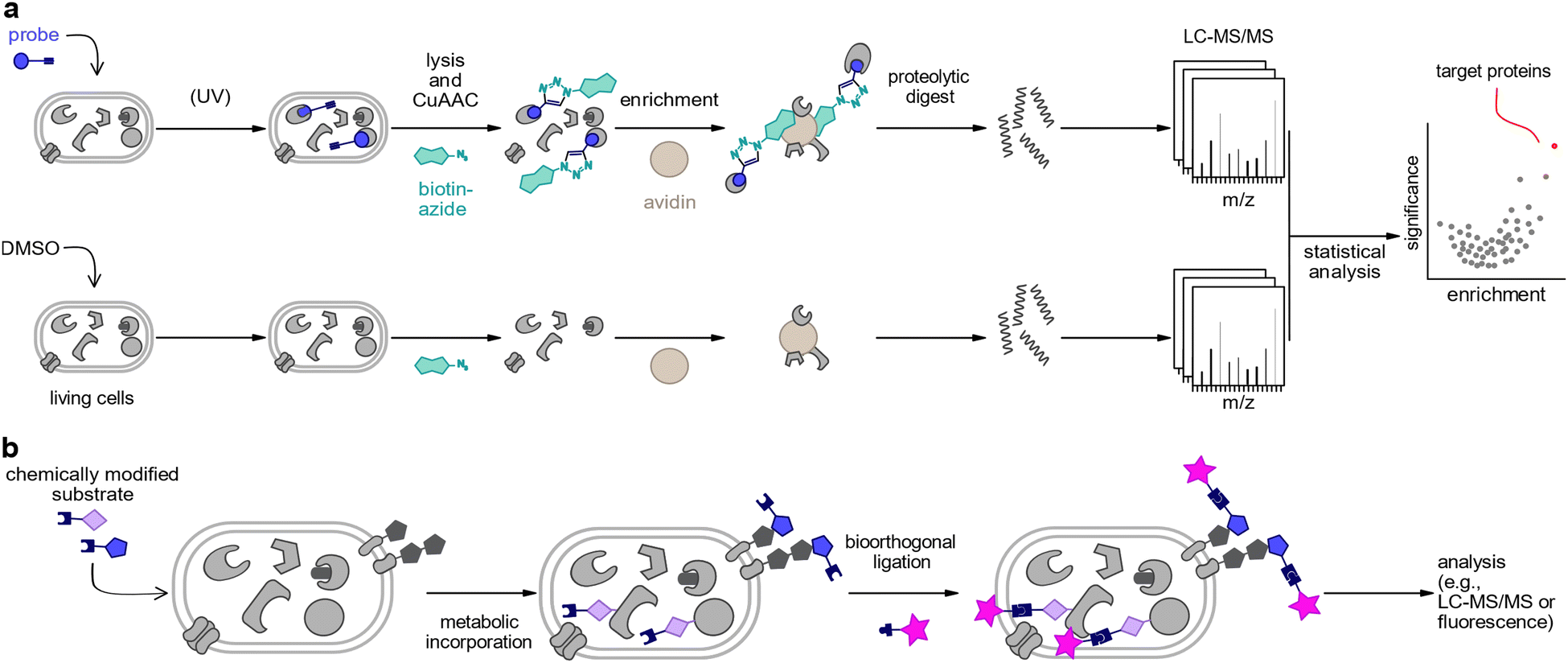 | ||
| Fig. 1 Chemical proteomics strategies for the investigation of host–pathogen interactions. (a) Basic concept of activity-based protein profiling (ABPP)/affinity-based protein profiling (AfBPP). CuAAC: copper(I)-catalysed azide alkyne cycloaddition. (b) General scheme for metabolic labelling. | ||
3. Host–microbe interactions via signalling molecules
Bacteria have lived in close association with eukaryotic hosts and, as a consequence, they have evolved specific receptors for small molecules such as eukaryotic hormones (Fig. 2(a)).74 Opportunistic bacterial pathogens can even exploit these signalling systems to adjust their expression of virulence genes to the host's stress status.75 For instance, dynorphin A, a mammalian peptide hormone that is released during host stress enhances the production of the toxin pyocyanin by the opportunistic pathogen Pseudomonas aeruginosa.76 To identify protein targets of dynorphin A in P. aeruginosa PAO1, Wright et al. devised a set of peptide photoprobes 1–5. They mainly consisted of the dynorphin A 1–13 sequence and carried a terminal alkyne as well as a benzophenone in 1 and 2, an aryl azide in 3, or a diazirine in 4 and 5.77 Probes 3 and 4 unravelled the histidine kinase ParS as target which was confirmed by dynorphin A mediated upregulation of a ParS-dependent bacterial defence response against antimicrobial peptides on the proteome level (Fig. 2(b)).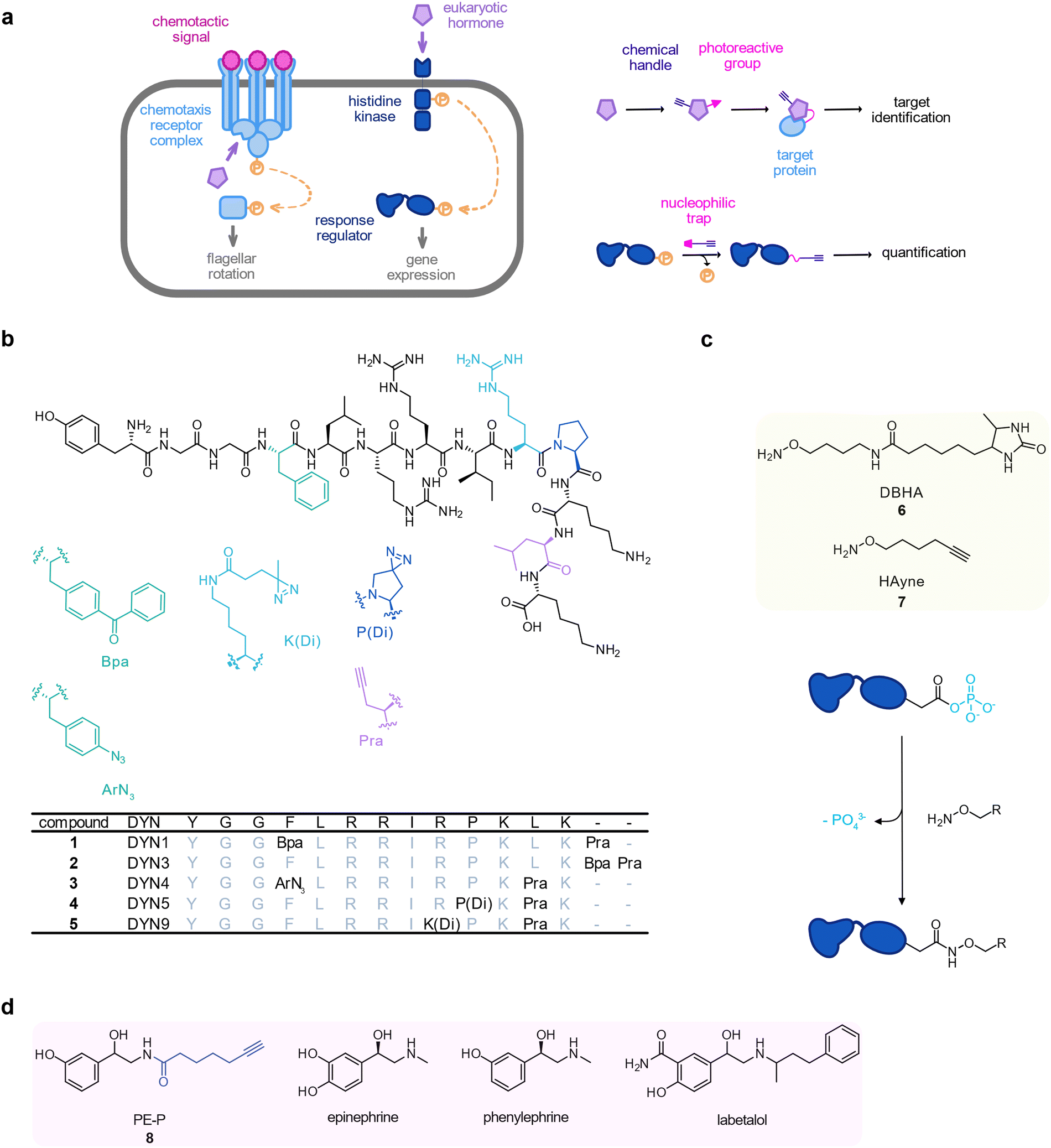 | ||
| Fig. 2 Investigation of hormone-based interkingdom signalling. (a) Schematic representation of bacterial sensing of eukaryotic hormones by the chemotaxis signalling complex (left) or a two-component system (right) and probes for target identification (top) or monitoring of two-component system activation (bottom). (b) Dynorphin A probes 1–5. Amino acids in the dynorphin A sequence were replaced by unnatural building blocks as indicated by the colours. (c) Hydroxylamine probes 6 (DHBA) and 7 (HAyne), (d) Catecholamine hormones and probe 8 (PE-P). | ||
The histidine kinase ParS forms a two-component system with its cognate response regulator ParR. In two-component systems, the most widespread type of bacterial signalling sensors,78 the histidine kinase ultimately transfers a phosphate group onto an aspartyl residue in the response regulator when it senses a signal in the periplasm, thereby activating the transcription of its target genes. Phosphoaspartates consist of a mixed carboxylic acid–phosphoric acid anhydride (acyl phosphate), which are highly prone to nucleophilic attack, and therefore, hydrolysis.79 Due to the short lifetime of phosphoaspartate modifications and their transient nature, it is hardly possible to directly detect them by common MS-based (phosphoproteomics) methods. In an approach termed reversed-polarity ABPP (RP-ABPP), Chang et al. reported a nucleophilic hydroxylamine probe 6 (desthiobiotin-containing O-alkyl hydroxylamine probe, DBHA) equipped with a desthiobiotin handle to capture phosphoaspartate modifications by converting them into stable hydroxamates.80 Allihn et al. used a refined, minimal clickable hydroxylamine alkyne probe 7 (HA-yne) to trap response regulators phosphorylated in the presence of dynorphin.817 was applied in live P. aeruginosa challenged with dynorphin and labelled proteins were ligated to isotopically labelled desthiobiotin azide (isoDTB) tags for peptide enrichment and quantification.82 As expected, ParR was phosphorylated in the presence of dynorphin, corroborating the results of the previous study. Interestingly, this approach revealed that another response regulator, CprR, responded to dynorphin treatment in an even stronger fashion. Similar to the ParRS system, CprR is part of a two-component system involved in the antimicrobial peptide defence system (Fig. 2(c)).
The catecholamine stress hormones epinephrine and norepinephrine (also known as adrenaline and noradrenaline) modulate a variety of virulence associated phenotypes in prokaryotes including enhanced motility of Vibrio campbelli.83–86 The bacterial adrenergic sensor of these hormones was recently revealed using photoaffinity labelling in a study by Weigert Muñoz et al.87 The catechol group is a widespread motif in bacterial siderophores to bind and scavenge iron. Catecholamines are also exploited by bacteria as so-called xenosiderophores to acquire iron from the host.86 To decipher the cellular receptors of these hormones, a probe was designed based on the adrenergic agonist phenylephrine, which, due to the lack of an intact catechol group, cannot act as a xenosiderophore. The probe still retained the enhanced motility phenotype but did not bind to iron-dependent proteins allowing to focus on the receptor targets. This inherently photoreactive probe, 8 (PE-P), enriched the chemotaxis protein CheW in live V. campbellii (Fig. 2(d)). Interestingly, binding of 8 at CheW was outcompeted not only by the parent compounds phenylephrine and epinephrine but also by the clinically applied adrenergic antagonist labetalol, corroborating that CheW is indeed an adrenergic receptor. Consistent with this, epinephrine and phenylephrine interfered with the chemotaxis of V. campbellii towards glucose.
Vice versa, bacterial signalling molecules can affect eukaryotic cell physiology (Fig. 3(a)). Quorum sensing denotes bacterial communication via diffusible small molecule signals, termed autoinducers, in order to coordinate cellular processes within their community or across species. Beyond that, autoinducers are known to influence eukaryotic cell physiology and to promote chronic inflammation and bacterial survival within the host.88–90N-acyl homoserine lactones (AHLs) are common signal molecules of Gram-negative bacteria,91 which induce the phosphorylation of MAPK p38 and eIF2α in mammalian cells and compromise the production of inflammation mediators.90 They furthermore attenuate TLR4-dependent innate immune responses to lipopolysaccharides (LPS) by disrupting NF-κB signalling in mammalian cells.89
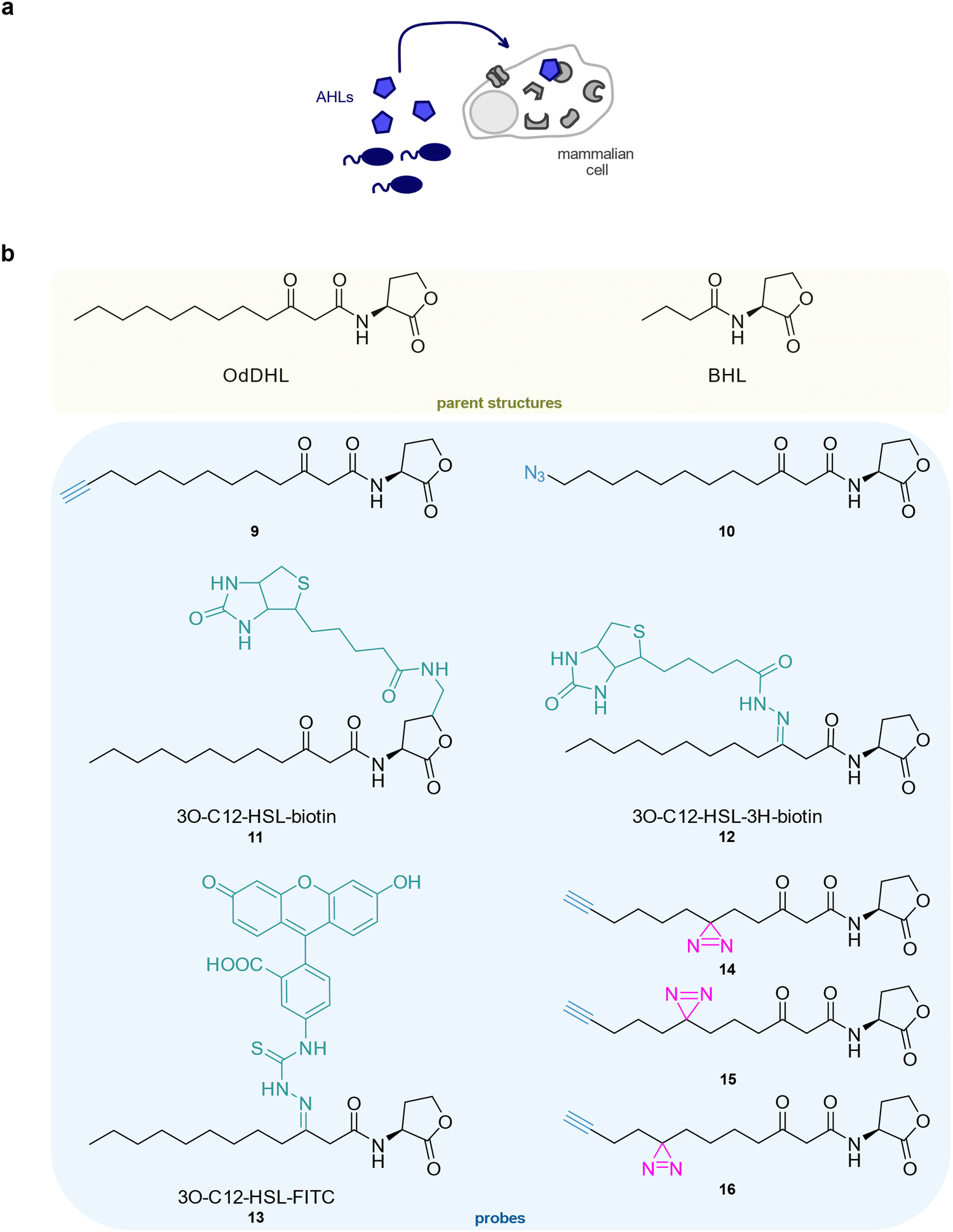 | ||
| Fig. 3 Investigation of autoinducer-mediated interkingdom signalling. (a) Targets of bacterial AHLs were investigated in human cells using different chemical probes. (b) Structures of AHL probes. BHL: N-butyryl homoserine lactone, OdDHL: N-(3-oxododecanoyl)-homoserine. | ||
An initial study from the Janda group reported a set of clickable AHL probes based on N-(3-oxododecanoyl)-homoserine lactone (OdDHL) from P. aeruginosa which were equipped with a terminal alkyne 9 or an azide 10 but lacked a photocrosslinker.92 Interestingly, 9 showed good activity in a P. aeruginosa autoinducer assay but induced only weak phosphorylation of MAPK p38 and eIF2α phosphorylation in mammalian cells. Interestingly, 10 was especially active in the latter system. The D-homoserine lactone enantiomer of 10 was inactive in both systems.90,92 At that stage, probes 9 and 10 were not used for target identification experiments. Another study from the Vikström lab used biotin-coupled OdDHL probes 11 and 12 to detect the IQ-motif containing GTPase-activating protein (IQGAP1) in Caco-2 cells as a putative protein target of OdHL.93 Interestingly, IQGAP1 could only be enriched by 12 but not by 11 in which the homoserine ring was modified. A fluorescein-coupled OdDHL probe 13 revealed the co-localisation of OdDHL and IQGAP1. By targeting IQGAP1, P. aeruginosa could induce remodelling of the host actin cytoskeleton to aid the infection process. Furthermore, the Meijler group designed three OdDHL diazirine probes 14–16 to enable the identification of protein targets via chemical proteomics.94–96 These probes bound to their native bacterial receptor LasR, and also elicited phosphorylation of the eukaryotic initiation factor 2a (eIF2a) and protein kinase p38.94,95 Although 14 and 15 induced the strongest responses in a P. aeruginosa QS reporter strain, 15 was most active in human cells, illustrating the context-dependence of biological activity. Finally, 14 was applied to identify OdDHL targets in human cells using stable isotope labelling by amino acids in cell culture (SILAC) labelling.96 The major vault protein (MVP) was identified as a target of OdDHL and the binding sites of 14 were within regions that could be involved in the interaction of MVP with lipid rafts. The translocation of MVP into lipid rafts was indeed promoted by OdDHL and it was proposed that these newly functionalised membranes can gauge apoptotic signalling in human cells. A recent study from the Meijler group reports chemical probes based on another P. aeruginosa signal, the short-chain N-butyryl homoserine lactone (BHL), which were applied for the identification of protein targets in P. aeruginosa.97 However, these probes have not yet been employed in an interkingdom signalling set-up (Fig. 3(b)).
4. Post-translational modifications between host and pathogens
4.1 AMPylation
Some pathogenic bacteria hijack human cellular processes by introducing effector proteins which post-translationally modify host proteins to alter their function, often targeting pathways that ultimately facilitate bacterial infection and replication in the host.98 Bacterial effector proteins that transfer an adenosine 5′-monophosphate (AMP) group onto a hydroxy group of host proteins have been discovered in important human pathogens such as VopS from Vibrio parahaemolyticus,99 IbpA from Histophilus somni,100 and DrrA from Legionella pneumophila.101 These so-called AMPylators target proteins from the family of Rho GTPases (among others), which typically can no longer interact with downstream effectors as a consequence.99–101 These processes can promote bacterial invasion by interfering with the host's actin skeleton dynamics (VopS, IbpA)99,100 or facilitate bacterial survival and replication inside the host cell (DrrA).102,103 Chemical AMP probes have enabled the identification of low abundance targets after enrichment in a global and unbiased fashion. Grammel et al. devised an alkynylated AMPylation probe, N6-propargyl adenosine-5′-triphosphate 17 (N6pATP),104 which was accepted by VopS, Fic2, and DrrA, and applicable in HeLa lysates, where the selective labelling of known targets such as Cdc42 was achieved. Follow-up studies employed 17 (N6pATP) in a cell-free protein array to detect novel substrates of bacterial AMPylators,105–107 or the human AMPylator FICD protein in cell lysate using mass spectrometric analysis of enriched proteins.108 The more sterically demanding N6-(6-amino)hexyl-ATP-5-carboxyl-fluorescein 18 (Fl-ATP) was applied to reveal VopS targets in whole cell extracts which were pulled down by an α-fluorescein antibody and analysed with mass spectrometry.109 However, these probes could not be used in live cells as the highly negatively charged triphosphate moiety impeded cell permeability.110,111 Kielkowski et al. devised two cell permeable pro-nucleotide probes, 19 (pro-N6pA) and 20 (pro-N6azA), where the negative charge of the phosphate was masked by a phosphoramidate.112,113 Metabolomics experiments showed that pro-N6pA is metabolised to the corresponding 17 (N6pATP) in the cell.113 A recent study by Rauh et al. applied 19 (pro-N6pA) to directly monitor AMPylation of human cells by the V. parahaemolyticus effector VopS after infection in live cells.114 This facilitated the tracking of AMPylation over time, identification of AMPylation sites, and the identification of both known and previously unknown Rho GTPase targeted by VopS. Infection with pathogens like P. aeruginosa PAO1 or E. coli CFT073, both of which carry Fic domain-containing proteins with only low similarity to the Fic domains of virulence effectors, did not result in 19 (pro-N6pA) labelling. This study therefore illustrates how only certain pathogens apply this infection strategy. Gulen et al. introduced co-substrate-linked enzymes as a novel sort of macromolecular ABPP-like probe for proteome profiling of AMPylated proteins.115 The bacterial nucleotide transferases IbpA and BepA were engineered to contain a cysteine in the ATP binding pocket at a position that enabled covalent binding to an electrophilic ATP analogue, termed thiol-reactive nucleotide derivatives (TreNDs). A probe 21 (TreND 1) tethered to IbpA was introduced into human cell lysate and the resulting ternary complexes were identified by affinity enrichment and mass spectrometry which revealed a set of hitherto unknown AMPylation targets (Fig. 4(a)).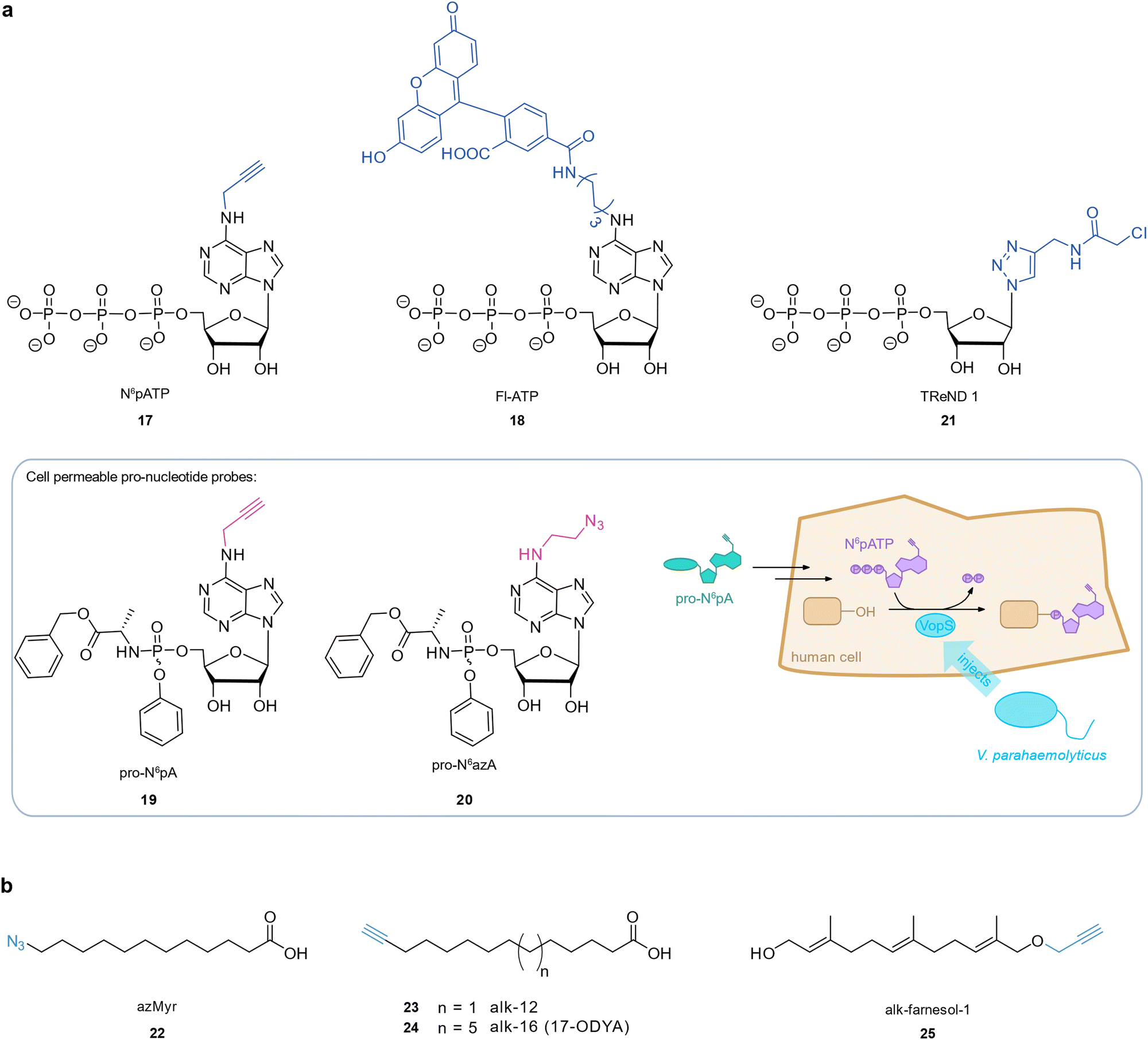 | ||
| Fig. 4 Investigation of host–pathogen interactions through post-translational protein modifications. (a) AMPylation probes. (b) Lipidation probes. | ||
4.2 Lipidation
Bacterial pathogens also target protein lipidation, a post-translational modification that serves as a membrane anchor.116,117 Bioorthogonal probes have emerged as powerful tools to study protein lipidation in an unbiased approach118 and have widely been used in the context of host–pathogen interactions.The Shigella flexneri effector protein IpaJ proteolytically cleaves N-myristoyl glycine off mammalian proteins.119 Metabolic labelling with myristic acid azide 22 (azMyr) in human cells transfected with IpaJ revealed a broad target spectrum of the bacterial protease.119 These targets could be identified in a later study, in which the authors used the corresponding myristic acid alkynyl-analogue 23 (alk-12),120 for metabolic labelling in human cells infected with S. flexneri.121 Proteomics analyses by mass spectrometry revealed that a suite of Golgi-associated ARF/ARL GTPases is targeted by the pathogen, which is in line with the Golgi destabilisation observed during S. flexneri infection.121 The Vibrio cholerae toxin RID acts as an Nε-fatty acyltransferase on human Rho GTPases with a strong preference for Rac1, a regulator of the actin cytoskeleton, disrupting related pathways.122 This was shown in cells metabolically labelled with 22 (alk-16) and transfected with RID from V. cholerae or infected with the bacteria to monitor the acylation of Rac1.122 Similarly, Liu et al. performed metabolic labelling of HeLa cells with 24 (alk-16) followed by transfection with the S. flexneri virulence factor IcsB or by infection with the pathogen. Labelled proteins were enriched and identified by mass spectrometry.123 IcsB was shown to acylate a great number of small GTPases of the Ras superfamily to disturb bacterial autophagy and facilitate its survival.123 These examples show how pathogens can modify the lipidation of host proteins. On the other hand, bacteria may exploit host lipidation enzymes to acylate their effector proteins for functional activation, stabilisation, and sub-cellular localisation. An alkynyl-farnesol reporter 25 (alk-farnesol-1)124 was applied in live HeLa cells transfected with Legionella pneumophila effector proteins to reveal their post-translational modification by human prenyltransferases.125 Prenylation was monitored by in-gel fluorescence scanning following immunoprecipitation of the Legionella proteins and ligation to a fluorescent dye. This approach revealed that multiple Legionella pneumophila effector proteins are prenylated by the host cell and, as a consequence, targeted to the membranes of host organelles.12524 has also served as tool to monitor protein palmitoylation.126Salmonella typhimurium effector proteins were shown to be palmitoylated by host proteins after being delivered into the host cell.127 In addition, mammalian cells transiently expressing the bacterial effectors as well as the putative palmitoylators were metabolically labelled with 24120 and modifications of bacterial effectors were read out as performed with probe 25.127 This study revealed palmitoylation as a critical trait for effector activity and localisation.127 Similarly, palmitoylation of the Legionella pneumophila effector proteins GobX and LpdA was shown by metabolic labelling of human cells transfected with the effectors.128,129 The Brucella effector PrpA was shown to be palmitoylated in an analogous approach (Fig. 4(b)).130
5. Interactions of the pathogen envelope with the host
The bacterial envelope is the interface between bacteria and their surroundings and is implicated in many interactions with the host.131 As many bacterial components are absent in human cells, metabolic labelling using probes mimicking bacterial cell wall building blocks can be performed within infected host cells.132,133 Furthermore, the bacterial cell wall is a major target of antibiotics and therefore of large scientific interest.2 The basic peptidoglycan structure consists of a conserved disaccharide backbone formed by the two aminosugars N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc).133,134 MurNAc is connected to a tetra- or pentapeptide typically containing a D-ala-D-ala motif at its fourth and fifth position. As bacterial D-alanine metabolism readily tolerates unnatural substrates, D-alanine derivatives serve as chemical reporters to directly track peptidoglycan in a selective manner.135,136A study from the Bertozzi lab applied D-ala analogues modified with an alkyne or azide bioorthogonal handle, R-propargylglycine 26 (alkDala) and R-2-amino-3-azidopropanoic acid 27 (azDala), to label newly synthesised peptidoglycan in replicating bacteria inside macrophages.136Listeria monocytogenes is an intracellular pathogen that uses a tail of polymerised host actin for motility after infection.137 Labelling studies with 26 revealed that the presence of host actin skewed bacterial division cycles to increase the proportion of freshly divided cells, which favours the formation of actin tails on L. monocytogenes. Thus, peptidoglycan labelling allowed to monitor how bacteria adjust their growth and division in a complex environment such as the host cell.
Wang et al. derivatised the peptidoglycan fragments γ-D-glutamyl-meso-diaminopimelic acid (iE-DAP) and muramyl-dipeptide (MDP) to generate photoaffinity probes, 28 (x-alk-MDP) and 29 (x-alk-iE-DAP), to detect their direct interaction with the mammalian intracellular immune receptors NOD1 and NOD2, respectively. 28 revealed the formation of a transient ternary complex between MDP, Arf6, and NOD2, which was no longer formed with a loss-of-function NOD2 mutant associated with Crohn's disease, suggesting this complex plays a role in the disease (Fig. 5(a)).138
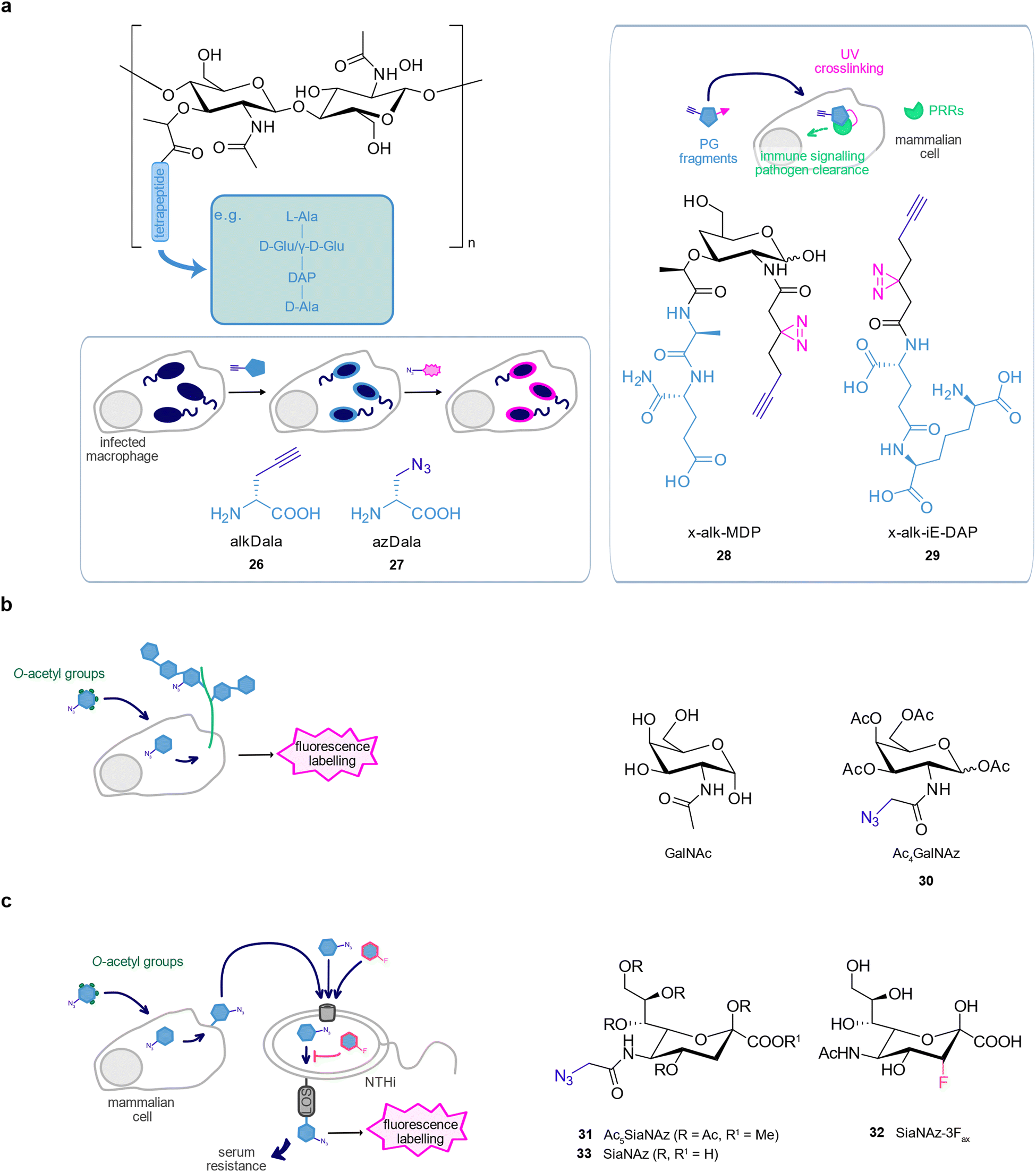 | ||
| Fig. 5 Sugar-based probes. (a) Peptidoglycan probes to label the bacterial cell wall. (b) GalNAc probes to label host glycoproteins (c) Sialic acid probes to monitor incorporation of host sialic acids by bacteria. | ||
A clickable sugar analogue, tetra-acetylated N-azidoacetylgalactosamine 30 (Ac4GalNAz), has been applied to monitor bacteria–host interactions by labelling host glycoproteins. Mucins are heavily glycosylated proteins and a key component of the intestinal mucus layer, where they form a barrier against invading pathogens. 30 carried acetylated hydroxy groups to facilitate cellular uptake that are removed by intracellular esterases and successfully labelled mucin-type O-linked glycoproteins.13930 has been used to follow the fate of mucins in live mice at different stages of infection with Helicobacter pylori, the causative agent of intestinal tract ulcers.140 Interestingly, H. pylori slowed the secretion of mucins to the cell surface, likely to impair pathogen clearing.140 In another study, labelling of transmembrane mucins at the surface of murine cornea with 30 revealed that P. aeruginosa preferentially colocalised with GalNAz-labelled regions (Fig. 5(b)).141
Human cells are covered by a glycocalyx, a dense layer of sugars attached to surface proteins and lipids. Pathogens use sialic acids – abundant constituents of the glycocalyx – to recognise and bind to their host.142 Beyond that, many bacteria remove sialic acid from human cells to incorporate it into their own cell surface. This molecular mimicry facilitates evasion of the immune response and promotes survival within the host.143 Scavenging of host sialic acid by non-typeable Haemophilus influenzae (NTHi) could be monitored by metabolic labelling in a coculture model with primary human bronchial epithelial cells (PHBEC).144,145 Mammalian cells only take up peracetylated sialic acids by passive diffusion whilst bacteria rely on active uptake of the unprotected sugar. Therefore, PHBEC cells were selectively labelled by feeding an acetylated azide sialic acid probe 31 (Ac5SiaNaz) which was then removed by a sialidase enzyme and incorporated by NTHi. The incorporation was monitored by CuAAC to biotin followed by staining with fluorescent streptavidin and flow cytometry.145 Interestingly, an analogous alkyne probe was resistant to removal from the mammalian glycans. Noteworthy, this pathway has been studied for its potential as an antiinfective target. The chemical analogue 3-fluorosialic acid 32 (SiaNAz-3Fax), for instance, proved to selectively inhibit NTHi LOS sialylation and abolish serum resistance.145 In another study, an unprotected azide sialic acid probe 33 (SiaNaz) was used to study a suite of potential inhibitors of LOS sialylation by quantifying the competition of labelling by 33 in NTHi.146 This is an example of how chemical biology methods can elucidate a potential antivirulence treatment (Fig. 5(c)).
Mycobacteria possess a unique membrane structure that is crucial for their interaction with the host. One key constituent is the non-mammalian disaccharide trehalose, which is as a precursor for essential cell wall glycolipids and other metabolites in mycobacteria. Backus et al. first developed a fluorescein-containing trehalose probe 34 (FITC-trehalose) that successfully labelled M. tuberculosis within macrophages (Fig. 6(a)).147 The Bertozzi group published azide-modified trehalose probes 35–38 that were incorporated into the mycomembrane of different mycobacteria148 as well as a solvatochromic trehalose probe 39 (DMN-Tre) whose fluorescence is activated upon being metabolically incorporated into the hydrophobic environment of the mycomembrane.149 DMN-Tre labelling proved superior compared to a non-fluorogenic analogue 40 (6-FlTre) as it produced significantly less background.149 A bifunctional trehalose probe 41 comprising both a terminal alkyne and azide group was designed by Pohane et al. It labels trehalose dimycolate (TDM) and arabinogalactan-linked mycolate (AGM) with its alkyne group under antigen 85 (Ag85) mycoloyltransferases catalysis while the resulting free trehalose azide probe labels trehalose monomycolate (TMM). Monitoring mycomembrane biosynthesis and trehalose recycling this way revealed that trehalose recycling efficiency increased upon carbon starvation with a concomitant increase in LpqY-SugABC expression.150 Dai et al. developed a cephalosphorinase-dependent green trehalose 42 (CDG-Tre) fluorogenic probe whose fluorescence is activated by the β-lactamase BlaC, which is uniquely expressed by Mycobacterium tuberculosis.151 As expected, the probe was modified with mycolic acid by mycobacterial enzymes and incorporated into the cell wall and therefore enabled selective fluorescence imaging of mycobacteria inside live macrophages.151 Altogether, trehalose-based probes constitute a very promising approach to monitor glycolipid dynamics, also in infection models, as trehalose is metabolically orthogonal to eukaryotic hosts152–155 and several derivatives of it have been published.152–155
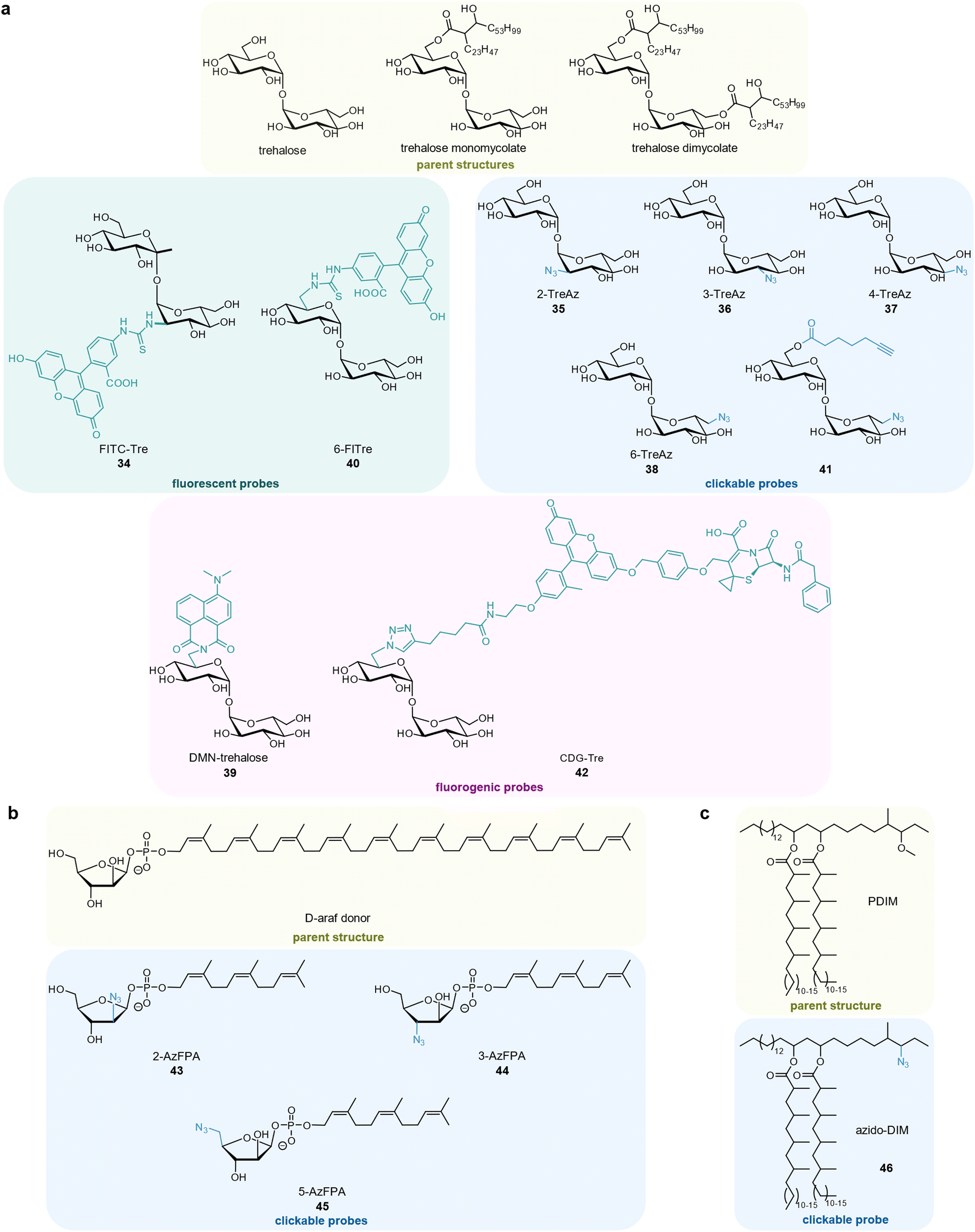 | ||
| Fig. 6 Mycomembrane components and corresponding probe derivatives. (a) Trehalose-based probes. (b) Arabinose-based probes. (c) Phthiocerol dimycocerosate-based probe. | ||
D-Arabinofuranose (D-Araf), is another essential component of the mycobacterial membrane that is absent in human cells. Mycobacteria enzymatically incorporate arabinose from arabinose-phospholipid donors by integral membrane glycosyltransferases rather than from free arabinose. The Kiessling group disclosed clickable azide derivatives 2-AzFPA 43, 3-AzFPA 44, and 5-AzFPA 45 mimicking the phospholipid precursor (Fig. 6(b)).156 2-AzFPA 43 was selectively incorporated into arabinofuranose-containing glycans in the cell surface of Msmeg and strain-promoted azide–alkyne cycloaddition (SPAAC) ligation to fluorescent dyes enabled its detection within infected macrophages.156
Another study from the Bertozzi lab used an azido-derivative of the mycobacterial virulence lipid phthiocerol dimycocerosate (PDIM) 46 (azido-DIM), to directly track it in an in vivo infection model of Mycobacterium marinum in live zebrafish (Fig. 6(c)).157 After ligating the azide handle to a fluorescent dye, it was observed that PDIM spread into the epithelial membranes of the host and that this process was dependent on host cholesterol. The spread of PDIM into host epithelial membranes was crucial for mycobacterial survival as it caused a suppression of the innate immune response. Treating the zebrafish with statins (inhibitors of cholesterol biosynthesis) significantly decreased mycobacterial infectivity.
6. Enzyme-mediated host–pathogen interactions monitored by fluorophosphonates
Fluorophosphonate probes have been widely used as tools to study the activity of serine proteases in the context of disease,158–161 however, there are only few reports of their use in infection models. Hatzios et al. used activity-based fluorophosphonate probes 47 and 48 to identify proteases that were active in animals or humans infected with Vibrio cholerae (Fig. 7(a) and (b)).162 They revealed that the serine protease secreted by V. cholerae, IvaP, reduced bacterial surface binding of intelectin, a host protein likely involved in microbial recognition. A follow-up study reported the cleavage of intelectin by IvaP.163 Similarly, Lentz et al. used a fluorophosphonate tetramethylrhodamine probe164 in S. aureus to show that the serine hydrolase FphB was activated by components of eukaryotic cell membranes and serum and that this enzyme was required for full S. aureus infectivity.165 Hence, this enzyme class provides promising targets for the development of selective inhibitors and the authors performed competitive labelling using a fluorescent fluorophosphonate probe to identify a selective inhibitor of FphB which reduced S. aureus infectivity in vivo (Fig. 7(c)).165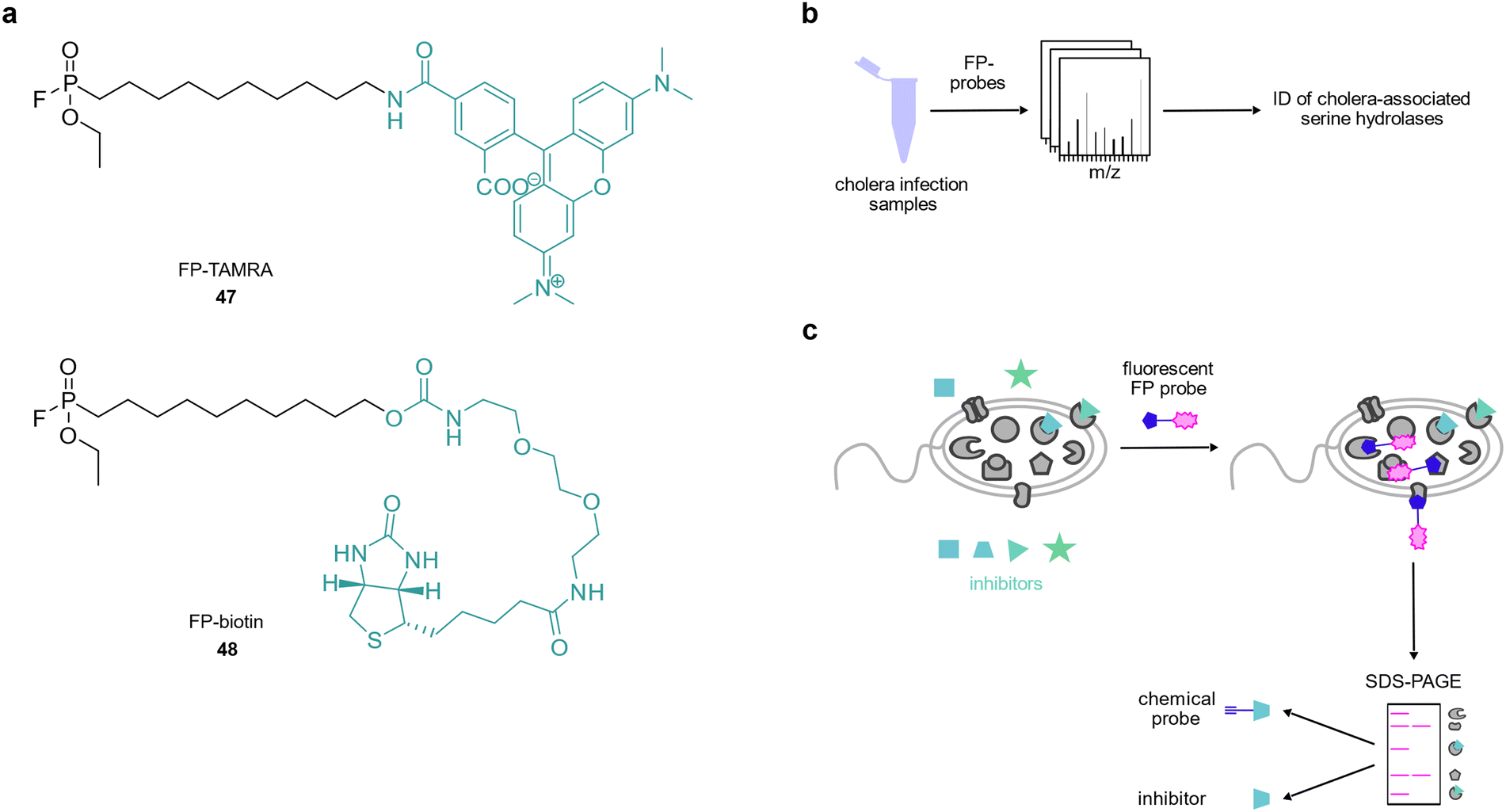 | ||
| Fig. 7 Investigation of hydrolases in the context of bacterial infection. (a) Fluorophosphonate probes. (b) Fluorophosphonate probes were applied to identify hydrolases in cholera infection samples and to (c) discover novel inhibitors of virulence-related hydrolases. | ||
7. Outlook
Chemical proteomics is a powerful tool to study the interaction of chemical compounds with proteins in a global and unbiased way which has helped elucidate the mode of action of a number of biologically active small molecules.37 However, there are limitations that necessitate special attention in the experimental design. One major challenge is that chemical proteomics studies often generate a suite of background hits which requires thorough control experiments to work out genuine targets that can be linked to the biological activity. This is especially important when photoreactive probes are used as they typically have high target promiscuity. Moreover, the incorporation of reactive groups or chemical handles into the probes may in some cases affect the biological activity and alter the target spectrum of the probes. This risk can be minimised by the application of multiple, chemically diverse probes and by performing careful structure–activity-relationship studies. Finally, although there are recent examples of small molecules targeting non-protein targets such as nucleic acids,166,167 chemical proteomics primarily focusses on proteins and might therefore miss targets such as nucleic acids or lipids.168 Nevertheless, chemical proteomics is an invaluable approach for determining interactions between small molecules and proteins as no prior knowledge on the identity of potential targets is required. Provided the probes are cell-permeable, their engagement with protein targets can be studied within live cells where proteins have native activity, expression levels, and localisation.Here, we have highlighted recent progress in the study of host–pathogen interactions achieved via chemical proteomics. This includes not only how bacteria sense host hormones such as dynorphin A and how the host responds to bacterial quorum sensing molecules, but also eukaryotic post-translational protein modifications exploited by microbes such as AMPylation, and lipidation, among others. The application of unnatural building blocks such as D-Ala analogues expands our understanding of the bacterial envelope which constitutes the interface of bacteria and their surroundings. These techniques have greatly benefitted from the development of bioorthogonal chemistries,169 the commercial availability of noncanonical biological buliding blocks,170 and advances in mass spectrometry techniques.171–174 Nevertheless, more efforts are still needed to resolve remaining questions. First, most reported studies represent a snapshot of host–pathogen interactions at certain infection time points but the interplay between host and pathogen is more complex and dynamic and may vary across different stages of infection.175,176 Therefore, monitoring these interactions in a time-resolved manner would provide a more complete view. In a pioneering study, Tao et al. reported a photoactivatable multifunctional chemical probe to crosslink bacterial and host proteins during Salmonella internalisation into macrophages.177 The photoactivatability allowed for time-resolved profiling of pathogen entry and revealed protein–protein interactions at different time points.177 Indeed, proteins involved in host actin polymerisation, which is important for pathogen internalisation, interacted with microbial proteins during early infection stages, while more nuclear proteins were identified in the late endocytosis process.177 Second, the majority of host–pathogen interaction studies has employed cellular systems rather than animal models. However, in vitro studies are not always suitable to reflect in vivo situations and animal studies would dramatically improve the physiological relevance of the results.178–180 In an intriguing approach, Waldor et al. have analysed microbe–host interactions at the bacterial envelope by biotinylation of proteins associated with the surface of V. cholerae isolated from an infant rabbit model.181 This approach revealed that numerous host proteins associate with V. cholerae and that their abundance on the pathogen surface is driven by the presence of cholera toxin. Some of the identified host proteins (AnxA1, LPO and ZAG) could also recognise distinct taxa of the murine intestinal microbiota. This suggests that the host factors may play a role in intestinal homeostasis – a discovery that would be elusive to in vitro studies.181 A challenge for in vivo studies is to detect small amounts of pathogen proteins. However, with the rapid technical development of mass spectrometry, the sensitivity of proteomic studies has significantly increased.171,182 For example, more than 1000 proteins can now be identified and quantified in single mammalian cells via single cell proteomics with elaborate isolation techniques, sample processing methods, and mass spectrometry instrumentation.183,184 These novel techniques may allow to minimise the required bacterial cell number in proteomic analysis for host–pathogen interaction studies, although bacteria possess much fewer proteins compared to mamalian cells.185 Finally, chemical proteomics approaches often rely on small molecules or small cellular building blocks since they are convenient to be modified. However, host–pathogen interactions are often mediated by macromolecules, an area of research that is currently underexplored. For example, some bacterial pathogens are capable of directly sensing and responding to host cytokines to facilitate their survival and transmission.186,187 IL-1β is reported to accelerate the growth of virulent E. coli,188 and human IFN-γ can activate a virulent phenotype of P. aeruginosa.189 Since the incorporation of unnatural amino acids into proteins or larger peptides has become more accessible by genetic code expansion or site-specific modification, the elucidation of protein-based host–pathogen interaction is now more easily accessible.190–195 Studying macromolecule-based communication could greatly benefit from chemical proteomics and would further eludicate another aspect of host–pathogen interactions from both bacterial and host perspectives.196
The antimicrobial resistance crisis requires novel approaches to treating bacterial infections. Targeting virulence pathways has been put forward as a promising strategy as it is expected to exert less selective pressure on pathogens compared to classical therapies. We have highlighted studies in which chemical probes have already been employed to identify chemical inhibitors, e.g., for the S. aureus hydrolase FphB or for sialylation of NTHi LOS146,165 but overall, to this date, there are not many reports of chemical inhibitors targeting host–pathogen interactions. This could be due to the fact that interkingdom communication is a very complex process and still not very well understood. We have summarised a number of studies that employed metabolic labelling to monitor, for instance, PTMs introduced on host proteins by bacterial pathogens. These approaches could be used in future studies to characterise potential antagonists of bacterial effector proteins involved in AMPylation or lipidation of host enzymes, for example. The interference with QS has already been investigated for some time as a potential antivirulence approach and several studies have successfully identified compounds that reduced virulence in important human pathogens by blocking interbacterial communication at different points in the signalling cascade.197–203 Interfering with interkingdom signalling has been explored in a study from the Sperandio group which reports LED209, an inhibitor targeting the two-component system QseBC.14 QseBC acts as a sensor for both the QS signal AI3 and the eukaryotic hormone epinephrine and its inhibition resulted in reduced bacterial virulence in animal studies.14 In general, two-component systems provide intriguing antivirulence targets as they are critical to many bacterial survival and pathogenicity mechanisms and are furthermore absent in mammalian cells. Indeed, chemical inhibition of two-component systems has been shown to result in reduced virulence in both in vitro and in vivo studies.204,205 Our review has delineated the potential of chemical proteomics to uncover novel targets involved in host–pathogen interactions and to elucidate underlying biological pathways.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. W. M. was supported by the German Academic Scholarship Foundation (Studienstiftung des Deutschen Volkes). W. Z. thanks the National Natural Science Foundation of China (22107075 to W. Z.), and the Natural Science Foundation of Top Talent of SZTU (20211061010013 to W. Z.) for financial support. S. A. S. acknowledges funding from Merck Future Insight Prize 2020, TUM innovation network NextGen, and SFB1371 microbiome signatures project 16.References
- C. Antimicrobial Resistance, Lancet, 2022, 399, 629–655 CrossRef.
- M. Lakemeyer, W. Zhao, F. A. Mandl, P. Hammann and S. A. Sieber, Angew. Chem., Int. Ed., 2018, 57, 14440–14475 CrossRef CAS.
- M. A. Fischbach and C. T. Walsh, Science, 2009, 325, 1089–1093 CrossRef CAS.
- K. Lewis, Cell, 2020, 181, 29–45 CrossRef CAS.
- K. E. Kortright, B. K. Chan, J. L. Koff and P. E. Turner, Cell Host Microbe, 2019, 25, 219–232 CrossRef CAS.
- S. Y. Jun, I. J. Jang, S. Yoon, K. Jang, K. S. Yu, J. Y. Cho, M. W. Seong, G. M. Jung, S. J. Yoon and S. H. Kang, Antimicrob. Agents Chemother., 2017, 61, e02629–16 CrossRef PubMed.
- R. Schuch, B. K. Khan, A. Raz, J. A. Rotolo and M. Wittekind, Antimicrob. Agents Chemother., 2017, 61, e02666–16 CrossRef CAS.
- M. A. B. Shabbir, M. Z. Shabbir, Q. Wu, S. Mahmood, A. Sajid, M. K. Maan, S. Ahmed, U. Naveed, H. Hao and Z. Yuan, Ann. Clin. Microbiol. Antimicrob., 2019, 18, 21 CrossRef.
- V. Thakur, A. Uniyal and V. Tiwari, Eur. J. Pharmacol., 2021, 903, 174151 CrossRef CAS.
- P. D. Tamma, S. E. Cosgrove and L. L. Maragakis, Clin. Microbiol. Rev., 2012, 25, 450–470 CrossRef CAS.
- A. DiGiandomenico and B. R. Sellman, Curr. Opin. Microbiol., 2015, 27, 78–85 CrossRef CAS PubMed.
- A. E. Clatworthy, E. Pierson and D. T. Hung, Nat. Chem. Biol., 2007, 3, 541–548 CrossRef CAS PubMed.
- M. Whiteley, S. P. Diggle and E. P. Greenberg, Nature, 2017, 551, 313–320 CrossRef CAS.
- D. A. Rasko, C. G. Moreira, R. Li de, N. C. Reading, J. M. Ritchie, M. K. Waldor, N. Williams, R. Taussig, S. Wei, M. Roth, D. T. Hughes, J. F. Huntley, M. W. Fina, J. R. Falck and V. Sperandio, Science, 2008, 321, 1078–1080 CrossRef CAS PubMed.
- A. Okada, Y. Gotoh, T. Watanabe, E. Furuta, K. Yamamoto and R. Utsumi, in Methods in Enzymology, ed. M. I. Simon, B. R. Crane and A. Crane, Academic Press, 2007, vol. 422, pp.386–395 Search PubMed.
- M. Sironi, R. Cagliani, D. Forni and M. Clerici, Nat. Rev. Genet., 2015, 16, 224–236 CrossRef CAS.
- A. Sukumaran, E. Woroszchuk, T. Ross and J. Geddes-McAlister, Can. J. Microbiol., 2021, 67, 213–225 CrossRef CAS PubMed.
- K. Mehta, H. P. Spaink, T. H. M. Ottenhoff, P. H. van der Graaf and J. G. C. van Hasselt, Trends Pharmacol. Sci., 2022, 43, 293–304 CrossRef CAS PubMed.
- S. H. E. Kaufmann, A. Dorhoi, R. S. Hotchkiss and R. Bartenschlager, Nat. Rev. Drug Discovery, 2018, 17, 35–56 CrossRef CAS PubMed.
- E. Torres-Sangiao, A. D. Giddey, C. Leal Rodriguez, Z. Tang, X. Liu and N. C. Soares, Front. Med., 2022, 9, 850374 CrossRef PubMed.
- A. T. Wright, L. A. Hudson and W. L. Garcia, Isr. J. Chem., 2023, 63, e202200099 CrossRef CAS.
- X. Yang, X. Zhao, V. Chen and H. C. Hang, RSC Chem. Biol., 2022, 3, 1397–1402 RSC.
- B. Liu, S. Zhuang, R. Tian, Y. Liu, Y. Wang, X. Lei and C. Wang, ACS Chem. Biol., 2022, 17, 2461–2470 CrossRef CAS PubMed.
- E. R. Forster, X. Yang, A. K. Tai, H. C. Hang and A. Shen, ACS Chem. Biol., 2022, 17, 3086–3099 CrossRef CAS.
- X. Yang, K. R. Stein and H. C. Hang, Nat. Chem. Biol., 2023, 19, 91–100 CrossRef CAS PubMed.
- G. F. Desrochers and J. P. Pezacki, Curr. Top. Microbiol. Immunol., 2019, 420, 131–154 CAS.
- M. Strmiskova, G. F. Desrochers, T. A. Shaw, M. H. Powdrill, M. A. Lafreniere and J. P. Pezacki, ACS Infect. Dis., 2016, 2, 773–786 CrossRef CAS.
- C. Schultz, S. E. Farley and F. G. Tafesse, J. Am. Chem. Soc., 2022, 144, 13987–13995 CrossRef CAS.
- C. R. Mansfield, M. E. Chirgwin and E. R. Derbyshire, Curr. Opin. Chem. Biol., 2023, 75, 102316 CrossRef CAS PubMed.
- M. Ritzefeld, M. H. Wright and E. W. Tate, Parasitology, 2018, 145, 157–174 CrossRef PubMed.
- K. P. Malarney and P. V. Chang, Isr. J. Chem., 2023, 63, e202200076 CrossRef CAS.
- R. Ramanathan and S. K. Hatzios, Isr. J. Chem., 2023, 63, e202200095 CrossRef CAS PubMed.
- M. H. Wright, Biochem. J., 2023, 480, 1445–1457 CrossRef.
- F. Meissner, J. Geddes-McAlister, M. Mann and M. Bantscheff, Nat. Rev. Drug Discovery, 2022, 21, 637–654 CrossRef CAS.
- M. Fonović and M. Bogyo, Expert Rev. Proteomics, 2008, 5, 721–730 CrossRef.
- B. F. Cravatt, A. T. Wright and J. W. Kozarich, Annu. Rev. Biochem., 2008, 77, 383–414 CrossRef CAS.
- M. H. Wright and S. A. Sieber, Nat. Prod. Rep., 2016, 33, 681–708 RSC.
- H. Deng, Q. Lei, Y. Wu, Y. He and W. Li, Eur. J. Med. Chem., 2020, 191, 112151 CrossRef CAS.
- J. Krysiak and S. A. Sieber, Methods Mol. Biol., 2017, 1491, 57–74 CrossRef CAS PubMed.
- S. Heinzlmeir and S. Müller, Drug Discovery Today, 2022, 27, 519–528 CrossRef CAS.
- J. Xu, X. Li, K. Ding and Z. Li, Chem. – Asian J., 2020, 15, 34–41 CrossRef CAS.
- C. Guo, Y. Chang, X. Wang, C. Zhang, P. Hao, K. Ding and Z. Li, Chem. Commun., 2019, 55, 834–837 RSC.
- X. Chen, Y. K. Wong, J. Wang, J. Zhang, Y. M. Lee, H. M. Shen, Q. Lin and Z. C. Hua, Proteomics, 2017, 17 DOI:10.1002/pmic.201600212.
- N. C. Sadler and A. T. Wright, Curr. Opin. Chem. Biol., 2015, 24, 139–144 CrossRef CAS PubMed.
- L. J. Keller, B. M. Babin, M. Lakemeyer and M. Bogyo, Curr. Opin. Chem. Biol., 2020, 54, 45–53 CrossRef CAS.
- T. Böttcher, M. Pitscheider and S. A. Sieber, Angew. Chem., Int. Ed., 2010, 49, 2680–2698 CrossRef.
- P. V. Chang, J. A. Prescher, E. M. Sletten, J. M. Baskin, I. A. Miller, N. J. Agard, A. Lo and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1821–1826 CrossRef CAS PubMed.
- N. J. Pedowitz and M. R. Pratt, RSC Chem. Biol., 2021, 2, 306–321 RSC.
- W. P. Heal, M. H. Wright, E. Thinon and E. W. Tate, Nat. Protoc., 2012, 7, 105–117 CrossRef CAS PubMed.
- S. A. Sieber, ChemBioChem, 2009, 10, 799–801 CrossRef CAS PubMed.
- X. Zhao, X. Yang and H. C. Hang, Biochemistry, 2022, 61, 2822–2834, DOI:10.1021/acs.biochem.1c00758.
- B. J. Ignacio, T. Bakkum, K. M. Bonger, N. I. Martin and S. I. van Kasteren, Org. Biomol. Chem., 2021, 19, 2856–2870 RSC.
- R. Gregor, S. David and M. M. Meijler, Chem. Soc. Rev., 2018, 47, 1761–1772 RSC.
- M. H. Wright, Proteomics, 2018, 18, e1700333 CrossRef PubMed.
- P. M. Jean Beltran, J. D. Federspiel, X. Sheng and I. M. Cristea, Mol. Syst. Biol., 2017, 13, 922 CrossRef PubMed.
- J. Ge and S. Q. Yao, Chem. Biol., 2015, 22, 434–435 CrossRef CAS PubMed.
- M. Coiras, E. Camafeita, M. R. López-Huertas, E. Calvo, J. A. López and J. Alcamí, Proteomics, 2008, 8, 852–873 CrossRef CAS PubMed.
- G. Kumar, R. Narayan and S. Kapoor, J. Med. Chem., 2020, 63, 15308–15332 CrossRef CAS PubMed.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- E. M. Sletten and C. R. Bertozzi, Acc. Chem. Res., 2011, 44, 666–676 CrossRef CAS PubMed.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Chang, I. A. Miller, A. Lo, J. A. Codelli and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16793–16797 CrossRef CAS PubMed.
- K. L. Kiick, E. Saxon, D. A. Tirrell and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 19–24 CrossRef CAS PubMed.
- E. Saxon and C. R. Bertozzi, Science, 2000, 287, 2007–2010 CrossRef CAS PubMed.
- M. Shahiduzzaman and K. M. Coombs, Front. Microbiol., 2012, 3, 308 CAS.
- Y. S. Ren, H. L. Li, X. H. Piao, Z. Y. Yang, S. M. Wang and Y. W. Ge, Biochem. Pharmacol., 2021, 194, 114798 CrossRef CAS PubMed.
- F. Feng, W. Zhang, Y. Chai, D. Guo and X. Chen, J. Pharm. Biomed. Anal., 2023, 223, 115107 CrossRef CAS PubMed.
- C. Le Sueur, H. M. Hammaren, S. Sridharan and M. M. Savitski, Curr. Opin. Chem. Biol., 2022, 71, 102225 CrossRef CAS PubMed.
- M. A. Hudson and S. W. Lockless, mBio, 2022, 13, e0224021 CrossRef PubMed.
- Y. Feng, G. De Franceschi, A. Kahraman, M. Soste, A. Melnik, P. J. Boersema, P. P. de Laureto, Y. Nikolaev, A. P. Oliveira and P. Picotti, Nat. Biotechnol., 2014, 32, 1036–1044 CrossRef CAS PubMed.
- L. Malinovska, V. Cappelletti, D. Kohler, I. Piazza, T. H. Tsai, M. Pepelnjak, P. Stalder, C. Dörig, F. Sesterhenn, F. Elsässer, L. Kralickova, N. Beaton, L. Reiter, N. de Souza, O. Vitek and P. Picotti, Nat. Protoc., 2023, 18, 659–682 CrossRef CAS PubMed.
- I. Piazza, N. Beaton, R. Bruderer, T. Knobloch, C. Barbisan, L. Chandat, A. Sudau, I. Siepe, O. Rinner, N. de Souza, P. Picotti and L. Reiter, Nat. Commun., 2020, 11, 4200 CrossRef PubMed.
- M. M. Kendall and V. Sperandio, mBio, 2016, 7, e01748 CrossRef CAS PubMed.
- L. R. Wu, O. Zaborina, A. Zaborin, E. B. Chang, M. Musch, C. Holbrook, J. R. Turner and J. C. Alverdy, Surg. Infect, 2005, 6, 185–195 CrossRef PubMed.
- O. Zaborina, F. Lepine, G. Xiao, V. Valuckaite, Y. Chen, T. Li, M. Ciancio, A. Zaborin, E. O. Petrof, J. R. Turner, L. G. Rahme, E. Chang and J. C. Alverdy, PLoS Pathog., 2007, 3, e35–e35 CrossRef PubMed.
- M. H. Wright, C. Fetzer and S. A. Sieber, J. Am. Chem. Soc., 2017, 139, 6152–6159 CrossRef CAS PubMed.
- P. M. Wolanin, P. A. Thomason and J. B. Stock, Genome Biol., 2002, 3, reviews3013.3011 CrossRef PubMed.
- P. V. Attwood, P. G. Besant and M. J. Piggott, Amino Acids, 2011, 40, 1035–1051 CrossRef CAS PubMed.
- J. W. Chang, J. E. Montgomery, G. Lee and R. E. Moellering, Angew. Chem., Int. Ed., 2018, 57, 15712–15716 CrossRef CAS PubMed.
- P. W. A. Allihn, M. W. Hackl, C. Ludwig, S. M. Hacker and S. A. Sieber, Chem. Sci., 2021, 12, 4763–4770 RSC.
- P. R. A. Zanon, L. Lewald and S. M. Hacker, Angew. Chem., Int. Ed., 2020, 59, 2829–2836 CrossRef PubMed.
- M. H. Karavolos, K. Winzer, P. Williams and C. M. A. Khan, Mol. Microbiol., 2013, 87, 455–465 CrossRef CAS PubMed.
- P. P. Freestone, S. M. Sandrini, R. D. Haigh and M. Lyte, Trends Microbiol., 2008, 16, 55–64 CrossRef CAS PubMed.
- M. Lyte, L. Vulchanova and D. R. Brown, Cell Tissue Res., 2011, 343, 23–32 CrossRef CAS PubMed.
- S. M. Sandrini, R. Shergill, J. Woodward, R. Muralikuttan, R. D. Haigh, M. Lyte and P. P. Freestone, J. Bacteriol., 2010, 192, 587–594 CrossRef CAS PubMed.
- A. Weigert Muñoz, E. Hoyer, K. Schumacher, M. Grognot, K. M. Taute, S. M. Hacker, S. A. Sieber and K. Jung, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2118227119 CrossRef PubMed.
- E. Beale, G. Li, M.-W. Tan and K. P. Rumbaugh, Appl. Environ. Microbiol., 2006, 72, 5135–5137 CrossRef CAS PubMed.
- V. V. Kravchenko, G. F. Kaufmann, J. C. Mathison, D. A. Scott, A. Z. Katz, D. C. Grauer, M. Lehmann, M. M. Meijler, K. D. Janda and R. J. Ulevitch, Science, 2008, 321, 259–263 CrossRef CAS PubMed.
- V. V. Kravchenko, G. F. Kaufmann, J. C. Mathison, D. A. Scott, A. Z. Katz, M. R. Wood, A. P. Brogan, M. Lehmann, J. M. Mee, K. Iwata, Q. Pan, C. Fearns, U. G. Knaus, M. M. Meijler, K. D. Janda and R. J. Ulevitch, J. Biol. Chem., 2006, 281, 28822–28830 CrossRef CAS PubMed.
- L. Kumar, S. K. S. Patel, K. Kharga, R. Kumar, P. Kumar, J. Pandohee, S. Kulshresha, K. Harjai and S. Chhibber, Molecules, 2022, 27, 7584 CrossRef CAS PubMed.
- A. L. Garner, J. Yu, A. K. Struss, C. A. Lowery, J. Zhu, S. K. Kim, J. Park, A. V. Mayorov, G. F. Kaufmann, V. V. Kravchenko and K. D. Janda, Bioorg. Med. Chem. Lett., 2011, 21, 2702–2705 CrossRef CAS PubMed.
- T. Karlsson, M. V. Turkina, O. Yakymenko, K. E. Magnusson and E. Vikström, PLoS Pathog., 2012, 8, e1002953 CrossRef PubMed.
- L. Dubinsky, A. Delago, N. Amara, P. Krief, J. Rayo, T. Zor, V. V. Kravchenko and M. M. Meijler, Chem. Commun., 2013, 49, 5826–5828 RSC.
- L. Dubinsky, L. M. Jarosz, N. Amara, P. Krief, V. V. Kravchenko, B. P. Krom and M. M. Meijler, Chem. Commun., 2009, 7378–7380, 10.1039/B917507E.
- J. Rayo, R. Gregor, N. T. Jacob, R. Dandela, L. Dubinsky, A. Yashkin, A. Aranovich, M. Thangaraj, O. Ernst, E. Barash, S. Malitsky, B. I. Florea, B. P. Krom, E. A. C. Wiemer, V. A. Kickhoefer, L. H. Rome, J. C. Mathison, G. F. Kaufmann, H. S. Overkleeft, B. F. Cravatt, T. Zor, K. D. Janda, R. J. Ulevitch, V. V. Kravchenko and M. M. Meijler, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2012529118 CrossRef CAS PubMed.
- A. Yashkin, J. Rayo, L. Grimm, M. Welch and M. M. Meijler, Chem. Sci., 2021, 12, 4570–4581 RSC.
- K. A. Chambers and R. A. Scheck, Nat. Chem. Biol., 2020, 16, 1043–1051 CrossRef CAS PubMed.
- M. L. Yarbrough, Y. Li, L. N. Kinch, N. V. Grishin, H. L. Ball and K. Orth, Science, 2009, 323, 269–272 CrossRef CAS PubMed.
- C. A. Worby, S. Mattoo, R. P. Kruger, L. B. Corbeil, A. Koller, J. C. Mendez, B. Zekarias, C. Lazar and J. E. Dixon, Mol. Cell, 2009, 34, 93–103 CrossRef CAS PubMed.
- M. P. Müller, H. Peters, J. Blümer, W. Blankenfeldt, R. S. Goody and A. Itzen, Science, 2010, 329, 946–949 CrossRef PubMed.
- I. Derré and R. R. Isberg, Infect. Immun., 2004, 72, 3048–3053 CrossRef PubMed.
- J. C. Kagan, M. P. Stein, M. Pypaert and C. R. Roy, J. Exp. Med., 2004, 199, 1201–1211 CrossRef CAS PubMed.
- M. Grammel, P. Luong, K. Orth and H. C. Hang, J. Am. Chem. Soc., 2011, 133, 17103–17105 CrossRef CAS PubMed.
- X. Yu, A. R. Woolery, P. Luong, Y. H. Hao, M. Grammel, N. Westcott, J. Park, J. Wang, X. Bian, G. Demirkan, H. C. Hang, K. Orth and J. LaBaer, Mol. Cell. Proteomics, 2014, 13, 3164–3176 CrossRef CAS PubMed.
- X. Yu, K. B. Decker, K. Barker, M. R. Neunuebel, J. Saul, M. Graves, N. Westcott, H. Hang, J. LaBaer, J. Qiu and M. P. Machner, J. Proteome Res., 2015, 14, 1920–1936 CrossRef CAS PubMed.
- X. Yu and J. LaBaer, Nat. Protoc., 2015, 10, 756–767 CrossRef CAS PubMed.
- M. Broncel, R. A. Serwa, T. D. Bunney, M. Katan and E. W. Tate, Mol. Cell. Proteomics, 2016, 15, 715–725 CrossRef CAS PubMed.
- D. M. Lewallen, C. J. Steckler, B. Knuckley, M. J. Chalmers and P. R. Thompson, Mol. BioSyst., 2012, 8, 1701–1706 RSC.
- Y. Mehellou, H. S. Rattan and J. Balzarini, J. Med. Chem., 2018, 61, 2211–2226 CrossRef CAS PubMed.
- M. Broncel, R. A. Serwa and E. W. Tate, ChemBioChem, 2012, 13, 183–185 CrossRef CAS PubMed.
- P. Kielkowski, I. Y. Buchsbaum, T. Becker, K. Bach, S. Cappello and S. A. Sieber, ChemBioChem, 2020, 21, 1285–1287 CrossRef CAS PubMed.
- P. Kielkowski, I. Y. Buchsbaum, V. C. Kirsch, N. C. Bach, M. Drukker, S. Cappello and S. A. Sieber, Nat. Commun., 2020, 11, 517 CrossRef CAS PubMed.
- T. Rauh, S. Brameyer, P. Kielkowski, K. Jung and S. A. Sieber, ACS Infect. Dis., 2020, 6, 3277–3289 CrossRef CAS PubMed.
- B. Gulen, M. Rosselin, J. Fauser, M. F. Albers, C. Pett, C. Krisp, V. Pogenberg, H. Schlüter, C. Hedberg and A. Itzen, Nat. Chem., 2020, 12, 732–739 CrossRef CAS PubMed.
- E. W. Tate, K. A. Kalesh, T. Lanyon-Hogg, E. M. Storck and E. Thinon, Curr. Opin. Chem. Biol., 2015, 24, 48–57 CrossRef CAS PubMed.
- K. F. Suazo, K.-Y. Park and M. D. Distefano, Chem. Rev., 2021, 121, 7178–7248 CrossRef CAS PubMed.
- E. Thinon and H. C. Hang, Biochem. Soc. Trans., 2015, 43, 253–261 CrossRef CAS PubMed.
- N. Burnaevskiy, T. G. Fox, D. A. Plymire, J. M. Ertelt, B. A. Weigele, A. S. Selyunin, S. S. Way, S. M. Patrie and N. M. Alto, Nature, 2013, 496, 106–109 CrossRef CAS PubMed.
- G. Charron, M. M. Zhang, J. S. Yount, J. Wilson, A. S. Raghavan, E. Shamir and H. C. Hang, J. Am. Chem. Soc., 2009, 131, 4967–4975 CrossRef CAS PubMed.
- N. Burnaevskiy, T. Peng, L. E. Reddick, H. C. Hang and N. M. Alto, Mol. Cell, 2015, 58, 110–122 CrossRef CAS PubMed.
- Y. Zhou, C. Huang, L. Yin, M. Wan, X. Wang, L. Li, Y. Liu, Z. Wang, P. Fu, N. Zhang, S. Chen, X. Liu, F. Shao and Y. Zhu, Science, 2017, 358, 528–531 CrossRef CAS PubMed.
- W. Liu, Y. Zhou, T. Peng, P. Zhou, X. Ding, Z. Li, H. Zhong, Y. Xu, S. Chen, H. C. Hang and F. Shao, Nat. Microbiol., 2018, 3, 996–1009 CrossRef CAS PubMed.
- G. Charron, L. K. Tsou, W. Maguire, J. S. Yount and H. C. Hang, Mol. BioSyst., 2011, 7, 67–73 RSC.
- S. S. Ivanov, G. Charron, H. C. Hang and C. R. Roy, J. Biol. Chem., 2010, 285, 34686–34698 CrossRef CAS PubMed.
- B. R. Martin, C. Wang, A. Adibekian, S. E. Tully and B. F. Cravatt, Nat. Methods, 2012, 9, 84–89 CrossRef CAS PubMed.
- S. W. Hicks, G. Charron, H. C. Hang and J. E. Galán, Cell Host Microbe, 2011, 10, 9–20 CrossRef CAS PubMed.
- Y. H. Lin, A. G. Doms, E. Cheng, B. Kim, T. R. Evans and M. P. Machner, J. Biol. Chem., 2015, 290, 25766–25781 CrossRef CAS PubMed.
- G. N. Schroeder, P. Aurass, C. V. Oates, E. W. Tate, E. L. Hartland, A. Flieger and G. Frankel, Infect. Immun., 2015, 83, 3989–4002 CrossRef CAS PubMed.
- J. M. Spera, F. Guaimas, M. M. Corvi, J. E. Ugalde and C. R. Roy, Infect. Immun., 2018, 86, e00402 CrossRef CAS PubMed.
- Y. Ramos, S. Sansone and D. K. Morales, PLoS Pathog., 2021, 17, e1009822 CrossRef CAS PubMed.
- M. S. Siegrist, B. M. Swarts, D. M. Fox, S. A. Lim and C. R. Bertozzi, FEMS Microbiol. Rev., 2015, 39, 184–202 CrossRef PubMed.
- A. R. Brown, R. A. Gordon, S. N. Hyland, M. S. Siegrist and C. L. Grimes, Cell Chem. Biol., 2020, 27, 1052–1062 CrossRef CAS PubMed.
- F. García-Del Portillo, Mol. Microbiol., 2020, 113, 613–626 CrossRef PubMed.
- E. Kuru, H. V. Hughes, P. J. Brown, E. Hall, S. Tekkam, F. Cava, M. A. de Pedro, Y. V. Brun and M. S. VanNieuwenhze, Angew. Chem., Int. Ed., 2012, 51, 12519–12523 CrossRef CAS PubMed.
- M. S. Siegrist, S. Whiteside, J. C. Jewett, A. Aditham, F. Cava and C. R. Bertozzi, ACS Chem. Biol., 2013, 8, 500–505 CrossRef CAS PubMed.
- P. Cossart, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 19484–19491 CrossRef CAS PubMed.
- Y.-C. Wang, N. P. Westcott, M. E. Griffin and H. C. Hang, ACS Chem. Biol., 2019, 14, 405–414 CrossRef CAS PubMed.
- H. C. Hang, C. Yu, D. L. Kato and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 14846–14851 CrossRef CAS PubMed.
- N. Navabi, M. E. V. Johansson, S. Raghavan, S. K. Lindén and B. A. McCormick, Infect. Immun., 2013, 81, 829–837 CrossRef CAS PubMed.
- A. L. Jolly, P. Agarwal, M. M. E. Metruccio, D. R. Spiciarich, D. J. Evans, C. R. Bertozzi and S. M. J. Fleiszig, FASEB J., 2017, 31, 2393–2404 CrossRef CAS PubMed.
- P. Burzyńska, Ł. F. Sobala, K. Mikołajczyk, M. Jodłowska and E. Jaśkiewicz, Biomolecules, 2021, 11, 831 CrossRef PubMed.
- M. P. Jennings, C. J. Day and J. M. Atack, Microbiology, 2022, 168, 001157 CrossRef CAS PubMed.
- E. Rossing, J. F. A. Pijnenborg and T. J. Boltje, Chem. Commun., 2022, 58, 12139–12150 RSC.
- T. Heise, J. D. Langereis, E. Rossing, M. I. de Jonge, G. J. Adema, C. Büll and T. J. Boltje, Cell Chem. Biol., 2018, 25, 1279–1285.e1278 CrossRef CAS PubMed.
- S. J. Moons, E. Rossing, J. J. A. Heming, M. Janssen, M. van Scherpenzeel, D. J. Lefeber, M. I. de Jonge, J. D. Langereis and T. J. Boltje, Bioconjugate Chem., 2021, 32, 1047–1051 CrossRef CAS PubMed.
- K. M. Backus, H. I. Boshoff, C. S. Barry, O. Boutureira, M. K. Patel, F. D’Hooge, S. S. Lee, L. E. Via, K. Tahlan, C. E. Barry, 3rd and B. G. Davis, Nat. Chem. Biol., 2011, 7, 228–235 CrossRef CAS PubMed.
- B. M. Swarts, C. M. Holsclaw, J. C. Jewett, M. Alber, D. M. Fox, M. S. Siegrist, J. A. Leary, R. Kalscheuer and C. R. Bertozzi, J. Am. Chem. Soc., 2012, 134, 16123–16126 CrossRef CAS PubMed.
- M. Kamariza, P. Shieh, C. S. Ealand, J. S. Peters, B. Chu, F. P. Rodriguez-Rivera, M. R. Babu Sait, W. V. Treuren, N. Martinson, R. Kalscheuer, B. D. Kana and C. R. Bertozzi, Sci. Transl. Med., 2018, 10, eaam6310 CrossRef PubMed.
- A. A. Pohane, D. J. Moore, I. Lepori, R. A. Gordon, T. O. Nathan, D. M. Gepford, H. W. Kavunja, B. M. Swarts and M. S. Siegrist, ACS Infect. Dis., 2022, 8, 2223–2231 CrossRef CAS PubMed.
- T. Dai, J. Xie, Q. Zhu, M. Kamariza, K. Jiang, C. R. Bertozzi and J. Rao, J. Am. Chem. Soc., 2020, 142, 15259–15264 CrossRef CAS PubMed.
- N. Banahene, H. W. Kavunja and B. M. Swarts, Chem. Rev., 2022, 122, 3336–3413 CrossRef CAS PubMed.
- F. P. Rodriguez-Rivera, X. Zhou, J. A. Theriot and C. R. Bertozzi, J. Am. Chem. Soc., 2017, 139, 3488–3495 CrossRef CAS PubMed.
- M. Carlier, E. Lesur, A. Baron, A. Lemétais, K. Guitot, L. Roupnel, C. Dietrich, G. Doisneau, D. Urban, N. Bayan, J.-M. Beau, D. Guianvarc’h, B. Vauzeilles and Y. Bourdreux, Org. Biomol. Chem., 2022, 20, 1974–1981 RSC.
- J. M. Groenevelt, L. M. Meints, A. I. Stothard, A. W. Poston, T. J. Fiolek, D. H. Finocchietti, V. M. Mulholand, P. J. Woodruff and B. M. Swarts, J. Org. Chem., 2018, 83, 8662–8667 CrossRef CAS PubMed.
- V. M. Marando, D. E. Kim, P. J. Calabretta, M. B. Kraft, B. D. Bryson and L. L. Kiessling, J. Am. Chem. Soc., 2021, 143, 16337–16342 CrossRef CAS PubMed.
- C. J. Cambier, S. M. Banik, J. A. Buonomo and C. R. Bertozzi, eLife, 2020, 9, e60648 CrossRef CAS PubMed.
- N. Jessani, Y. Liu, M. Humphrey and B. F. Cravatt, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 10335–10340 CrossRef CAS PubMed.
- D. A. Bachovchin, T. Ji, W. Li, G. M. Simon, J. L. Blankman, A. Adibekian, H. Hoover, S. Niessen and B. F. Cravatt, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20941–20946 CrossRef CAS PubMed.
- A. Di Venere, E. Dainese, F. Fezza, B. C. Angelucci, N. Rosato, B. F. Cravatt, A. Finazzi-Agrò, G. Mei and M. Maccarrone, Biochem. Biophys. Acta, 2012, 1821, 1425–1433 CAS.
- T. Bisogno, A. Mahadevan, R. Coccurello, J. W. Chang, M. Allarà, Y. Chen, G. Giacovazzo, A. Lichtman, B. Cravatt, A. Moles and V. Di Marzo, Br. J. Pharmacol., 2013, 169, 784–793 CrossRef CAS PubMed.
- S. K. Hatzios, S. Abel, J. Martell, T. Hubbard, J. Sasabe, D. Munera, L. Clark, D. A. Bachovchin, F. Qadri, E. T. Ryan, B. M. Davis, E. Weerapana and M. K. Waldor, Nat. Chem. Biol., 2016, 12, 268–274 CrossRef CAS PubMed.
- M. Howell, D. G. Dumitrescu, L. R. Blankenship, D. Herkert and S. K. Hatzios, J. Biol. Chem., 2019, 294, 9888–9900 CrossRef CAS PubMed.
- M. P. Patricelli, D. K. Giang, L. M. Stamp and J. J. Burbaum, Proteomics, 2001, 1, 1067–1071 CrossRef CAS PubMed.
- C. S. Lentz, J. R. Sheldon, L. A. Crawford, R. Cooper, M. Garland, M. R. Amieva, E. Weerapana, E. P. Skaar and M. Bogyo, Nat. Chem. Biol., 2018, 14, 609–617 CrossRef CAS PubMed.
- M. D. Disney, J. Am. Chem. Soc., 2019, 141, 6776–6790 CrossRef CAS PubMed.
- S. Y. Yang, P. Lejault, S. Chevrier, R. Boidot, A. G. Robertson, J. M. Y. Wong and D. Monchaud, Nat. Commun., 2018, 9, 4730 CrossRef PubMed.
- I. V. L. Wilkinson, G. C. Terstappen and A. J. Russell, Drug Discovery Today, 2020, 25, 1998–2005 CrossRef CAS PubMed.
- C. G. Parker and M. R. Pratt, Cell, 2020, 180, 605–632 CrossRef CAS PubMed.
- D. de la Torre and J. W. Chin, Nat. Rev. Genet., 2021, 22, 169–184 CrossRef CAS PubMed.
- M. Tajik, M. Baharfar and W. A. Donald, Trends Biotechnol., 2022, 40, 1374–1392 CrossRef CAS PubMed.
- D. D. Vallejo, C. Rojas Ramirez, K. F. Parson, Y. Han, V. V. Gadkari and B. T. Ruotolo, Chem. Rev., 2022, 122, 7690–7719 CrossRef CAS PubMed.
- X. Tian, H. P. Permentier and R. Bischoff, Mass Spectrom. Rev., 2021, 42, 546–576, DOI:10.1002/mas.21709.
- A. Revesz, H. Hever, A. Steckel, G. Schlosser, D. Szabo, K. Vekey and L. Drahos, Mass Spectrom. Rev., 2021, e21763, DOI:10.1002/mas.21763.
- T. Pacchiarotta, A. M. Deelder and O. A. Mayboroda, Bioanalysis, 2012, 4, 919–925 CrossRef CAS PubMed.
- E. K. Jo, Exp. Mol. Med., 2019, 51, 1–3 CrossRef CAS PubMed.
- Y. Zhang, D. S. Kao, B. Gu, R. Bomjan, M. Srivastava, H. Lu, D. Zhou and W. A. Tao, Angew. Chem., Int. Ed., 2020, 59, 2235–2240 CrossRef CAS PubMed.
- D. G. Russell, Cell. Microbiol., 2019, 21, e13075 CrossRef CAS PubMed.
- A. Grote and A. M. Earl, Curr. Opin. Microbiol., 2022, 70, 102197 CrossRef CAS PubMed.
- J. P. Newson, M. S. Gaissmaier, S. C. McHugh and W. D. Hardt, Curr. Opin. Microbiol., 2022, 70, 102224 CrossRef CAS PubMed.
- A. Zoued, H. Zhang, T. Zhang, R. T. Giorgio, C. J. Kuehl, B. Fakoya, B. Sit and M. K. Waldor, Nat. Chem. Biol., 2021, 17, 1199–1208 CrossRef CAS PubMed.
- Y. Bian, C. Gao and B. Kuster, Expert Rev. Proteomics, 2022, 19, 153–164 CrossRef CAS PubMed.
- N. Slavov, Science, 2020, 367, 512–513 CrossRef CAS PubMed.
- R. T. Kelly, Mol. Cell. Proteomics, 2020, 19, 1739–1748 CrossRef CAS PubMed.
- D. Cohen, J. A. Dickerson, C. D. Whitmore, E. H. Turner, M. M. Palcic, O. Hindsgaul and N. J. Dovichi, Annu. Rev. Anal. Chem., 2008, 1, 165–190 CrossRef CAS PubMed.
- M. Ahmed, J. Mackenzie, L. Tezera, R. Krause, B. Truebody, D. Garay-Baquero, A. Vallejo, K. Govender, J. Adamson, H. Fisher, J. W. Essex, S. Mansour, P. Elkington, A. J. C. Steyn and A. Leslie, Commun. Biol., 2022, 5, 1317 CrossRef PubMed.
- G. Luo, D. W. Niesel, R. A. Shaban, E. A. Grimm and G. R. Klimpel, Infect. Immun., 1993, 61, 830–835 CrossRef CAS PubMed.
- R. Porat, B. D. Clark, S. M. Wolff and C. A. Dinarello, Science, 1991, 254, 430–432 CrossRef CAS PubMed.
- L. Wu, O. Estrada, O. Zaborina, M. Bains, L. Shen, J. E. Kohler, N. Patel, M. W. Musch, E. B. Chang, Y. X. Fu, M. A. Jacobs, M. I. Nishimura, R. E. Hancock, J. R. Turner and J. C. Alverdy, Science, 2005, 309, 774–777 CrossRef CAS PubMed.
- W. Zhou and A. Deiters, Curr. Opin. Chem. Biol., 2021, 63, 123–131 CrossRef CAS PubMed.
- K. Lang and J. W. Chin, Chem. Rev., 2014, 114, 4764–4806 CrossRef CAS PubMed.
- M. N. Maas, J. C. J. Hintzen and J. Mecinovic, Chem. Commun., 2022, 58, 7216–7231 RSC.
- J. Park, S. Lee, Y. Kim and T. H. Yoo, Bioorg. Med. Chem., 2021, 30, 115946 CrossRef CAS PubMed.
- N. L. Kjaersgaard, T. B. Nielsen and K. V. Gothelf, ChemBioChem, 2022, 23, e202200245 CrossRef CAS PubMed.
- N. H. Fischer, M. T. Oliveira and F. Diness, Biomater. Sci., 2022, 11, 719–748, 10.1039/d2bm01237e.
- M. Srivastava, Y. Zhang, J. Chen, D. Sirohi, A. Miller, Y. Zhang, Z. Chen, H. Lu, J. Xu, R. J. Kuhn and W. Andy Tao, Nat. Commun., 2020, 11, 3896 CrossRef PubMed.
- N. Amara, R. Mashiach, D. Amar, P. Krief, S. A. Spieser, M. J. Bottomley, A. Aharoni and M. M. Meijler, J. Am. Chem. Soc., 2009, 131, 10610–10619 CrossRef CAS PubMed.
- C. Schütz and M. Empting, Beilstein J. Org. Chem., 2018, 14, 2627–2645 CrossRef PubMed.
- G. D. Geske, J. C. O’Neill, D. M. Miller, M. E. Mattmann and H. E. Blackwell, J. Am. Chem. Soc., 2007, 129, 13613–13625 CrossRef CAS PubMed.
- M. Prothiwa, F. Englmaier and T. Böttcher, J. Am. Chem. Soc., 2018, 140, 14019–14023 CrossRef CAS PubMed.
- S. Rütschlin and T. Böttcher, Chem. – Eur. J., 2020, 26, 964–979 CrossRef PubMed.
- C. A. Lowery, N. T. Salzameda, D. Sawada, G. F. Kaufmann and K. D. Janda, J. Med. Chem., 2010, 53, 7467–7489 CrossRef CAS PubMed.
- T. J. Polaske, C. G. Gahan, K. E. Nyffeler, D. M. Lynn and H. E. Blackwell, Cell Chem. Biol., 2021, 29, 605–614.e4, DOI:10.1016/j.chembiol.2021.12.005.
- M. Goswami, A. Espinasse and E. E. Carlson, Chem. Sci., 2018, 9, 7332–7337 RSC.
- C. N. Tsai, C. R. MacNair, M. P. T. Cao, J. N. Perry, J. Magolan, E. D. Brown and B. K. Coombes, Cell Chem. Biol., 2020, 27, 793–805.e797 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |