
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
CO reduction by calcium and ytterbium hydride complexes with a bulky monodentate carbazolyl ligand†
Alexander
Hinz
 *,
Lucas
Winkler
and
Xiaofei
Sun
*,
Lucas
Winkler
and
Xiaofei
Sun
Karlsruhe Institute of Technology (KIT) Institute of Inorganic Chemistry (AOC) Engesserstr. 15, Gebäude 30.45, 76131 Karlsruhe, Germany. E-mail: alexander.hinz@kit.edu
First published on 9th September 2024
Abstract
The bulky monodentate carbazolyl ligand 1,8-bis(3,5-ditertbutylphenyl)-3,6-ditertbutylcarbazole (dtbpCbz) was employed in the synthesis of monomeric heteroleptic amido carbazolyl complexes of Ca and Yb. For both central metal atoms, dimeric hydride complexes [(dtbpCbz)Ca(benzene)H]2, [(dtbpCbz)Ca(THF)H]2, [(dtbpCbz)Yb(benzene)H]2 and [(dtbpCbz)Yb(THF)H]2 were obtained, which show remarkably poor solubility in organic solvents. The characteristic hydride 1H NMR resonance of [(dtbpCbz)Ca(benzene)H]2 was observed at 2.07 ppm, and for the first time, characteristic vibrational modes of the Ca2H2 and Yb2H2 moiety are discussed. Despite their poor solubility, the hydride complexes could be reacted with CO to yield the corresponding ethenediolate complexes.
Recent years have seen increased interest in the development of group 2 metal hydrides because of the high abundance and low toxicity of alkaline earth elements, in particular with calcium.1,2 Due to their similarity in ionic radii, Yb(II) frequently shows similar coordination behaviour. However, difficulties arise from the tendency of these compounds to disproportionate, and therefore, stabilisation by specific ligand systems is required. The addition of multidentate neutral ligands and use of sterically demanding monoanionic ligands was demonstrated by Okuda who employed the TACD (1,4,7,10-tetramethyl-1,4,7,10-tetraaminocyclododecane) to obtain the cationic dinuclear calcium hydrides [(TACD)2Ca2H2]2+ (Scheme 1A) and [(TACD)2Ca2H3]+.3 The most frequently used approach to access heavy alkaline earth hydrides is the utilisation of multidentate monoanionic ligands such as β-diketiminates (BDI). After Harder's seminal report on the dimeric complex [{BDI}Ca(THF)H]2 (B),4 many applications for calcium hydrides were discovered, including stoichiometric and catalytic reactions in alkene hydrogenation5 and imine hydrosilylation6 as well as reduction of arenes7 and alkylation of benzene.8 A variety of calcium hydrides with multidentate ligands are known to date.
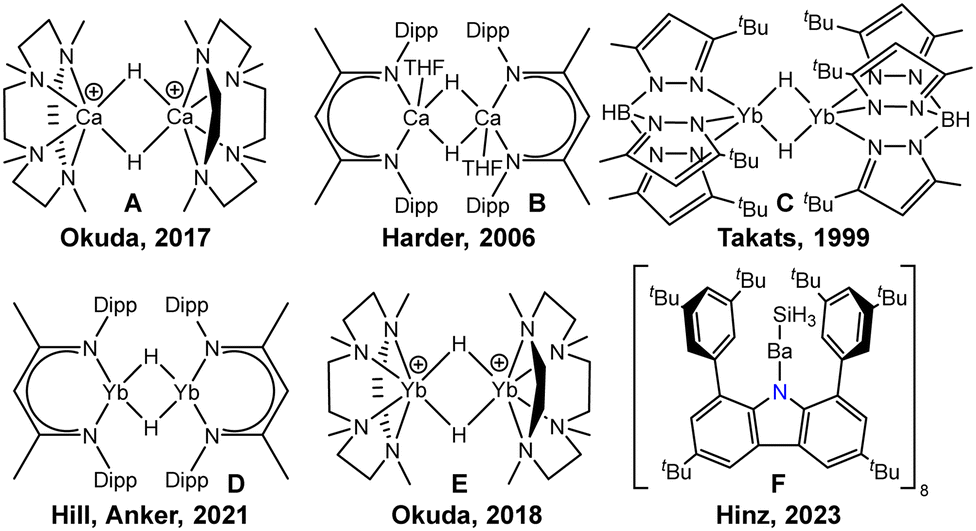 | ||
| Scheme 1 Selected alkaline earth metal hydrides and bis(imino)carbazolate F (Dipp = 2,6-diisopropylphenyl, Ad = adamantyl). | ||
Analogous ytterbium(II) hydrides are generally expected to show similar properties due to nearly identical ionic radii of Yb(II) and Ca(II). The development of ytterbium(II) hydrides was spearheaded by Takats, who reported on the synthesis of a tris(pyrazolyl)borate-stabilised Yb(II) hydride complex C in 1999 with a new synthesis in 2002.9,10 A bulkier tris(pyrazolyl)borate ligand was introduced to the field by Cheng in 2021 who prepared the first monomeric Yb(II) hydride complex.11,12 Harder studied the Yb(II) analogues of the BDI complex B to elucidate similarities and differences of the two species.13 Remarkably, the group of Hill and Anker observed BDI-substituted Yb(II) hydrides without co-ligands (D) in the alkylation of arenes14 and subsequently investigated the reduction chemistry with arenes and cyclopentadienes.15,16 The groups of Xiang and Chen also employed an amine-decorated tetradentate BDI ligand to synthesise Yb(II) hydrides.17 In 2012, Trifonov prepared the first amidinate-stabilised Yb(II) hydride complex and observed double addition of the complex to diphenyl acetylene.18 Subsequently, the group studied the reactivity of the complex towards various other substrates.19 Moreover, Okuda employed neutral tetradentate amine ligands to stabilise cationic Yb(II) hydrides in 2018 (E).20
Our earlier contributions include the synthesis of a bulky carbazolyl ligand to stabilise very low-coordinated environments in main group chemistry,21,22 and enabled efficient luminescence in non-covalently bound complexes.23 Our previous attempts of generating a barium hydride complex with this ligand instead afforded a barium silanide complex (F).24 In this contribution we describe the synthesis of carbazolyl hydrido complexes of calcium and ytterbium and their reactivity towards carbon monoxide.‡
The carbazolyl amido precursor complexes was prepared by treating the carbazole dtbpCbz–H with bis-amido compounds of the calcium or ytterbium. In both cases, the starting material is the THF-containing [M(N{SiMe3}2)2(THF)2] (M = Ca, Yb). The transamination reaction proceeded readily and was complete within hours. After workup of the product, the carbazolyl amido complexes [(dtbpCbz)Ca(N{SiMe3}2)(THF)] (1Ca, Scheme 2) and [(dtbpCbz)Yb(N{SiMe3}2)(THF)] (1Yb) were obtained as greenish-yellow and orange material, respectively. The remaining coordinated molecule of THF was removed by prolonged drying at 120 °C in dynamic vacuum to yield [(dtbpCbz)Ca(N{SiMe3}2)] (2Ca) which shows similar appearance as 1Ca. However, with 1Yb this desolvation reaction did not proceed selectively and 2Yb was not isolated. The NMR spectra for the complexes 1Ca, 2Ca, and 1Yb indicate averaged C2v symmetry on the NMR time scale, which is derived from the magnetic equivalency of both aryl-tertbutyl groups and each of the arene ortho-positions. The complexes were both observed exclusively as monomeric compounds in the solid state, which is in contrast to the analogous barium compound.24
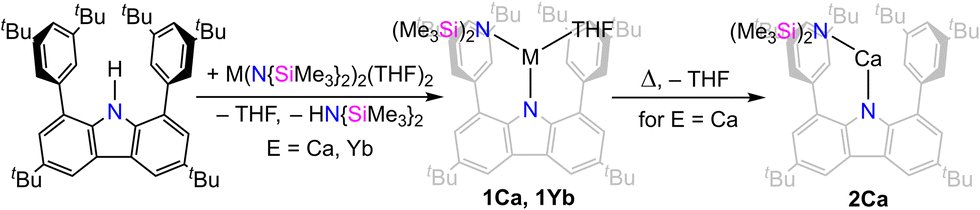 | ||
| Scheme 2 Synthesis of calcium and ytterbium carbazolyl amido complexes. | ||
The molecular structures of the amido complexes 1Ca and 1Yb (Fig. 1) show the metal bent out of the central C4N plane of the carbazolyl ligand by (47.4° to 56.0°). The NMN moieties themselves are bent as well, with the angle varying between 131° and 145°. Table 1 summarises bond distances of 1Ca and 1Yb. All M–N distances in 1Ca are shorter than the equivalent ones in 1Yb, with the exception of the shortest M–Carene contact. Generally, the M–Namide bonds are shorter than the M–Ncarb bonds. Upon removal of the THF ligand of 1Ca, 2Ca was obtained. The structure changes in terms of Ca coordination sphere, as there is now a near-linear N–Ca–C angle involving an agostic interaction with one of the silyl carbon atoms (Fig. 1, N1–Ca1–C49 170.64(6)°) while the N–Ca–N angle is still obtuse (122.80(5)°). At the same time, the Ca–N contacts contract (2.2502(15), 2.2820(12) Å), while the Ca–Carene contacts remain largely unchanged (shortest: 2.904(2) Å).
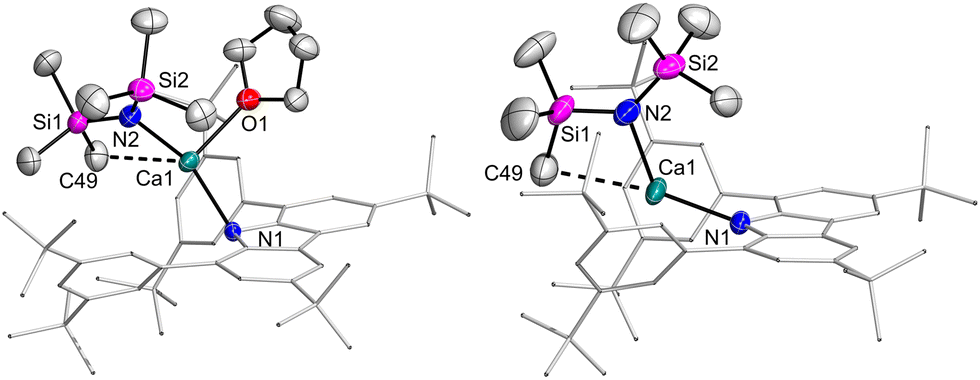 | ||
| Fig. 1 Molecular structures of Ca amides 1Ca (left) and 2Ca (right). Thermal ellipsoids at 50% probability. | ||
| M–Ncarb | M–Namide | M–Carene contacts | |
|---|---|---|---|
| 1Ca | 2.3243(11) | 2.2579(14) | C12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.9072(13) :2.9072(13) |
| C35:2.9286(13) |
|||
| C11:3.1278(14) |
|||
| 1Yb | 2.383(4) | 2.311(4) | C35:2.861(4) |
| C12:2.943(4) |
|||
| C11:3.092(4) |
With the goal of preparing the corresponding hydride complexes the alkaline earth metal carbazolyl amido complexes were treated with phenyl silane (PhSiH3, Scheme 3).
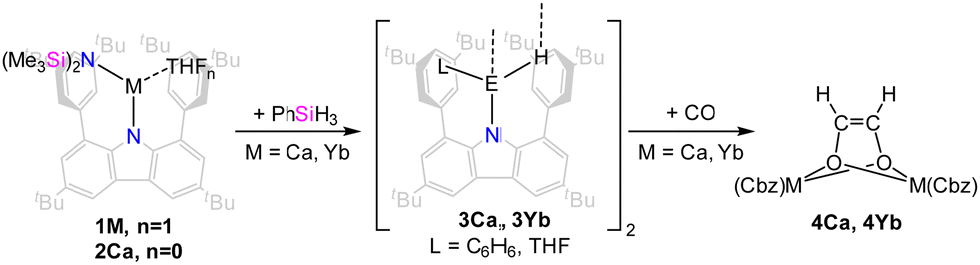 | ||
| Scheme 3 Synthesis of calcium and ytterbium(II) hydride complexes and reaction with CO. | ||
After addition of phenyl silane, crystalline materials were obtained within one day. When 1Ca was used for this metathesis reaction in benzene, the THF co-ligand was retained, and the dimeric hydride complex [(dtbpCbz)Ca(THF)H]2 (3Ca·THF) was obtained. In contrast, for the synthesis of [(dtbpCbz)Ca(C6H6)H]2 (3Ca·C6H6), the THF-free precursor 2Ca was employed. Both of these complexes are poorly soluble in aliphatic and aromatic hydrocarbon solvents and decompose in THF. The 1H NMR resonance of the hydride of 3Ca·C6H6 is found at 2.07 ppm which is at higher field than known calcium hydride complexes (3.07 to 4.71 ppm) but agrees reasonably well with the computed value of 1.49 ppm (see SI 4 for details, ESI†).4,25–28 The strong shielding of this resonance is attributed to the interaction with the coordinated benzene molecule and, to a lesser extent, the flanking arenes of the carbazolyl ligand. Removal of the benzene molecule in the model leads to a predicted 1H NMR resonance at 3.87, while removal of the flanking arene results in a predicted 1H NMR shift of 2.00 ppm.
Both 3Ca·C6H6 and 3Ca·THF comprise the dimeric, bis-hydride-bridged structural motif (Fig. 2) observed for other Ca hydrides. Each of the Ca atoms is coordinated to one additional donor, which is either THF or benzene. The molecular structures show Ca⋯Ca distances of 3.4495(6) and 3.4632(9) Å, which agree well with other Ca2H2 complexes. In all complexes the Ca atoms show contacts to coordinated arenes in the range of 2.8–3.0 Å.
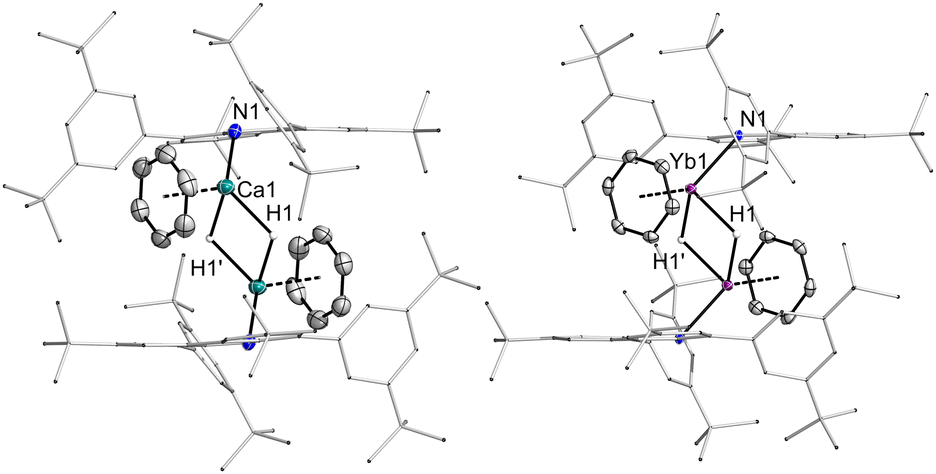 | ||
| Fig. 2 Molecular structures of hydride complexes 3Ca·C6H6 and 3Yb·C6H6. Thermal ellipsoids at 50% probability. | ||
In an analogous manner, ytterbium(II) hydride complexes could be prepared. However, when 1Yb was employed in the reaction with phenyl silane in benzene, mixed benzene and THF coordination was found. Thus, to obtain 3Yb·THF, the metathesis reaction had to be conducted in n-hexane. For the preparation of 3Yb·C6H6, 2Yb was generated in situ by dissolution of 1Yb in Et2O and evaporation of all volatiles, and then redissolution in benzene with addition of phenyl silane but the yield was lower than for the other hydride complexes. A likely cause is partial decomposition of 1Yb upon removal of the THF ligand. NMR data of 3Yb·C6H6 and 3Yb·THF could not be obtained due to the low solubility of the compounds.
The molecular structures of the ytterbium(II) hydride complexes 3Yb·C6H6 and 3Yb·THF are similar to the calcium analogs. The Yb⋯Yb (3.4676(9), 3.4896(3) Å) and Yb–N distances (2.379(2) 2.336(6) Å) are slightly longer than their Ca counterparts. In 3Yb·C6H6, the Yb-benzene contacts range from 2.804 to 2.943 Å, while the corresponding contacts in 3Ca·C6H6 are observed between 2.859(2) and 3.038(2) Å. Apparently, the Ca ion is slightly smaller and harder in these complexes, but for Yb the interaction with the π-scaffold of arenes is more efficient.
Three characteristic vibrations of 3Ca·C6H6 and 3Yb·C6H6 of high intensity were observed in the otherwise nearly identical IR spectra (Fig. 3, 3Ca·C6H6δ1 1069, δ2 785, δ3 556 cm−1; 3Yb·C6H6δ1 1073, δ2 797, δ3 550 cm−1; see SI 4.1, ESI†). Based on DFT calculations, the three vibrational bands illustrated in Fig. 3, δ1 (symmetric in-plane deformation), δ2 (antisymmetric in-plane deformation) and δ3 (symmetric out-of-plane deformation) were identified and exclusively involve the Ca2H2 or Yb2H2 core of the molecule. For corresponding calcium deuteride, 3Ca-D·C6H6 (see SI 2.8, ESI†), the three vibrations are shifted to smaller energies (obs. δ1 774, δ2 571, δ3 404 cm−1; calc. δ1 829, δ2 601, δ3 416 cm−1). To date, similar identifications of vibrational modes were only made for a magnesium hydride complex by Jones and Stasch who assigned a Mg–H stretch to a vibration at 1403 cm−1.29 Surprisingly, the vibrational bands of the Yb complex are observed at very similar energies as the Ca complex and no heavy atom effect is observable. Two further vibrational that involve the Ca2H2 or Yb2H2 moiety were found in the DFT model (SI 4.1, ESI†). However, they have only weak predicted intensity and could not be observed experimentally. In the Raman spectra, no clear assignment was possible.
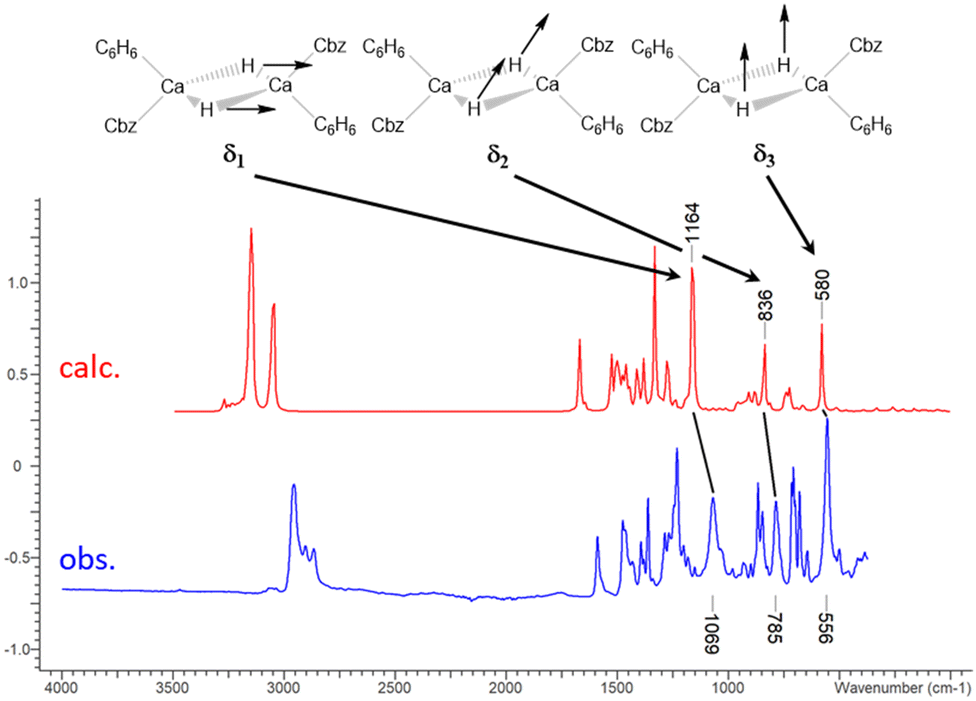 | ||
| Fig. 3 Observed vibrational modes of the hydride core of 3Ca. | ||
Reactions of calcium hydrides with carbon monoxide as well as carbonyl complexes are well studied.30–32 Subsequently, the reactivity of the hydride complexes towards CO was investigated (Scheme 3). Suspensions of the hydride complexes in n-hexane or benzene were exposed to 1 bar of CO. The reactions proceeded and intermittently, the solution became more strongly coloured, until the crystals of the reaction product, the corresponding ethenediolate complexes [(dtbpCbz)2M2Ln(O2C2H2)] (4, L = THF or benzene, n = 2,3) formed. Quality of the crystals strongly depends on the solvate and solvent employed, and crystals suitable for single-crystal X-ray structure elucidation could only be obtained from 3Ca·THF in n-hexane and from 3Yb·THF in benzene. In situ NMR observation of the reaction mixture of 3Ca with CO allowed the identification of the ethenediolate moiety with 1H and 13C NMR resonances at 2.39 and 69.46 ppm, which are both at higher field compared to the related complexes by Hill and Okuda.30,31 However, for the corresponding reaction with 3Yb only decomposition products were observed in solution which likely involve Yb(III) species as all lines were significantly broadened. However, crystallographic analysis corroborate the formation of ethenediolates in both cases (Fig. 4). IR analysis of 4b confirmed the disappearance of the bands assigned to the Yb2H2 core.
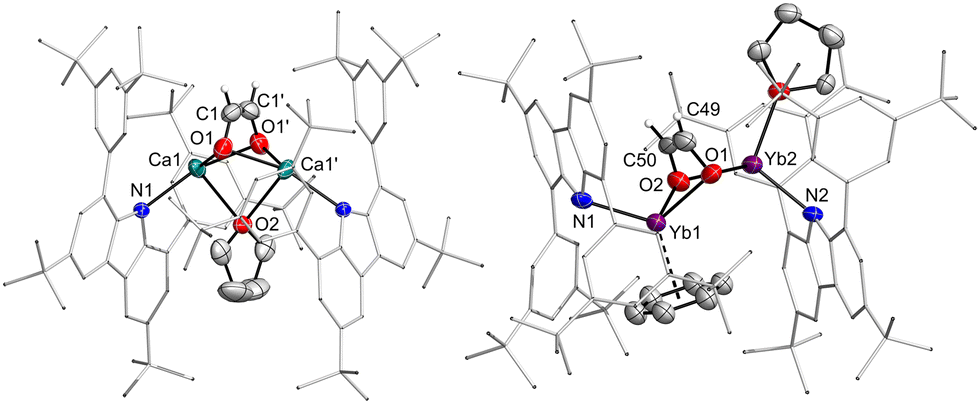 | ||
| Fig. 4 Molecular structure of the ethenediolate complexes 4Ca and 4Yb. Thermal ellipsoids at 50% probability. | ||
Structurally, the ethenediolate complex 4Ca is characterised by a Ca–Ca distance of 3.3613(7) Å. The Ca–O contacts to the bridging dianion amount to 2.2935(14) and 2.2817(13) Å, while the Ca–C contacts are found to be 2.6438(19) and 2.644(2) Å. Within the dianion, there is no bond alternation as the O–C and C–C bonds are 1.356(2) and 1.310(4) Å long, respectively. The Ca–N distance is 2.3434(12) Å. In 4Yb the Yb–Yb distance is considerably longer (3.690(5) Å), but also, there is a second solvent molecule coordinated in the compound. The Yb–O contacts range from 2.320(7) to 2.372(7) Å, with longer Yb–C contacts between 2.628(10) and 2.680(11) Å. The Yb-arene contacts are found between 2.796(14) and 2.889(14) Å. The geometry of the dianion is again found to be without bond alternation (O–C 1.367(13), 1.370(14), C–C 1.377(16) Å). The Yb–N distances are found to be 2.360(8) and 2.378(7) Å.
In conclusion, we have demonstrated the stabilisation of dimeric calcium and ytterbium(II) hydrido complexes with monodentate carbazolyl co-ligands. For the first time, characteristic vibrations of the M2H2 core could be observed. The dimeric complexes are surprisingly insoluble which hampered their NMR-spectroscopic characterisation, but nonetheless, the 1H NMR resonance of the calcium hydride complex could be detected at 2.07 ppm. Despite the poor solubility, the hydride compounds proved to be reactive towards CO to yield the corresponding ethenediolate complexes. The potential of the carbazolide ligand for the stabilisation of other hydride complexes is under investigation.
Financial support by Deutsche Forschungsgemeinschaft (DFG) via the Collaborative Research Centre “4f for Future” (CRC 1573, project A5) and the Emmy Noether Programme (HI 2063/1-1) is gratefully acknowledged. We thank Prof. Frank Breher and Prof. Peter Roesky for continuous support. Dr Ralf Köppe and Dr Adrian Hauser are acknowledged for measuring the Raman spectra. This work was carried out with the support of the Karlsruhe Nano Micro Facility (KNMF), a Helmholtz Research Infrastructure at Karlsruhe Institute of Technology (KIT). We acknowledge support by the state of Baden-Württemberg through bwHPC and DFG through grant no. INST 40/575-1 FUGG (JUSTUS 2 cluster).
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data can be obtained from CCDC with deposition numbers 2145430 (1Ca), 2145431 (1Ca·C6H14), 2145432 (2Ca), 2145437 (3Ca·THF), 2145438 (3Ca·C6H6), 2372499 (1Yb), 2372500 (3Yb·THF), 2372501 (3Yb·C6H6), 2372502 (4Ca), 2372503 (4Yb).Conflicts of interest
There are no conflicts to declare.Notes and references
- H. Bauer, M. Alonso, C. Färber, H. Elsen, J. Pahl, A. Causero, G. Ballmann, F. De Proft and S. Harder, Nat. Catal., 2018, 1, 40–47 CrossRef CAS.
- J. Spielmann and S. Harder, Chem. – Eur. J., 2007, 13, 8928–8938 CrossRef CAS PubMed.
- D. Schuhknecht, C. Lhotzky, T. P. Spaniol, L. Maron and J. Okuda, Angew. Chem., Int. Ed., 2017, 56, 12367–12371 CrossRef CAS PubMed.
- H. Elsen, C. Fischer, C. Knüpfer, A. Escalona and S. Harder, Chem. – Eur. J., 2019, 25, 16141–16147 CrossRef CAS PubMed.
- M. S. Hill, A. Wilson, C. Dinoi, M. Mahon, L. Maron and E. Richards, Angew. Chem., Int. Ed., 2019, 07, 1232–1237 Search PubMed.
- A. S. S. Wilson, M. S. Hill, M. F. Mahon, C. Dinoi and L. Maron, Science, 2017, 358, 1168–1171 CrossRef CAS PubMed.
- G. M. Ferrence, R. McDonald and J. Takats, Angew. Chem., Int. Ed., 1999, 38, 2233–2237 CrossRef CAS.
- G. M. Ferrence and J. Takats, J. Organometalic Chem., 2002, 647, 84–93 CrossRef CAS.
- X. Shi, P. Deng, T. Rajeshkumar, L. Zhao, L. Maron and J. Cheng, Chem. Commun., 2021, 57, 10047–10050 RSC.
- X. Shi, T. Rajeshkumar, L. Maron and J. Cheng, Chem. Commun., 2022, 58, 1362–1365 RSC.
- C. Ruspic, J. Spielmann and S. Harder, Inorg. Chem., 2007, 46, 5320–5326 CrossRef CAS PubMed.
- G. M. Richardson, I. Douair, S. A. Cameron, J. Bracegirdle, R. A. Keyzers, M. S. Hill, L. Maron and M. D. Anker, Nat. Commun., 2021, 12, 3147 CrossRef CAS PubMed.
- G. M. Richardson, I. Douair, S. A. Cameron, L. Maron and M. D. Anker, Chem. – Eur. J., 2021, 27, 13144–13148 CrossRef CAS PubMed.
- G. M. Richardson, J. Howarth, M. J. Evans, A. J. Edwards, S. A. Cameron and M. D. Anker, J. Coord. Chem., 2022, 75, 1954–1966 CrossRef CAS.
- Q. Wen, B. Feng, L. Xiang, X. Leng and Y. Chen, Inorg. Chem., 2021, 60, 13913–13919 CrossRef CAS PubMed.
- I. V. Basalov, D. M. Lyubov, G. K. Fukin, A. S. Shavyrin and A. A. Trifonov, Angew. Chem., Int. Ed., 2012, 51, 3444–3447 CrossRef CAS PubMed.
- I. V. Basalov, D. M. Lyubov, G. K. Fukin, A. V. Cherkasov and A. A. Trifonov, Organometallics, 2013, 32, 1507–1516 CrossRef CAS.
- D. Schuhknecht, K. N. Truong, T. P. Spaniol, L. Maron and J. Okuda, Chem. Commun., 2018, 54, 11280–11283 RSC.
- A. Hinz, Chem. – Eur. J., 2019, 25, 3267–3271 CrossRef CAS PubMed.
- A. Hinz, Angew. Chem., Int. Ed., 2020, 59, 19065–19069 CrossRef CAS PubMed.
- M. Kaiser, M. P. Müller, F. Krätschmer, M. Rutschmann and A. Hinz, Inorg. Chem. Front., 2023, 10, 2987–2994 RSC.
- X. Sun and A. Hinz, Inorg. Chem., 2023, 62, 10249–10255 CrossRef CAS PubMed.
- B. Rösch, T. X. Gentner, J. Eyselein, J. Langer, H. Elsen and S. Harder, Nature, 2021, 592, 717–721 CrossRef PubMed.
- A. Causero, G. Ballmann, J. Pahl, H. Zijlstra, C. Färber and S. Harder, Organometallics, 2016, 35, 3350–3360 CrossRef CAS.
- X. Shi, G. Qin, Y. Wang, L. Zhao, Z. Liu and J. Cheng, Angew. Chem., Int. Ed., 2019, 58, 4356–4360 CrossRef CAS PubMed.
- B. Maitland, M. Wiesinger, J. Langer, G. Ballmann, J. Pahl, H. Elsen, C. Färber and S. Harder, Angew. Chem., Int. Ed., 2017, 56, 11880–11884 CrossRef CAS PubMed.
- S. P. Green, C. Jones and A. Stasch, Angew. Chem., Int. Ed., 2008, 47, 9079–9083 CrossRef CAS PubMed.
- M. D. Anker, C. E. Kefalidis, Y. Yang, J. Fang, M. S. Hill, M. F. Mahon and L. Maron, J. Am. Chem. Soc., 2017, 139, 10036–10054 CrossRef CAS PubMed.
- D. Schuhknecht, T. P. Spaniol, Y. Yang, L. Maron and J. Okuda, Inorg. Chem., 2020, 59, 9406–9415 CrossRef CAS PubMed.
- J. M. Parr and M. R. Crimmin, Angew. Chem., Int. Ed., 2024, 62, e202219203 CrossRef PubMed.
- J. M. Parr, A. J. P. White and M. R. Crimmin, Chem. Commun., 2023, 59, 9840–9843 RSC.
- A. N. Selikhov, M. A. Bogachev, Y. V. Nelyubina, G. Y. Zhigulin, S. Y. Ketkov and A. A. Trifonov, Inorg. Chem. Front., 2024, 11, 4336–4346 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2145430–2145432, 2145437, 2145438 and 2372499–2372503. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc03714f |
| ‡ During the preparation of this work, Trifonov and co-workers also reported calcium and ytterbium hydride complexes with the carbazolyl ligand and their role in pyridine hydrosilylation. |
| This journal is © The Royal Society of Chemistry 2024 |