
Visible-light-enabled cascade cross-dehydrogenative-coupling/cyclization to construct α-chromone substituted α-amino acid derivatives†
Zhi-Qiang
Zhu
 *,
Jia-Yu
Hu
,
Zong-Bo
Xie
and
Zhang-Gao
Le
*
*,
Jia-Yu
Hu
,
Zong-Bo
Xie
and
Zhang-Gao
Le
*
Jiangxi Province Key Laboratory of Synthetic Chemistry, School of Chemistry and Materials Science, East China University of Technology, Nanchang, 330013, China. E-mail: zhuzq@ecut.edu.cn; zhgle@ecut.edu.cn; Fax: (+86) 0791-83896550
First published on 24th November 2023
Abstract
Organophotocatalytic cascade cross-dehydrogenative-coupling/cyclization reaction of o-hydroxyarylenaminones with α-amino acid derivatives for the construction of α-chromone substituted α-amino acid derivatives was developed. Various N-arylglycine esters, amides and dipeptides underwent the cascade cyclization reaction well with o-hydroxyarylenaminones to afford the corresponding 3-aminoalkyl chromones in good to excellent yields. This approach consists of visible-light-promoted oxidation of α-amino acid derivatives, the Mannich reaction, and intramolecular nucleophilic cyclization under acidic conditions, and features a wide reaction scope, a simple operation and mild reaction conditions, which may have the potential to be used for the synthesis of bioactive molecules.
Chromones are common motifs abundant in many natural products, pharmaceuticals and materials.1 Particularly, C3-substituted chromones have recently received considerable attention due to the diverse physiological and biological activities, including anti-inflammatory, anticancer, antioxidant, antimicrobial, anti-HIV, etc.2 Moreover, 3-substituted chromones have been employed as useful building blocks in many research areas such as organic synthesis, biochemistry, and medicinal chemistry.3 Consequently, tremendous efforts have been devoted to constructing various C3-functionalized chromones.4 Conventional methods for 3-substituted chromones are mainly focused on the transition-metal-catalyzed cross-coupling reactions of halochromones as well as direct C–H functionalization of chromones.4b,5 In recent years, cascade vinyl C–H functionalization and chromone annulation of o-hydroxyphenylenaminones has emerged as a powerful and reliable strategy to synthesize various C3-substituted chromones.6 Although significant achievements have been attained, cascade cross-dehydrogenative-coupling of o-hydroxyarylenaminones with various coupling partners and subsequent chromone annulation have scarcely been reported in the literature. In 2020, Wan's group reported transient halogenation-mediated chromone annulation for the synthesis of 3-vinyl chromones via the domino C–H alkenylation and cyclization of o-hydroxyarylenaminones.5b
Cross-dehydrogenative-coupling reactions are a valuable synthetic route to forging new chemical bonds because this type of reaction avoids functional group interconversion and has the advantages of being atom economic and environmentally friendly.7 α-Amino acids are often seen as green reactants, which are more readily available and are widely used to construct complex organic molecules.8 Intensive interest and research have been aroused on direct cross-dehydrogenative-coupling of α-amino acid derivatives with a variety of nucleophiles to afford α-substituted α-amino acid derivatives in the past few years.9 Considering that the introduction of the chromone skeleton into α-amino acid derivatives is significant in the structural modification of drugs and the application of organic synthesis, and as our continuing research interest in photocatalytic C–H transformation,10 we herein report visible-light-enabled cascade cross-dehydrogenative-coupling/cyclization of o-hydroxyarylenaminones with glycine derivatives for the synthesis of diverse α-chromone substituted α-amino acid derivatives.
At the outset, o-hydroxyarylenaminone 1a and N-4-methylphenylglycine ester 2a were selected as model substrates to test the feasibility of the designed process. The results are summarized in Table 1; after a systematic survey of various parameters, such as photosensitizers, solvents, and additives, the optimal conditions were determined as follows: 1a (0.2 mmol, 1.0 equiv.), 2a (0.4 mmol, 2.0 equiv.), Na2-eosin Y (5 mol%), and citric acid·H2O (2.0 equiv.) in EtOH (2 mL, 0.1 M) under the irradiation of 18 W blue LEDs (456 nm) in air at room temperature for 24 h (entry 1, Table 1). There was no improvement in the reaction yields when photosensitizers other than Na2-eosin Y were used (entries 2–4, Table 1). Various solvents such as THF and DMSO were evaluated, and EtOH was found to be the best for the yield of 3aa (entries 5 and 6, Table 1). The structure of 3aa was unambiguously confirmed by X-ray (CCDC: 2237743†) as well as NMR analysis. Further screening of the reaction conditions revealed that the other additives and oxidants led to lower yields of 3aa (entries 7–9, Table 1). The experimental results indicated that visible-light, citric acid·H2O and air were critical to the desired coupling cyclization (entries 10 and 11, Table 1). It was noteworthy that the desired cyclization product 3aa was formed in 38% yield only in the presence of citric acid·H2O under the irradiation of visible-light (entry 12, Table 1). The ultraviolet-visible (UV-Vis) absorption spectrum of individual reaction partners and that of combined reaction components were recorded, which indicated the formation of an EDA complex (see Fig. S5 in the ESI†). A 63% yield of product 3aa was isolated under the illumination of green LEDs.
|
|
||
|---|---|---|
| Entry | Variations from standard conditions | Yielda,b (%) |
| a Reaction conditions: 1a (0.2 mmol, 1.0 equiv.), 2a (0.4 mmol, 2.0 equiv.), photosensitizer (5 mol%), solvent (2 mL, 0.1 M), additive (2.0 equiv.) at room temperature under the irradiation of 18 W light (456 nm) for 24 h in air. b Isolated yields. | ||
| 1 | None | 75 |
| 2 | Ru(bpy)3Cl2·6H2O instead of Na2-eosin Y | 26 |
| 3 | 4CzPIN instead of Na2-eosin Y | 9 |
| 4 | Rose bengal instead of Na2-eosin Y | 21 |
| 5 | THF instead of EtOH | 31 |
| 6 | DMSO instead of EtOH | 24 |
| 7 | Na2HPO4 instead of citric acid·H2O | 20 |
| 8 | Acetic acid instead of citric acid·H2O | 57 |
| 9 | Under N2 | 0 |
| 10 | In the dark | trace |
| 11 | No citric acid·H2O | 15 |
| 12 | No Na2-eosin Y | 38 |
| 13 | Green LEDs instead of blue LEDs | 63 |
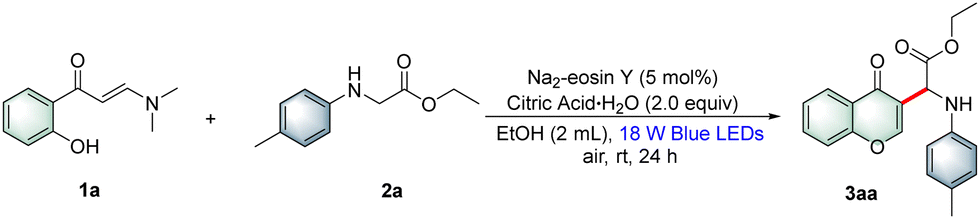
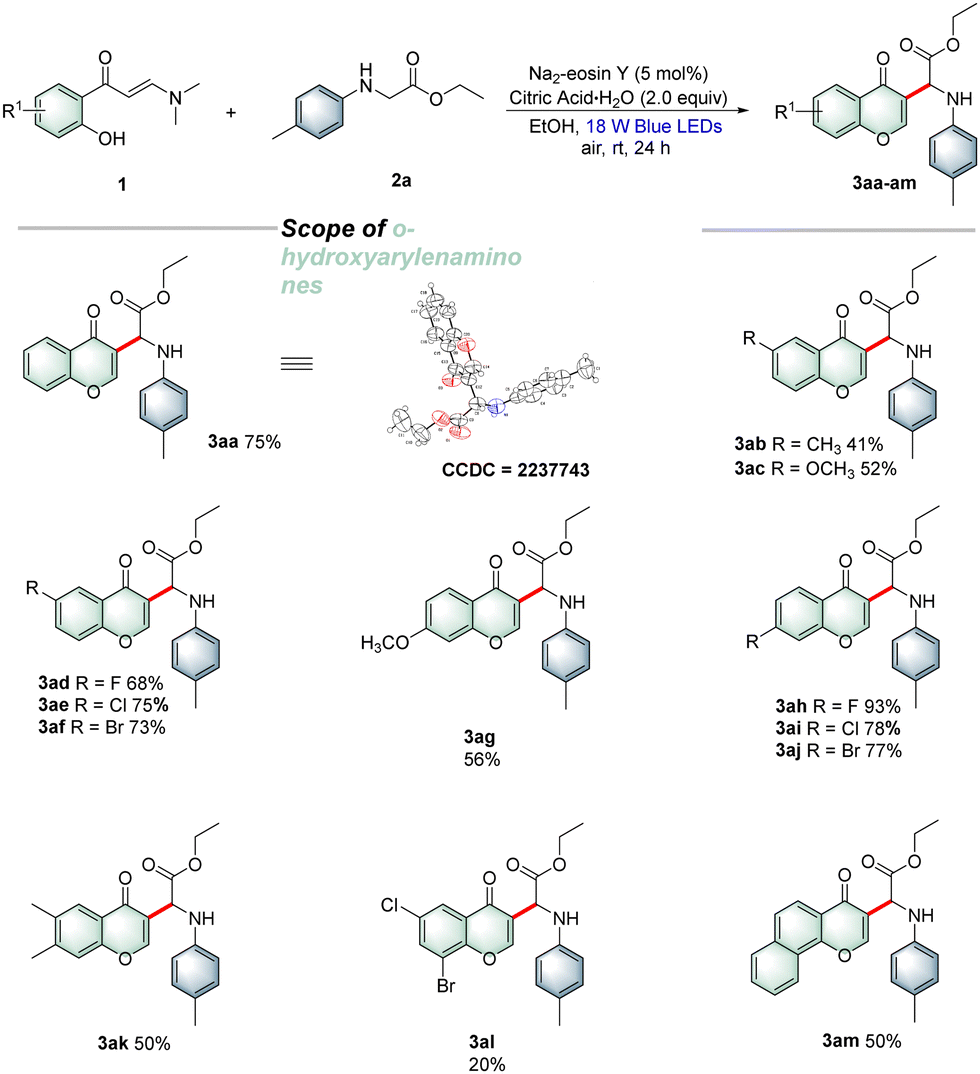
With the optimal reaction conditions established, a wide array of o-hydroxyarylenaminones 1 were prepared and subjected to reaction with N-4-methylphenylglycine ester 2a under the standard reaction conditions to explore the substrate scope. As shown in Table 2, various o-hydroxyphenyl enaminones 1b–f bearing different substituents R1 (–Me, –OMe, –F, –Cl, and –Br) at the C5-position were engaged in this protocol efficiently to afford the corresponding products 3ab–af in 41–75% yields. Similarly, o-hydroxyarylenaminones 1 possessing different substituents R1 (–OMe, –F, –Cl, and –Br) at the C4-position were examined, and the desired products 3ag–aj were isolated in good to high yields. The experimental results indicated that chromones bearing electron-withdrawing groups on the benzene rings were more favorable for the transformation than those bearing electron-donating groups. Furthermore, the disubstituted o-hydroxyarylenaminones 1k and 1l were also tolerated and gave the target products 3ak and 3al in 50% and 20% yields, respectively. As expected, fused aryl substrate 1m underwent the desired coupling cyclization smoothly to afford the desired product 3am with a satisfactory yield. Encouraged by the results presented above, we next investigated the reactions of o-hydroxyphenylenaminone 1a with various α-amino acid derivatives 2 under the current reaction conditions. As outlined in Table 3, a broad range of α-amino acid derivatives 2 underwent cyclization reactions readily with o-hydroxyphenylenaminone 1a, providing the corresponding products 3ba–ja in 47–70% yields. N-Arylglycine esters 2c–e bearing electron-rich (–Me, –OMe and 3,4-Me) substituents on the benzene ring reacted more efficiently than those bearing electron-deficient substituents 2f–j. Remarkably, several N-arylglycine esters, such as methyl, isopropyl, tert-butyl, and benzyl esters 2k–n, were also compatible with the reaction, producing the corresponding products 3ka–na in 40–67% yields. Interestingly, glycine amide 2o and dipeptide 2p were also suitable for this transformation, delivering the target products 3oa and 3pa in 43% and 48% yields, respectively. In addition, we also successfully realized the conversion of tertiary amine with 30% isolated yield by changing the reaction conditions appropriately.
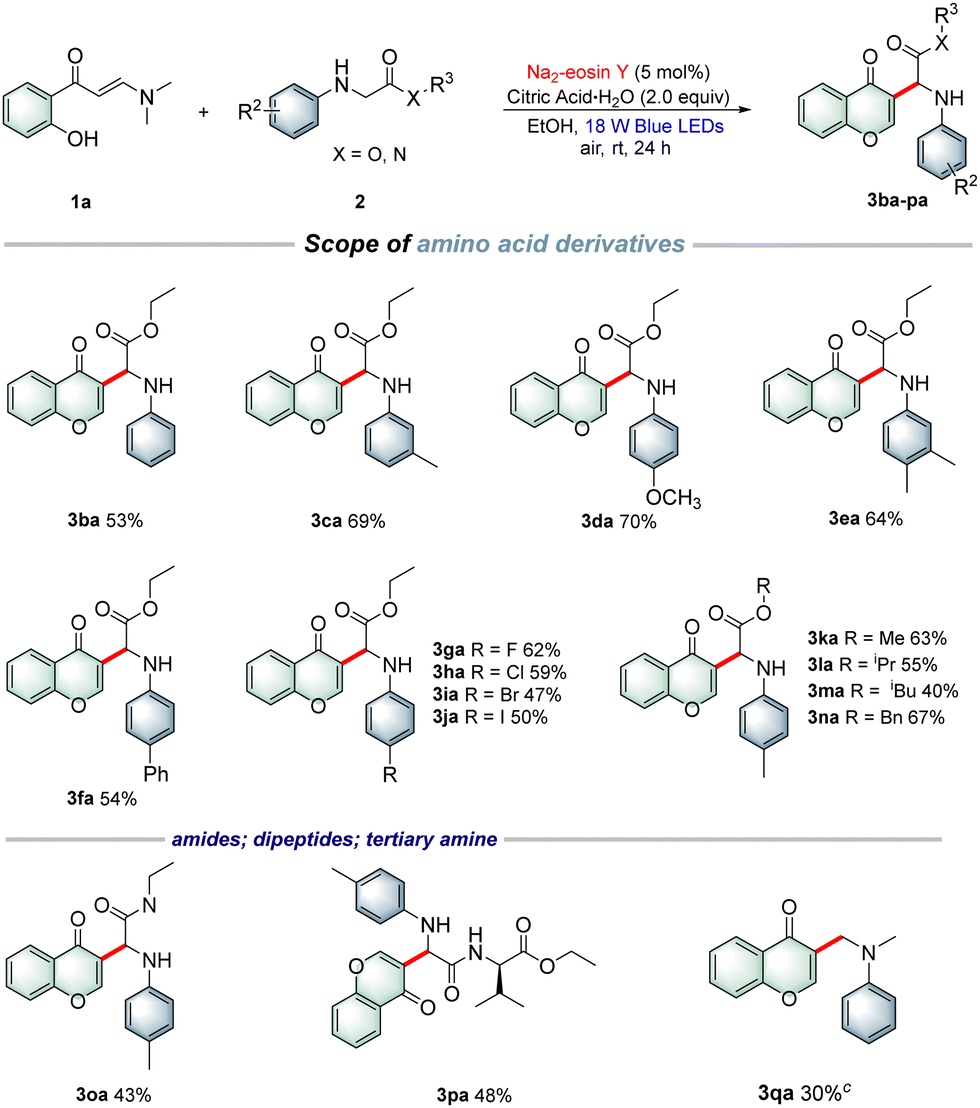
To demonstrate the applicability of this protocol, a gram-scale synthesis was conducted under the standard conditions, giving the desired product 3aa in 63% yield (Scheme 1a). Furthermore, a set of control experiments were carried out to explore this photocatalytic reaction mechanism. Radical-trapping experiments indicated that the corresponding radical adducts and imine intermediate were detected by HRMS analysis. Thus, a free-radical pathway was probably involved in the process (Scheme 1b). Next, imine intermediate 6a was prepared and then reacted with o-hydroxyphenylenaminone 1a to obtain 3aa in 52% yield (Scheme 1c). Furthermore, the light on–off experiment results revealed that the photocatalytic cross-dehydrogenative-coupling/cyclization reaction required continuous visible-light irradiation (Scheme 1d). Moreover, fluorescence quenching experiments with the Na2-eosin Y photocatalyst as well as cyclic voltammetry (CV) experiments to study the redox potential of substrates were also performed (see the ESI†), and the results suggested that N-arylglycine ethyl ester 2a was preferentially oxidized by Na2-eosin Y in the excited state, and thus the reaction might proceed through a reductive quenching pathway.
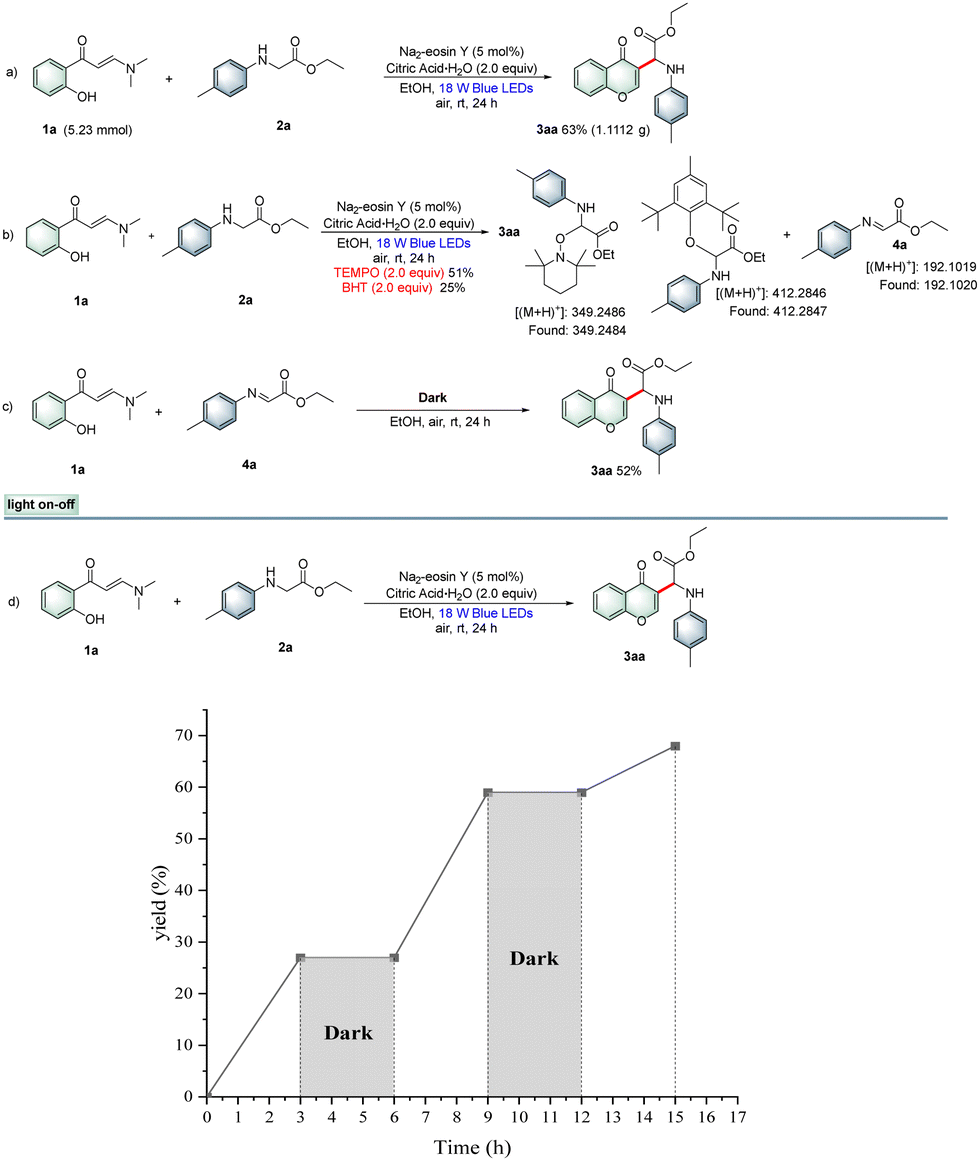 | ||
| Scheme 1 Control experiments. | ||
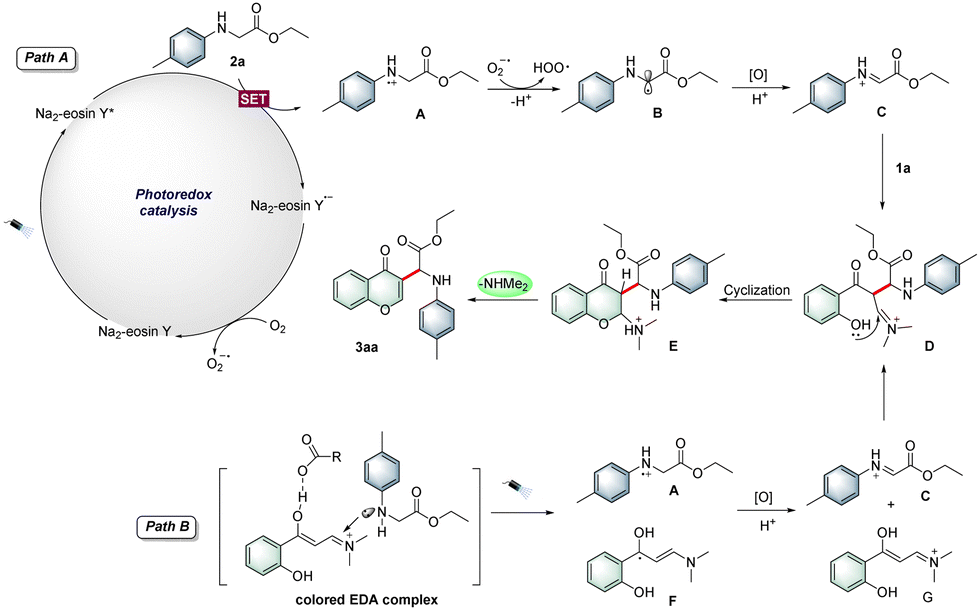
On the basis of the abovementioned experimental results and literature survey,10a,b a plausible mechanism for the photocatalytic reaction is proposed in Scheme 2. Initially, upon irradiation, Na2-eosin Y was excited to produce the excited-state Na2-eosin Y*, which underwent single electron transfer (SET) with N-4-methylphenylglycine ester 2a to produce the radical cation A as well as Na2-eosin Y˙−. At the same time, Na2-eosin Y˙− was oxidized by molecular oxygen to generate the ground-state Na2-eosin Y and superoxide radical anion (O2−˙). Then, the radical intermediate A resulted in the formation of aminoalkyl radical B by the superoxide radical (O2−˙). In the presence of citric acid·H2O, the intermediate B was then oxidized and protonated to afford the imine intermediate C, which interacted with o-hydroxyarylenaminone 1a to form the iminium intermediate D (path A). On the other hand, a ternary colored EDA complex was formed in the presence of 1a, 2a and citric acid·H2O.11 Upon irradiation, SET occurred between 1a and 2a under acidic conditions, followed by the formation of radical cation A and radical F, and they were further oxidized to the corresponding iminium/imine species, which then transformed into the key intermediate D (path B). Finally, intermediate D underwent intramolecular nucleophilic cyclization to deliver the desired product 3aa.
 | ||
| Scheme 2 Plausible mechanism. | ||
In conclusion, we have developed a novel visible-light-enabled cascade cross-dehydrogenative-coupling/cyclization of o-hydroxyarylenaminones with α-amino acid derivatives for the synthesis of structurally diverse α-chromone substituted α-amino acid derivatives. A plausible reaction mechanism has also been proposed on the basis of control experiments. This synthetic protocol features broad substrate scopes and good functional group tolerance under mild conditions, which provides a new and practical approach for the construction of various α-chromone substituted α-amino acid derivatives. Further investigations of reaction development and synthetic applications of this methodology are in progress in our laboratory.
Financial support from the National Natural Science Foundation of China (21966003), the Natural Science Foundation of Jiangxi Province (20232BAB203001), and the Open Fund of Jiangxi Province Key Laboratory of Synthetic Chemistry (JXSC202206) is gratefully acknowledged.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) A. Gaspar, M. J. Matos, J. Garrido, E. Uriarte and F. Borges, Chem. Rev., 2014, 114, 4960 CrossRef CAS; (b) B. M. Trost, E. Gnanamani, C. A. Kalnmals, C.-I. J. Hung and J. S. Tracy, J. Am. Chem. Soc., 2019, 141, 1489 CrossRef CAS; (c) J. Cui, N. Kumagai, T. Watanabe and M. Shibasaki, Chem. Sci., 2020, 11, 7170 RSC; (d) A. Nazhand, A. Durazzo, M. Lucarini, R. Romano, M. A. Mobilia, A. A. Izzo and A. Santini, Nat. Prod. Res., 2020, 34, 137 CrossRef CAS PubMed; (e) D. R. Joshi and I. Kim, J. Org. Chem., 2021, 86, 10235 CrossRef CAS; (f) V. M. Patil, N. Masand, S. Verma and V. Masand, Chem. Biol. Drug Des., 2021, 98, 943 CrossRef CAS.
- (a) A. Danda, N. Kesava-Reddy, C. Golz, C. Strohmann and K. Kumar, Org. Lett., 2016, 18, 2632 CrossRef CAS; (b) J. Lei, Y. Li, L.-J. He, Y.-F. Luo, D.-Y. Tang, W. Yan, H.-K. Lin, H.-Y. Li, Z.-Z. Chen and Z.-G. Xu, Org. Chem. Front., 2020, 7, 987 RSC; (c) C. Zhao, B. H. Shah, H. Li, X. Wu and Y. J. Zhang, Asian J. Org. Chem., 2021, 10, 545 CrossRef CAS; (d) Y. Dai, W. Meng, X. Feng and H. Du, Chem. Commun., 2022, 58, 1558 RSC.
-
(a) L. Albrecht, A. Cruz
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Acosta, A. Fraile, A. Albrecht, J. Christensen and K. A. Jørgensen, Angew. Chem., Int. Ed., 2012, 51, 9088 CrossRef CAS;
(b) D. Jiang, J. Chen, N. Zan, C. Li, D. Hu and B. Song, J. Agric. Food Chem., 2021, 69, 12126 CrossRef CAS;
(c) L. Meng, X. Chang, Z. Lin and J. Wang, Org. Biomol. Chem., 2021, 19, 2663 RSC;
(d) D.-G. Guo, H.-J. Wang, Y. Zhou and X.-L. Liu, Org. Biomol. Chem., 2022, 20, 4681 RSC.
Acosta, A. Fraile, A. Albrecht, J. Christensen and K. A. Jørgensen, Angew. Chem., Int. Ed., 2012, 51, 9088 CrossRef CAS;
(b) D. Jiang, J. Chen, N. Zan, C. Li, D. Hu and B. Song, J. Agric. Food Chem., 2021, 69, 12126 CrossRef CAS;
(c) L. Meng, X. Chang, Z. Lin and J. Wang, Org. Biomol. Chem., 2021, 19, 2663 RSC;
(d) D.-G. Guo, H.-J. Wang, Y. Zhou and X.-L. Liu, Org. Biomol. Chem., 2022, 20, 4681 RSC. - (a) D. Kang, K. Ahn and S. Hong, Asian J. Org. Chem., 2018, 7, 1136 CrossRef CAS; (b) S. Tian, T. Luo, Y. Zhu and J.-P. Wan, Chin. Chem. Lett., 2020, 31, 3073 CrossRef CAS; (c) J. Duan, Z. Xiong, Y. Zhou, W. Yao, X. Li, M. Zhang and Z. Wang, Org. Lett., 2021, 23, 8007 CrossRef CAS.
- (a) L. Klier, T. Bresser, T. A. Nigst, K. Karaghiosoff and P. Knochel, J. Am. Chem. Soc., 2012, 134, 13584 CrossRef CAS PubMed; (b) L. Fu, Z. Xu, J.-P. Wan and Y. Liu, Org. Lett., 2020, 22, 9518 CrossRef CAS; (c) H. Choi, M. Min, Q. Peng, D. Kang, R. S. Paton and S. Hong, Chem. Sci., 2016, 7, 3900 RSC; (d) Y. Guo, Y. Xiang, L. Wei and J.-P. Wan, Org. Lett., 2018, 20, 3971 CrossRef CAS PubMed.
- (a) S. Mkrtchyan and V. O. Iaroshenko, Chem. Commun., 2020, 56, 2606 RSC; (b) S. Mkrtchyan and V. O. Iaroshenko, J. Org. Chem., 2021, 86, 4896 CrossRef CAS PubMed; (c) H.-Y. Liu, J.-R. Zhang, G.-B. Huang, Y.-H. Zhou, Y.-Y. Chen and Y.-L. Xu, Adv. Synth. Catal., 2021, 363, 1656 CrossRef CAS; (d) Z.-W. Wang, Y. Zheng, Y.-E. Qian, J.-P. Guan, W.-D. Lu, C.-P. Yuan, J.-A. Xiao, K. Chen, H.-Y. Xiang and H. Yang, J. Org. Chem., 2022, 87, 1477 CrossRef CAS PubMed; (e) L. Lu, X. Zhao, W. Dessie, X. Xia, X. Duan, J. He, R. Wang, Y. Liu and C. Wu, Org. Biomol. Chem., 2022, 20, 1754 RSC.
- (a) T. Tian, Z.-P. Li and C.-J. Li, Green Chem., 2021, 23, 6789 RSC; (b) H. Wang, X. Gao, Z. Lv, T. Abdelilah and A. Lei, Chem. Rev., 2019, 119, 6769 CrossRef CAS.
- (a) J. B. Hedges and K. S. Ryan, Chem. Rev., 2020, 120, 3161 CrossRef CAS; (b) L. Xiao, Z. Zhu, Z. Xie and Z. Le, Chin. J. Org. Chem., 2019, 39, 2345 CrossRef.
- (a) Z.-H. Wang, P.-S. Gao, X. Wang, J.-Q. Gao, X.-T. Xu, Z. He, C. Ma and T.-S. Mei, J. Am. Chem. Soc., 2021, 143, 15599 CrossRef CAS; (b) Z.-H. Yuan, H. Xin, J.-Q. Tao, Y.-J. Ma, X.-H. Duan and L.-N. Guo, Org. Chem. Front., 2022, 9, 3546 RSC; (c) Y. Gao, J. Liu, C. Wei, Y. Li, K. Zhang, L. Song and L. Cai, Nat. Commun., 2022, 13, 7450 CrossRef CAS; (d) L. Poletti, D. Ragno, O. Bortolini, F. Presini, F. Pesciaioli, S. Carli, S. Caramori, A. Molinari, A. Massi and G. Di Carmine, J. Org. Chem., 2022, 87, 7826 CrossRef CAS PubMed and references therein.
- (a) Z.-Q. Zhu, D. Guo, J.-J. Ji, X. Zhu, J. Tang, Z.-B. Xie and Z.-G. Le, J. Org. Chem., 2020, 85, 15062 CrossRef CAS; (b) Z.-Q. Zhu, S. Liu, Z.-Y. Hu, Z.-B. Xie, J. Tang and Z.-G. Le, Adv. Synth. Catal., 2021, 363, 2568 CrossRef CAS; (c) Z.-Q. Zhu, J.-Y. Hu, Z.-B. Xie, J. Tang and Z.-G. Le, Adv. Synth. Catal., 2022, 364, 2169 CrossRef CAS.
- C. Wang, R. Qi, H. Xue, Y. Shen, M. Chang, Y. Chen, R. Wang and Z. Xu, Angew. Chem., Int. Ed., 2020, 59, 7461 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2237743. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc04107g |
| This journal is © The Royal Society of Chemistry 2024 |