
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal ion-driven formation of 2H-silolides and -germolides†‡
Chenghuan
Liu
 ,
Marc
Schmidtmann
and
Thomas
Müller
*
,
Marc
Schmidtmann
and
Thomas
Müller
*
Institute of Chemistry, Carl von Ossietzky University Oldenburg, Carl von Ossietzky-Str. 9-11, D-26129 Oldenburg, Federal Republic of Germany. E-mail: thomas.mueller@uni-oldenburg.de
First published on 30th August 2024
Abstract
The synthesis of potassio-1H-silolides and -germolides substituted with an amidinate-stabilized silylene is reported. Both anions undergo a thermal rearrangement to the 2H-isomer yielding cyclic sila- and germavinyl anions. The reaction is driven by complex formation with metal ions.
1H-Siloles and -germoles (1-sila- and 1-germacyclopenta-2,4-dienes) are well established classes of heterocycles.1,2 In contrast, their 2H-isomers were only discussed as short-living intermediates during pyrolysis experiments3,4 and the parent compounds were identified in hydrocarbon matrices at 77 K.5,6 The anionic 1H-silol-1-ides and 1H-germol-1-ides show a versatile structural chemistry.7 The results of DFT and ab initio computations suggest that their planar, aromatic conformation I is only a transition state (Fig. 1).8,9 Their ground state structure shows a trigonal pyramidal coordination of the tetrel element and the π-system of the heterocycle is localized (II) similar to that of the isoelectronic phospholes.10 The energy difference between both structures is however small (e.g. 16–25 kJ mol−1 for the parent 1H-silolide).9,10 Experimental results demonstrate that the nature of the coordinating metal cation as well as the effects of the substituents at the five-membered ring influence this delicate equilibrium. Paired with alkali metal cations, silolides and germolides show both conformations of the anion, I and II,11–15 while transition metal complexes usually favour η5-coordination to the planar anion I.16,17 We were interested in combining the flexible coordination chemistry of the tetrolides with a strong σ-donor ligand, such as a tricoordinated, amidinate-stabilized silylene18 in compounds such as III. We found a surprising preference of the trigonal planar conformation for Si(II)-substituted silolides (III, E = Si) and an unprecedented anionic 1H-/2H-tetrole rearrangement (IV/V, E = Si, Ge), which is driven by metal ion coordination and provides access to isolable cyclic sila- and germavinyl anions V. This rearrangement resembles the known transformation between 1H-/2H-phospholes (Fig. 1).19,20
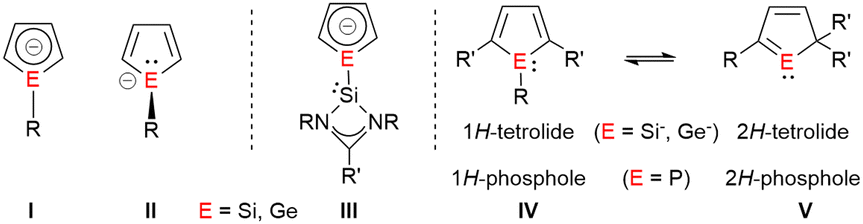 | ||
| Fig. 1 Molecular structures of 1-tetrolides I and II; planar, silylene-substituted tetrolides III and 1H/2H-phosphole and tetrolide rearrangements IV → V. | ||
Treatment of dipotassium silacyclopentadiendiide K2[1] with amidinate-stabilized silylene chloride 2 gives the expected potassium salt of the silylene-substituted silole anion [3]− (Scheme 1). Its formation was confirmed by NMR spectroscopy and by single crystal X-ray diffraction (sc-XRD) analysis.
 | ||
| Scheme 1 Synthesis of potassium mono-silylene-silolides K[3]. | ||
Separated resonances for both silyl groups and for each atom of the silole ring in the 1H, 13C and 29Si NMR spectra indicate a non-planar coordination environment of the silylene silicon atom Si2, a perpendicular conformation around the newly formed Si1–Si2 bond in solution and a hindered rotation around this bond. The 29Si NMR spectrum shows for the two trimethylsilyl groups two sharp singlets at δ29Si = −15.1 and −12.7. Two 29Si NMR resonances at higher frequencies were assigned to the amidinate- (δ29Si(2) = 35.6) and silole silicon atom (δ29Si(1) = 70.5). Due to missing correlations in the 1H29Si HMBC spectra, we assigned these two signals based on the results of NMR chemical shift calculations. (δ29Sicalc(2) = 33, δ29Sicalc(1) = 75, see Table S11, ESI‡). The deshielding of the silole silicon atom Si1 differs significantly from that reported for the solvent separated potassium silolide [K(18-c-6)][4]11 but is comparable to that reported for the ion-paired lithium silolide [Li(THF)][5] with a similar substitution pattern at the silole ring (Fig. 2).13 This suggests for K[3] in benzene solution an ion-paired structure similar to that reported for [Li(THF)][5] with the silole silicon atom Si1 in an almost trigonal planar coordination environment and a silole ring with a delocalized electronic situation. To enforce ion separation, we performed the synthesis of K[3] in the presence of 18-crown-6 (18-c-6) ether and isolated after recrystallization from diethylether the silolide [K(18-c-6)Et2O][3] (Scheme 1). Although we anticipated a significant change of the 29Si NMR chemical shift of the silole silicon atom Si1, we detected only a moderate low-frequency shift for this signal of Δδ29Si(1) = −11.6 (δ29Si(1) = 58.9). This is still far low-field shifted compared to the potassium silolide [K(18-c-6)][4] but is in the 29Si NMR chemical shift range of planar silole monoanions with a delocalized π-electron system (e.g. [K(THF)2][6] and [Li(THF)][5], Fig. 2).11,13,14
 | ||
| Fig. 2 NMR and structural parameters of alkali silolides (bond lengths in pm, ∑α(Si): sum of the bond angles around silicon). | ||
Red crystals of potassium silolide [K(THF)3][3] suitable for sc-XRD analysis were obtained and Fig. 3a shows the molecular structure of the contact ion pair. It confirms the formation of the silole anion-silylene linkage (Si1–Si2 = 237.2 pm). The planes of the silole ring and the silicon amidinate ring are twisted by 69°. The potassium atom is placed above the silole ring (K–C separations: 302–323 pm, K–Si: 334.2 pm). The silole ring itself is virtually planar with the silicon atom only 9 pm above the plane spanned by the four carbon atoms. The inner-cyclic CC bonds are almost equal (142.1–143.7 pm) and the lengths of the Si–C bonds (182 pm) fall in between regular SiC single and double bonds (Si![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C: 174 pm, Si–C: 191 pm).21 The coordination environment of the silole silicon atom is almost trigonal planar as indicated by the sum of the bond angles around silicon, ∑α(Si), of 353.2°. These structural features indicate a delocalized structure of the monoanion [3]− like those reported for the silolides [5]− and [6]− in their respective alkali metal salts (Fig. 2).13,14 The results of the sc-XRD analysis of the crystals obtained from [K(18-c-6)][3] confirms the separation of the ions through complexation of the potassium with the crown ether (K–Si > 644 pm, Fig. 3c). Surprisingly, also the cation-separated silolide [3]− features a silicon atom in a trigonal planar coordination environment (∑α(Si1) = 359.8°), integrated in a planar silole ring. The inner-cyclic CC bonds of the silole ring are almost equal (142.0–143.3 pm) and the Si–C bonds are short (181–182 pm) indicating a delocalized structure of the silolide [3]−. This is the first example of a silole monoanion with a delocalized structure without the supporting coordination of a transition or alkali metal ion to the silole ring (see ESI,‡ Fig. S7).
C: 174 pm, Si–C: 191 pm).21 The coordination environment of the silole silicon atom is almost trigonal planar as indicated by the sum of the bond angles around silicon, ∑α(Si), of 353.2°. These structural features indicate a delocalized structure of the monoanion [3]− like those reported for the silolides [5]− and [6]− in their respective alkali metal salts (Fig. 2).13,14 The results of the sc-XRD analysis of the crystals obtained from [K(18-c-6)][3] confirms the separation of the ions through complexation of the potassium with the crown ether (K–Si > 644 pm, Fig. 3c). Surprisingly, also the cation-separated silolide [3]− features a silicon atom in a trigonal planar coordination environment (∑α(Si1) = 359.8°), integrated in a planar silole ring. The inner-cyclic CC bonds of the silole ring are almost equal (142.0–143.3 pm) and the Si–C bonds are short (181–182 pm) indicating a delocalized structure of the silolide [3]−. This is the first example of a silole monoanion with a delocalized structure without the supporting coordination of a transition or alkali metal ion to the silole ring (see ESI,‡ Fig. S7).
 | ||
| Fig. 3 (a) Molecular structure of [K(THF)3]3, (b) side view of part of [K(THF)3]3 in the crystal, (c) molecular structure of [K(18-c-6)(Et2O)]3, (d) side view of part of [K(18-c-6)(Et2O)]3 in the crystal (hydrogen atoms are omitted; thermal ellipsoids at 50% probability). Selected atom distances [pm] and angles [°]: [K(THF)3]3: Si1–Si2 237.15(7), Si1–C1 182.09(17), Si1–C4 181.81(18), C1–C2 143.09(26), C2–C3 142.11(29), C3–C4 143.71(22), K–Si1 334.24(7), C1–Si1–Si2 122.73(6), C4–Si1–Si2 134.38(6), C1–Si1–C4 96.09(8); [K(18-c-6)(Et2O)]3: Si1–Si2 233.37(8), Si1–C1 181.69(16), Si1–C4 181.19(1), C1–C2 143.32(15), C2–C3 142.00(17), C3–C4 143.25(2), C1–Si1–Si2 118.94(4), C4–Si1–Si2 144.22(4), C1–Si1–C4 96.77(5). | ||
Towards mild oxidants, the silole anion [3]− follows the expected reactivity pattern. [K(18-c-6)(Et2O)][3] forms with elemental tellurium the potassium disilatelluride [K(18-c-6)][7] (Scheme 2). Its identity and structure were confirmed by NMR spectroscopy and sc-XRD analysis. The 29Si NMR spectrum of [K(18-c-6)][7] shows four singlets at low frequencies (δ29Si = −0.7 (Si1), −9.9, −10.0 (SiMe3) and −32.2 (Si2)) and the 125Te{1H} NMR spectrum two resonances at δ125Te = −802.0 and −1233.9 as expected for two different SiTe units. The molecular structure of [K(18-c-6)][7] shows Si1 and Si2 in a distorted tetrahedral coordination environment (Fig. 4a). K and Te atoms are well separated (K–Te1 = 358.3 pm, K–Te2 = 517 pm). The Si–Te bond lengths are 239.4 pm (Si2–Te2) and 242.4 pm (Si1–Te1), slightly longer than reported for related silicon tellurides (234–237 pm, see ESI,‡ Fig. S5) featuring tetracoordinated silicon atoms.
 | ||
| Scheme 2 Reaction of [K(18-c-6)(Et2O)][3] with elemental tellurium. | ||
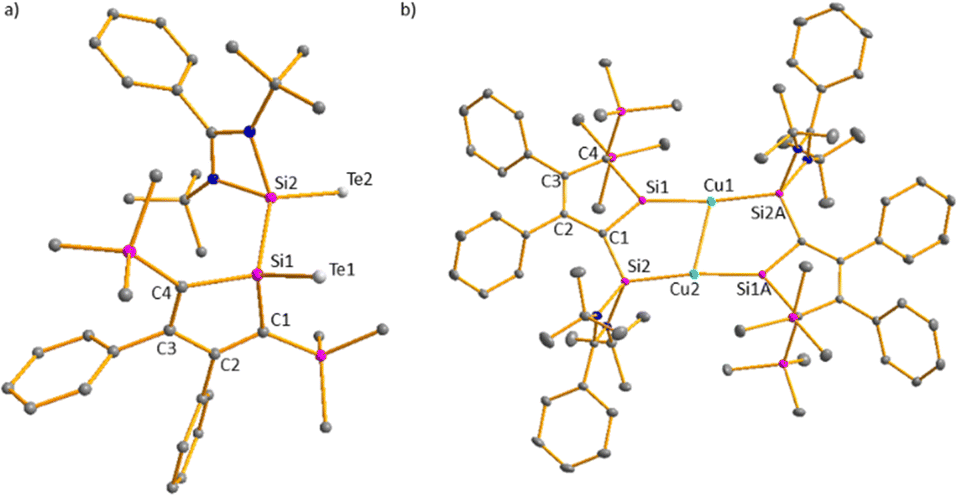 | ||
| Fig. 4 (a) Molecular structure anion [7]− in the crystal of [K(18-c-6)][7] and (b) molecular structure of Cu2[8]2 in the crystal (hydrogen atoms are omitted; thermal ellipsoids at 50% probability). Selected atom distances [pm] and angles [°], [7]−: Si1–Te1 242.43(4), Si2–Te2 239.37(4), Si1–Si2 239.24(5), Si1–C1 189.97(11), Si–C4 188.66(12), C1–C2 136.58(15), C2–C3 150.03(17), C3–C4 136.42(14), C1–Si1–Si2 106.98(4), C4–Si1–Si2 110.85(4), C1–Si1–C4 93.27(5). Cu2[8]2: Si1–Cu1 228.46(5), Si2A–Cu1 229.59(5), Si1–C1 178.32(6), Si–C4 192.20(7), Si2–C1 181.69(7), Cu1–Cu2 266.71(4), C1–C2 145.66(9), C2–C3 138.10(8), C3–C4 151.96(8), Si1–Cu1–Si2A 172.71(7), Si1–Cu1–Cu2 77.98(7), Si2A–Cu1–Cu2 108.11(7), C4–Si1–Cu1 129.19(18), C1–Si1–Cu1 137.12(2), C1–Si1–C4 93.70(2), Si1–C1–C2 110.03(4), Si1–C1–Si2 107.86(3), Si2–C1–C2 141.76(4), C1–Si2–Cu2 108.10(18). | ||
Subsequently, we reacted [K(18-c-6)(Et2O)][3] with CuI to test its coordination ability (Scheme 3). Surprisingly, the NMR data indicate a symmetric structure of the product. The 29Si NMR spectrum of the product shows three singlets at δ29Si = 238.9, 15.1 and −12.7. The two low frequency resonances are in the typical region for SiMe3 groups and Cu(I) complexes of amidinate-stabilized silylenes (see ESI,‡ Fig. S6). The strongly low-field shifted resonance of the silole silicon atom Si1 is remarkable and indicates a very different electronic environment for this atom.
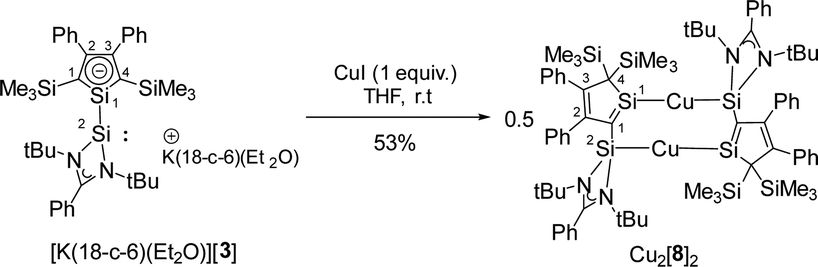 | ||
| Scheme 3 Reaction of [K(18-c-6)(Et2O)][3] with CuI. | ||
The molecular structure of the product Cu2[8]2 (Fig. 4b) was obtained from a sc-XRD analysis. It exhibits a dimeric structure with an almost planar (Si2–C1–Si1–Cu)2 eight-membered ring as its central element. The Cu(I) atoms are each coordinated by a silole ring and by a silylene in a linear coordination mode. While the structural parameters of this eight-membered ring differ not significantly from data reported for related compounds (ESI,‡ Fig. S6), the structure of the silole ring changed compared to the precursor. One trimethylsilyl group migrated from C1 to C4 forming a bissilylmethylene group and the silylene moved to the C1 position forming a cyclic silavinyl anion with Si–C1 distance of 178.32 pm in the range of a SiC double bond (174 pm).21 Remarkably, the exocyclic Si2–C1 bond is relatively short as well (181.7 pm). Thus, upon complexation of the silolide [3]− with Cu(I), a 1H-silol-1-ide/2H-silol-1-ide rearrangement took place (Scheme 4). 1H-Silole/2H-silole transformations of neutral molecules have been previously taken into account to rationalise the course of pyrolysis reactions and has been observed in matrix isolation studies.3–6 More relevant for the here reported anionic rearrangement are the well-established tautomerisations between the isoelectronic 1H- and 2H-phospholes studied by Mathey.19,20 These rearrangements are symmetry allowed suprafacial [1,5]-H shifts22 and the here reported transformation [3]− → [8]− is a series of three consecutive [1,5] silicon shifts (Scheme 4).
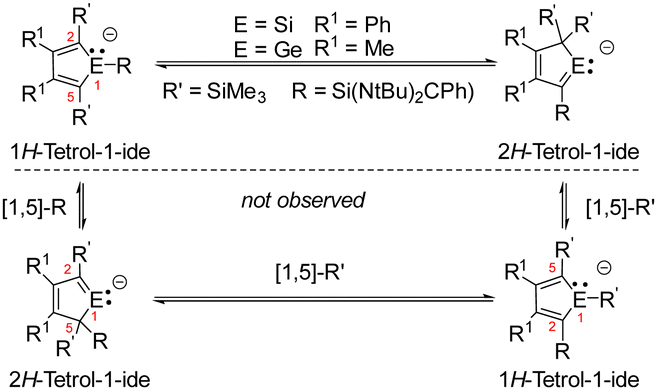 | ||
| Scheme 4 The observed rearrangements [3]− → [8]− and [10]− → [11]− as a series of three [1,5]-sigmatropic silicon shifts. | ||
In order to show the generality of this rearrangement, we tested the reaction of the amidinated silylene chloride 2 with dipotassium germacyclopentadiendiide K2[9] (Scheme 5). As germanium compared to silicon prefers low coordination states, we assumed that the thermodynamic driving force for the formation of the 2H-germol-1-ide is larger than its silicon analogue. Indeed, the rearrangement took place instantaneously and the potassium 2H-germolide [K(18-c-6)][11] was formed and isolated in 33% yield of the crystalline material. We assume that 1H-germolide [10]− is formed first and rearranges under the applied conditions to yield the 2H-germolide [11]−. Indicative of the formation of [11]− are two sets of signals for the methyl groups at C2 and C3 and one set of signals for the trimethylsilyl groups at C4 in the NMR spectra, and a low field shifted resonance of C1 in the 13C NMR spectrum δ13C(C1) = 210.2 (atom numbering, Fig. 5). The anticipated molecular structure was finally confirmed by the results of a sc-XRD analysis of [K(18-c-6)][11] (Fig. 5). Despite the coordination of the potassium ion to the crown ether, it is separated from the two tetrel atoms by only 348 pm (Ge1) and 354 pm (Si1). This is in both cases significant less than the sum of the van der Waals radii (Si/K 485 pm; Ge/K 486 pm).23 The central features of the molecular structure of the germolide [11]− are the central planar five-membered germol ring and the Si-amidinate ring, which are oriented almost orthogonal (dihedral 89.55°). The Ge–C1 bond is short (187.4 pm) indicating multiple bonding and also the exocyclic C1–Si1 bond is shorter (183.0 pm) than expected for a Si–C single bond (191 pm). The innercyclic C1–C2 bond (144.9 pm) is a short single bond and the C2C3 bond (136.4 pm) a long double bond. The structural data confirms the expected π-conjugation between the GeC1 bond and the C2C3 bond, and suggests π/σ*-conjugation (hyperconjugation) between the GeC1 bond and the SiN2 group. This is supported by the results of a NBO analysis for [11]− that indicates the delocalization of the polarized Ge–C1 π-bond towards the silylene silicon atom and towards the C2–C3 π-bond (Fig. 5 and ESI,‡ Fig. S11). Results of DFT computations (SCIPCM(C6H6)/M062X/6-311+G(d,p)) suggest that the [10]− → [11]− rearrangement is a series of three 1H-/2H-germolide interconversions via sigmatropic [1,5] silicon shifts (Scheme 5(Scheme 4) and Fig. S9, ESI‡). The barrier for the rate determining step is ΔG‡ = 86 kJ mol−1 and the rearrangement is practically thermo-neutral for the isolated molecules (ΔG = −3 kJ mol−1). Only explicit consideration of the cation/anion interaction by optimizing the structure of both ion pairs [K(18-c-6)][10] and [K(18-c-6)][11] resulted in a significant thermodynamic driving force (ΔG = −50 kJ mol−1; ESI,‡ Fig. S10). Thus, the coordination of the potassium ion is needed to drive the equilibrium [10]− → [11]− to the product. The corresponding rearrangement of the 1H-silolide [3]− to give the 2H-silolide [8]− is predicted to be endergonic by ΔG = 27 kJ mol−1 (ESI,‡ Table S10) in qualitative agreement with the experiment and therefore complexation to a stronger Lewis acid is needed to drive the reaction towards the 2H-silolide [8]−.
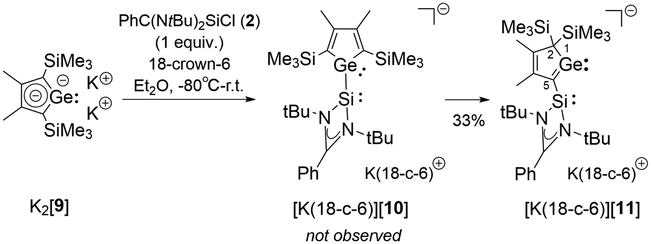 | ||
| Scheme 5 Synthesis of potassium 5-silylene-2H-germol-1-ide [K(18-c-6)][11]. | ||
 | ||
| Fig. 5 Left: Molecular structure of [K(18-c-6)][11] in the crystal (hydrogen atoms are omitted; thermal ellipsoids at 50% probability). Selected atom distances [pm] and angles [°]: Si1–C1 183.04(2), Ge1–C1 187.39(15), Ge1–C4 207.41(2), C1–C2 144.90(27), C2–C3 136.42(26), C3–C4 150.70(22), K–Ge1 348.20(6), K–Si1 353.79(6), Si1–C1–C2 135.9, Si1–C1–Ge1 111.30(7), Ge1–C1–C2 112.74(1), C1–Ge1–C4 87.46(6). Right: Surface diagram of the NBO (π(GeC1)) and of the NLMO (π(GeC1)) showing the delocalization of the GeC1 bond (M062X/6-311+G(d,p), isosurface at 0.04). | ||
1H-Tetrolides, substituted with tricoordinated silylenes were synthesized via salt metathesis reaction starting from dipotassio tetroldiides. Different to all other known 1H-tetrolides, the silolide [3]− adopts a planar structure even as a separated ion pair. The 1H-germolide [10]− undergoes a series of sigmatropic [1,5]-silicon shifts to give 2H-germolide [11]−. This anionic rearrangement closely resembles the established tautomeric 1H/2H-heterole equilibrium known from the isoelectronic phospholes.19,20 A related rearrangement was reported by Sekiguchi and co-workers for a trisilacyclopentadienide.24
Investigation, data curation, formal analysis, validation: C. L.; conceptualization, writing, visualization, funding acquisition: C. L, T. M; project administration, supervision, methodology, resources: T. M.; XRD: M. S.
Data availability
The data supporting this article have been included as part of the ESI.‡ Crystallographic data for [K(THF)3][3], [K(18-c-6)Et2O][3], [K(18-c-6)][7], Cu2[8]2, and [K(18-c-6)][11] has been deposited at the CCDC (2368313–2368316 and 2368318) and can be obtained from [https://www.ccdc.cam.ac.uk/].Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Y. Corey, in Advances in Organometallic Chemistry, ed. A. F. Hill and M. J. Fink, Academic Press, 2011, vol. 59, pp. 1–180 Search PubMed.
- Y. Adachi and J. Ohshita, Main Group Strategies towards Functional Hybrid Materials, 2017 Search PubMed.
- J. P. Beteille, M. P. Clarke, I. M. T. Davidson and J. Dubac, Organometallics, 1989, 8, 1292–1299 CrossRef CAS.
- D. Lei, Y. S. Chen, B. H. Boo, J. Frueh, D. L. Svoboda and P. P. Gaspar, Organometallics, 1992, 11, 559–563 CrossRef CAS.
- V. N. Khabashesku, V. Balaji, S. E. Boganov, O. M. Nefedov and J. Michl, J. Am. Chem. Soc., 1994, 116, 320–329 CrossRef CAS.
- V. N. Khabashesku, S. E. Boganov, D. Antic, O. M. Nefedov and J. Michl, Organometallics, 1996, 15, 4714–4724 CrossRef CAS.
- M. Saito and M. Yoshioka, Coord. Chem. Rev., 2005, 249, 765–780 CrossRef CAS.
- J. R. Damewood, Jr., J. Org. Chem., 1986, 51, 5028–5029 CrossRef.
- B. Goldfuss and P. V. R. Schleyer, Organometallics, 1995, 14, 1553–1555 CrossRef CAS.
- C. Fekete, I. Kovács, L. Könczöl, Z. Benkő and L. Nyulászi, Struct. Chem., 2014, 25, 377–387 CrossRef CAS.
- W. P. Freeman, T. D. Tilley, L. M. Liable-Sands and A. L. Rheingold, J. Am. Chem. Soc., 1996, 118, 10457–10468 CrossRef CAS.
- S.-B. Choi and P. Boudjouk, J. Chem. Soc., Dalton Trans., 2000, 841–844 RSC.
- C. Fekete, I. Kovács, L. Nyulászi and T. Holczbauer, Chem. Commun., 2017, 53, 11064–11067 RSC.
- C. R. W. Reinhold, M. Schmidtmann, B. Tumanskii and T. Müller, Chem. – Eur. J., 2021, 27, 12063–12068 CrossRef CAS.
- Z. Dong, M. Schmidtmann and T. Müller, Chem. – Eur. J., 2019, 25, 10858–10865 CrossRef CAS.
- J. M. Dysard and T. D. Tilley, J. Am. Chem. Soc., 2000, 122, 3097–3105 CrossRef CAS.
- W. P. Freeman, J. M. Dysard, T. D. Tilley and A. L. Rheingold, Organometallics, 2002, 21, 1734–1738 CrossRef CAS.
- C.-W. So, H. W. Roesky, J. Magull and R. B. Oswald, Angew. Chem., Int. Ed., 2006, 45, 3948–3950 CrossRef CAS.
- F. Mathey, F. Mercier, C. Charrier, J. Fischer and A. Mitschler, J. Am. Chem. Soc., 1981, 103, 4595–4597 CrossRef CAS.
- C. Charrier, H. Bonnard, G. De Lauzon and F. Mathey, J. Am. Chem. Soc., 1983, 105, 6871–6877 CrossRef CAS.
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 12770–12779 CrossRef.
- S. M. Bachrach, J. Org. Chem., 1993, 58, 5414–5421 CrossRef CAS.
- M. Mantina, A. C. Chamberlin, R. Valero, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. A, 2009, 113, 5806–5812 CrossRef CAS.
- H. Yasuda, V. Y. Lee and A. Sekiguchi, J. Am. Chem. Soc., 2009, 131, 6352–6353 CrossRef CAS.
Footnotes |
| † This work is dedicated to Prof. Yitzhak Apeloig on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available: Pdf-file with experimental and computational details, xyz-file with all optimized molecular structures. CCDC 2368313–2368316 and 2368318. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc03984j |
| This journal is © The Royal Society of Chemistry 2024 |