
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolyoxometalate-incorporated NiFe-based oxyhydroxides for enhanced oxygen evolution reaction in alkaline media†
Yuyan
Qiao‡
abc,
Yanqiu
Pan‡
c,
Wenjun
Fan
 *a,
Guifa
Long
*d and
Fuxiang
Zhang
*a
*a,
Guifa
Long
*d and
Fuxiang
Zhang
*a
aState Key Laboratory of Catalysis, iChEM, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, 116023, China. E-mail: wjfan@dicp.ac.cn; fxzhang@dicp.ac.cn
bJiangxi Province Engineering Research Center of Ecological Chemical Industry, Jiujiang University, Jiujiang, 332005, China
cSchool of Chemical Engineering, Dalian University of Technology, Dalian, 116024, China
dGuangxi Key Laboratory of Chemistry and Engineering of Forest Products, School of Chemistry and Chemical Engineering, Guangxi University for Nationalities, Nanning, 530008, China. E-mail: gflong@gxmzu.edu.cn
First published on 10th September 2024
Abstract
NiFe-based oxyhydroxides are promising electrocatalysts for the oxygen evolution reaction (OER) in alkaline media, but further enhancing their OER performance remains a significant challenge. Herein, we in situ incorporated polyoxometalates into NiFe oxyhydroxides to form a homogeneous/heterogeneous hybrid material, which induces the electronic interaction between Ni, Fe and Mo sites, as revealed by a variety of characterization experiments and theoretical calculations. The resulting hybrid electrocatalyst delivers a low overpotential of 203 mV at 10 mA cm−2 and a TOF of 2.34 s−1 at 1.53 V in alkaline media. This work presents a critical step towards developing high-performance OER catalysts by constructing metal–POM hybrids.
Solar-to-hydrogen conversion through electrochemical water splitting is a promising technology for utilizing renewable energy resources.1,2 However, the overall efficiency of water splitting is hampered by the sluggish kinetics of the anodic oxygen evolution reaction (OER). Although IrO2 and RuO2 have shown promising catalytic activity for the OER, their scarcity and high costs remain major obstacles to their large-scale applications.3,4 A large number of strategies have been developed to optimize the catalytic performance of OER electrocatalysts.5,6 The catalytic activity can be modified by designing nanostructured electrocatalysts,7 heteroatom doping,8 strain effects,9 and defect engineering.10 The stability of catalysts can be improved by in situ growing catalysts on conductive substrates,11 synthesizing carbon composite catalysts,12 or incorporating stabilizers,13etc. Despite significant advancements in this field, the development of non-precious metal electrocatalysts with high activity and stability remains a formidable challenge.
Among the critical factors contributing to this challenge are low intrinsic activity and limited utilization of active sites. By contrast, homogeneous catalysts, with their tunable structures at the molecular level, exhibit ultrahigh intrinsic activity.14,15 However, the poor stability and recyclability dramatically hinder their practical application. Thus, constructing efficient and stable catalysts by leveraging the advantages of both homogeneous and heterogeneous catalysts is highly desired.16 Polyoxometalate (POM)-based materials, with rich redox chemistry and tunable structures, have attracted widespread attention as potential candidates in electrocatalysis.17 Typically, phosphomolybdic acid (H3PMo12O40, PMA) has a classical Keggin structure with 36 oxygen atoms exposed and is an ideal platform for anchoring sites.18 The POM-based materials have been verified as promising molecular water oxidation catalysts under close-to-neutral pH conditions.19 Nevertheless, POM-based compounds face the problems of poor stability and conductivity in electrochemical processes. To address this, heterogenization of POM with conductive substrates or construction of electron transfer between the POM matrix and the active sites is urgently necessary for accelerating the OER process and increasing the stability.
Herein, by adopting the cluster-nuclei assembly strategy, we controllably synthesized a carbon nanotube (CNT) supported hybrid NiFe oxyhydroxide–PMA catalyst (denoted as NiFe–PMA/CNTs) on a carbon fiber paper (CFP) substrate for the alkaline oxygen evolution reaction (OER). CNTs can control the morphology of catalysts, increasing the surface area of the electrode and accelerate the charge transfer between the electrode and catalysts. The preparation of the NiFe–PMA/CNTs/CFP electrode follows a two-step synthesis approach (Fig. S1, ESI†). Typically, CNTs were first in situ grown on CFP by chemical vapor deposition to obtain CNTs/CFP electrodes (Fig. S2, ESI†), on which PMA clusters were incorporated with inorganic NiFeOx by a solvothermal method to obtain the target catalyst. For comparison, the control samples of CNT supported NiFeOx (NiFe/CNTs), CNT supported PMA (PMA/CNTs) or bare CNTs were prepared (see the ESI† for details).
The as-obtained NiFe–PMA/CNT catalyst (Fig. 1a) displays a typical three-dimensional nanostructure similar to the CNTs/CFP, which can potentially supply a large number of tunnels for gas release. A closer look at the catalyst by transmission electron microscopy (TEM) and aberration-corrected high-angle annular dark field scanning transmission electron microscopy (AC HAADF-STEM) reveals the coverage of small nanoparticles/clusters on carbon nanotubes (Fig. 1b, c inset and Fig. S3, ESI†), indicating the tight connection between CNTs and the catalyst.20 The lattice fringe with a spacing of 0.34 nm is ascribed to the (002) plane of graphite-2H (Fig. 1b), while no prominent lattice fringe assigned to the NiFe or PMA species could be found, suggesting the amorphous character of the catalyst. Consistent results can be found in the X-ray diffraction (XRD) patterns of the catalysts in Fig. S4 (ESI†), where no signals of Ni(Fe)OOH or PMA can be detected in the XRD patterns for NiFe–PMA/CNTs. The homogeneous distribution of Ni, Fe, Mo, P and O elements was confirmed by energy dispersive X-ray (EDX) elemental mapping (Fig. 1d–i). Furthermore, the elemental contents of transition metals for different electrocatalysts were examined by inductively coupled plasma-atomic emission spectroscopy (ICP-AES) and the Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe:Mo ratio in NiFe–PMA/CNTs was determined to be 1:1:0.6 (Table S1, ESI†). Fourier transform infrared (FTIR) spectra show that the characteristic peaks assigned to PMA at 1070, 966, 869, and 791 cm−1 are well preserved for the NiFe–PMA/CNT electrocatalyst (Fig. S5, ESI†).21 The shift of the corresponding characteristic peaks verifies the strong interaction between PMA, carbon materials, and NiFe (oxy)hydroxide catalysts.
:Fe:Mo ratio in NiFe–PMA/CNTs was determined to be 1:1:0.6 (Table S1, ESI†). Fourier transform infrared (FTIR) spectra show that the characteristic peaks assigned to PMA at 1070, 966, 869, and 791 cm−1 are well preserved for the NiFe–PMA/CNT electrocatalyst (Fig. S5, ESI†).21 The shift of the corresponding characteristic peaks verifies the strong interaction between PMA, carbon materials, and NiFe (oxy)hydroxide catalysts.
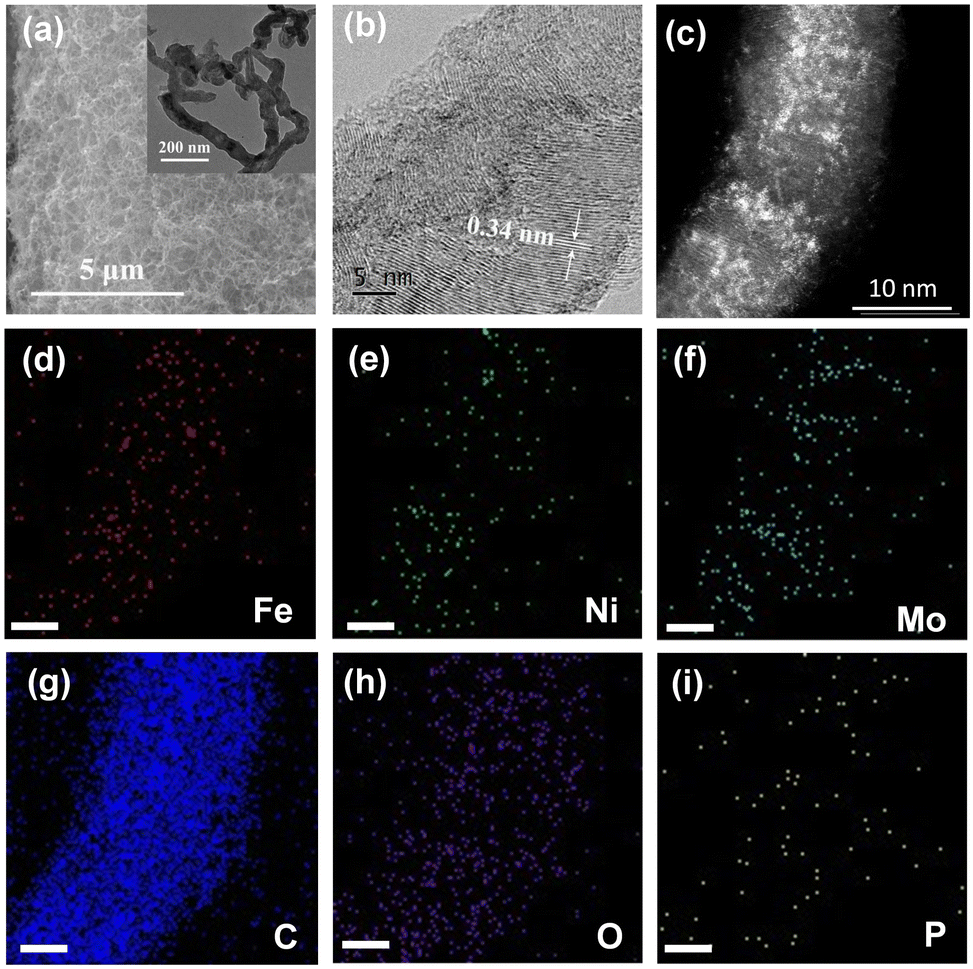 | ||
| Fig. 1 Morphological characterization of NiFe–PMA/CNTs. (a) SEM; the inset shows the TEM of the catalyst, (b) HRTEM of one nanotube, (c) AC HAADF-STEM image of NiFe–PMA/CNTs, and (d)–(i) the corresponding EDX elemental mapping, scale bar: 5 nm. | ||
The chemical states of the NiFe–PMA/CNT catalyst (Fig. S6 and S7, ESI†) were analyzed by X-ray photoelectron spectroscopy (XPS). The high-resolution spectrum of Fe 2p3/2 in NiFe/CNTs exhibits a prominent peak at 711.0 eV, corresponding to the Fe3+ species (Fig. 2a). By contrast, the main peak of Fe 2p3/2 for NiFe–PMA/CNTs experiences a slightly negative shift of ∼0.2 eV, suggesting the interaction between PMA and Fe element. A similar negative shift in the binding energy of Ni 2p3/2 can be found in NiFe–PMA/CNTs as compared with the NiFe/CNTs (Fig. 2b). It is worth noting that Ni3+ (858.0 eV and 857.4 eV) peaks appear in the Ni 2p spectrum of these two samples, respectively, which is known to be beneficial for prompting the formation of intermediate species (*OOH) for the OER.22 Accordingly, the binding energy of O 1s in NiFe–PMA/CNTs was found to be higher than that in PMA/CNTs (Fig. 2c), suggesting the electron transfer from O to Ni and Fe sites, which is further supported by the theoretical calculation results (Fig. 4).
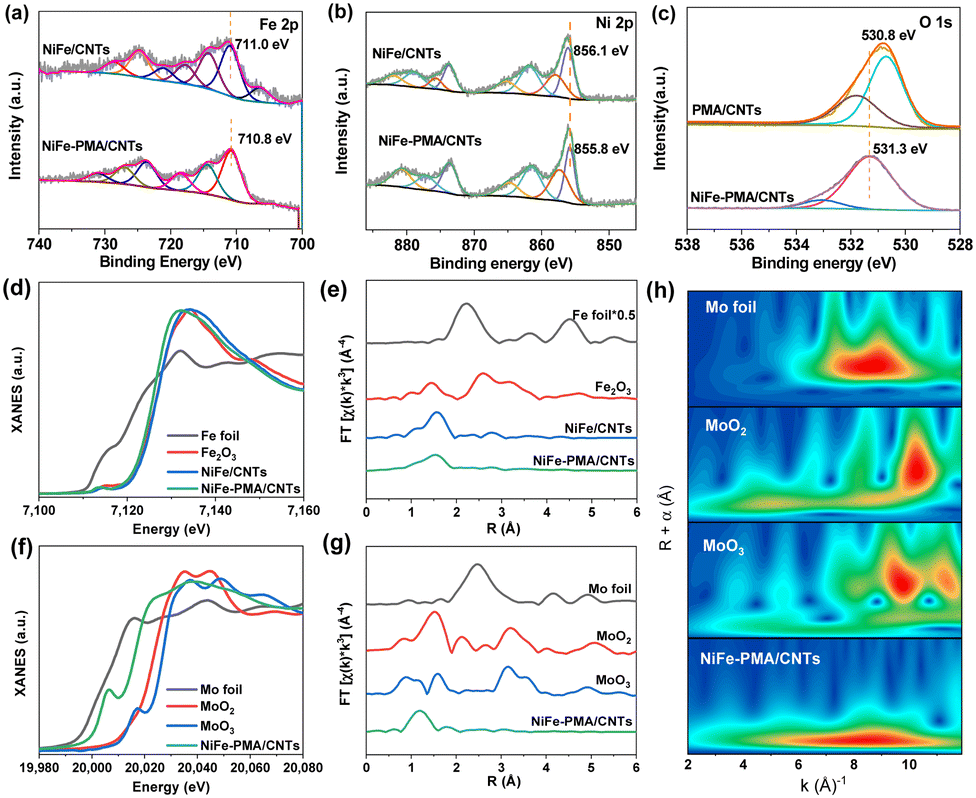 | ||
| Fig. 2 (a) Fe 2p and (b) Ni 2p XPS spectra of NiFe/CNTs and NiFe–PMA/CNTs, respectively. (c) O 1s of NiFe–PMA/CNTs and PMA/CNTs. (d) and (f) Normalized K-edge XANES spectra, and (e) and (g) the corresponding k3-weighted EXAFS spectra of NiFe/CNTs, NiFe–PMA/CNTs and reference samples for Fe and Mo (without phase correction), respectively. (h) Wavelet transform of EXAFS for Mo foil, MoO2, MoO3, and NiFe–PMA/CNTs. | ||
The local coordination structure of the NiFe–PMA/CNT electrocatalyst was further verified by X-ray absorption fine structure (XAFS). For NiFe–PMA/CNTs, the Fe K-edge X-ray absorption near-edge structure (XANES) spectra exhibit similar absorption edge of NiFe–PMA/CNTs, NiFe/CNTs and Fe2O3, indicating that the oxidation state of catalysts is around +3 (Fig. 2d). However, the Fe absorption edge in NiFe–PMA/CNTs is slightly lower than that of the NiFe/CNTs. This result suggests the prominent interaction between NiFe and PMA in which electrons are transferred from PMA to Fe, in accordance with the XPS result (Fig. 2a and c). The Fourier transform extended XAFS (FT-EXAFS) curves in Fig. 2e reveal the existence of the Fe–O peak at about 1.5 Å (Fig. 2e), which is further supported by the fitting results obtained from NiFe/CNTs and NiFe–PMA/CNTs (Fig. S8 and Table S2, ESI†). The Mo K-edge absorption of NiFe–PMA/CNTs indicates the oxidation valence of Mo is between +0 and +4 (Fig. 2f). The Mo–O coordination is validated by FT-EXAFS data and the corresponding wavelet transform (WT) (Fig. 2g, h and Table S3, ESI†).23,24
The electrocatalytic OER activity of NiFe–PMA/CNTs was evaluated in 1 M KOH. Remarkably, the NiFe–PMA/CNT catalyst exhibits the highest catalytic OER activity as compared with the NiFe/CNT catalyst and the other control samples (Fig. 3a and Fig. S9, ESI†), underscoring the synergistic effect between PMA and NiFeOx species. Specifically, the overpotentials at 10 mA cm−2 and 50 mA cm−2 reach 203 mV and 246 mV for the NiFe–PMA/CNT catalyst (Fig. 3b), respectively. When considering all metals as active sites, the TOF value of the NiFe–PMA/CNT catalyst at 1.53 V (vs. RHE) is 2.34 s−1, also superior to those of NiFe/CNTs (0.20 s−1) and PMA/CNTs (0.03 s−1). Notably, the high OER activity of the NiFe–PMA/CNT catalyst surpasses those of most non-noble metal electrocatalysts in alkaline solution (Table S4, ESI†). The high OER activity is also reflected by the much lower Tafel slope (Fig. 3c), revealing the fast reaction kinetics.
 | ||
| Fig. 3 (a) LSV polarization curves of NiFe–PMA/CNTs, NiFe/CNTs, and IrO2 in 1 M KOH. (b) OER overpotentials at 50 mA cm−2 for the various electrocatalysts in 1 M KOH. (c) Tafel plots of NiFe–PMA/CNTs, NiFe/CNTs, and IrO2 in 1 M KOH. (d) Plots of current densities at 0.1 V (vs. Hg/HgO) versus scan rates of typical samples, respectively. (e) Nyquist plots of typical samples in 1 M KOH at 1.48 V (vs. RHE) with the frequency range from 0.1 to 100 KHz. (f) Chronopotentiometry curve of NiFe–PMA/CNTs at 10 mA cm−2 in 1 M KOH. | ||
To investigate the effect of PMA on the catalytic activity of NiFe–PMA/CNTs, the double layer capacitance (Cdl) and the electrochemically active surface area (ECSA) were evaluated. As shown in Fig. S10 (ESI†), the calculated Cdl value of NiFe–PMA/CNTs is 29.8 mF cm−2, which is smaller than that of NiFe/CNTs (Cdl = 50.0 mF cm−2), indicating the higher intrinsic activity of NiFe–PMA/CNTs (Fig. 3d). The Nyquist plots at 1.48 V (vs. RHE) and related fitting results of EIS spectra were applied to compare the interface kinetics of comparative samples (Fig. 3e and Table S5, ESI†). NiFe–PMA/CNTs showed relatively lower interface charge transfer resistance (Rct = 1.0 Ω) than NiFe-C/CNTs (Rct = 2.2 Ω), suggesting their enhanced interface kinetics.
To further evaluate the long-term stability of NiFe–PMA/CNTs, chronopotentiometric (CP) measurements were carried out at 10 mA cm−2 in 1.0 M KOH electrolyte. As depicted in Fig. 3f, the overpotential exhibits marginal change during a 100 h test, demonstrating excellent stability for NiFe–PMA/CNTs. During this process, the faradaic efficiency of the NiFe–PMA/CNT electrocatalyst was around 100% (Fig. S11, ESI†), indicating the pure water oxidation reaction process. Additionally, the metal content in the electrolyte after the electrochemical reaction (Table S6, ESI†) shows insignificant changes and thus manifests the robustness of the NiFe–PMA/CNT catalyst. In comparison, NiFe/CNTs also show comparative stability for 100 h (Fig. S12, ESI†). Based on the analysis of characterization studies such as SEM, TEM, XRD and XPS before and after stability, NiFe–PMA/CNTs and NiFe/CNTs show non-considerable changes in the structures and compositions (Fig. S13–S16, ESI†).
To further clarify the interaction of PMA and NiFe in the NiFe–PMA–CNT catalyst, density functional theory (DFT) calculations were performed. The calculation model was built based on the experimental characterization studies. The optimized structures of PMA/CNTs, NiFe/CNTs and NiFe–PMA/CNTs are shown in Fig. S17 (ESI†) and Fig. 4a. The electronic structure of the NiFe–PMA/CNT catalyst was studied using the differential charge density (Fig. 4b). It is revealed that electrons move from PMA to NiO or FeO by metal–O bonds, resulting in increased charge density of Ni and Fe, which is consistent with the results from XPS and XAFS (Fig. 2). The charge transfers between PMA and NiFe demonstrate the robust interaction existing between them. Then, the charge transfers between the PMA and NiFe were investigated further by Bader charge analysis. As shown in Fig. 4c, the charges of Ni and Fe on NiFe–PMA/CNTs (9.01 and 6.63, respectively) are higher than those on NiFe/CNTs (8.81 and 6.37, respectively). The oxidation states of Ni and Fe from the Bader charge analysis were calculated and are depicted in Fig. 4d. It manifests that the Ni and Fe atoms in NiFe–PMA/CNTs (0.99 and 1.37, respectively) have lower chemical valence states than NiFe/CNTs (1.19 and 1.63, respectively), which is consistent with XPS and XAFS results. Accordingly, the redistribution of electron density can promote the absorption of metal centers to intermediate species and optimize the catalytic performances of electrocatalysts.25,26
 | ||
| Fig. 4 (a) Optimized structures of the NiFe–PMA/CNT catalyst. The balls in grey, red, pink, blue, violet and cyan represent C, O, P, Ni, Fe and Mo atoms, respectively. (b) The 3D differential charge density of the NiFe–PMA/CNT catalyst. The green symbols represent a decrease in charge density, while the yellow symbols represent an increase in charge density. (c) The charge of Ni and Fe atoms on NiFe/CNTs and NiFe–PMA/CNTs from Bader charge analysis. (d) The chemical valence state of Ni and Fe atoms on NiFe/CNTs and NiFe–PMA/CNTs from Bader charge analysis. | ||
In summary, PMA was introduced as a promoter to enhance NiFe oxyhydroxides for the OER in alkaline media. The constructed homogeneous/heterogeneous hybrid catalyst exhibits superior catalytic activity for the OER, with a low overpotential of 203 mV at 10 mA cm−2 and a high TOF of 2.34 s−1 at 1.53 V. The incorporation of PMA induces the electronic interaction between Ni, Fe and Mo sites, as revealed by a variety of characterization studies and DFT calculations, which regulate the electronic structure of NiFe, and improve the catalytic performances of the hybrid electrocatalyst. The introduction of PMA not only enhances the activity of the NiFe catalyst but also maintains the high stability of the catalyst for the OER, delivering a high stability of 100 h at 10 mA cm−2 without prominent decay. This work provided a new pathway for the synthesis and application of metal–POM hybrids for high-performance electrocatalysis.
This work was supported by the National Key Research and Development Program of China (No. 2020YFA0406102), the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No: XDB0600100), the National Natural Science Foundation of China (21925206 and 22332005), the Dalian Supports High-level Talent Innovation and Entrepreneurship Projects (2020RD06), the DICP project (I202439) and the Liaoning Revitalization Talents Program (XLYC1807241). The authors gratefully acknowledge 1W1B beamline of the Beijing Synchrotron Radiation Facility (BSRF), Beijing, China, for providing the beam time.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.References
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 0003 CrossRef CAS
.
- J. Kibsgaard and I. Chorkendorff, Nat. Energy, 2019, 4, 430–433 CrossRef
- M. Elmaalouf, M. Odziomek, S. Duran, M. Gayrard, M. Bahri, C. Tard, A. Zitolo, B. Lassalle-Kaiser, J.-Y. Piquemal, O. Ersen, C. Boissiere, C. Sanchez, M. Giraud, M. Faustini and J. Peron, Nat. Commun., 2021, 12, 3935 CrossRef CAS PubMed
- C. Wang, Q. Geng, L. Fan, J.-X. Li, L. Ma and C. Li, Nano Res. Energy, 2023, 2, e9120070 CrossRef
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed
- F. Lyu, Q. Wang, S. M. Choi and Y. Yin, Small, 2019, 15, 1804201 CrossRef PubMed
- C. Wang, H. Yang, Y. Zhang and Q. Wang, Angew. Chem., Int. Ed., 2019, 58, 6099–6103 CrossRef CAS
- Y. Sun, R. Li, X. Chen, J. Wu, Y. Xie, X. Wang, K. Ma, L. Wang, Z. Zhang, Q. Liao, Z. Kang and Y. Zhang, Adv. Energy Mater., 2021, 11, 2003755 CrossRef CAS
- B. You, M. T. Tang, C. Tsai, F. Abild-Pedersen, X. Zheng and H. Li, Adv. Mater., 2019, 31, 1807001 CrossRef PubMed
- D. Yan, Y. Li, J. Huo, R. Chen, L. Dai and S. Wang, Adv. Mater., 2017, 29, 1606459 CrossRef PubMed
- C. Andronescu, S. Barwe, E. Ventosa, J. Masa, E. Vasile, B. Konkena, S. Moller and W. Schuhmann, Angew. Chem., Int. Ed., 2017, 56, 11258–11262 CrossRef CAS PubMed
- X. Zhao, P. Pachfule, S. Li, J. R. J. Simke, J. Schmidt and A. Thomas, Angew. Chem., Int. Ed., 2018, 57, 8921–8926 CrossRef CAS
- Y. Zhao, X. Jia, G. Chen, L. Shang, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung, D. O'Hare and T. Zhang, J. Am. Chem. Soc., 2016, 138, 6517–6524 CrossRef CAS PubMed
- L. D. Wickramasinghe, R. Zhou, R. Zong, P. Vo, K. J. Gagnon and R. P. Thummel, J. Am. Chem. Soc., 2015, 137, 13260–13263 CrossRef CAS PubMed
- L. Wang, L. Duan, R. B. Ambre, Q. Daniel, H. Chen, J. Sun, B. Das, A. Thapper, J. Uhlig, P. Dinér and L. Sun, J. Catal., 2016, 335, 72–78 CrossRef CAS
- S. H. Talib, Z. Lu, X. Yu, K. Ahmad, B. Bashir, Z. Yang and J. Li, ACS Catal., 2021, 11, 8929–8941 CrossRef CAS
- K. Xia, K. Yamaguchi and K. Suzuki, Angew. Chem., Int. Ed., 2023, 62, e202214506 CrossRef CAS PubMed
- B. Zhang, H. Asakura, J. Zhang, J. Zhang, S. De and N. Yan, Angew. Chem., Int. Ed., 2016, 55, 8319–8323 CrossRef CAS
- F. M. Toma, A. Sartorel, M. Iurlo, M. Carraro, P. Parisse, C. Maccato, S. Rapino, B. Rodriguez Gonzalez, H. Amenitsch, T. Da Ros, L. Casalis, A. Goldoni, M. Marcaccio, G. Scorrano, G. Scoles, F. Paolucci, M. Prato and M. Bonchio, Nat. Chem., 2010, 2, 826–831 CrossRef CAS
- G. Y. Lee, I. Kim, J. Lim, M. Y. Yang, D. S. Choi, Y. Gu, Y. Oh, S. H. Kang, Y. S. Nam and S. O. Kim, J. Mater. Chem. A, 2017, 5, 1941–1947 RSC
- Q. Zhang, C. Yue, Q. Pu, T. Yang, Z. Wu and Y. Zhang, ACS Omega, 2019, 4, 9041–9048 CrossRef CAS
- M. Gao, W. Sheng, Z. Zhuang, Q. Fang, S. Gu, J. Jiang and Y. Yan, J. Am. Chem. Soc., 2014, 136, 7077–7084 CrossRef CAS PubMed
- S. V. B. Billy, B. Bardin, M. Neurock Robert and J. Davis, J. Phys. Chem. B, 1998, 102, 10817–10825 CrossRef
- Z. Cai, P. Wang, J. Zhang, A. Chen, J. Zhang, Y. Yan and X. Wang, Adv. Mater., 2022, 34, 2110696 CrossRef CAS
- I. C. Man, H.-Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martinez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Norskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS
- C. Liang, R. R. Rao, K. L. Svane, J. H. L. Hadden, B. Moss, S. B. Scott, M. Sachs, J. Murawski, A. M. Frandsen, D. J. Riley, M. P. Ryan, J. Rossmeisl, J. R. Durrant and I. E. L. Stephens, Nat. Catal., 2024, 7, 763–775 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc03874f |
| ‡ These two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |