
Total synthesis of highly oxygenated phomopsol B via acid-induced etherification to construct a bridged structure†
Yuichiro
Kawamoto
 *,
Ayaka
Nishitani
,
Yukari
Yoshimura
,
Toyoharu
Kobayashi
and
Hisanaka
Ito
*
*,
Ayaka
Nishitani
,
Yukari
Yoshimura
,
Toyoharu
Kobayashi
and
Hisanaka
Ito
*
School of Life Sciences, Tokyo University of Pharmacy and Life Sciences, Hachioji, Tokyo 192-0392, Japan. E-mail: ykawa@toyaku.ac.jp; itohisa@toyaku.ac.jp
First published on 18th November 2023
Abstract
The first total synthesis of phomopsol B has been achieved in a racemic form. The synthesis comprises a deacetylative cyclization to construct a bicyclic skeleton followed by primary alcohol-assisted dihydroxylation, ether cyclization to construct a dioxabicyclo [3.2.1] skeleton and γ-lactone formation based on oxidation by TEMPO.
Wu and coworkers reported the isolation of phomopsol A (1) and phomopsol B (2) from the Thai mangrove endophytic fungus Phomopsis sp. xy211 (Fig. 1). The molecular structures of these compounds were elucidated by NMR spectroscopic analysis, and 1 was found to be a polyketide-derived alkaloid-N-oxide whereas 2 was a highly oxidized polyketide. The absolute configuration of the latter was established by single-crystal XRD analysis with Cu Kα radiation. Compound 1 has a 3,4-dihydro-2H-indeno[1,2-b]pyridine 1-oxide skeleton while compound 2 possesses a unique 6/6/5-fused tricyclic 3,5-dihydro-2H-2,5-methanobenzo[e][1,4]-dioxepine framework. Compounds 1 and 2 were tested for their neuroprotective effects against corticosterone induced injury. The former exhibited neuroprotective activity in a dose-dependent manner based on the results of an MTT assay using PC12 cells. The unprecedented structure and bioactivity of these compounds are of interest and so it would be beneficial to devise efficient approaches to the synthesis of both. Herein we report the first total synthesis of 2.
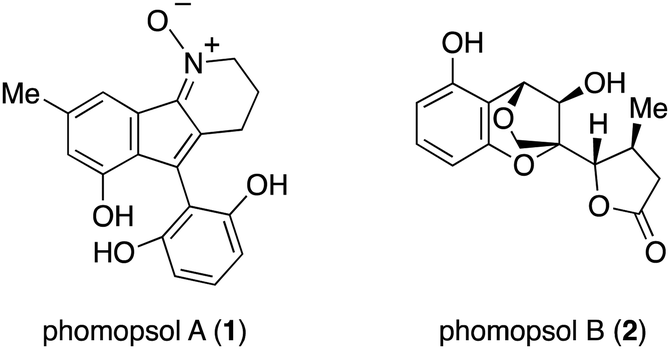 | ||
| Fig. 1 Chemical structure of phomopsols A and B. | ||
Our retrosynthetic analysis of the total synthesis of 2 is outlined in Scheme 1. It is envisioned that the formation of the γ-lactone moiety would be carried out at the late stage. The dioxabicyclo [3.2.1] skeleton of compound 3 would be constructed from compound 4 by means of acid-promoted ether cyclization. Compound 4 could in turn be prepared from compound 5, which may be accessed via Sonogashira coupling involving bromoalkene 6 and alkyne 7.
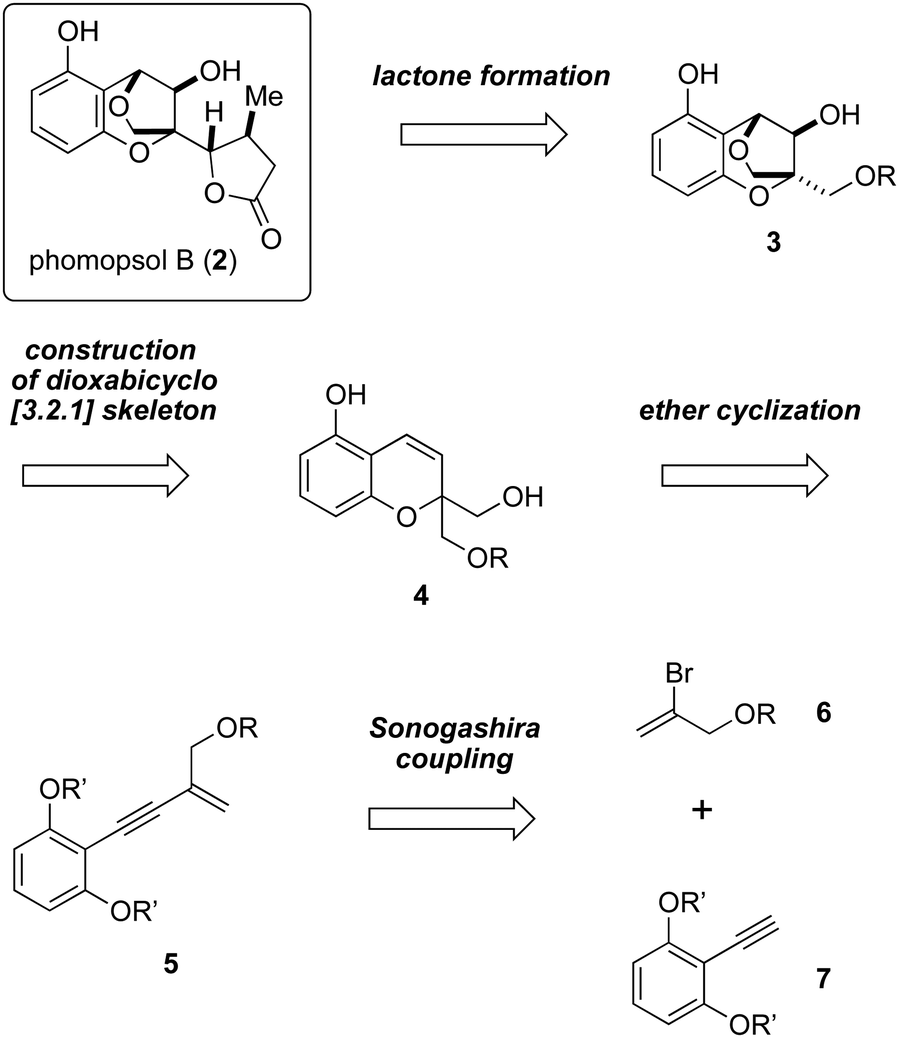 | ||
| Scheme 1 Retrosynthetic analysis of 2. | ||
The development of the present synthesis commenced with the preparation of bicyclic compound 14, as shown in Scheme 2. This compound was derived from alkyne 82 in five steps through Sonogashira coupling3 with bromoalkene 10,4 epoxidation of the alkene, partial reduction of the alkyne and deacetylative ether cyclization. Thus, compound 8 was converted into compound 9 by acetylation of phenolic hydroxy groups. The carbon chain was extended via Sonogashira coupling with bromoalkene 10 to give enyne 11. Epoxidation of enyne 11 proceeded readily to afford compound 12.5 A partial reduction of the alkyne moiety in compound 12 provided the Z-isomer of compound 13 by employing 2,4,6-triisopropylbenzenesulfonyl hydrazide6 in the presence of NaHCO3. Upon treatment of compound 13 with K2CO3 in MeOH, deacetylation followed by intramolecular SN2 attack on the epoxide successfully took place to furnish compound 14.
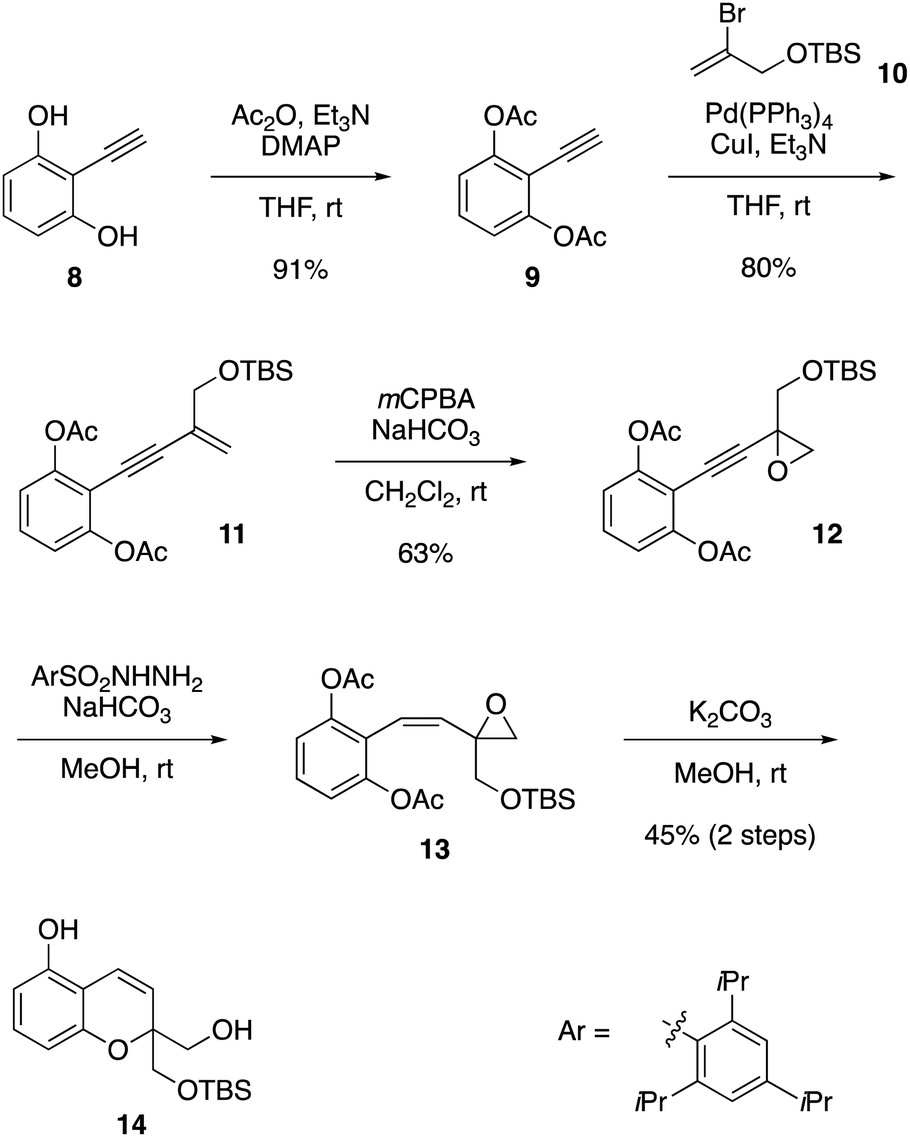 | ||
| Scheme 2 Construction of bicyclic skeleton 14. | ||
With the bicyclic compound 14 in hand, the next challenge was to construct a unique dioxabicyclo [3.2.1] skeleton fused to an aromatic ring (Scheme 3). Compound 14 was subjected to dihydroxylation using a catalytic amount of OsO4 with NMO to give a mixture of product 15 with desired stereochemistry and its diastereomer 15′ in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio.7 In this process, the desired isomer 15 was produced through oxidation of the olefin moiety on the same face as the primary alcohol. Exposure of the resulting 15 to Amberlyst® 15 induced etherification to construct the bridged structure. On this basis, a reaction mechanism can be proposed as follows. Treatment with Amberlyst® 15 generates benzylic cation 17, for which the non-aromatic resonance form 18 is also possible. The primary alcohol can subsequently trap the benzylic cation in 17 in an intramolecular fashion to give compound 16.8 In this manner, the dioxabicyclo [3.2.1] framework fused to a benzene ring and having the desired stereochemistry was synthesized from bicyclic compound 14 in only two steps.
:1 ratio.7 In this process, the desired isomer 15 was produced through oxidation of the olefin moiety on the same face as the primary alcohol. Exposure of the resulting 15 to Amberlyst® 15 induced etherification to construct the bridged structure. On this basis, a reaction mechanism can be proposed as follows. Treatment with Amberlyst® 15 generates benzylic cation 17, for which the non-aromatic resonance form 18 is also possible. The primary alcohol can subsequently trap the benzylic cation in 17 in an intramolecular fashion to give compound 16.8 In this manner, the dioxabicyclo [3.2.1] framework fused to a benzene ring and having the desired stereochemistry was synthesized from bicyclic compound 14 in only two steps.
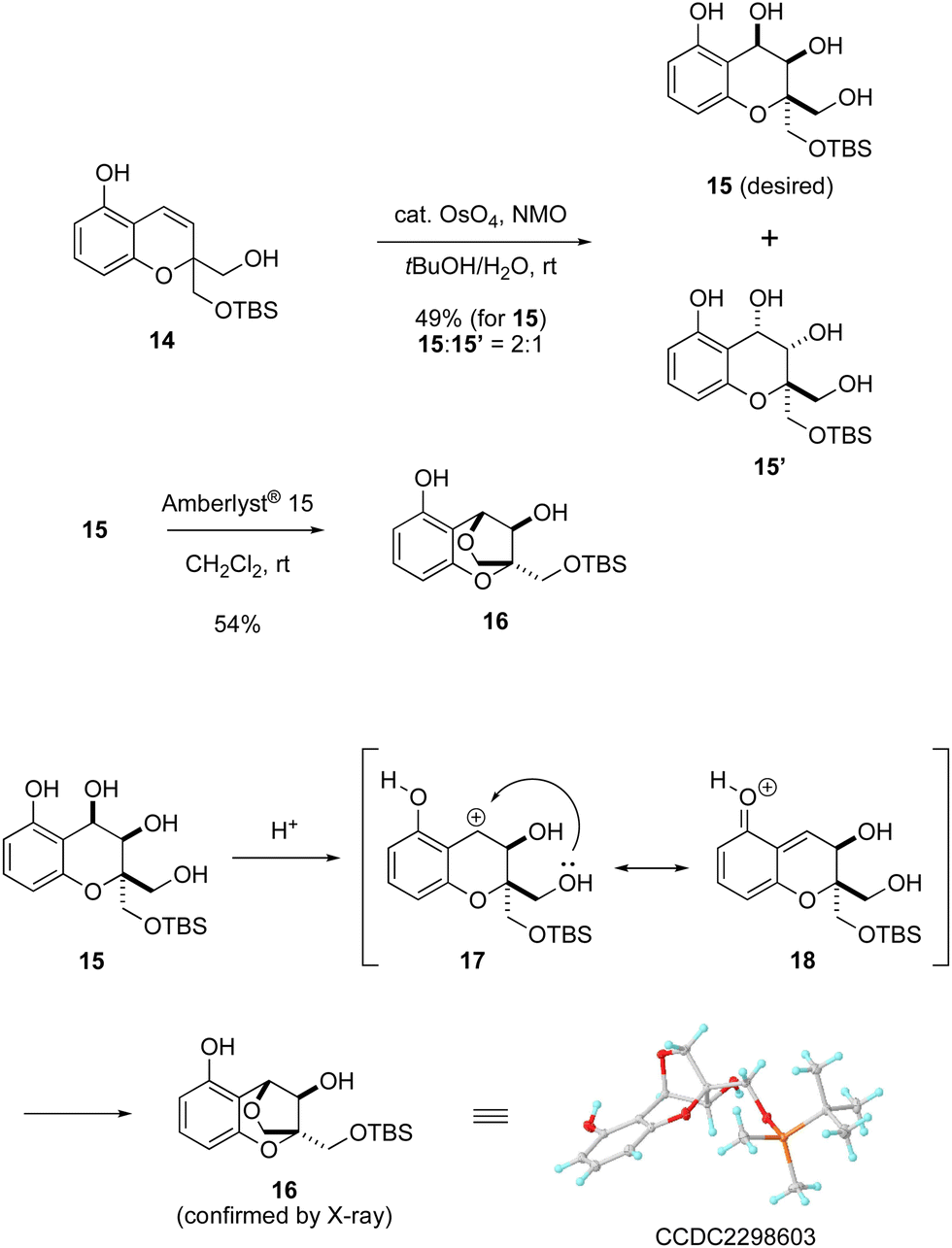 | ||
| Scheme 3 Construction of tricyclic skeleton 16. | ||
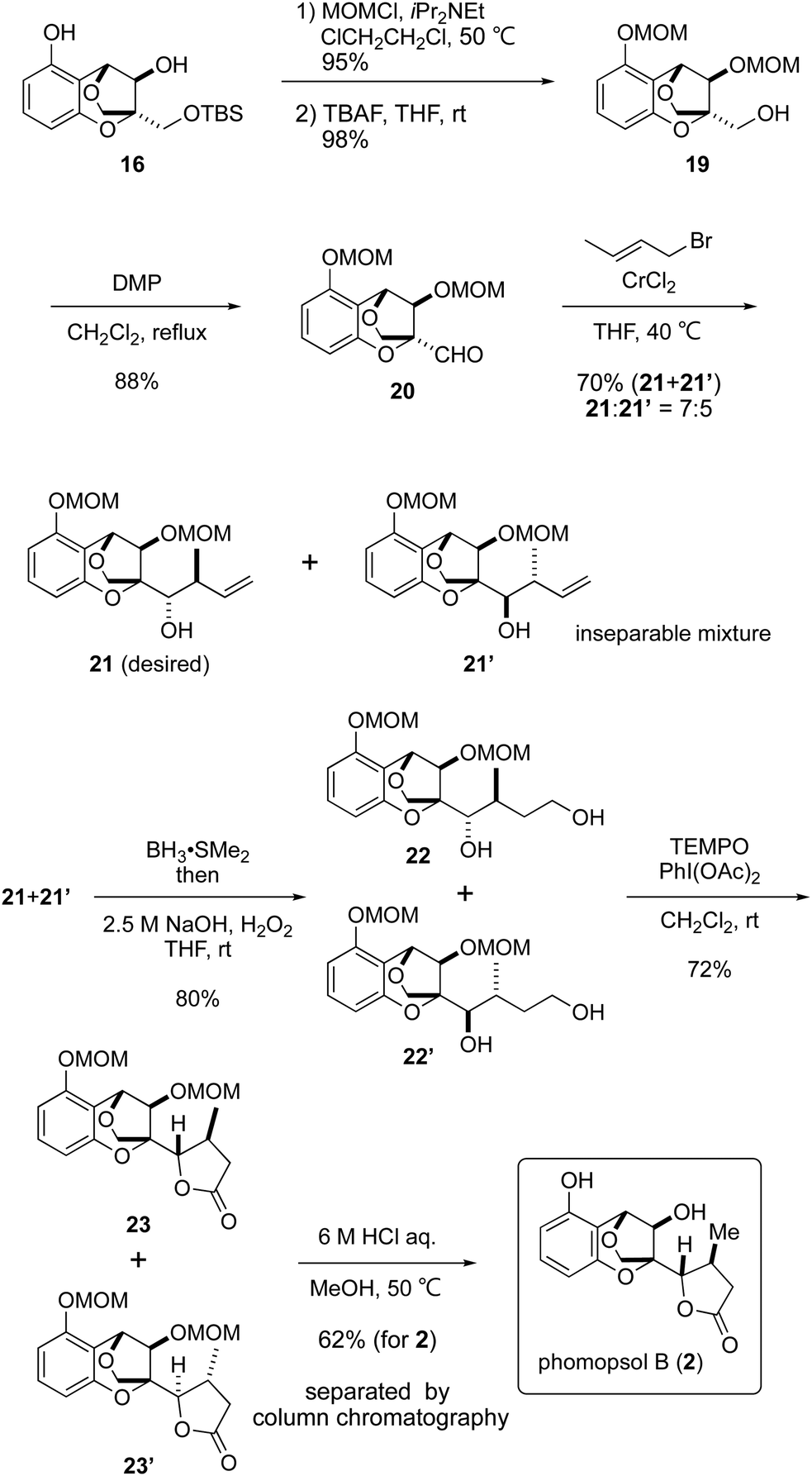
Following the construction of the requisite 16, the formation of the γ-lactone was attempted (Scheme 4). The secondary alcohol and phenolic hydroxy group in 16 were initially protected as MOM ethers and the TBS group was removed with TBAF to liberate the primary alcohol. Dess–Martin oxidation9 of the primary alcohol in compound 19 afforded aldehyde 2010 in an excellent yield, after which attempts were made to achieve crotylation of the latter. After various trials, the combination of crotyl bromide and chromium(II) chloride11 gave an acceptable result, forming the desired product 21 and undesired 21′ in a 7:5 ratio as an inseparable mixture.12
 | ||
| Scheme 4 Synthetic scheme toward natural product 2. | ||
The mixture of compounds 21 and 21′ was transformed into 22 and 22′via hydroboration13 of the terminal olefin. TEMPO oxidation with PhI(OAc)214 as a co-oxidant led to γ-lactones 23 and 23′via the corresponding lactols. Finally, cleavage of the MOM ethers under acidic conditions was successful to isolate phomopsol B (2) after separation from another isomer, accomplishing a total synthesis of 2. Both the 1H and 13C NMR spectra of synthetic phomopsol B were identical with those previously reported in the literature.1
In conclusion, the racemic total synthesis of phomopsol B (2) was completed from alkyne 8 as the starting material. The most significant step in our synthesis is Amberlyst-induced etherification to construct the bridged structure fused to the aromatic ring. Our synthetic method also incorporates deacetylative ether cyclization, dihydroxylation assisted by a primary alcohol and efficient lactone formation with TEMPO to afford the target molecule.
Conflicts of interest
There are no conflicts to declare.Notes and references
- W.-S. Li, H.-B. Hu, Z.-H. Huang, R.-J. Yan, L.-W. Tian and J. Wu, Org. Lett., 2019, 21, 7919 CrossRef CAS PubMed.
- Compound 8 was synthesized in three steps from resorcinol: see ESI† in detail.
- K. Sonogashira, Y. Tohda and N. Hagiwara, Tetrahedron Lett., 1975, 50, 4467 CrossRef.
- (a) WO2020070651A1 Search PubMed; (b) M. Charpenay, A. Boudhar, C. Hulot, G. Blond and J. Suffert, Tetrahedron, 2013, 69, 7568 CrossRef CAS.
- Asymmetric epoxidation using the corresponding allyl alcohol (removed TBS group) was attempted, but it resulted in unsatisfactory yield and poor enantioselectivity: see ESI† in detail (page S5).
- (a) J. Hanus and J. Vorisek, J. Collect. Czech. Chem. Commun., 1929, 1, 223 CrossRef CAS; (b) E. J. Corey, W. L. Mock and D. J. Pasto, Tetrahedron Lett., 1961, 2, 347 CrossRef; (c) N. J. Cusack, C. B. Reese, A. C. Risius and B. Roozepeikar, Tetrahedron, 1976, 32, 2157 CrossRef CAS.
- Hydroxyl group-directed epoxidation with compound 14 was also conducted, but with low yield. In addition, exposure of the resultant epoxide to some Lewis acids failed to construct the desired bridged structure: see ESI† in detail (page S7).
- The structure of compound 16 was determined by X-ray crystallographic analysis (CCDC 2298603†).
- (a) D. B. Dess and J. C. Martin, J. Org. Chem., 1983, 48, 4155 CrossRef CAS; (b) D. B. Dess and J. C. Martin, J. Am. Chem. Soc., 1991, 113, 7277 CrossRef CAS.
- Aldehyde 20 included inseparable hydrated product.
- G. Fronza, C. Fuganti, P. Grasselli, G. Pedrocchi-Fantoni and C. Zirotti, Chem. Lett., 1984, 335 CrossRef CAS.
- Relative stereochemistry of 21 and 21′ was confirmed by NOESY experiment of the corresponding acetonide after MOM deprotection; see ESI†.
- (a) H. C. Brown and R. B. C. Subba, J. Am. Chem. Soc., 1956, 78, 5694 CrossRef CAS; (b) H. C. Brown and R. B. C. Subba, J. Am. Chem. Soc., 1958, 80, 1552 CrossRef CAS.
- (a) P. L. Anelli, C. Biffi, F. Montanari and S. Quici, J. Org. Chem., 1987, 52, 2559 CrossRef; (b) J. B. Eppi and T. S. Widlanski, J. Org. Chem., 1999, 64, 293 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2298603. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc05130g |
| This journal is © The Royal Society of Chemistry 2024 |