
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSuperoxide-responsive quinone methide precursors (QMP-SOs) to study superoxide biology by proximity labeling and chemoproteomics†
Hinyuk
Lai
a and
Clive Yik-Sham
Chung
 *abc
*abc
aSchool of Biomedical Sciences, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Pokfulam Road, Hong Kong SAR, P. R. China. E-mail: cyschung@hku.hk
bDepartment of Pathology, School of Clinical Medicine, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Pokfulam Road, Hong Kong SAR, P. R. China
cCentre for Oncology and Immunology, Hong Kong Science Park, Hong Kong SAR, China
First published on 7th August 2024
Abstract
Superoxide is a reactive oxygen species (ROS) with complex roles in biological systems. It can contribute to the development of serious diseases, from aging to cancers and neurodegenerative disorders. However, it can also serve as a signaling molecule for important life processes. Monitoring superoxide levels and identifying proteins regulated by superoxide are crucial to enhancing our understanding of this growing field of redox biology and signaling. Given the high reactivity and very short lifetime of superoxide compared to other ROS in biological systems, proteins redox-modified by superoxide should be in close proximity to where superoxide is generated endogenously, i.e. superoxide hotspots. This inspires us to develop superoxide-specific quinone methide-based precursors, QMP-SOs, for proximity labeling of proteins within/near superoxide hotspots to image superoxide and profile proteins associated with superoxide biology by chemoproteomics. QMP-SOs specifically react with superoxide to generate an electrophilic quinone methide intermediate, which subsequently reacts with nucleophilic amino acids to induce a covalent tag on proteins, as revealed by liquid chromatography-mass spectrometry (LC-MS) and shotgun MS experiments. The alkyne handle on the covalent tag enables installation of fluorophores onto the tagged proteins for fluorescence imaging of superoxide in cells under oxidative stress. By establishing a chemoproteomics platform, QMP-SO-TMT, we identify DJ-1 and DLDH as proteins associated with superoxide biology in liver cancer cells treated with menadione. This work should provide insights into the crosstalk between essential cellular events and superoxide redox biology, as well as the design principles of quinone methide-based probes to study redox biology through proximity labeling and chemoproteomics.
Introduction
Superoxide (O2˙−) is a primary source of reactive oxygen species (ROS) in living systems and is known to participate in pathological processes such as innate immunity.1–4 Recent research has shown that superoxide, along with other ROS such as hydrogen peroxide and the hydroxyl radical, plays important roles in life processes and can act as a signaling molecule when produced in a controlled manner.5–9 It can modulate the functions/activities of biomolecules through redox post-translational modifications,1,10,11 thus mediating a variety of cellular signals. On the other hand, dysregulated production of superoxide can lead to oxidative stress, contributing to the development of serious diseases such as cancer, aging and neurodegenerative disorders.12–14 Therefore, monitoring superoxide levels in biological systems and profiling biomolecules associated with redox signaling is critical to enhance our understanding of superoxide biology and promote health.Superoxide-specific fluorescent probes have been developed to detect superoxide in biological samples.15–24 The “turn-on” fluorescence from these probes after reaction with superoxide enables the monitoring of superoxide through fluorescence imaging with high spatiotemporal resolution.17–19 However, these probes cannot directly determine the biomolecules that are redox-modified by superoxide (Fig. 1a). To identify proteins with oxidative post-translational modifications, chemoproteomic probes have been demonstrated to be promising tools,10,25–32 with dimedone probes for profiling proteins with sulfenic acid modification as an example. These chemoproteomics probes are powerful in studying proteins with oxidative modifications, but do not show specificity toward modifications mediated by a particular ROS. Therefore, they provide no information about which ROS mediates these modifications, and how these oxidative modifications are initiated and regulated. In fact, the molecular mechanisms underlying the initiation, transduction, and regulation of superoxide redox biology, as well as the identity of proteins involved, remain insufficiently understood. This is because, unlike other cellular signaling pathways, superoxide redox biology is primarily mediated by reversible oxidations.1,11 These unstable modifications are highly dynamic and hence difficult to be detected by conventional biochemical/biological experiments.
 | ||
| Fig. 1 Superoxide-responsive quinone methide precursors, QMP-SOs, to study superoxide biology. (a) Examples of reported superoxide probes and their applications in studying superoxide. (b) Chemical structures of QMP-SOs reported in this study. The alkyne handle enables conjugation to a fluorophore or desthiobiotin through CuAAC for detection and analysis. (c) Schematic cartoon illustrating the working principle of QMP-SOs for proximal protein labeling within or adjacent to superoxide-rich compartments, i.e. superoxide hotspots. This allows monitoring superoxide levels in biological samples by fluorescence imaging and identification of proteins associated with superoxide biology by liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS). | ||
It is noteworthy that superoxide is highly reactive, short-lived, and has a small diffusion radius in biological systems,3,33 while other ROS, such as H2O2, have higher biostability.3 This suggests that the proteins redox-modified by superoxide should be in close proximity to where superoxide is generated endogenously,34,35 inspiring us to develop new chemical probes to induce proximity labeling of proteins in superoxide-rich compartments, i.e. superoxide hotspots, to profile proteins associated with superoxide biology. The chemical probes should tag onto nearby proteins for analysis without diffusing away from the superoxide hotspots to prevent off-target labeling. Quinone methide, which is highly electrophilic and reacts readily with nucleophilic amino acids on proteins,36–38 should be an ideal moiety for proximity labeling. Herein, we report the design and synthesis of superoxide-responsive quinone methide precursors, QMP-SOs (Fig. 1b). LC-MS experiments revealed the good reactivity and selectivity of QMP-SO toward superoxide over other ROS in aqueous buffer solutions. Shotgun MS confirmed the ability of QMP-SOs to induce covalent tags onto proteins in a superoxide-dependent manner. Further applications of QMP-SOs in fluorescence imaging enabled the detection of superoxide in cells under oxidative stress. Importantly, QMP-SOs could be coupled with tandem mass tags (TMTs) to perform a mass spectrometry (MS)-based chemoproteomics experiment, QMP-SO-TMT. This method allowed the profiling of proteins regulated by superoxide in liver cancer cells treated with menadione, which induced the production of mitochondrial superoxide. DJ-1 and DLDH are two of the proteins identified by QMP-SO-TMT, providing insights into the interplay of cell survival, autophagy, cell death and cell metabolism with superoxide redox biology.
Results and discussion
Design, synthesis and characterization of QMP-SOs
QMP-SOs are superoxide-specific probes capable of studying superoxide biology through fluorescence imaging and chemoproteomics. The probes contain a superoxide-responsive trigger which remains intact in the absence of superoxide.15–24 Upon exposure to superoxide, the trigger would be activated and cleaved off, forming a 4-hydrobenzyl carbamate intermediate. Subsequent self-immolation through the 1,6-elimination reaction39,40 generates the highly electrophilic quinone methide, which reacts readily with nucleophilic amino acids on proximal proteins and hence induces covalent tags onto the proteins (Fig. 1c). Further functionalization of the probes with an alkyne handle allows downstream analysis of the tagged proteins through the installation of a fluorophore or desthiobiotin by a copper catalyzed azide–alkyne cycloaddition (CuAAC) reaction.41We successfully synthesized QMP-SO-1 and three alkyne-containing probes, namely QMP-SO-OTf-alkyne, QMP-SO-C3-alkyne and QMP-SO-C5-alkyne (Fig. 1b and Schemes S1–S3 in the ESI†). The QMP-SO-OTf-alkyne contains a triflate group as the superoxide-responsive trigger,17 while the others utilize diphenylphosphonate as the trigger.18 The alkyne probes were synthesized by first installing the alkyne moiety onto the meta-position of 3,4-dihydroxybenzaldehyde through a nucleophilic substitution reaction, followed by incorporating the trigger onto the para-position. Subsequent aldehyde reduction, activation of the hydroxyl group by disuccinimidyl carbonate and reaction with amine-functionalized glycol yielded the final products (Scheme S2, ESI†). All the compounds have been successfully characterized by 1H, 13C, 19F and/or 31P NMR (ESI), and LC-MS.
Reactivity of QMP-SOs with superoxide in vitro
LC-MS experiments of aqueous solutions of QMP-SOs (Fig. 2a) revealed a fast consumption of the probes in the presence of superoxide in vitro. QMP-SO-C3-alkyne and QMP-SO-C5-alkyne completely reacted with KO2 in 15 and 60 min, respectively (Fig. 2b–d), while QMP-SO-OTf-alkyne showed a slower reaction with KO2. We also observed a larger consumption of QMP-SO-C3-alkyne and QMP-SO-C5-alkyne with superoxide generated by the enzymatic reaction of xanthine oxidase and xanthine (Fig. 2e–g), compared to the reaction of QMP-SO-OTf-alkyne under the same experimental conditions (Fig. 2f and g). These results suggest that QMP-SOs with a diphenylphosphonate trigger should be more sensitive than those with a triflate group for superoxide detection.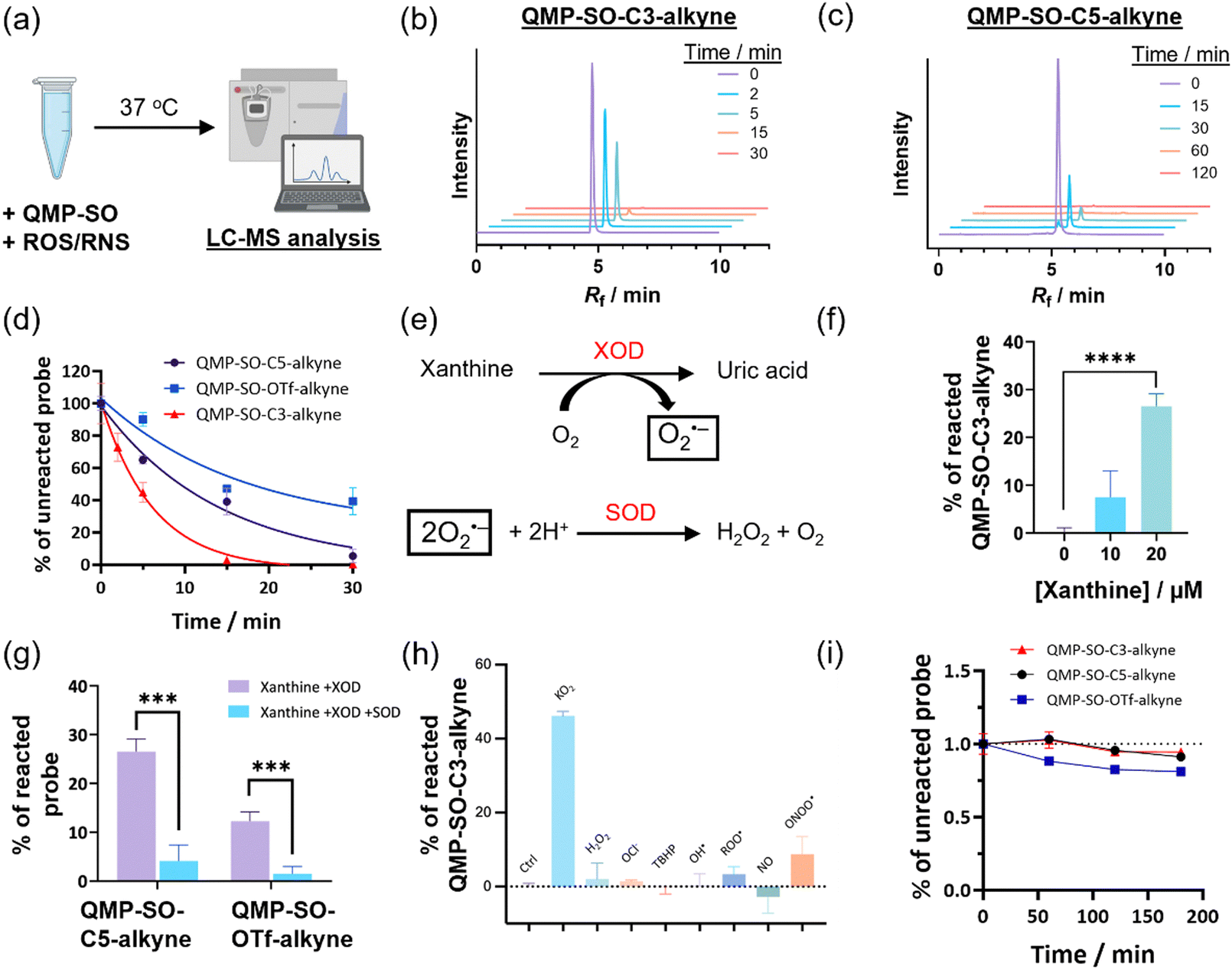 | ||
Fig. 2 LC-MS experiment to study the reaction kinetics of QMP-SO. (a) Schematic cartoon illustrating the LC-MS experiments to study the reactivity of QMP-SO toward redox-active species in aqueous buffer solution. (b) and (c) Selected-ion chromatograms (SICs), corresponding to the molecular ions from the QMP-SO-C3-alkyne and QMP-SO-C5-alkyne, respectively, were measured from aliquots of the reaction mixture of the QMP-SO-C3-alkyne/QMP-SO-C5-alkyne and KO2 in the PBS-MeOH mixture (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) at the indicated time. (d) Percentage of unreacted QMP-SOs in the solution mixture with KO2. (e) Equations illustrating the generation of superoxide (O2˙−) by the xanthine and xanthine oxidase (XOD) system, and the scavenging of superoxide by superoxide dismutase (SOD). (f) Percentage of the QMP-SO-C5-alkyne reacted in the solution mixture containing xanthine and XOD. (g) Percentage of QMP-SO reacted in the presence of xanthine, XOD and/or SOD. (h) Percentage of QMP-SO-C3-alkyne reacted after incubation with the redox-active species for 30 min in the aqueous buffer solution. (i) Stability of QMP-SOs in the PBS-MeOH mixture (4:1, v/v). Quantified data were shown in average ± SD. Statistical analysis using a two-tailed Student's t-test. ***p < 0.001 and ****p < 0.0001. :1, v/v) at the indicated time. (d) Percentage of unreacted QMP-SOs in the solution mixture with KO2. (e) Equations illustrating the generation of superoxide (O2˙−) by the xanthine and xanthine oxidase (XOD) system, and the scavenging of superoxide by superoxide dismutase (SOD). (f) Percentage of the QMP-SO-C5-alkyne reacted in the solution mixture containing xanthine and XOD. (g) Percentage of QMP-SO reacted in the presence of xanthine, XOD and/or SOD. (h) Percentage of QMP-SO-C3-alkyne reacted after incubation with the redox-active species for 30 min in the aqueous buffer solution. (i) Stability of QMP-SOs in the PBS-MeOH mixture (4:1, v/v). Quantified data were shown in average ± SD. Statistical analysis using a two-tailed Student's t-test. ***p < 0.001 and ****p < 0.0001. | ||
QMP-SOs showed good specificity toward superoxide, as indicated by a significant decrease in probe consumption in the presence of superoxide dismutase (SOD), a known superoxide scavenger3,34 (Fig. 2e–g). The reactivity of QMP-SOs toward superoxide was not sensitive to changes in the pH of the aqueous buffer solution (Fig. S1, ESI†). No significant reaction was observed between QMP-SO-C3-alkyne and other ROS/reactive nitrogen species (RNS; Fig. 2h), except that peroxynitrite could slightly react with QMP-SO-C3-alkyne. Yet, the probe consumption by peroxynitrite was less than 5-fold compared to that found in the solution mixture with superoxide, and the working concentration of peroxynitrite in the LC-MS experiment was 100 μM which is much higher than its physiologically relevant concentrations.42 This suggests that the interference from peroxynitrite in studying superoxide in biological samples by QMP-SOs should be minimal. In addition, QMP-SOs demonstrated good stability in aqueous buffer solution without superoxide (Fig. 2i). All these results highlight the good reactivity and selectivity of QMP-SOs toward superoxide.
Superoxide-induced labeling onto BSA and cell lysates by QMP-SOs
After characterizing the fast reaction of QMP-SOs with superoxide, we sought to investigate their ability to induce covalent tags onto proteins in a superoxide-dependent manner. An aqueous solution of a model protein, BSA, was first incubated with QMP-SO-C5-alkyne, KO2 and/or SOD. The subsequent CuAAC reaction enabled the installation of a fluorophore onto the alkyne handle on the tagged BSA by QMP-SOs (Fig. 3a). A stronger in-gel fluorescence was observed in the BSA incubated with QMP-SO-C5-alkyne and KO2, while co-incubation with SOD significantly reduced the in-gel fluorescence intensity (Fig. 3b). A similar increase in the in-gel fluorescence intensity was also observed in BSA incubated with xanthine oxidase/xanthine which can generate superoxide catalytically (Fig. 3c). These results indicate that QMP-SOs could react with superoxide and covalently modify BSA.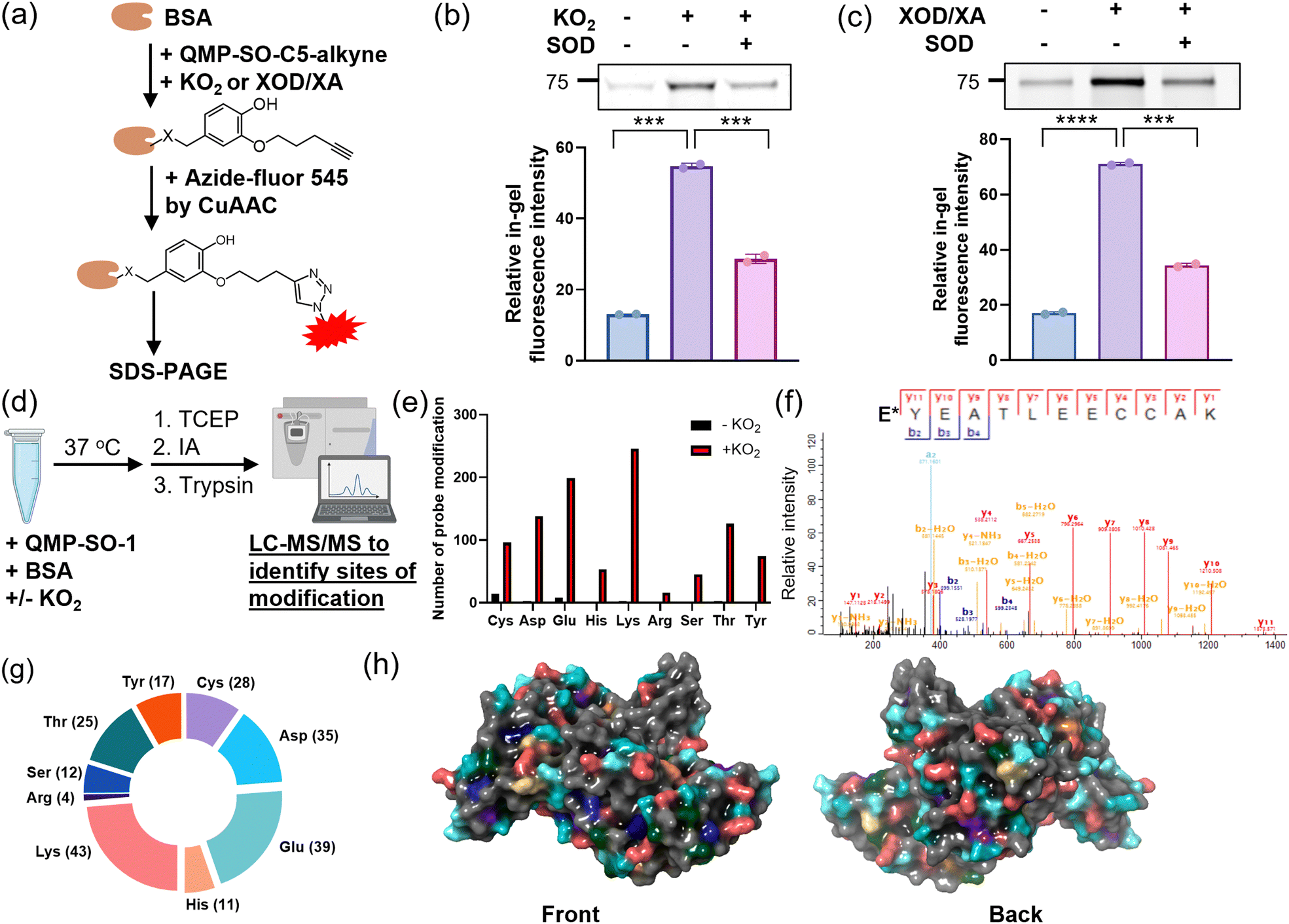 | ||
| Fig. 3 Gel-based chemoproteomics and LC-MS/MS experiments to study covalent modifications on proteins induced by QMP-SOs. (a) Schematic cartoon illustrating the gel-based chemoproteomics experiments to identify QMP-SO-C5-alkyne-induced modifications on a model protein, bovine serum albumin (BSA). (b) In-gel fluorescence from BSA incubated with the QMP-SO-C5-alkyne, KO2 and/or SOD in an aqueous buffer solution. (c) In-gel fluorescence from BSA incubated with QMP-SO-C5-alkyne, XOD/xanthine and/or SOD in an aqueous buffer solution. (d) Schematic cartoon illustrating the LC-MS/MS experiments to identify QMP-SO-1-induced modifications on BSA. (e) The number of QMP-SO-1-induced modifications on BSA in PBS with or without addition of KO2. (f) Representative MS/MS revealing the covalent modification of Glu (E*) of BSA by QMP-SO-1. (g) The number of modified amino acids on BSA by QMP-SO-1. (h) The sites of QMP-SO-1-induced modifications on BSA, with the same labeling color as that in (g). Quantified data were shown in average ± SD (n = 2 replicates/group). Statistical analysis using a two-tailed Student's t-test. ***p < 0.001 and ****p < 0.0001. | ||
To identify the covalent modification on BSA by QMP-SOs, shotgun MS experiments were performed by LC-MS/MS to study the tryptic digested peptides from the solution mixture of BSA, QMP-SO-1 and/or KO2 (Fig. 3d). Covalent modifications with a adduct mass of the 4-(hydroxyphenyl)methylene group were detected on nucleophilic amino acids, including Cys, Asp, Glu, His, Lys, Arg, Ser, Thr and Tyr, in the solution mixtures with KO2 (Fig. 3e, f and Fig. S2–S4, and ESI†). This supports the formation of a quinone methide from the reaction of QMP-SOs with superoxide, resulting in a subsequent electrophilic attack on BSA to form the 4-(hydroxyphenyl)methylene modification (Fig. 1c). Also, we observed a significant increase in the number of covalent modifications on BSA upon addition of KO2 (>27-fold; Fig. 3e), suggesting that the protein modifications induced by QMP-SO-1 were superoxide-dependent. It is noteworthy that probe modifications were found on a large number of nucleophilic amino acids (Fig. 3g and Fig. S5, ESI†), particularly on the protein surface (Fig. 3h). This broad reactivity with amino acids illustrates that QMP-SO should allow good tagging on almost all possible proteins of interest in a superoxide-dependent manner, regardless of the protein primary sequence. This is an important feature for profiling proteins associated with superoxide biology.
Next, we proceeded to examine the performance of QMP-SOs in labeling proteins in HepG2 cell lysates, which are more complex biological samples. A turn-on in-gel fluorescence was found in the lysates treated with potassium superoxide, while co-incubation of the cell lysates with SOD resulted in a significant decrease in the in-gel fluorescence intensity (Fig. 4a and d and Fig. S6, ESI†). This indicates the successful superoxide-induced tagging of proteins by QMP-SO-C3-alkyne and QMP-SO-C5-alkyne, as well as their high specificity toward superoxide.
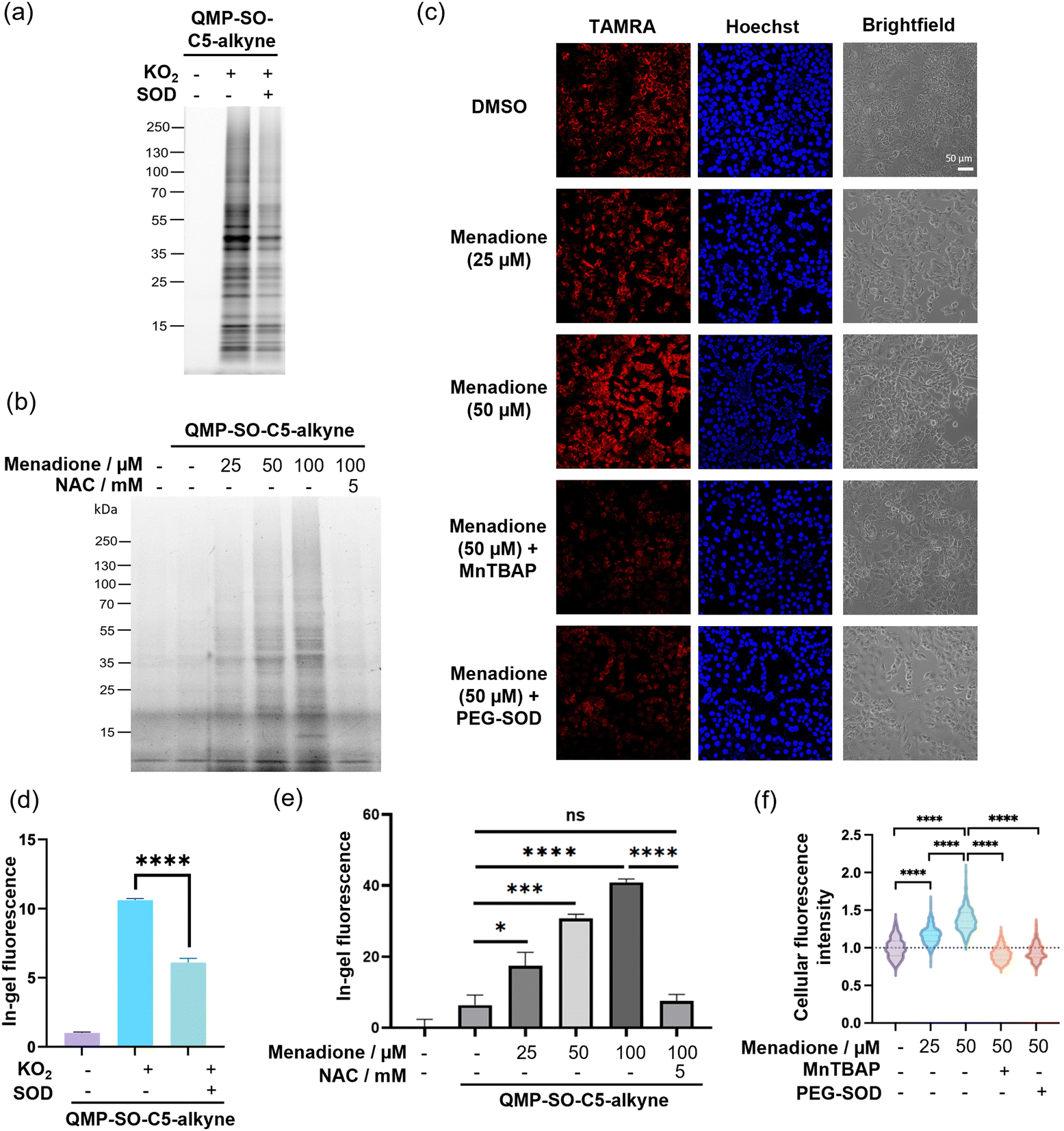 | ||
| Fig. 4 Gel-based chemoproteomics and confocal fluorescence imaging of HepG2 cell lysates/live cells probed with QMP-SO-C5-alkyne. (a) HepG2 cell lysates (50 μg) in PBS were incubated with QMP-SO-C5-alkyne (10 μM), KO2 (500 μM) and/or SOD (75 mU mL−1) for 2 h. Excess reagents were removed by acetone precipitation at −20 °C overnight, and the precipitated proteins were re-dissolved in PBS. The labeled proteins were then reacted with azide-fluor 545 (25 μM) using a copper-catalyzed azide–alkyne cycloaddition (CuAAC) reaction, boiled with sampling buffer and read out by in-gel fluorescence after SDS-PAGE. (b) Live HepG2 cells were incubated with the QMP-SO-C5-alkyne (10 μM), menadione and/or N-acetylcysteine (NAC; 5 mM) for 3 h in complete medium. The cells were washed with PBS and lysed by sonication in PBS. After protein assay and normalization, the cell lysates were reacted with azide-fluor 545 (25 μM) by CuAAC. The labeled proteins were then boiled with sampling buffer, separated by SDS-PAGE and visualized by their in-gel fluorescence. (c) Live HepG2 cells were co-incubated with QMP-SO-C5-alkyne (10 μM) and the indicated reagents for 3 h in complete medium. The cells were washed with PBS, fixed by pre-chilled methanol, permeabilized by PBS + 0.3 vol% Triton X-100, and reacted with azide-fluor 545 (25 μM) by CuAAC at room temperature in the dark for 1 h. The stained cells were washed with PBS and further incubated with Hoechst (8.2 μM) for 15 min. The cells were then washed with PBS and imaged by confocal fluorescence microscopy. (d) and (e) Quantification of the in-gel fluorescence intensity from (a) and (b) respectively. n = 3 replicates/group. (f) Quantification of the confocal fluorescence imaging experiment shown in (c). n = 30 cells from 3 different biological replicates/group. Quantified data were shown in average ± SD. Statistical analysis using a two-tailed Student's t-test. *p < 0.05, ***p < 0.001 and ****p < 0.0001. ns = not significant. | ||
Fluorescence imaging for superoxide detection in cancer cells by QMP-SOs
Menadione is known to induce endogenous production of superoxide, particularly in mitochondria. This is primarily through a one-electron reduction of menadione by complex I in the mitochondrial electron transport chain, resulting in the formation of semiquinone which can readily react with molecular oxygen to form superoxide.43 We attempted to utilize QMP-SO-C5-alkyne for monitoring dynamic changes in superoxide levels in live HepG2 cells treated with menadione and/or NAC. The live cells treated with menadione and QMP-SO-C5-alkyne were lysed by probe sonication, followed by reaction with azide-fluor 545 through CuAAC and SDS-PAGE. A dose-dependent increase in the in-gel fluorescence intensity was found in HepG2 cells treated with menadione and QMP-SO-C5-alkyne, while co-incubation with the antioxidant NAC abolished the enhanced fluorescence intensity (Fig. 4b and e). This can be explained by the high local concentrations of superoxide generated in HepG2 cells upon menadione treatment, resulting in superoxide-mediated covalent tagging of proximal proteins by QMP-SO-C5-alkyne. We confirmed that the QMP-SO-C5-alkyne was not toxic to HepG2 cells during the 4 h incubation, with >90% viable cells in the 40 μM treatment as revealed by the MTT assay (Fig. S7, ESI†). Also, a diminished in-gel fluorescence intensity was found in cells treated with the SOD mimetic, MnTBAP44 (Fig. S8, ESI†), supporting the specific response of the QMP-SO-C5-alkyne toward superoxide.Superoxide levels in HepG2 cells were also monitored by confocal fluorescence imaging experiments. The live cells were incubated with QMP-SO-C5-alkyne, menadione, MnTBAP (SOD mimetic) and/or cell-permeable PEG-SOD. The treated cells were fixed with pre-chilled methanol and permeabilized using PBS with 0.3 vol% Triton-X100. Probe-labeled proteins were then reacted with azide-fluor 545 through CuAAC, and the fixed cells were stained with Hoechst and imaged by confocal fluorescence microscopy. Stronger fluorescence was found in cells treated with increasing concentrations of menadione, while a significant decrease in the fluorescence intensity was observed in cells treated with the superoxide scavengers MnTBAP and PEG-SOD, respectively (Fig. 4c and f). This illustrates that QMP-SO-induced protein labeling is superoxide-dependent and QMP-SO is highly sensitive to dynamic changes in cellular superoxide levels, enabling superoxide monitoring by confocal fluorescence imaging.
QMP-SO-enabled chemoproteomics to profile proteins associated with superoxide redox biology
HepG2 cells can withstand oxidative stress induced by menadione treatment at a low dosage/for a short period of time, while higher doses/prolonged incubation results in substantial cell death. However, the proteins that govern the cellular signals for survival, metabolism and cell death under menadione treatment remain underexplored. In view of the large amount of superoxide generated from menadione treatment, superoxide could redox-modify proteins and modulate various cellular processes. We attempted to identify these proteins associated with superoxide biology using the MS-based chemoproteomics platform, QMP-SO-TMT (Fig. 5a). Based on the high reactivity and small diffusion radius of superoxide,3,33 proteins in close proximity to superoxide-rich compartments (superoxide hotspots) should be potential targets that are redox-regulated by superoxide to transduce cellular signals.34,35 By using QMP-SO-C5-alkyne to induce a covalent tag onto proximal proteins in superoxide hotspots, we successfully enriched and profiled 15 proteins with a 2-fold enrichment and statistical significance in the menadione-treated samples (Fig. 5b and Supplementary Data 2, ESI†). GO analysis revealed that these profiled proteins were located in the mitochondria/mitochondrial matrix (Fig. 5c). This is in agreement with reported studies showing that mitochondrial superoxide is generated in menadione-treated cells. More importantly, these proteins were associated with the response to oxidative stress (Fig. 5d), and 6 of them have been annotated to the oxidation–reduction process (GOBP: 0055114; Fig. 5b). All these results highlight the success of the QMP-SO-C5-alkyne in tagging proteins within or near superoxide hotspots to profile proteins associated with redox biology by the TMT-based MS experiments.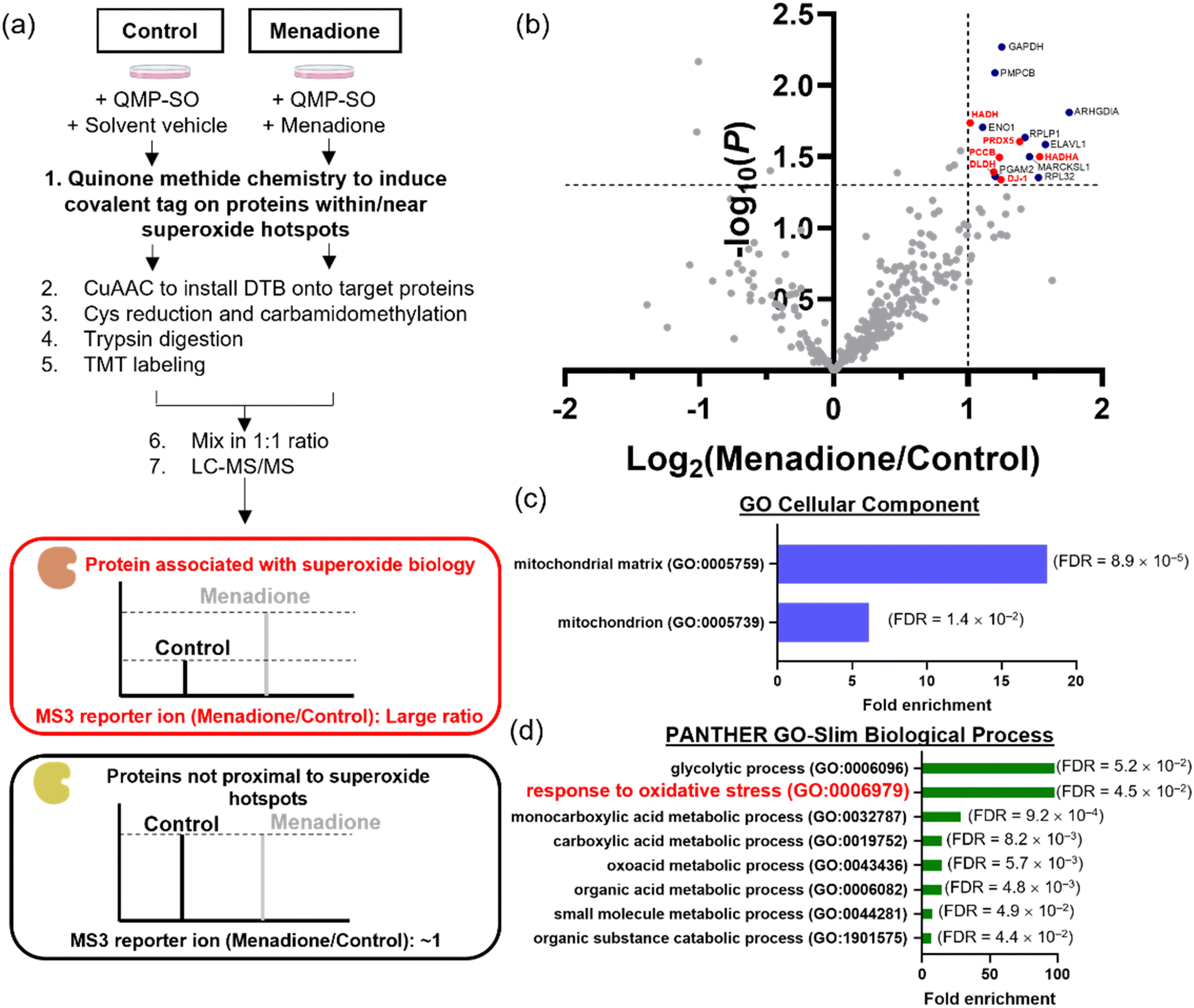 | ||
| Fig. 5 MS-based chemoproteomics experiment to identify proteins labeled by QMP-SOs in HepG2 cells. (a) Schematic cartoon illustrating the workflow of the MS experiment to identify proteins labeled by QMP-SO-C5-alkyne. (b) Volcano plot showing the MS result from HepG2 cells treated with menadione (100 μM) for 2 h. The red dots are the proteins enriched in the menadione-treated sample (>2-fold) with statistical significance (p < 0.05) and annotation to the oxidation–reduction process (Gene ontology biological process: 0055114). (c) Gene ontology (GO) analysis of the cellular component of the enriched proteins. (d) PANTHER GO-Slim analysis of the biological process of the enriched proteins. | ||
DJ-1 is one of the enriched proteins in the QMP-SO-TMT experiment. It is known to activate proliferative signals through Erk1/2 and PI3K/Akt pathways and modulate the autophagy process.45–48 Interestingly, we found an increase in DJ-1 Cys106 oxidation, as detected by a specific antibody for DJ-1 Cys106 sulfenylation/sulfinylation, in HepG2 cells treated with menadione (50 μM) for the first 2 h (Fig. 6a). Co-treatment with the SOD mimetic, MnTBAP, resulted in a significant decrease in the oxidized form of DJ-1 (Fig. 6a), suggesting that the oxidation of DJ-1 should be primarily mediated by superoxide. An increase in the phosphorylation levels of MEK, Erk1/2 and Akt were also observed in cells treated with menadione for the first 2 h (Fig. 6b), indicating the activation of MEK/Erk and Akt to transduce survival signals through the oxidation of DJ-1. In addition, we observed a decrease in p62 level and a concomitant increase in LC3BII/LC3BI in cells treated with menadione for 2 h (Fig. 6b), revealing the activation of autophagy to cope with cellular oxidative stress. It is noteworthy that co-incubation of cells with antioxidant NAC could recuse cells from all these changes induced by menadione (Fig. 6b). This illustrates the importance of oxidative modifications on DJ-1 in activating the pro-survival and autophagy signals. On the other hand, a longer treatment with menadione resulted in a decrease in DJ-1 Cys106 sulfenylation/sulfinylation (Fig. 6c), suggesting overoxidation and hence inactivation of DJ-1. This led to a decrease in the pro-survival signal, as evidenced by the decrease in phosphorylation levels of MEK, Erk1/2 and Akt (Fig. 6c). A decrease in the PARP level was also found in cells treated with menadione for 4 h, pointing to its cleavage and induction of apoptosis. Genetic knockdown of DJ-1 in HepG2 cells by siRNA was found to abolish the activation of proliferation signals after 2 h of menadione treatment, as indicated by no significant increase in p-Erk. Instead, early induction of apoptosis was observed (Fig. S9, ESI†). All these results demonstrate the critical role of oxidative modifications and DJ-1 activity in governing pro-survival and pro-apoptotic events in cells treated with menadione, with superoxide being one of the primary ROS mediating these oxidative modifications (Fig. 6d).
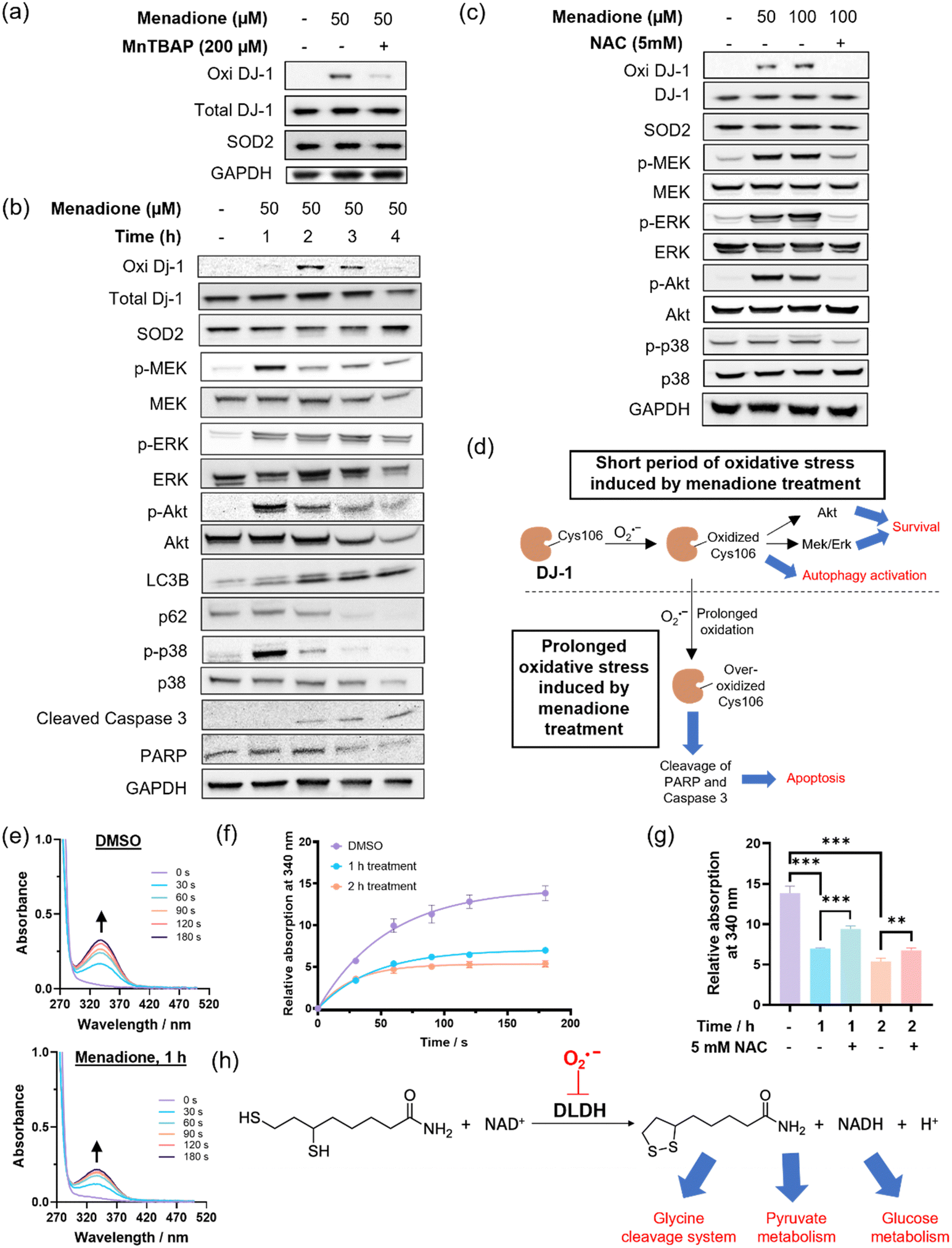 | ||
| Fig. 6 DJ-1 and DLDH are proteins regulated by superoxide in HepG2 cells treated with menadione. (a) Immunoblotting of cells treated with menadione and SOD mimetic, MnTBAP, for 2 h at the indicated concentrations. (b) and (c) Immunoblotting of cells treated with menadione at the indicated concentrations and time intervals, in the absence or presence of NAC. (d) Schematic cartoon illustrating the roles of DJ-1 in regulating cell survival and cell death signals in cells facing superoxide stress from menadione treatment. (e) DLDH activity assay by monitoring the UV-vis absorption spectra of the mitochondria extract from cells treated with DMSO or menadione, dihydrolipoamide (3 mM), EDTA (1.5 mM) and NAD+ (3 mM) in potassium phosphate (100 mM, pH 8.0). (f) Changes in UV-vis absorption at 340 nm over time from the solution mixture of mitochondria extract, dihydrolipoamide, EDTA and NAD+. (g) UV-vis absorption at 340 nm of the solution mixture after incubation for 180 s. (h) Schematic cartoon illustrating the roles of DLDH and its oxidation in governing lipoamide dehydrogenation. Quantified data were shown in average ± SD (n = 3 replicates/group). Statistical analysis using a two-tailed Student's t-test. **p < 0.01, ***p < 0.001 and ****p < 0.0001. | ||
DLDH is another protein target identified in the MS-based chemoproteomics experiment (Fig. 5b). It is important for energy metabolism as a component of the glycine cleavage system and a E3 component in the mitochondrial pyruvate dehydrogenase (PDH) and α-ketoglutarate dehydrogenase (α-KGDH) complexes.47,49 We collected mitochondrial extracts from cells treated with solvent vehicle or menadione (Fig. S10, ESI†), and assayed the DLDH activity in the solution mixture of the mitochondrial extract, dihydrolipoamide and NAD+ according to the experimental setup reported in literature.50,51 We observed a significant increase in UV-vis absorption at 340 nm over time, owing to the production of NADH from the enzymatic reaction of DLDH (Fig. 6e). Notably, mitochondrial extracts from cells treated with menadione showed a slower increase in UV-vis absorption at 340 nm compared to the control sample (Fig. 6f and g), indicating lower DLDH activity in extracts from menadione-treated cells. Since the addition of the antioxidant NAC can partly restore the enzymatic activity with statistical significance (Fig. 6g) and DLDH is a protein target identified by the superoxide-specific QMP-SO-C5-alkyne, the lower DLDH activity in extracts from menadione-treated cells should originate from superoxide-mediated oxidation of DLDH (Fig. 6h), probably on Cys45 and Cys50 which are in the active site of DLDH and known to be redox-sensitive.52 In this study, we uncover the role of superoxide in modulating cellular metabolism through the redox-modification of DLDH.
Conclusions
We have presented the design, synthesis and applications of QMP-SOs, a new class of superoxide-specific probes capable of fluorescence imaging of superoxide levels and chemoproteomic profiling of protein targets associated with superoxide redox biology.LC-MS, shotgun MS and cell-based experiments using superoxide inducers and scavengers have demonstrated the specific reaction of QMP-SOs with superoxide to generate a highly reactive intermediate, quinone methide, for the subsequent labeling of proximal proteins. This enables us to introduce an alkyne handle onto proteins in close proximity to where superoxide is endogenously generated in cells, i.e. superoxide hotspots, thus allowing the CuAAC reaction for the installation of fluorophores and desthiobiotin onto the labeled proteins for fluorescence imaging and chemoproteomics analysis.
Due to the high reactivity, short lifetime and small diffusion radius of superoxide in biological systems,3,33 proteins proximal to or within superoxide hotspots are potential targets regulated by superoxide redox biology. With the broad reactivity of quinone methide with nucleophilic amino acids on proteins revealed by the shotgun MS experiment, QMP-SOs are perfect candidates for labeling proteins in a superoxide-dependent and primary sequence-independent manner. Coupled with TMT-based MS experiments, we have successfully developed a chemoproteomics platform, QMP-SO-TMT, for profiling proteins proximal to or within superoxide hotspots in cells treated with menadione, as shown in the GO analysis that many of the profiled proteins are in the mitochondrial matrix where superoxide is generated upon menadione treatment.
DJ-1 is one of the profiled proteins in menadione-treated HepG2 cells using QMP-SO-TMT. Immunoblotting experiments indicate the superoxide-mediated sulfenylation/sulfinylation of DJ-1 Cys106 in HepG2 cells treated with menadione for a short period of time. This is accompanied by the activation of proliferative MEK/Erk and Akt pathways, as well as the autophagy process. Prolonged treatment with menadione triggers the over-oxidation of DJ-1 Cys106, leading to the inactivation of DJ-1 and the decrease in MEK/Erk and Akt signals. This favors pro-apoptotic events, as evidenced by the cleavage of PARP and subsequent cell death. Our QMP-SOs and the chemoproteomics platform QMP-SO-TMT have successfully uncovered the superoxide-mediated redox regulations of DJ-1 and its important roles in governing cell survival and cell death under oxidative stress.
We have also identified DLDH using QMP-SO-TMT in menadione-treated HepG2 cells. A significant decrease in DLDH activity has been found in HepG2 cells treated with menadione, as compared to the cells treated with solvent vehicle. The recuse effect from NAC treatment suggests that the loss of DLDH activity in menadione-treated cells should originate from the oxidation of DLDH by mitochondrial superoxide formed upon menadione treatment. This finding illustrates the connection between superoxide redox biology and cell metabolism through oxidation-sensitive proteins such as DLDH.
QMP-SOs undergo 1,6-elimination to generate a para-quinone methide intermediate after reacting with superoxide for protein labeling. This mechanism is distinct from the reported quinone methide-based H2O2 probes,36,37 which form ortho-quinone methide through 1,4-elimination of 2-fluoromethylphenol upon reaction with H2O2. We anticipate that our design is highly modular and can introduce different functionalities onto the molecules by using different building blocks for the carbamate linkage. For example, organelle-targeting moieties can be incorporated onto the probes to enable imaging and profiling of superoxide-associated proteins with subcellular resolution. This capability is particularly crucial for studying superoxide because it has a small diffusion radius and the signaling regulated by superoxide is likely highly localized.
This study should provide insights into the development of molecular probes for studying redox biology through proximity labeling of proteins in a superoxide-dependent manner. Furthermore, it should motivate further research on redox biology through fluorescence imaging and chemoproteomics. In a broader context, the present work establishes the relevance of superoxide redox biology, which remains underexplored, to essential cellular processes such as cell proliferation, cell metabolism and autophagy.
Experimental
Chemical synthesis and characterization
Synthetic scheme, experimental details for chemical synthesis and characterization data of the compounds can be found in the ESI.†Cell culture
HepG2 cells were cultured in DMEM supplemented with 10% FBS and 1% PS (complete DMEM medium) and maintained at 37 °C with 5% CO2. The cells were subcultured when 80% confluence was reached.LC-MS experiments to investigate the reactivity and selectivity of QMP-SOs
A stock solution of QMP-SOs in DMSO was diluted with a PBS/MeOH solution mixture (1:1, v/v) to a final concentration of 100 μM. The stock solution of ROS was freshly prepared and added to the compound solution at a final concentration of 500 μM. The solution mixture was incubated at 37 °C for 60 min. After incubation, the reaction mixture (50 μL) was diluted with PBS/MeOH (1:1, v/v; 450 μL) and 10 μL of the diluted solution was subjected to LC-MS analysis with a Waters Autopurification System using a SunFire C18 HPLC column (50 × 4.6 mm with 5 μm diameter particles, Waters). Separation was achieved by gradient elution from 5% to 100% MeCN in water (constant 0.1 vol% formic acid) over 4 min, isocratic elution with 100% MeCN (with 0.1 vol% formic acid) from 4 to 8 min, and returning to 5% MeCN in water (with 0.1 vol% formic acid) and equilibrated for 2 min. The selected ion chromatogram, with m/z corresponding to the molecular ion of QMP-SO, was extracted. The data were analysed using MassLynx™ software by determining the area under the curve. The peak area was recorded to calculate the percentage change of QMP-SO after ROS incubation. The ROS stock solution was prepared as follows: O2˙− was generated from KO2. The concentration was determined by UV absorption at 256 nm (molar extinction coefficient = 2686 M−1 cm−1) using UV-vis absorption spectrometry. OCl− was generated from NaOCl (∼4% w/w) purchased from Macklin. ˙OH was generated through the Fenton reaction using (NH4)2Fe(SO4)2·6H2O solution and H2O2. (CH3)3COO˙ was generated from the Fenton reaction between (NH4)2Fe(SO4)2·6H2O and (CH3)3COOH. NO˙ was generated from NONOate solution diluted in 10 mM NaOH. The concentration was determined by UV absorption at 252 nm (molar extinction coefficient = 8400 M−1 cm−1) using UV-vis absorption spectroscopy. Peroxynitrite was purchased from Calbiochem. The concentration was determined by UV-vis absorption at 302 nm (molar extinction coefficient = 1670 M−1 cm−1) using UV-vis absorption spectroscopy.
Reaction of QMP-SOs with O2˙− generated from a xanthine/xanthine oxidase (XOD) system
For O2˙− generated from xanthine and xanthine oxidase, XOD was premixed with the QMP-SO-C5-alkyne at final concentrations of 0.08 U mL−1 and 5 μM respectively, before the addition of xanthine at the indicated concentration. For samples co-incubated with SOD, SOD was premixed with XOD at a final concentration of 75 mU mL−1 before the addition of xanthine. After 30 min of incubation, an aliquot of the reaction mixture (10 μL) was sent for LC-MS analysis as described previously. Selected ion chromatograms were extracted and analyzed by integrating the area under curve.In-gel fluorescence assay of protein labeling on BSA using QMP-SO-1
50 μL of BSA (2 mg mL−1) in PBS was incubated with QMP-SO-C5-alkyne (10 μM), KO2 (500 μM) or xanthine oxidase (0.08 U mL−1)/xanthine (5 μM) in the presence or absence of SOD (150 mU mL−1) for 1 h. After 1 h of incubation at room temperature, proteins were precipitated with pre-chilled acetone (300 μL) at −20 °C for 4 h. The samples were centrifuged at 5000g at 4 °C for 10 min, and the supernatant was discarded. The protein pellets were washed with pre-chilled 0.01 M HCl/90% acetone and then methanol, and re-suspended in 200 μL of PBS. The protein samples were further diluted in PBS (sample:PBS = 1:49, v/v). A master mix of CuAAC was prepared from azide-fluor 545 (1 mM), copper(II) sulfate (50 mM), THPTA (50 mM) and freshly prepared sodium ascorbate (250 mM) and added to 50 μL of the lysates with final concentrations of azide-fluor 545, copper(II) sulfate, THPTA and sodium ascorbate in the solution mixture of 5 μM, 1 mM, 3 mM and 5 mM, respectively. The solution was incubated in the dark at room temperature for 1 h, quenched with a 4× reducing Laemmli SDS sample loading buffer and boiled at 90 °C for 5 min. The samples were then separated by molecular weight on precast FuturePAGE 4–20% gels and imaged using ChemiDoc MP (Bio-Rad Laboratories, Inc) for measuring in-gel fluorescence. The protein loading was determined using a Pierce™ Silver Stain Kit (Thermo Fisher Scientific, #24612).
In-gel fluorescence assay of protein labeling in HepG2 cell lysates using QMP-SO-C5-alkyne
HepG2 cell lysates were prepared by probe sonication in PBS. 50 μL of cell lysates (2 mg mL−1) in PBS were incubated with QMP-SO-C5-alkyne (10 μM) and indicated concentrations of KO2 (0, 250 or 500 μM) in the presence or absence of GSH (10 mM) or SOD (75 mU mL−1) for 1 h. After 1 h of incubation at room temperature, the proteins were precipitated with pre-chilled acetone (300 μL) at −20 °C for 4 h. The samples were centrifuged at 5000g at 4 °C for 10 min, and the supernatant was discarded. The protein pellets were washed twice with pre-chilled 0.01 M HCl/90% acetone and then methanol, and re-suspended in 50 μL of PBS. The azide-fluor 545 was conjugated to QMP-SO-labeled proteins through the CuAAC reaction described previously. The solution was incubated in the dark at room temperature for 1 h, quenched with a 4× reducing Laemmli SDS sample loading buffer and boiled at 90 °C for 5 min. Samples were then separated by molecular weight using precast FuturePAGE 4–20% gels and imaged using ChemiDoc MP for measuring in-gel fluorescence. SimplyBlue™ SafeStain was applied to measure the protein loading.In-gel fluorescence assay of protein labeling in live HepG2 cells using the QMP-SO-C5-alkyne
HepG2 cells were cultured on 90 mm dishes under a humidified atmosphere of 5% CO2 at 37 °C in complete DMEM. At 70% confluency, the cells were incubated in complete DMEM containing QMP-SO-C5-alkyne (30 μM) and indicated concentrations of menadione in the presence or absence of NAC (5 mM) or MnTBAP (200 μM) for 4 h. For MnTBAP-treated conditions, the cells were pre-incubated with MnTBAP for 12 h before the 4 h-treatment with QMP-SO-alkyne, menadione and MnTBAP. The cells were washed twice with PBS and lysed by sonication in PBS on ice. The protein concentrations were measured using BCA. 50 μL of the lysates were labeled with azide-fluor 545 using the CuAAC reaction according to previously described procedures. The solution was incubated in the dark at room temperature for 1 h, quenched with a 4× reducing Laemmli SDS sample loading buffer and boiled at 90 °C for 5 min. The proteins were separated by molecular weight using precast FuturePAGE 4–20% gels and scanned using ChemiDoc MP for measuring in-gel fluorescence. SimplyBlue™ SafeStain was applied to measure the protein loading.MTT cell viability assay of the QMP-SO-C5-alkyne
HepG2 cells were cultured on 96-well plates under a humidified atmosphere of 5% CO2 at 37 °C in complete DMEM medium. At 70% confluency, the cells were then incubated with the DMSO solvent control or QMP-SO-C5-alkyne at indicated concentrations for 4 h. After 4 h, 10 μL of MTT solution in PBS (5 mg mL−1) was added to the cells for a final concentration of 0.25 mg mL−1. The cells were incubated in the dark at 37 °C with 5% CO2 for 4 h, and then lysed with 100 μL of SDS solution in PBS (0.5 g mL−1 with 0.01 M HCl). The plates were kept in the dark overnight, and cell viability was assayed by measuring the absorption at 580 nm on a PerkinElmer Victor 3 (Molecular Devices).Confocal fluorescence imaging of HepG2 cells labeled with the QMP-SO-C5-alkyne
HepG2 cells (3 × 104 cells) were cultured on the 8-well Nunc Lab-Tek chambered slide system under a humidified atmosphere of 5% CO2 in air at 37 °C for 48 h in complete DMEM. At 70% confluency, the cells were incubated in complete DMEM containing the QMP-SO-C5-alkyne (30 μM) and indicated concentrations of menadione and in the presence or absence of MnTBAP (200 μM) or PEG-SOD (300 U mL−1) for 4 h. For the experiments with MnTBAP and PEG-SOD, the cells were pre-incubated with MnTBAP or PEG-SOD for 12 h before the 4 h-treatment with the QMP-SO-C5-alkyne, menadione and MnTBAP/PEG-SOD. After treatment, the cells were washed with PBS and fixed with pre-chilled methanol at −20 °C for 10 min. After fixation, the cells were washed with PBS and permeabilised with 0.3 vol% Triton X-100 at room temperature for 30 min. Next, the cells were washed with PBS and incubated with CuAAC master-mix solution with CuSO4, THPTA, azide-fluor 545 and sodium ascorbate at final concentrations of 100, 500, 20 and 5000 μM, respectively. The cells were incubated in the dark for 1 h, then washed with PBS and stained with Hoechst 33342 in PBS (8.2 μM) at room temperature for 15 min. The cells were washed thrice and then imaged in PBS using a Zeiss LSM880 with an Airyscan 2 confocal microscope.Shotgun MS to investigate covalent modification of BSA with QMP-SO-1
1 μL of QMP-SO-1 (50 mM) and 20 μL of KO2 (5 mM) were added to 80 μL of BSA (10 mg mL−1) in PBS and incubated at 37 °C for 1 h. KO2 was replaced with an equal volume of DMSO in the control sample. After incubation, the proteins were precipitated with pre-chilled acetone (600 μL) at −20 °C overnight. The samples were centrifuged at a maximum speed of 4 °C for 10 min, and the supernatant was discarded. The protein pellets were washed with pre-chilled 0.01 M HCl/90% acetone and then resuspended in 100 μL of 8M urea in PBS. The protein concentration was measured with BCA and normalised to 1.67 mg mL−1. 15 μL of the diluted samples was aliquoted out, allowing 25 μg of the proteins for further MS preparation. 20 μL of 1× ProteaseMax was added to the samples and vortexed vigorously for 15 s. Then, 58.5 μL of ammonium bicarbonate (0.1 M) was added. The 10 μL of TCEP (110 mM) were then added to the samples and incubated at 60 °C for 30 min. Next, 10 μL of iodoacetamide (IA, 150 mM) was added and the solution mixture was incubated at 37 °C for 30 min. After that, the samples were mixed with 1.2 μL of 5× ProteaseMax and vortexed. Sequencing-grade trypsin (20 μg; Promega) was reconstituted in 40 μL of trypsin buffer and 1.5 μL of the trypsin solution was added to each sample. Samples were incubated at 37 °C overnight. Samples were then acidified with a final concentration of 5% formic acid and centrifuged at 13200g for 30 min. The supernatant was collected, desalted using C18 StageTips, and sent for LC-MS/MS analysis on a commercial C18 column (75 μm i.d. × 50 cm length × 2 μm particle size) coupled to a NanoTrap column (75 μm i.d. × 2 cm length × 3 μm particle size) with an Orbitrap Fusion Tribid Lumos mass spectrometer (ThermoFisher).
Chromatographic separation was carried out using a linear gradient of increasing buffer B (80% MeCN and 0.1% formic acid) and declining buffer (0.1% formic acid) at 300 nL min−1. Buffer B was increased to 27.5% B in 88 min and ramped to 44% B in the next 16 min, followed by a quick ramp to 95%. An isocratic gradient of 95% buffer B was added in 5 min with a decrease of buffer B to 3% and then the column was re-equilibrated. MS data were collected in the m/z range of 350–1500. A data-dependent top speed method with a time interval of 3 s between every survey scan was operated during which higher-energy collisional dissociation (HCD) was used. Spectra were obtained at an MS2 resolution of 30000 with a custom normalized AGC target of 200% and the maximum ion injection time (IT) of 60 ms, an isolation width of 1.6 m/z, and a normalized collisional energy of 30%. Preceding precursor ions targeted for HCD were dynamically excluded for 20 s.
The data were searched against the UniProt BSA database (UP00000913) using MaxQuant v2.0.3.0,53 specified with trypsin digestion (allowed up to 3 missed cleavages) and cysteine carbamidomethylation (+57.02146) as a static modification. The search also allowed up to 5 variable modifications for methionine oxidation (+15.99491), N-terminal acetylation (+42.01056), quinone methide modification (Cysteine, +49.072; Serine, threonine, aspartic acid, glutamic acid, histidine, tyrosine, arginine and lysine, +106.0419). The peptide false discovery rate (FDR) was set to 1%.
QMP-SO-TMT chemoproteomics experiment
HepG2 cells were cultured on 150 mm dishes under a humidified atmosphere of 5% CO2 at 37 °C for 48 h in complete DMEM medium. At a cell confluency of 70%, the cells were incubated in complete DMEM medium containing QMP-SO-C5-alkyne (30 μM) and menadione (100 μM) for 4 h. Menadione was replaced with DMSO in the control sample. The cells were washed twice with PBS and lysed by sonication in PBS on ice. The protein concentrations were measured by BCA. A master-mix solution for the CuAAC reaction was prepared with CuSO4, THPTA, DTB-PEG-azide and sodium ascorbate at final concentrations of 1 mM, 3 mM, 100 μM and 5 mM, respectively. 360 μL of the CuAAC master mix solution was added to 2.5 mL of cell lysates (2 mg mL−1) for DTB-PEG-azide conjugation through the CuAAC reaction. After 1 h of incubation at room temperature, proteins were precipitated with pre-chilled acetone (18 mL) at −20 °C overnight. The samples were centrifuged at 5000g at 4 °C for 10 min, and the supernatant was discarded. The protein pellet was washed twice with methanol and resuspended in 1.2% SDS in PBS (w/v) with heating at 80 °C for 5 min. The samples were centrifuged, and the supernatant was transferred to a PBS solution containing Pierce™ Streptavidin Agarose beads (20349; Thermo Scientific) with a final concentration of SDS equal to 0.2% (w/v). The samples and beads were incubated at 4 °C with rotation overnight. The beads were then washed with PBS and water and re-dispersed in 6M urea in PBS. The samples were reduced by TCEP (1 mM) at 65 °C for 20 min, followed by alkylation with iodoacetamide (18 mM) at 37 °C for 30 min in the dark. The beads were centrifuged at 1400g for 2 min, washed with PBS and re-suspended in 2M urea in PBS. The proteins on the beads were then digested by sequencing grade trypsin (Promega) at 37 °C overnight. After tryptic digestion, the beads were centrifuged at 1400g for 2 min, and the supernatant was collected. The beads were washed twice with 100 μL of PBS and centrifuged, and then the supernatant was collected. The combined fractions gave a total volume of 400 μL of tryptic digested peptides. The samples were dried under a vacuum concentrator and redissolved in 70 μL of TEAB (50 mM). TMT-6plex reagents were prepared according to the manufacturer's instructions. 30 μL of acetonitrile and 1.5 μL of the TMT reagent in acetonitrile were added to each sample and the solution mixtures were incubated at room temperature for 1 h. Then, 0.6 μL of 5% hydroxylamine was added to the samples and incubated for 15 min. The resulting samples were mixed at equal volumes, desalted using C18 StageTips, and sent for LC-MS/MS analysis on a commercial C18 column (75 μm i.d. × 50 cm length × 2 μm particle size) coupled to a NanoTrap column (75 μm i.d. × 2 cm length × 3 μm particle size) with an Orbitrap Fusion Tribid Lumos mass spectrometer (Thermo Fisher).Chromatographic separation was carried out using buffer A (0.1% formic acid) and buffer B (80% MeCN and 0.1% formic acid) with a flow rate of 300 nL min−1. Buffer B was increased to 27.5% with a linear gradient in 164 min, followed by a further increase to 44% in 52 min. Then, there were a quick ramp of buffer B to 95% in 2 min, an isocratic gradient of 95% buffer B in 7 min and a quick decrease to 3%, where it was held and the column was re-equilibrated. An Orbitrap Fusion was operated in the data-dependent mode for both MS2 and MS3. MS1 scan was acquired using an Orbitrap mass analyzer with a resolution of 120000 at an m/z ratio of 400. The Top speed instrument method was used for MS2 and MS3. For MS2, the isolation width was set at 0.5 Da and isolated precursors were fragmented by CID at a normalized collision energy (NCE) of 35% and analyzed in the ion trap using “turbo” scan. Following the acquisition of each MS2 spectrum, a synchronous precursor selection (SPS) MS3 scan was collected for the top 10 most intense ions in the MS2 spectrum. SPS-MS3 precursors were fragmented by higher energy collision-induced dissociation (HCD) at an NCE of 60% and analyzed using an Orbitrap at a resolution of 50000.
The data were searched against the UniProt human database (UP000005640) using the TMT10-MS3 workflow in MSFragger 3.8,54 specified with trypsin digestion (allowed up to 2 missed cleavages) and cysteine carbamidomethylation (+57.02146) as a static modification. The search also allowed up to 5 variable modifications for methionine oxidation (+15.9949) and N-terminal acetylation (+42.0106). TMT-6 reporter ion annotations were assigned with quantification at MS3. Proteins with zero reporter ion intensities for 2 out of the 3 replicate runs in either the control group or treated group were filtered out. The reporter ion ratios for other peptides were calculated, and peptides with invalid values were filtered out. The Gene Ontology Cellular Compartment (GOCC) and PANTHER GO-Slim Biology Process of the identified proteins were determined using Perseus v2.0.9.0.55 and Panther 18.0.56,57
Data availability
The mass spectrometry proteomics data have been deposited at the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD052283 and PXD052284. The data supporting this article have been included as part of the ESI.†Author contributions
H. L.: conceptualization, data curation, formal analysis, investigation, methodology, validation, visualization, and writing – review & editing; C. Y.-S. C.: conceptualization, formal analysis, funding acquisition, methodology, supervision, visualization, writing – original draft, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. Y.-S. C. acknowledges support from the Early Career Scheme (27315922) from University Grants Committee, and the Seed Fund for Basic Research (2202100627) and the Enhanced New Staff Start-up Research Grant from the University Research Committee and Li Ka Shing Faculty of Medicine, the University of Hong Kong. This work was also supported by the Centre for Oncology and Immunology under the Health@InnoHK Initiative funded by the Innovation and Technology Commission, The Government of Hong Kong SAR, China. H. L. acknowledges the receipt of a Postgraduate Studentship administered by The University of Hong Kong. We acknowledge the support from the Imaging and Flow Cytometry Core and Proteomics and Metabolomics Core, from the Centre for PanorOmic Sciences (CPOS), Li Ka Shing Faculty of Medicine, The University of Hong Kong, on confocal fluorescence microscopy imaging, flow cytometry and MS experiments. We also thank Ms Bonnie Yan and Mr Ivan Lai, at the Department of Chemistry and Department of Microbiology, the University of Hong Kong, for their help in NMR experiments.References
- B. C. Dickinson and C. J. Chang, Nat. Chem. Biol., 2011, 7, 504–511 CrossRef CAS.
- K. M. Holmström and T. Finkel, Nat. Rev. Mol. Cell Biol., 2014, 15, 411–421 CrossRef.
- C. C. Winterbourn, Nat. Chem. Biol., 2008, 4, 278–286 CrossRef CAS.
- C. Nathan and A. Cunningham-Bussel, Nat. Rev. Immunol., 2013, 13, 349–361 CrossRef CAS PubMed.
- T. Nishi, N. Shimizu, M. Hiramoto, I. Sato, Y. Yamaguchi, M. Hasegawa, S. Aizawa, H. Tanaka, K. Kataoka, H. Watanabe and H. Handa, J. Biol. Chem., 2002, 277, 44548–44556 CrossRef CAS PubMed.
- M. J. Morgan and Z. Liu, Cell Res., 2011, 21, 103–115 CrossRef CAS PubMed.
- J. Buzek, Nucleic Acids Res., 2002, 30, 2340–2348 CrossRef CAS PubMed.
- T. Suzuki, A. Muramatsu, R. Saito, T. Iso, T. Shibata, K. Kuwata, S. Kawaguchi, T. Iwawaki, S. Adachi, H. Suda, M. Morita, K. Uchida, L. Baird and M. Yamamoto, Cell Rep., 2019, 28, 746–758 CrossRef CAS PubMed.
- J. Kwon, S.-R. Lee, K.-S. Yang, Y. Ahn, Y. J. Kim, E. R. Stadtman and S. G. Rhee, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16419–16424 CrossRef CAS PubMed.
- L. J. Alcock, M. V. Perkins and J. M. Chalker, Chem. Soc. Rev., 2018, 47, 231–268 RSC.
- C. E. Paulsen and K. S. Carroll, Chem. Rev., 2013, 113, 4633–4679 CrossRef CAS PubMed.
- R. A. Cairns, I. S. Harris and T. W. Mak, Nat. Rev. Cancer, 2011, 11, 85–95 CrossRef CAS.
- C. Gorrini, I. S. Harris and T. W. Mak, Nat. Rev. Drug Discovery, 2013, 12, 931–947 CrossRef CAS.
- C. Soto, Nat. Rev. Neurosci., 2003, 4, 49–60 CrossRef CAS PubMed.
- K. M. Robinson, M. S. Janes and J. S. Beckman, Nat. Protoc., 2008, 3, 941–947 CrossRef CAS PubMed.
- J. Zielonka, J. Vasquez-Vivar and B. Kalyanaraman, Nat. Protoc., 2008, 3, 8–21 CrossRef CAS PubMed.
- J. J. Hu, N.-K. Wong, S. Ye, X. Chen, M.-Y. Lu, A. Q. Zhao, Y. Guo, A. C.-H. Ma, A. Y.-H. Leung, J. Shen and D. Yang, J. Am. Chem. Soc., 2015, 137, 6837–6843 CrossRef CAS PubMed.
- J. Zhang, C. Li, R. Zhang, F. Zhang, W. Liu, X. Liu, S. M.-Y. Lee and H. Zhang, Chem. Commun., 2016, 52, 2679–2682 RSC.
- L. Wu, A. C. Sedgwick, X. Sun, S. D. Bull, X.-P. He and T. D. James, Acc. Chem. Res., 2019, 52, 2582–2597 CrossRef CAS PubMed.
- Z. Zhang, J. Fan, Y. Zhao, Y. Kang, J. Du and X. Peng, ACS Sens., 2018, 3, 735–741 CrossRef CAS PubMed.
- J. J. Gao, K. H. Xu, B. Tang, L. L. Yin, G. W. Yang and L. G. An, FEBS J., 2007, 274, 1725–1733 CrossRef CAS PubMed.
- Z. H. Yu, C. Y.-S. Chung, F. K. Tang, T. F. Brewer and H. Y. Au-Yeung, Chem. Commun., 2017, 53, 10042–10045 RSC.
- J. Chan, S. C. Dodani and C. J. Chang, Nat. Chem., 2012, 4, 973–984 CrossRef CAS PubMed.
- H. Xiao, W. Zhang, P. Li, W. Zhang, X. Wang and B. Tang, Angew. Chem., Int. Ed., 2020, 59, 4216–4230 CrossRef CAS PubMed.
- C. E. Paulsen, T. H. Truong, F. J. Garcia, A. Homann, V. Gupta, S. E. Leonard and K. S. Carroll, Nat. Chem. Biol., 2011, 8, 57–64 CrossRef PubMed.
- Y. H. Seo and K. S. Carroll, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 16163–16168 CrossRef CAS PubMed.
- V. Gupta, J. Yang, D. C. Liebler and K. S. Carroll, J. Am. Chem. Soc., 2017, 139, 5588–5595 CrossRef CAS PubMed.
- Y. Shi and K. S. Carroll, Acc. Chem. Res., 2020, 53, 20–31 CrossRef CAS PubMed.
- J. Meng, L. Fu, K. Liu, T. Caiping, W. Ziyun, J. Youngeun, R. B. Ferreira, K. S. Carroll, B. T. Keith and J. Yang, Nat. Commun., 2021, 12, 1415 CrossRef CAS.
- J. Zivanovic, E. Kouroussis, J. B. Kohl, B. Adhikari, B. Bursac, S. Schott-Roux, D. Petrovic, J. L. Miljkovic, D. Thomas-Lopez, Y. Jung, M. Miler, S. Mitchell, V. Milosevic, J. E. Gomes, M. Benhar, B. Gonzalez-Zorn, I. Ivanovic-Burmazovic, R. Torregrossa, J. R. Mitchell, M. Whiteman, G. Schwarz, S. H. Snyder, B. D. Paul, K. S. Carroll and M. R. Filipovic, Cell Metab., 2020, 31, 207 CrossRef CAS.
- T.-Y. Koo, H. Lai, D. K. Nomura and C. Y.-S. Chung, Nat. Commun., 2023, 14, 3564 CrossRef CAS PubMed.
- L. Fu, K. Liu, J. He, C. Tian, X. Yu and J. Yang, Antioxid. Redox Signaling, 2020, 33, 1061–1076 CrossRef CAS PubMed.
- D. I. Brown and K. K. Griendling, Free Radical Biol. Med., 2009, 47, 1239–1253 CrossRef CAS PubMed.
- Y. Wang, R. Branicky, A. Noë and S. Hekimi, J. Cell Biol., 2018, 217, 1915–1928 CrossRef CAS PubMed.
- K. Chen, M. T. Kirber, H. Xiao, Y. Yang and J. F. Keaney, J. Cell Biol., 2008, 181, 1129–1139 CrossRef CAS PubMed.
- H. Zhu, T. Tamura, A. Fujisawa, Y. Nishikawa, R. Cheng, M. Takato and I. Hamachi, J. Am. Chem. Soc., 2020, 142, 15711–15721 CrossRef CAS PubMed.
- H. Iwashita, E. Castillo, M. S. Messina, R. A. Swanson and C. J. Chang, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2018513118 CrossRef CAS.
- S. Uchinomiya, T. Nagaura, M. Weber, Y. Matsuo, N. Zenmyo, Y. Yoshida, A. Tsuruta, S. Koyanagi, S. Ohdo, N. Matsunaga and A. Ojida, J. Am. Chem. Soc., 2023, 145, 8248–8260 CrossRef CAS PubMed.
- W. Wang and C. Alexander, Angew. Chem., Int. Ed., 2008, 47, 7804–7806 CrossRef CAS PubMed.
- A. Alouane, R. Labruère, T. Le Saux, F. Schmidt and L. Jullien, Angew. Chem., Int. Ed., 2015, 54, 7492–7509 CrossRef CAS PubMed.
- A. E. Speers, G. C. Adam and B. F. Cravatt, J. Am. Chem. Soc., 2003, 125, 4686–4687 CrossRef CAS PubMed.
- C. Szabó, H. Ischiropoulos and R. Radi, Nat. Rev. Drug Discovery, 2007, 6, 662–680 CrossRef PubMed.
- D. N. Criddle, S. Gillies, H. K. Baumgartner-Wilson, M. Jaffar, E. C. Chinje, S. Passmore, M. Chvanov, S. Barrow, O. V. Gerasimenko, A. V. Tepikin, R. Sutton and O. H. Petersen, J. Biol. Chem., 2006, 281, 40485–40492 CrossRef CAS PubMed.
- Y. L. Chua, E. Dufour, E. P. Dassa, P. Rustin, H. T. Jacobs, C. T. Taylor and T. Hagen, J. Biol. Chem., 2010, 285, 31277–31284 CrossRef CAS PubMed.
- J. Cao, M. Ying, N. Xie, G. Lin, R. Dong, J. Zhang, H. Yan, X. Yang, Q. He and B. Yang, Antioxid. Redox Signaling, 2014, 21, 1443–1459 CrossRef CAS PubMed.
- J. Drechsel, F. A. Mandl and S. A. Sieber, ACS Chem. Biol., 2018, 13, 2016–2019 CrossRef CAS PubMed.
- J. Blackinton, M. Lakshminarasimhan, K. J. Thomas, R. Ahmad, E. Greggio, A. S. Raza, M. R. Cookson and M. A. Wilson, J. Biol. Chem., 2009, 284, 6476–6485 CrossRef CAS PubMed.
- F. Billia, L. Hauck, D. Grothe, F. Konecny, V. Rao, R. H. Kim and T. W. Mak, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6085–6090 CrossRef CAS PubMed.
- O. Chalifoux, B. Faerman and R. J. Mailloux, J. Biol. Chem., 2023, 299, 105399 CrossRef CAS PubMed.
- D. W. Bak and E. Weerapana, Methods Mol. Biol., 2019, 1967, 211–227 CrossRef CAS PubMed.
- A. P. Håkansson and A. W. Smith, J. Biol. Chem., 2007, 282, 29521–29530 CrossRef PubMed.
- X. Yang, J. Song and L.-J. Yan, Antioxidants, 2019, 8, 32 CrossRef PubMed.
- S. Tyanova, T. Temu and J. Cox, Nat. Protoc., 2016, 11, 2301–2319 CrossRef CAS PubMed.
- A. T. Kong, F. V. Leprevost, D. M. Avtonomov, D. Mellacheruvu and A. I. Nesvizhskii, Nat. Methods, 2017, 14, 513–520 CrossRef CAS PubMed.
- S. Tyanova, T. Temu, P. Sinitcyn, A. Carlson, M. Y. Hein, T. Geiger, M. Mann and J. Cox, Nat. Methods, 2016, 13, 731–740 CrossRef CAS PubMed.
- H. Mi, A. Muruganujan, X. Huang, D. Ebert, C. Mills, X. Guo and P. D. Thomas, Nat. Protoc., 2019, 14, 703–721 CrossRef CAS PubMed.
- P. D. Thomas, D. Ebert, A. Muruganujan, T. Mushayahama, L.-P. Albou and H. Mi, Protein Sci., 2022, 31, 8–22 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cb00111g |
| This journal is © The Royal Society of Chemistry 2024 |